

Background: Peptidylglycine α-hydroxylating monooxygenase catalyzes the generation of C-terminal carboxamides of peptide hormones, neurotransmitters, and growth factors for biological activation.
Results: We compare several possible reaction mechanisms by means of quantum mechanics/molecular mechanics calculations.
Conclusion: Our results suggest that the most likely abstracting species is [CuOOH]2+.
Significance: Our computational study proposes a new mechanism, which explains the experimental observations.
Keywords: Computer Modeling, Copper, Enzyme Catalysis, Metalloproteins, Post-translational Modification, Kinetic Isotope Effects, PHM, QM/MM Simulation, Quantum Tunneling, Reaction Mechanism
Abstract
Peptidylglycine α-hydroxylating monooxygenase is a noninteracting bicopper enzyme that stereospecifically hydroxylates the terminal glycine of small peptides for its later amidation. Neuroendocrine messengers, such as oxytocin, rely on the biological activity of this enzyme. Each catalytic turnover requires one oxygen molecule, two protons from the solvent, and two electrons. Despite this enzyme having been widely studied, a consensus on the reaction mechanism has not yet been found. Experiments and theoretical studies favor a pro-S abstraction of a hydrogen atom followed by the rebinding of an OH group. However, several hydrogen-abstracting species have been postulated; because two protons are consumed during the reaction, several protonation states are available. An electron transfer between the copper atoms could play a crucial role for the catalysis as well. This leads to six possible abstracting species. In this study, we compare them on equal footing. We perform quantum mechanics/molecular mechanics calculations, considering the glycine hydrogen abstraction. Our results suggest that the most likely mechanism is a protonation of the abstracting species before the hydrogen abstraction and another protonation as well as a reduction before OH rebinding.
Introduction
Copper-containing enzymes play a critical role in the nervous system of mammals. These enzymes catalyze the generation of C-terminal carboxamides of peptide hormones, neurotransmitters (e.g. oxytocin), and growth factors. Therefore, these enzymes are targets of drug design for human diseases such as autoimmune disorder, depression, or cancer.
Peptidylglycine α-amidating monooxygenase (PAM)3 is a bifunctional enzyme whose task is the post-translational C-terminal amidation of small peptides. Unexpectedly, the amidation is not done by transferring an NH2 group; nature's clever trick is the N-oxidative cleavage of a glycine-extended prohormone instead. Many neuroendocrine messengers are small peptides post-translationally amidated by PAM.
PAM contains two components, a monooxygenase and a lyase, that are separated either by endoproteolytic cleavage or independent gene expression (1, 2). The peptidylglycine α-hydroxylating monooxygenase (PHM; EC 1.14.17.3) is a bicopper enzyme that hydroxylates the terminal glycine of small neurologically active peptides, such as oxytocin, substance P, thyrotropin, and calcitonin (1, 2). The second part of PAM is peptidyl α-hydroxylglycine-α-amidating lyase (PAL; EC 4.3.2.5), and transforms the products of PHM into the final amidated peptides and glyoxylate. PAL requires a metal center to function. It may as well use other metals apart from copper (3). Fig. 1 shows details of the reaction. Because of the relationship of some amidated peptides with cell and tumor growth (2, 4, 5), inhibitors of PAM have been proven to reduce the growth of tumors (4, 6).
FIGURE 1.
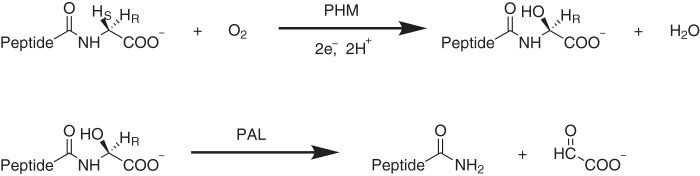
Global reaction catalyzed by PAM as follows: (top) stereospecific hydroxylation of glycine catalyzed by PHM and (bottom) lysis catalyzed by PAL.
The two copper atoms in PHM are noninteracting. They are in two different subunits of the protein separated by a solvent-exposed gap of 11 Å (7). Both coppers are essential for the protein to function (8), and they cycle through cupric and cuprous oxidation states during the catalysis.
These coppers are denoted in literature as CuH and CuM (sometimes CuA and CuB, respectively), due to their bonds to neighboring amino acids. CuH is bonded to three histidine residues (His-107, His-108, and His-172 in PHM from Rattus norvegicus), whereas CuM is bonded to two histidines and one methionine (His-242, His-244, and Met-314). In the crystal structure of the precatalytic complex (7) CuM adopts a tetrahedral coordination, with the methionine sulfur and histidine Nϵ atoms at three of the coordination positions and one oxygen atom from O2 at the remaining one. CuH adopts a trigonal planar one, with Nδ of the three histidines at the three coordination positions (7). Molecular oxygen is bound to CuM, and the substrate is located close to CuM as well, so this copper atom is part of the reaction active site. The role of the other copper atom (CuH) is not known, but there is evidence that it works as an electron reservoir and transfers electrons to CuM during the reaction (7, 9–12).
The overall reaction of PHM consists of a stereospecific hydroxylation of the pro-S hydrogen (13), using molecular O2 as oxygen source (14). Additionally, two electrons and two protons are consumed during the reaction (Fig. 1). Ascorbate is the best (but not the only possible) reductant (15). In the protein resting state, both copper atoms are in the +2 oxidation state and are reduced by ascorbate to +1 (16). Molecular oxygen then binds to CuM as the substrate binds to the protein (7).
Several mechanisms have been suggested for the next step, the hydroxylation, but a completely satisfactory mechanism is still an open question. Consensus has been reached in the fact that a neutral H• atom is abstracted from glycine (2, 17–19). However, several abstracting species have been considered. Prigge et al. (7) suggested CuM+–O2− ↔ CuM2+– O22− as the abstracting species based on the crystal structure. Gas phase density functional theory calculations with a truncated model of the protein by Chen and Solomon (19, 20) suggested a Cu(II)-superoxo species to be responsible for the abstraction of the glycine hydrogen. This mechanism was ruled out by Crespo et al. (21) because including the environment of the protein in a QM/MM fashion enlarged the energy barrier up to 84 kJ mol−1 (gas-phase calculations by Chen and Solomon (19, 20) gave a value of ∼60 kJ mol−1 for the energy barrier). Instead, Crespo et al. (21) suggested Cu-oxo as a possible species, because of the small energy barrier involved, similar to the reaction mechanism suggested for the homologous dopamine β-monooxygenase (EC 1.14.17.1) by Kamachi et al. (22). A hydroxo species has been ruled out by Chen and Solomon (19), by Crespo et al. (21), and by Kamachi et al. (22) in dopamine β-monooxygenase.
Francisco et al. (23, 24) found a large value of the KIE (a primary hydrogen/deuterium kinetic isotope effect of 10.6 ± 0.8 at 37 °C and pH 6), and they concluded that atom tunneling should be important in the hydrogen abstraction (24). Their findings imply two things as follows: first, the energy barrier for H• abstraction is large and thin enough to enhance atom tunneling; second, the hydrogen abstraction is at least partially rate-limiting, competing with the loss of the substrate step of the catalytic cycle. This experimental finding contradicts the mechanism suggested by Crespo et al. (21), because a very small energy barrier does not lead to a significant KIE. Klinman and co-workers (10, 25) suggested a Cu(II)-superoxo mechanism, similar to the one proposed by Solomon and co-workers (19, 20), based on experimental indications but without computational backup. Based on recent QM/MM calculations (12), it was argued that the high barrier for the Cu(II)-superoxo case is a spurious effect of the lack of relaxation of the solvent.
There are several proposals for the next step, the OH rebinding. They can be divided into two main groups as follows: direct OH rebinding from the copper-oxygen moiety (9, 19, 21, 22) or an inner sphere alcohol intermediate (10, 12, 26). Because OH rebinding is not rate-limiting, experimental evidence of that step is hard to obtain. Theoretical studies mainly point toward direct rebinding (19, 21, 22). McIntyre et al. (26) obtained indirect evidence of an inner sphere alcohol intermediate by considering the unnatural substrate benzaldehyde iminooxyacetic acid, which undergoes dealkylation independently of PAL. QM/MM calculations suggest that the alcohol intermediate is necessary for dealkylation of this substrate, but it involves a very large energy barrier (larger than 200 kJ mol−1). Recently Meliá et al. (12) did a computational investigation of a similar mechanism where a hydroxide ion is already coordinated to the copper.
There have been experimental studies of the hydrogen peroxide reactivity of PHM. Based on the interchangeability of O2 and H2O2, it was postulated (27) that an intermediate electronically equivalent to CuM(I)-O2 exists. Recently, Rudzka et al. (28) obtained the crystal structure of PHM with H2O2 (without substrate) and compared the geometric parameters of the active site with those obtained by means of QM/MM optimization of several protonation states for the oxygen species. They conclude that CuM(II)-O22− is the species that best fits with the crystal structure (as compared with CuM(II)-H2O2 and CuM(II)-OOH−).
In this work we present a systematic theoretical study of the reaction mechanism, taking all possible abstracting species into account. Two protons are consumed during the reaction, but it is not known in which order compared with hydrogen atom abstraction from the substrate. An electron transfer from CuH to CuM at some point of the reaction is likely. This means that the hydrogen abstraction might occur after zero, one, or two protons, and/or an extra electron was delivered to the cofactor. We tested all six resulting possibilities as follows: [CuO2]+, [CuO2]0, [CuOOH]2+, [CuOOH]+, [CuO]3+ + H2O, and [CuO]2+ + H2O (Table 1). The emerging and most likely mechanism has not been proposed in the literature previously.
TABLE 1.
Possible H-abstracting species
| 0 e− | 1 e− (CuH → CuM) | |
|---|---|---|
| 0 H+ | [CuO2]+ | [CuO2]0 |
| 1 H+ | [CuOOH]2+ | [CuOOH]+ |
| 2 H+ | [CuO]3+ + H2O | [CuO]2+ + H2O |
CALCULATION DETAILS
The reaction mechanism was simulated using a QM/MM (29, 30) approach. The crystal structure at 1.85 Å resolution (7) was used as a starting point for our calculations (Protein Data Bank code 1SDW). The slow substrate N-acetyl-diiodotyrosyl-d-threonine was replaced by a Tyr-Gly dipeptide because of its structural similarity. The model substrate was truncated at its N terminus (Tyr) by an acetyl group. The C terminus of Gly remained deprotonated. Protonation states of protein residues were assigned based on the REDUCE code (31) corresponding to pH 7. The N and C termini of the protein were truncated by an acetyl group and remained deprotonated, respectively. The protein was solvated in a pre-equilibrated box of TIP3P water (32) using VMD (33) version 1.9. For molecular dynamics simulations, we used the CHARMM22 force field (34). Copper, O2, and OOH were parameterized from analogy to existing moieties, and partial charges were obtained from the natural population analysis as implemented in Turbomole (see below). Both His-242 and His-244, which are ligands of CuM, were modeled and protonated at the Nδ atoms, which are not bound to copper. Topology and parameter files for Cu/O2 and Cu/OOH can be found in the supplemental material.
To obtain realistic initial geometries, the system was equilibrated in molecular dynamics (MD) simulations using NAMD (35), with the crystal structure as the initial geometry. We performed separate MD runs with [CuO2]+ and [CuOOH]2+ as abstracting species (see under “Results and Discussion”). The temperature was kept at 300 K by a Langevin thermostat; a time step of 1 fs was chosen. All bond lengths involving hydrogen atoms were constrained. For the protein equilibration, cycles of minimization of 2000 steps followed by MD runs of 50,000 steps were performed, where the protein atoms were restrained (restraint parameters were lowered for each cycle: frozen protein atoms, 5.0, 2.0, and 0.1 kcal mol−1 Å−2). This protocol allows water molecules to penetrate cavities within the protein. Then a free MD run of 4 ns was carried out; the Langevin piston Nosé-Hoover method was used to keep the system at 300 K and 1 bar.
A snapshot was taken from the [CuO2]+ trajectory. The frame with the shortest distance between the abstracted hydrogen and an oxygen atoms from the abstracting species was chosen, because this offers the highest probability of a low barrier. This snapshot was the starting point for our QM/MM calculations, which were carried out with the ChemShell suite (36–38). ChemShell interfaces several QM an MM codes, taking care of the communication between them. It also handles geometry optimization, by means of the robust and flexible geometry optimizer DL-FIND (39). The QM part consisted of CuM and the side chain of its three ligating residues (His-242, His-244, and Met-314), the abstracting species, the glycine substrate, and its amide bond (Fig. 2). In several calculations, additional solvent waters that may have catalytic importance were included in the QM part. In total, between 41 and 58 atoms were considered as QM. The whole protein and all waters within 15 Å of the active site were included in the QM/MM calculations (between 6475 and 6495 atoms, depending on the snapshot, see below). All residues (or water molecules) with at least one atom closer than 8 Å to CuM or closer than 4 Å to the abstracting species or substrate were allowed to relax. This ensures that we optimize the first two solvation layers of the active site. With respect to the QM/MM interface, we used an additive scheme with electrostatic embedding. Hydrogen link atoms, with the charge shift scheme were used for truncation of C–C bonds.
FIGURE 2.
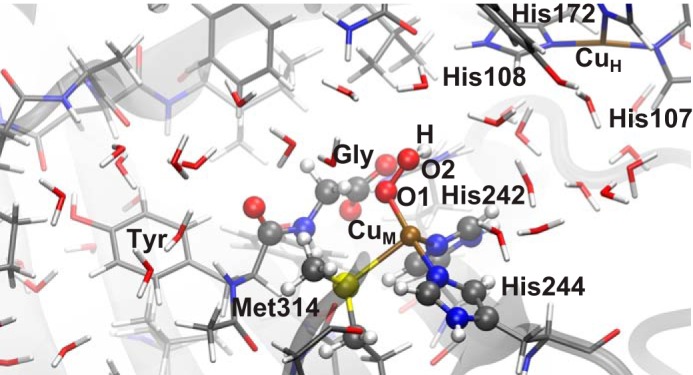
Solvent-exposed active site of PHM. The reactant state with [CuOOH]2+ as the abstracting species is shown. The small dipeptide Tyr-Gly is used as a model substrate in our calculations. QM atoms are shown as balls. Atom colors are as follows: white (hydrogen), gray (carbon), blue (nitrogen), red (oxygen), yellow (sulfur), and brown (copper).
The optimization of all structures was carried out using DL-FIND (39), which is included in ChemShell. Reactant and product geometries were optimized by means of the L-BFGS method (40, 41). Transition states were obtained by means of the dimer method (42) as implemented in DL-FIND (43), where the two starting geometries were taken from a potential energy surface scan, using chemically intuitive reaction coordinates. Hybrid-delocalized internal coordinates (44) were used in all minimizations and dimer calculations. In case of divergence of dimer transition state searches, nudged elastic band calculations (45), as implemented in DL-FIND (46), were employed. The transition states were refined by dimer calculations starting from the climbing image and the image with highest energy of the nudged elastic band path. The latter procedure was mainly necessary to find transition states for the OH rebinding mechanism (see under “Results and Discussion”). This approach led us to converged transition states with only one imaginary frequency.
The snapshot of the molecular dynamics run with [CuO2]+ in the active site was used to model and compare different possible mechanisms. Different oxidation states were modeled by adding electrons to the system. A primary protonation was modeled by placing an additional proton that was bound to the O2 atom. The addition of a second proton releases a water molecule and forms a copper-oxo species (21). In the mechanism we found to be most likely, the abstracting species is [CuOOH]2+. Thus, we ran another MD simulation with that moiety in the active site and took two more snapshots, again considering a short O–H distance as the guide for choosing the snapshot. They are separated in time by 0.44 ns and thus should be statistically independent.
For the MM part of our QM/MM system, we used DL_POLY (47) and the same CHARMM force field as we used in the classical MD simulations. For the quantum treatment of the QM part, we employed density functional theory. The exchange correlation functional is unrestricted BP86 (48, 49) as implemented in turbomole version 6.03 (50), in combination with resolution of the identity (51). It has been shown by our group in previous work (52) that enzymatic geometries obtained with BP86 and with B3LYP coincide very well. Recently, Meliá et al. (12) showed that the choice of the exchange correlation functional does not alter the energy barriers in PHM too much; the pure functionals (PBE), hybrid functionals (B3LYP), and meta-GGA functionals (M06) barriers differ by less than 10 kJ mol−1. We used the def2-SVP basis set (53) for the comparison of mechanisms. The most likely mechanism, with [CuOOH]2+ as abstracting species, was reoptimized using the def2-TZVP (54, 55) basis set.
Nuclear quantum effects were taken into account. Harmonic zero point energy corrections were calculated for our proposed mechanism but ignored for the comparison of mechanisms. Contributions of atom tunneling were treated in an approximate manner. The crossover temperature Tc, defined in Equation 1, can be interpreted as the temperature below which tunneling significantly contributes to the reaction rate (56)
 |
where ħ is Planck's constant, kB is Boltzmann's constant, and ωb is the magnitude of the imaginary frequency of the unstable mode at the saddle point. Because Tc is well below room temperature in all relevant cases, tunneling is comparatively weak, and therefore simple approximations for the tunneling contribution can be used. We used transition state theory with all vibrational degrees of freedom approximated by harmonic oscillators. Along the reaction coordinate, a symmetric Eckart barrier (57) was fitted to the reaction path to match the barrier height and ωb. The quantum mechanical flux through that barrier, including all tunneling and nonclassical reflection contributions, is calculated analytically. The rate was corrected by the ratio of the classical rate and the analytic rate via the Eckart barrier. Note that all degrees of freedom perpendicular to the transition mode are still treated as harmonic oscillators. Both the pro-S and pro-R α-deuterated glycyl substrate were used in our reaction rate calculations in order to compare with experimental primary and secondary KIEs (23), respectively.
Two-point difference Hessians of all QM atoms were calculated to obtain zero point energy and Eckart tunneling corrections. QM waters were taken out from the Hessian calculations because their chemical environment does not change too much from the reactant to the transition state. Moreover, including QM waters would result in the occurrence of soft modes, which may lead to numerical instabilities. In any case, a single imaginary frequency was obtained for all transition states and only real ones in reactants or products, thus validating the approximations made for the Hessian calculations.
Thermal averages of reaction barriers (58, 59) were calculated by using Equation 2,
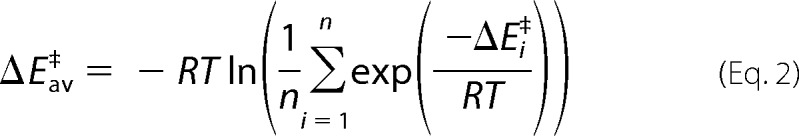 |
with ΔEav‡ being the activation barriers of the individual snapshots, T being the absolute temperature, and R the gas constant.
RESULTS AND DISCUSSION
First Step, Hydrogen Abstraction
The first goal of our calculations was to identify the species responsible for hydrogen abstraction. As indicated in the Introduction, KIE measurements show that hydrogen abstraction is the rate-limiting chemical step. The crystal structure was obtained for the [CuO2]+ form, which indicates that this form is reasonably stable. It might directly refer to the hydrogen-abstracting species. Alternatively, prior to hydrogen abstraction, [CuO2]+ can be protonated once or twice or reduced by an electron transfer from CuH. This leaves a minimum of six possibilities for the hydrogen-abstracting species (Table 1). We tested all of them. From the theoretical point of view, the stability of the different species is hard to predict, which would require the calculation of pKa values or reduction potentials. Both quantities require a lot of sampling. However, we are aided by experimental knowledge. The barrier has to be large enough to explain the experimental KIE but small enough to be consistent with the experimental rate. Actually, barrierless hydrogen abstractions should be discarded, as they will not exhibit any KIEs. Apart from the barrier, the creation of a full picture to understand the reaction mechanism will be guided by various chemical properties and the thorough comparison of experimental as well as theoretical findings.
For the reaction to proceed, the abstraction of a hydrogen atom from the substrate has to have a lower barrier than the abstraction of a proton from any surrounding water molecule. These conditions turn out to be sufficient to find a unique state that represents the most likely active species (Fig. 9).
FIGURE 9.
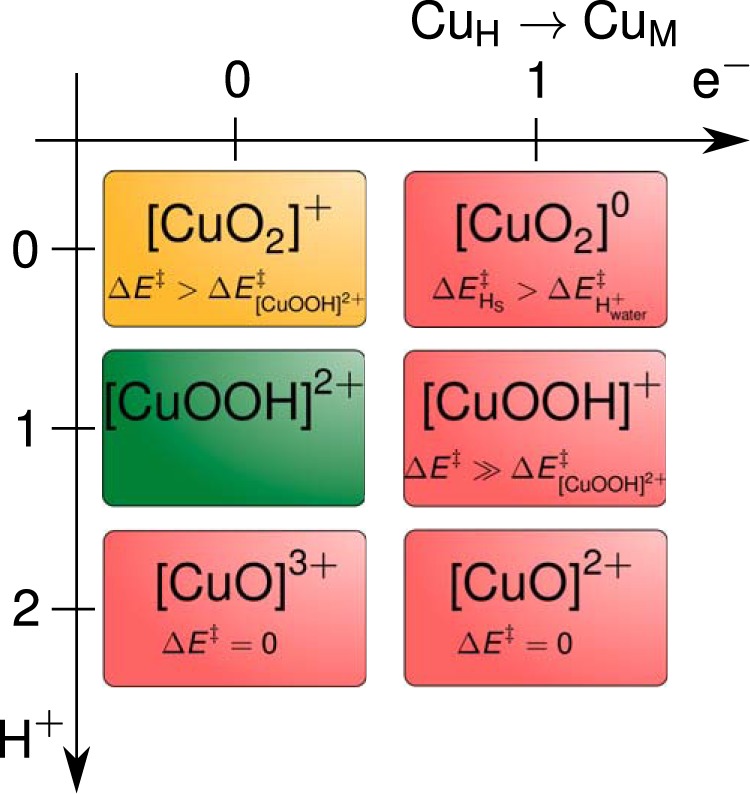
Possible abstracting species. Green denotes the most likely mechanism. The ones colored in red are not plausible (the arguments are specified in the text), the one in yellow is plausible, but our mechanism has a lower energy barrier.
Generally, we observed that the barrier for proton abstraction from surrounding water decreases by reduction (addition of electrons) as expected, because the copper moiety becomes a stronger base. The barrier for hydrogen atom abstraction from the substrate was found to decrease with increasing protonations of the abstracting species. For example, in both cases in the third row (2 H+) of Table 1, the reaction is spontaneous.
[CuOOH]2+, 1 H+ 0 e−
The resulting most likely hydrogen-abstracting species is [CuOOH]2+. It will be discussed first, before covering the results of the alternatives and arguing why they are unlikely.
Although an accurate calculation of the protonation event of the abstracting species [CuO2]+ + H+ → [CuOOH]2+ is computationally intricate, one can assume a quite low pKa value for the [CuO2]+ species. Recent results by Peterson et al. (60) on a copper superoxo species in a different environment also suggest a low pKa. Because an accurate calculation of pKa values is always tricky, we consider for the [CuO2]+ species the range of 0 < pKa < 4. This means that the free energy difference between [CuO2]+ + H+ and [CuOOH]2+ is between 20 and 40 kJ mol−1, which follows from ΔG = −RT ln 10(pKA − pH). Even if the energy difference between the unprotonated and protonated species is as large as 40 kJ mol−1, the low barrier for the hydrogen abstraction by [CuOOH]2+ as compared with [CuO2]+ makes the former a better candidate for the hydrogen abstraction than the latter.
We calculated mechanisms for three different geometrical configurations that are displayed in Figs. 3 and 4, a, d, and g, respectively. The first one (snapshot 1) is taken from an MD simulation of [CuO2]+ with one proton added in the subsequent QM/MM calculations (Fig. 4a). The other two (snapshot 2 and 3) are taken from an MD run with [CuOOH]2+ in the active center (Fig. 4, d and g, respectively).
FIGURE 3.

Schematic view of the structure of the reactant, transition state (TS), and intermediate of the three snapshots. Important bond lengths (in Å) are included in the scheme.
FIGURE 4.

Geometries of the reactant (a, d, and g), transition state (b, e, and h), and intermediate state (c, f, and i) of snapshots 1 (a–c), 2 (d–f), and 3 (g–i) studied in our proposed mechanism. QM atoms are represented by balls and sticks. The remaining part of the substrate and residues His-242, His-244, and Met-314 are represented as sticks. For atom colors, see Fig. 2.
In the reactant state (Figs. 3 and 4, a, d, and g), copper is coordinated in a trigonal-bipyramidal manner in snapshot 1. OOH and Nϵ of His-244 are the axial ligands, and S of Met-314, Nϵ of His-242, and a water molecule are the equatorial ones. Compared with the tetrahedral coordination in the crystal structure, an additional water molecule completes the coordination of copper. Another water molecule would be available in the QM part for further coordination to copper, but it stays in the bulk. Geometric data are given in Fig. 3 and in Table 2. In snapshot 2, copper is coordinated in a square-planar manner, including one water molecule, with S of Met forming an additional axial ligand with a large distance (2.748 Å) from copper. Finally, in snapshot 3, copper is in a stretched octahedral environment, including two water molecules. S and the water ligand in the trans position to it are more distant from copper than the other ligands. These coordination shells are roughly preserved during the mechanism of hydrogen abstraction. However, in snapshots 2 and 3 the Cu–S bond is broken during the hydrogen abstraction process. Therefore, the Cu–S distance increases from 2.748 to 3.878 Å in snapshot 2 and from 2.805 to 3.135 Å in snapshot 3 (Fig. 3 and Table 2).
TABLE 2.
Geometric data of [CuOOH]2+
Distances are in Å; angles are in degrees; RS is reactant state; TS is transition state 1; IS is intermediate state; s is singlet; t is triplet; Nc is nitrogen ligand cis to O; Nt is nitrogen ligand trans to O.
| Snapshot 1 |
Snapshot 2 |
Snapshot 3 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RS-s | RS-t | TS-s | IS-s | RS-s | RS-t | TS-s | IS-s | RS-s | RS-t | TS-s | IS-s | |
| Cu–O1 | 1.978 | 1.992 | 2.090 | 2.195 | 1.864 | 1.958 | 2.026 | 2.171 | 1.878 | 1.973 | 2.005 | 2.106 |
| O1–O2 | 1.348 | 1.340 | 1.421 | 1.471 | 1.365 | 1.290 | 1.420 | 1.454 | 1.373 | 1.378 | 1.422 | 1.461 |
| O2–Hα | 2.191 | 2.222 | 1.173 | 0.984 | 2.065 | 2.108 | 1.157 | 1.044 | 1.976 | 2.106 | 1.170 | 1.023 |
| C–Hα | 1.100 | 1.100 | 1.434 | 3.249 | 1.106 | 1.102 | 1.455 | 2.715 | 1.109 | 1.102 | 1.431 | 2.468 |
| Cu–O1–O2 | 115.4 | 119.4 | 118.2 | 114.4 | 105.7 | 105.8 | 113.0 | 117.4 | 112.8 | 118.5 | 119.5 | 125.2 |
| O1–O2–H | 106.8 | 105.3 | 104.3 | 101.7 | 104.4 | — | 103.9 | 104.3 | 106.0 | 108.1 | 104.0 | 102.4 |
| Cu–S | 2.460 | 2.554 | 2.463 | 2.477 | 2.748 | 2.581 | 2.714 | 3.878 | 2.805 | 2.784 | 2.836 | 3.135 |
| Cu–Nt | 1.974 | 1.992 | 2.005 | 1.988 | 1.939 | 1.964 | 1.984 | 2.066 | 1.957 | 2.031 | 1.986 | 2.006 |
| Cu–Nc | 2.172 | 2.134 | 2.120 | 2.140 | 2.030 | 2.213 | 2.048 | 1.981 | 2.101 | 2.074 | 2.098 | 2.077 |
| Cu–OH2 | 2.129 | 2.121 | 2.091 | 2.149 | 2.096 | 2.257 | 2.073 | 2.009 | 2.175 | 2.167 | 2.155 | 2.153 |
| Cu–OH2 2 | 2.377 | 2.354 | 2.321 | 2.402 | 2.353 | |||||||
| Nc–Cu–OH2 | 119.1 | 118.7 | 120.0 | 116.7 | 167.5 | 164.1 | 162.7 | 169.2 | 177.6 | 178.2 | 173.0 | 173.9 |
In the reactant state of each snapshot, the proton of the hydroperoxide points toward a water molecule. Thus, proton abstraction from such a water molecule via a Grotthuss chain could be a plausible route for protonation. In snapshot 2, this water molecule is quite close to the C-terminal carboxylate group of the glycine substrate. The latter may be part of the proton delivery channel. Actually, the geometry shown in Fig. 4d for the singlet is isoenergetic with a protonated carboxylic acid and a [CuO2]+ species, with an energy barrier lower than 3.5 kJ mol−1 between the minima (Fig. 3). So, at room temperature, the extra proton will be moving back and forth from the carboxylic acid to the abstracting species (see supplemental material). The triplet structure is only stable with the proton transferred to the carboxylate, which accounts for the comparably short O–O distance of 1.290 Å in Table 2. The orientation of the water molecules in snapshots 1 and 3, however, is not favorable for this proton jump to happen (Fig. 4, a and g).
The reactant state, [CuOOH]2+, can formally be described by either Cu3+ OOH−, in which both components would be singlet, or Cu•2+ OOH•0, in which both components are doublets, which can couple to a singlet or a triplet. In a density functional description, the latter singlet would be represented by an open-shell singlet (spin contamination). In snapshots 1 and 2, the triplet state is slightly lower in energy than the singlet (the latter is closed-shell in snapshot 2 but open-shell in snapshot 1). In snapshot 3, the singlet state is lower by 25.4 kJ mol−1 (Table 3). These small energy differences are below the accuracy that can be expected from density functional theory for singlet-triplet splittings. The pure singlet states of snapshots 2 and 3 can only be described by Cu3+ OOH−, although the triplet states as well as the open-shell singlet of snapshot 1 are probably best described by Cu•2+ OOH•0 or a mixture. Thus, the copper atom already reduced the dioxygen moiety by one or two internal reduction equivalents even before hydrogen is abstracted or the system is further reduced by external electrons. However, it should be noted that an assignment of formal oxidation states from electronic structure calculations is generally ambiguous.
TABLE 3.
The three snapshots of [CuOOH]2+ (* indicates without zero point energy (ZPE) corrections)
| Snapshot 1 |
Snapshot 2 |
Snapshot 3 |
||||
|---|---|---|---|---|---|---|
| Singlet | Triplet | Singlet | Triplet | Singlet | Triplet | |
| ΔES-T (kJ mol−1) | 0 | −5.1 | 0 | −7.6 | 0 | +25.4 |
| ΔE‡* (kJ mol−1) | 47.8 | 76.9 | 46.0 | 82.6 | 37.2 | 41.2 |
| ΔEr* (kJ mol−1) | +8.6 | +29.9 | +27.0 | +36.5 | −2.6 | −21.8 |
| ΔE‡ (kJ mol−1) | 33.1 | 63.7 | 31.2 | 64.3 | 21.8 | 28.9 |
| ΔEr (kJ mol−1) | +8.3 | +32.2 | +20.7 | +25.8 | −5.8 | −23.6 |
| ωb (cm−1) | 991.3i | 1512.0i | 825.1i | 1321.7i | 926.6i | 1357.1i |
| Tc (K) | 227.0 | 346.2 | 188.9 | 302.6 | 212.2 | 310.8 |
| Primary KIE at 37 °C | 7.7 | 20.7 | 6.2 | 14.6 | 6.2 | 10.7 |
| Secondary KIE at 37 °C | 1.09 | 1.08 | 1.05 | 1.06 | 1.09 | 1.08 |
| Primary KIE (exp.) (23) | 10.6 ± 0.8 | |||||
| Secondary KIE (exp.) (23) | 1.20 ± 0.03 | |||||
The reaction is an abstraction of a radical hydrogen atom. The transition states are noticeably higher (by 24.0, 29.0, and 29.4 kJ mol−1 in snapshots 1, 2, and 3, respectively) in energy in the triplet channel (ferromagnetic coupling between the spin on copper and the emerging spin on Cα) than in the antiferromagnetic open-shell singlet channel (Table 3). This can qualitatively be explained by superexchange-like interaction between the carbon radical and the copper spin center. Therefore, the reaction will take place via the singlet channel. This finding agrees with previous calculations (21, 61), although the abstracting species used there was different. Our thermally averaged reaction barrier is 24.4 kJ mol−1.
After the hydrogen abstraction, in the intermediate state, the active site is a [CuOHOH]2+ biradical with copper in the Cu2+ form (d9) and a carbon radical at the glycine-α position, which is stabilized by both neighboring groups as follows: the carboxylate and the amide bond (Fig. 5). The intermediate state is an open-shell singlet in all snapshots. Although the corresponding triplet structures are not much higher in energy, all three have a negative band gap, the highest occupied α orbital is higher than the lowest unoccupied β orbital. They are not relevant for the reaction mechanism.
FIGURE 5.

Spin density (contour ± 0.005 atomic units, orange, positive value; green, negative value) of the intermediate state of snapshot 3. The radical at the substrate is delocalized.
The energy barriers as well as the primary and secondary KIEs at 37 °C are shown in Table 3. Triplet values for KIEs are given for the sake of completeness only. For the singlet channel, the Tc is small enough to justify the approximation by an Eckart barrier to estimate tunneling at 37 °C. KIEs are slightly lower than the ones found experimentally (10.6 ± 0.8 at the same temperature (23)) but still higher than the one expected when the zero-point energy, but not tunneling, is taken into account (∼5), so tunneling does account for a significant part of the KIEs observed. Calculated secondary KIEs are also somewhat smaller than the ones found experimentally (∼1.08 and 1.20, respectively).
For the reaction to proceed, it is necessary that the barrier for hydrogen abstraction from the substrate is lower than the barrier for proton abstraction from water by the reactive species. The energy barrier for proton abstraction is higher than 90 kJ mol−1 (snapshot 2). Thus, [CuOOH]2+ has a higher affinity for a hydrogen from glycine than a proton from water.
We have thus seen that our proposed [CuOOH]2+ species can account for the glycine hydrogen abstraction. Its energy barrier is reasonably low, and KIEs for this step are similar to those observed experimentally. It also agrees with the experimental finding that there exists an intermediate electronically equivalent to Cu(I)-O2 (27).
Other abstracting species were considered as well. The two protons can be taken from the environment at any time (before or after the hydrogen abstraction). The electron transfer from CuH to CuM can occur at any stage as well. This means that there is a total of six possible abstracting species that need to be considered.
Note that we have used a different basis set for the calculations shown above for [CuOOH]2+ (def2-TZVP) than used for the comparison of the other abstracting species (def2-SVP). However, the energy differences depend only weakly on the basis set. As an example, the energy barrier for [CuOOH]2+ in snapshot 2 is 54.0 kJ mol−1 using a def2-SVP basis set to be compared with 46.0 kJ mol−1 obtained with def2-TZVP (Table 3). The zero point energy corrections were neglected in this comparison. The numerical value of these corrections for an hydrogen atom abstraction is around 10–15 kJ mol−1. Taking them into account does not change any of the arguments given below.
[CuO2]+, 0 H+ 0 e−
This is the abstracting species first proposed by Chen and Solomon (19) based on gas phase calculations. It assumes that hydrogen abstraction occurs prior to any protonation or electron transfer. However, as pointed out by Crespo et al. (21), the energy barrier is very high when the protein environment is taken into account. They obtained 104 and 84 kJ mol−1 for the triplet and singlet configuration that is very similar to our numbers, 110.5 kJ mol−1 in the triplet state and 72.6 kJ mol−1 in the open-shell singlet. The reaction rate would be very low, so [CuOOH]2+ is a more plausible species for hydrogen abstraction than [CuO2]+.
[CuO2]0, 0 H+ 1 e−
This species was first suggested by Prigge et al. (7). One electron is transferred from CuH to the abstracting species before the reaction proceeds. Two possible spin configurations are possible, doublet and quadruplet. Because the quadruplet configuration has an energy 219.8 kJ mol−1 higher than the doublet, we conclude that the reaction would proceed through the doublet configuration.
The abstraction of a proton from water has a smaller energy barrier (∼20 kJ mol−1) than abstracting the hydrogen from glycine (∼140 kJ mol−1), so this cannot be the abstracting species. We use nudged elastic band calculations without a climbing image because of the large barrier differences in this case, so the barrier values are approximate. Abstracting one H+ from the environment turns [CuO2]0 into [CuOOH]+, which will be discussed next.
[CuOOH]+,: 1 H+ 1 e−
The energy barrier for the glycine hydrogen abstraction by [CuOOH]+ is 168.4 kJ mol−1. It is so high that this reaction will not take place at room temperature, so we can discard it. This species was also discarded by Chen and Solomon (19), Crespo et al. (21), and Kamachi et al. (22) in dopamine β-monooxygenase.
[CuO + H2O]3+, 2 H+ 0 e−
Crespo et al. (21) showed that two protonations lead to a dissociation of the abstracting species into a water molecule and a copper-oxo species that potentially can abstract the hydrogen from glycine.
In this highly oxidized and protonated case, copper has a formal oxidation state of +5. So this is highly unlikely to be stable enough to be a realistic abstracting species. QM/MM optimization leads to a spontaneous hydrogen abstraction and OH rebinding. A spontaneous reaction cannot explain the large KIE measured experimentally.
[CuO + H2O]2+, 2 H+ 1 e−
We obtain the same results as Crespo et al. (21); [CuO]2+ spontaneously abstracts the hydrogen from glycine. The OH rebinding is also spontaneous. As in the previous case, a spontaneous reaction cannot explain the large KIE measured experimentally, so we can exclude it as the abstracting species.
At this point we should point out the similarity of this case with our suggested mechanism. Although in our proposal the hydrogen abstraction is carried out by the singly protonated [CuOOH]2+ species, the OH rebinding implies a second protonation and an electron transfer to the active site of our model, just as in this case. One may ask when the second protonation is more likely, before or after the hydrogen abstraction. Chemical intuition suggests that the second protonation is easier once the H• from glycine is abstracted (because of the charge stabilization). Our calculations also point in that direction.
[CuO +H2O]+, 2 H+ 2 e−
Kamachi et al. (22) and Crespo et al. (21) claimed this to be a possible abstracting species. It would be the result of a protonation of [CuO2]+, an electron transfer from CuH, and a hydrogen abstraction from a neighboring residue (62). The resulting species dissociates to a water molecule and [CuO]+. Tyr-79 and Tyr-318 were considered to be a potential source of the hydrogen atom (36, 63), but Francisco et al. (36) found no significant change in the reaction rate after mutagenic experiments. An alternative source of hydrogen has not been suggested up to date.
We include this species only to compare with previous calculations on [CuO]+. Two spin states are possible. The reaction is spontaneous in the singlet channel, and it has a very small energy barrier (2.0 kJ mol−1) in the triplet state. Crespo et al. (21) also obtained a small barrier for the [CuO]+ species. Even if the reaction proceeds through the triplet channel, the KIE will be very small, because of the low barrier, so we can also deduce that this is not the reaction mechanism.
Second Step, OH Rebinding
We have shown that the first and rate-limiting part of the reaction (the hydrogen abstraction) only proceeds after one proton was taken up from the environment. The hydrogen abstraction results in [CuOHOH]2+. Still, another proton is going to be consumed by the end of the reaction, as well as one electron is going to be transferred from CuH to CuM. This can happen either before or after the OH rebinding step. We have tested these possibilities.
We have found that the second protonation and the electron transfer actually occur before the OH rebinding. In the following we will describe the details of this OH rebinding mechanism.
[CuOHOH2]3+, 2 H+ 1 e−
We have first simulated the extra proton take-up. This entails one question. Where should the extra proton be placed? By turning several waters into hydronium ions, we have found that it usually ends up at the C-terminal carboxylate of the substrate. This happens in snapshots 2 and 3. However, in snapshot 1, the proton is absorbed by the [CuOHOH]2+ moiety, which dissociates into [CuOH]3+ + H2O. In all cases, the distance between this extra proton and O2 is very small (0.99, 1.58, and 1.56 Å for snapshots 1, 2, and 3, respectively).
We have checked the possibility of the reaction to take place after this extra proton is delivered. The reaction can indeed proceed after the protonation. The energy barriers are smaller than in the hydrogen abstraction step (the thermally averaged barrier is 14.6 kJ mol−1). This means that the hydrogen abstraction is the rate-limiting step, and thus, this mechanism is in agreement with experimental evidence (24). Table 4 shows the reaction energy and the barriers for the three snapshots; Fig. 7 shows the geometries of the double protonated intermediate state (Fig. 7, a, d, and g), the OH rebinding transition state (Fig. 7, b, e, and h), and the products (Fig. 7, c, f, and i) for snapshots 1 (Fig. 7, a–c), 2 (Fig. 7, d–f), and 3 (Fig. 7, g–i). All this geometrical information is summarized in Fig. 6. We should warn here that, because a proton is irreversibly delivered after the hydrogen abstraction, there is barely a reason to worry about the energy barrier of the backward reaction in the first step (hydrogen abstraction).
TABLE 4.
OH rebinding energies for the reaction for the three snapshots after a second proton is taken from the environment
For spontaneous reactions, ΔEr* are the differences between the initial and final geometry of the optimization. (*, without ZPE corrections).
| Snapshot | [CuOHOH2]3+, 2H+ 0 e− |
[CuOHOH2]2+, 2H+ 1 e− |
||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | |
| ΔE‡* (kJ mol−1) | 11.9 | 35.9 | 39.7 | 0 | ∼ 0 | 26.7 |
| ΔEr* (kJ mol−1) | −140.7 | −210.7 | −113.2 | −194.9 | −257.7 | −238.1 |
| ΔE‡ (kJ mol−1) | 10.7 | 21.6 | 21.9 | 0 | ∼0 | 20.2 |
FIGURE 7.

Geometries of the double protonated intermediates (a, d, and g), transition state (b, e, and h), and product (c, f, and i) of snapshots 1 (a–c), 2 (d–f), and 3 (g–i). No extra electron is considered. See Fig. 4 for the representation.
FIGURE 6.

Schematic view of the structure of the double protonated intermediate, transition state (TS2), and product of the three snapshots. Important bond lengths (in Å) are included in the scheme.
In the following, we will point out the differences found between the three calculated snapshots. In snapshot 1, after the proton take-up, the energy barrier for OH rebinding is very small. This is because the O–O bond is already broken, and the product water molecule is already formed. The 10.7 kJ mol−1 energy barrier comes, to a large extent, from the breaking of the Cu–O1 bond.
In snapshots 2 and 3, however, the O–O bond is not yet broken after the proton take-up. This is mainly because the carboxylic acid of the substrate is protonated, instead of the [CuOHOH]2+ species (Fig. 6). However, the structure adopts a favorable form for water desorption, as can be seen in Fig. 7, d and g. The energy barrier for this case is larger than for snapshot 1, because the O–O bond still needs to be broken. In the case of snapshot 2, this energy barrier is 21.6 kJ mol−1, and for snapshot 3 it is 21.9 kJ mol−1. It is worth commenting that in snapshot 3 the Cu–O bond is significantly weakened after the proton adsorption (Cu–O distance, 3.11 Å), although the effect is weaker in snapshot 2 (Cu–O distance 2.18 Å). The reason for this may be related to the Cu–S bond. In snapshot 2, the Cu–S bond is broken, whereas in snapshot 3 it is not. This suggests that the presence (absence) of the Cu–S bond is related to the ability of copper atom to keep the hydrogen peroxide molecule bound once the hydrogen from glycine is abstracted. However, taking into account the strong hydrogen bond between the O2 and the hydrogen from the carboxylic acid, as well as hydrogen bonds with other solvent atoms, and a weak (but not negligible) Cu–O interaction, we conclude that our active site is stable enough to be able to hydroxylate the glycine radical.
However, if we simulate an electron transfer from CuH to CuM after the proton addition, by adding an electron to our QM subsystem, then the OH rebinding is spontaneous for snapshots 1 and 2. For snapshot 3, however, the reaction is not spontaneous, although the energy barrier is smaller than for the case with no electron transfer (Table 4). Thus, we conclude that the OH rebinding step happens after a proton is taken up from the environment and an electron is transferred from the CuH atom.
At this point, we should point out the difficulty of simulating a proton adsorption. A rigorous study of the OH rebinding step should include much larger statistics; this means several snapshots and pKa calculation of the different proton acceptors, which is far beyond the scope of this work. Here, our emphasis lies on a plausible mechanism for the OH rebinding. Such a detailed study may help to reach a consensus in the OH rebinding mechanism in PHM and should be studied in the future. The three snapshots studied here lead to different reaction paths, but all of them explain the experimental evidence (OH rebinding is not rate-limiting). The differences between our snapshots are a good indication that we are mapping different regions of the configuration space, which means that we are able to capture a large part of the statistics with a small number of snapshots. To sum up, we have shown evidence that the OH rebinding follows the second protonation and electron transfer from CuH. The OH rebinding with no extra protons or electrons will be described in below.
[CuOHOH]2+, 1 H+ 0 e−
In this case, no extra protons or electrons are taken from the environment after the hydrogen abstraction and before OH rebinding. We have calculated the energy barrier of this, and we have found that the energy barrier is too high (69.5 kJ mol−1). It is important to note here that the backward energy barrier is 13.2 kJ mol−1 (thermal average). This means that backward reaction is favored over OH rebinding and that the OH rebinding step would be rate-limiting. This is against experimental evidence, so the reaction will not proceed this way.
Summary
PHM is prototypical for monooxygenases with noninteracting copper atoms. Until now, several mechanisms have been proposed from the theoretical and experimental side, but they disagree with the experimental evidence or with the theoretical calculations. In this study, we compare several possible mechanisms on equal footing. We propose a mechanism that is consistent with our theoretical calculations and agrees well with the experimental results.
In our mechanism, the solvent-exposed [CuO2]+ species is protonated. The resulting [CuOOH]2+ species is able to abstract the glycine pro-S hydrogen atom in the singlet configuration with a small energy barrier (thermal average at 300 K of 24.4 kJ mol−1). Even though the barrier is small, significant KIEs (6.2–7.7) are found within the Eckart barrier approximation.
This proposal is in agreement with experimental results; the energy barrier is not very large and is smaller than a water proton abstraction by [CuOOH]2+. The KIEs found imply that hydrogen is transferred partly by quantum tunneling, although tunneling is not the main cause of the catalytic effect. Moreover, our proposed abstracting species is in agreement with the experimental conclusion that there should be an intermediate electronically equivalent to Cu(I)-O2 (27).
Another protonation and an electron transfer from CuH makes the OH rebinding step an (almost) spontaneous process and thus not rate-limiting. In Fig. 8, we show an overview of our proposed mechanism.
FIGURE 8.
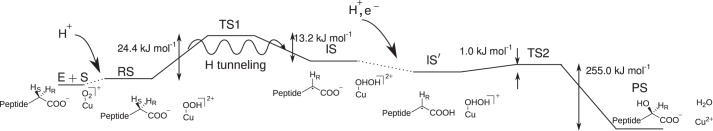
Mechanism proposed in this paper. See text for details.
According to general reaction stoichiometry (Fig. 1) and experimental findings, one can think of other abstracting species (Table 1 and Fig. 9). Two electrons are provided by ascorbate at the beginning of the reaction cycle and reduce the CuH(II) and CuM(II) to CuH(I) and CuM(I) (16). However, an electron transfer from CuH to CuM is likely to happen at some point during the reaction (7, 9–12). Moreover, two protons are taken from the environment. All of this may happen before the hydrogen abstraction. We have studied the resulting six possible abstracting species, and we have found that only two reaction mechanisms are in agreement with experimental evidence (Fig. 9). Although [CuO2]+ is a plausible abstracting species, its energy barrier is high compared with the one from the favored form.
This work was supported by the Alexander von Humboldt Foundation, Landesstiftung Baden-Württemberg, and Cluster of Excellence in Simulation Technology Grant EXC 310/1 (University of Stuttgart).

This article contains supplemental topology and parameter files and a movie.
- PAM
- peptidylglycine α-amidating monooxygenase
- PHM
- peptidylglycine α-hydroxylating monooxygenase
- PAL
- peptidyl α-hydroxylglycine-α-amidating lyase
- QM/MM
- quantum mechanics/molecular mechanics
- KIE
- kinetic isotope effect
- MD
- molecular dynamics.
REFERENCES
- 1. Klinman J. P. (1996) Mechanisms whereby mononuclear copper proteins functionalize organic substrates. Chem. Rev. 96, 2541–2562 [DOI] [PubMed] [Google Scholar]
- 2. Prigge S. T., Mains R. E., Eipper B. A., Amzel L. M. (2000) New insights into copper monooxygenases and peptide amidation: structure, mechanism and function. Cell. Mol. Life Sci. 57, 1236–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bell J., Ash D. E., Snyder L. M., Kulathila R., Blackburn N. J., Merkler D. J. (1997) Structural and functional investigations on the role of zinc in bifunctional rat peptidylglycine α-amidating enzyme. Biochemistry 36, 16239–16246 [DOI] [PubMed] [Google Scholar]
- 4. Saldise L., Martínez A., Montuenga L. M., Treston A., Springall D. R., Polak J. M., Vázquez J. J. (1996) Distribution of peptidylglycine α-amidating mono-oxygenase (PAM) enzymes in normal human lung and in lung epithelial tumors. J. Histochem. Cytochem. 44, 3–12 [DOI] [PubMed] [Google Scholar]
- 5. Martinez A., Treston A. M. (1996) Where does amidation take place? Mol. Cell. Endocrinol. 123, 113–117 [DOI] [PubMed] [Google Scholar]
- 6. Iwai N., Avis I., Scott F., Quinn K., Cuttitta F., Mulshine J., Treston A. (1993) Novel tumor cell growth inhibitors acting via autocrine loops which are dependent on peptidyl-α-amidating enzyme. Proc. Annu. Meet. Am. Assoc. Cancer Res. 34, 266 [Google Scholar]
- 7. Prigge S. T., Eipper B. A., Mains R. E., Amzel L. M. (2004) Dioxygen binds end-on to mononuclear copper in a precatalytic enzyme complex. Science 304, 864–867 [DOI] [PubMed] [Google Scholar]
- 8. Kulathila R., Consalvo A. P., Fitzpatrick P. F., Freeman J. C., Snyder L. M., Villafranca J. J., Merkler D. J. (1994) Bifunctional peptidylglycine α-amidating enzyme requires 2 copper atoms for maximum activity. Arch. Biochem. Biophys. 311, 191–195 [DOI] [PubMed] [Google Scholar]
- 9. Prigge S. T., Kolhekar A. S., Eipper B. A., Mains R. E., Amzel L. M. (1999) Substrate-mediated electron transfer in peptidylglycine α-hydroxylating monooxygenase. Nat. Struct. Biol. 6, 976–983 [DOI] [PubMed] [Google Scholar]
- 10. Evans J. P., Ahn K., Klinman J. P. (2003) Evidence that dioxygen and substrate activation are tightly coupled in dopamine β-monooxygenase. Implications for the reactive oxygen species. J. Biol. Chem. 278, 49691–49698 [DOI] [PubMed] [Google Scholar]
- 11. de la Lande A., Martí S., Parisel O., Moliner V. (2007) Long distance electron-transfer mechanism in peptidylglycine α-hydroxylating monooxygenase: a perfect fitting for a water bridge. J. Am. Chem. Soc. 129, 11700–11707 [DOI] [PubMed] [Google Scholar]
- 12. Meliá C., Ferrer S., Řezáč J., Parisel O., Reinaud O., Moliner V., de la Lande A. (2013) Investigation of the hydroxylation mechanism of noncoupled copper oxygenases by ab initio molecular dynamics simulations. Chem. Eur. J. 19, 17328–17337 [DOI] [PubMed] [Google Scholar]
- 13. Ramer S. E., Cheng H., Palcic M. M., Vederas J. C. (1988) Formation of peptide amides by peptidylglycine . α-amidating monooxygenase: a new assay and stereochemistry of hydrogen loss. J. Am. Chem. Soc. 110, 8526–8532 [Google Scholar]
- 14. Noguchi M., Seino H., Kochi H., Okamoto H., Tanaka T., Hirama M. (1992) The source of the oxygen atom in the α-hydroxyglycine intermediate of the peptidylglycine α-amidating reaction. Biochem. J. 283, 883–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eipper B. A., Mains R. E., Glembotski C. C. (1983) Identification in pituitary tissue of a peptide α-amidation activity that acts on glycine-extended peptides and requires molecular oxygen, copper, and ascorbic acid. Proc. Natl. Acad. Sci. U.S.A. 80, 5144–5148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Freeman J. C., Villafranca J. J., Merkler D. J. (1993) Redox cycling of enzyme-bound copper during peptide amidation. J. Am. Chem. Soc. 115, 4923–4924 [Google Scholar]
- 17. Bradbury A. F., Mistry J., Roos B. A., Smyth D. G. (1990) 4-Phenyl-3-butenoic acid, an in vivo inhibitor of peptidylglycine hydroxylase (peptide amidating enzyme) Eur. J. Biochem. 189, 363–368 [DOI] [PubMed] [Google Scholar]
- 18. Katopodis A. G., May S. W. (1990) Novel substrates and inhibitors of peptidylglycine α-amidating monooxygenase. Biochemistry 29, 4541–4548 [DOI] [PubMed] [Google Scholar]
- 19. Chen P., Solomon E. I. (2004) Oxygen activation by the noncoupled binuclear copper site in peptidylglycine α-hydroxylating monooxygenase. Reaction mechanism and role of the noncoupled nature of the active site. J. Am. Chem. Soc. 126, 4991–5000 [DOI] [PubMed] [Google Scholar]
- 20. Chen P., Solomon E. I. (2004) O2 activation by binuclear Cu sites: noncoupled versus exchange coupled reaction mechanisms. Proc. Natl. Acad. Sci. U.S.A. 101, 13105–13110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Crespo A., Martí M. A., Roitberg A. E., Amzel L. M., Estrin D. A. (2006) The catalytic mechanism of peptidylglycine α-hydroxylating monooxygenase investigated by computer simulation. J. Am. Chem. Soc. 128, 12817–12828 [DOI] [PubMed] [Google Scholar]
- 22. Kamachi T., Kihara N., Shiota Y., Yoshizawa K. (2005) Computational exploration of the catalytic mechanism of dopamine β-monooxygenase: modeling of its mononuclear copper active sites. Inorg. Chem. 44, 4226–4236 [DOI] [PubMed] [Google Scholar]
- 23. Francisco W. A., Merkler D. J., Blackburn N. J., Klinman J. P. (1998) Kinetic mechanism and intrinsic isotope effects for the peptidylglycine α-amidating enzyme reaction. Biochemistry 37, 8244–8252 [DOI] [PubMed] [Google Scholar]
- 24. Francisco W. A., Knapp M. J., Blackburn N. J., Klinman J. P. (2002) Hydrogen tunneling in peptidylglycine α-hydroxylating monooxygenase. J. Am. Chem. Soc. 124, 8194–8195 [DOI] [PubMed] [Google Scholar]
- 25. Klinman J. P. (2006) The copper-enzyme family of dopamine β-monooxygenase and peptidylglycine α-hydroxylating monooxygenase: resolving the chemical pathway for substrate hydroxylation. J. Biol. Chem. 281, 3013–3016 [DOI] [PubMed] [Google Scholar]
- 26. McIntyre N. R., Lowe E. W., Jr., Merkler D. J. (2009) Imino-oxy acetic acid dealkylation as evidence for an inner-sphere alcohol intermediate in the reaction catalyzed by peptidylglycine α-hydroxylating monooxygenase. J. Am. Chem. Soc. 131, 10308–10319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bauman A. T., Yukl E. T., Alkevich K., McCormack A. L., Blackburn N. J. (2006) The hydrogen peroxide reactivity of peptidylglycine monooxygenase supports a Cu(II)-superoxo catalytic intermediate. J. Biol. Chem. 281, 4190–4198 [DOI] [PubMed] [Google Scholar]
- 28. Rudzka K., Moreno D. M., Eipper B., Mains R., Estrin D. A., Amzel L. M. (2013) Coordination of peroxide to the Cu(M) center of peptidylglycine α-hydroxylating monooxygenase (PHM): structural and computational study. J. Biol. Inorg. Chem. 18, 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Warshel A., Karplus M. (1972) Calculation of ground and excited state potential surfaces of conjugated molecules. I. formulation and parametrization. J. Am. Chem. Soc. 94, 5612 [Google Scholar]
- 30. Warshel A., Levitt M. (1976) Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 103, 227–249 [DOI] [PubMed] [Google Scholar]
- 31. Word J. M., Lovell S. C., Richardson J. S., Richardson D. C. (1999) Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 285, 1735–1747 [DOI] [PubMed] [Google Scholar]
- 32. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926 [Google Scholar]
- 33. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 34. MacKerell A. D., Bashford D., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 35. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Francisco W. A., Blackburn N. J., Klinman J. P. (2003) Oxygen and hydrogen isotope effects in an active site tyrosine to phenylalanine mutant of peptidylglycine α-hydroxylating monooxygenase: mechanistic implications. Biochemistry 42, 1813–1819 [DOI] [PubMed] [Google Scholar]
- 37. Sherwood P., de Vries A. H., Guest M. F., Schreckenbach G., Catlow C. A., French S. A., Sokol A. A., Bromley S. T., Thiel W., Turner A. J., Billeter S., Terstegen F., Thiel S., Kendrick J., Rogers S. C., Casci J., Watson M., King F., Karlsen E., Sj M., Fahmi A., Schäfer A., Lennartz C. (2003) QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis. J. Mol. Struct. THEOCHEM 632, 1–28 [Google Scholar]
- 38. Metz S., Kästner J., Sokol A. A., Keal T. W., Sherwood P. (2014) Chemshell–a modular software package for QM/MM simulations. Comput. Mol. Sci. 4, 101–110 [Google Scholar]
- 39. Kästner J., Carr J. M., Keal T. W., Thiel W., Wander A., Sherwood P. (2009) DL-FIND: an open-source geometry optimizer for atomistic simulations. J. Phys. Chem. A 113, 11856–11865 [DOI] [PubMed] [Google Scholar]
- 40. Nocedal J. (1980) Updating Quasi-Newton matrices with limited storage. Math. Comput. 35, 773 [Google Scholar]
- 41. Liu D. C., Nocedal J. (1989) On the limited memory BFGS method for large scale optimization. Math. Prog. 45, 503–528 [Google Scholar]
- 42. Henkelman G., Jónsson H. (1999) A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 111, 7010 [Google Scholar]
- 43. Kästner J., Sherwood P. (2008) Superlinearly converging dimer method for transition state search. J. Chem. Phys. 128, 014106 [DOI] [PubMed] [Google Scholar]
- 44. Billeter S. R., Turner A. J., Thiel W. (2000) Linear scaling geometry optimization and transition state search in hybrid delocalised internal coordinates. Phys. Chem. Chem. Phys. 2, 2177 [Google Scholar]
- 45. Jónsson H., Mills G., Jacobsen K. W. (1998) in Classical and Quantum Dynamics in Condensed Phase Simulations (Berne B. J., Ciccotti G., Coker D. F., eds) pp. 385–404, World Scientific Publishing, Singapore [Google Scholar]
- 46. Goumans T. P., Catlow C. R., Brown W. A., Kästner J., Sherwood P. (2009) An embedded cluster study of the formation of water on interstellar dust grains. Phys. Chem. Chem. Phys. 11, 5431–5436 [DOI] [PubMed] [Google Scholar]
- 47. Smith W., Yong C., Rodger P. (2002) DL_POLY: Application to molecular simulation. Mol. Simul. 28, 385–471 [Google Scholar]
- 48. Becke A. D. (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 [DOI] [PubMed] [Google Scholar]
- 49. Perdew J. (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter 33, 8822–8824 [DOI] [PubMed] [Google Scholar]
- 50. University of Karlsruhe and Forschungszentrum Karlsruhe GmbH (2009) TURBOMOLE, Version 6.0.2, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, Germany [Google Scholar]
- 51. Von Arnim M., Ahlrichs R. (1998) Performance of parallel TURBOMOLE for density functional calculations. J. Comput. Chem. 19, 1746 [Google Scholar]
- 52. Rommel J. B., Kästner J. (2011) The fragmentation-recombination mechanism of the enzyme glutamate mutase studied by QM/MM simulations. J. Am. Chem. Soc. 133, 10195–10203 [DOI] [PubMed] [Google Scholar]
- 53. Schäfer A., Horn H., Ahlrichs R. (1992) Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571 [Google Scholar]
- 54. Weigend F., Häser M., Patzelt H., Ahlrichs R. (1998) RI-MP2: optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 294, 143–152 [Google Scholar]
- 55. Weigend F., Ahlrichs R. (2005) Balanced basis sets of split valence, triple ζ valence and quadruple ζ valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 [DOI] [PubMed] [Google Scholar]
- 56. Gillan M. J. (1987) Quantum-classical crossover of the transition rate in the damped double well. J. Phys. C. 20, 3621 [Google Scholar]
- 57. Eckart C. (1930) The penetration of a potential barrier by electrons. Phys. Rev. 35, 1303 [Google Scholar]
- 58. Lonsdale R., Harvey J. N., Mulholland A. J. (2010) Compound I reactivity defines alkene oxidation selectivity in cytochrome P450cam. J. Phys. Chem. B. 114, 1156–1162 [DOI] [PubMed] [Google Scholar]
- 59. Kästner J., Sherwood P. (2010) The ribosome catalyses peptide bond formation by providing high ionic strength. Mol. Phys. 108, 293 [Google Scholar]
- 60. Peterson R. L., Ginsbach J. W., Cowley R. E., Qayyum M. F., Himes R. A., Siegler M. A., Moore C. D., Hedman B., Hodgson K. O., Fukuzumi S., Solomon E. I., Karlin K. D. (2013) Stepwise protonation and electron-transfer reduction of a primary copper-dioxygen adduct. J. Am. Chem. Soc. 135, 16454–16467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de la Lande A., Parisel O., Gérard H., Moliner V., Reinaud O. (2008) Theoretical exploration of the oxidative properties of a (tren Me1)CuO2]+ adduct relevant to copper monooxygenase enzymes: insights into competitive dehydrogenation versus hydroxylation reaction pathways. Chem. Eur. J. 14, 6465–6473 [DOI] [PubMed] [Google Scholar]
- 62. Tian G., Berry J. A., Klinman J. P. (1994) Oxygen-18 kinetic isotope effects in the dopamine β-monooxygenase reaction: Evidence for a new chemical mechanism in non-heme, metallomonooxygenase. Biochemistry 33, 226–234 [DOI] [PubMed] [Google Scholar]
- 63. Farrington G. K., Kumar A., Villafranca J. J. (1990) Active site labeling of dopamine β-hydroxylase by two mechanism-based inhibitors: 6-hydroxybenzofuran and phenylhydrazine. J. Biol. Chem. 265, 1036–1040 [PubMed] [Google Scholar]