

Background: Ler alleviates H-NS-mediated gene silencing.
Results: Ler·DNA binding properties and Ler effects on H-NS·DNA binding are investigated using magnetic tweezers and atomic force microscopy.
Conclusion: Ler binds noncooperatively to stiffen and fold DNA, and it can replace prebound H-NS in the conditions tested.
Significance: The findings provide new insights into the molecular mechanism of anti-silencing by Ler.
Keywords: Atomic Force Microscopy, DNA-binding Protein, Gene Regulation, Protein-DNA Interaction, Single Molecule Biophysics, H-NS, Ler, Magnetic Tweezers
Abstract
The locus of enterocyte effacement-encoded regulator (Ler) of enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC) functions to activate transcription of virulence genes silenced by the histone-like nucleoid-structuring protein (H-NS). Despite its important role in the bacterial gene regulation, the binding mode of Ler to DNA and its mechanism in alleviating genes repressed by H-NS are largely unknown. In this study, we use magnetic tweezers to demonstrate that Ler binds extended DNA through a largely noncooperative process, which results in DNA stiffening and DNA folding depending on protein concentration. We also show that Ler can replace prebound H-NS on DNA over a range of potassium and magnesium concentrations. Our findings reveal the DNA binding properties of Ler and shed light to further understand the anti-silencing activity of Ler.
Introduction
Gene regulation is an essential feature in bacteria that allows the expression of the proteins needed in a certain environment. In enteropathogenic Escherichia coli and enterohemorrhagic E. coli, the genes responsible for their pathogenic activity are encoded in the locus of enterocyte effacement (LEE) pathogenicity island, which consists of five major operons, LEE1 to LEE5. Under normal conditions, these operons are silenced by a histone-like nucleoid-structuring protein (H-NS),4 a global transcriptional silencer, especially for laterally acquired genes (1–4). The process of antisilencing involves LEE-encoded regulator (Ler), a protein encoded in LEE1, which is the key regulator that functions to activate the transcription of other major operons in the pathogenicity islands (5). The regulation of ler in the LEE1 operon itself is multifactorial and responsive to environmental changes (6). Together, these proteins are the major regulators controlling the expression of virulence genes in enterohemorrhagic E. coli and enteropathogenic E. coli.
H-NS consists of the N terminus that serves as the oligomerization domain and the C terminus that serves as the DNA-binding domain (7–10). It has been demonstrated that H-NS has two distinct DNA-binding modes. At low magnesium concentration (<2 mm), H-NS predominantly binds cooperatively along the DNA track resulting in the formation of a rigid nucleoprotein filament (11, 12). At higher magnesium concentrations (>5 mm), H-NS binding instead results in the formation of DNA hairpins via H-NS-mediated DNA bridging (12, 13). In the intermediate range of magnesium concentration, both DNA stiffening and DNA bridging co-exist (12). Interestingly, rigid nucleoprotein filaments have recently been shown to be shared by many gene-silencing H-NS-like proteins identified from various bacteria such as MvaT from Pseudomonas aeruginosa (14), StpA from E. coli (15), and Lsr2 from Mycobacterium tuberculosis (16). Based on these findings, it has been proposed that the ability of H-NS and H-NS-like proteins to silence genes is likely based on their ability to form rigid nucleoprotein filaments, which may block RNA polymerases from accessing the promoters or block translocation of RNA polymerases (17). Consistently, in vitro studies have shown that point mutations in the oligomerization domains of E. coli H-NS and P. aeruginosa MvaT that resulted in the loss of gene silencing functions also resulted in loss of the formation of the rigid nucleoprotein filaments (14, 17). The importance of the rigid nucleoprotein filaments is highlighted by a recent in vitro study showing that SsrB, an H-NS antagonizing protein, could replace the H-NS nucleoprotein complex at a low magnesium concentration, where H-NS nucleoprotein complex predominantly adopts rigid filamentous structures (18).
Ler shares 24% amino acid identity and 44% amino acid similarity to H-NS. Surprisingly, rather than a gene silencer, its main role has been characterized as an antagonizing protein that relieves H-NS-mediated transcriptional silencing (19). The C-terminal domain, which is known as the DNA-binding domain, bears much similarity to that of H-NS, whereas the N terminus and linker region are more divergent compared with H-NS (20–23). Despite the differences between the two N termini, Ler is also able to form dimers and higher order oligomers in solution (20). Despite its similarity to H-NS, experiments have demonstrated that Ler can neither substitute nor interact directly with H-NS (24, 25).
Ler and H-NS are important transcriptional regulators of the LEE pathogenicity islands. Both proteins bind overlapping DNA regions, where H-NS serves to repress the gene expression and Ler counters the H-NS-mediated gene silencing. It has been suggested that Ler relieves H-NS silencing by binding with a higher affinity and disrupting the H-NS nucleoprotein complexes (20, 21). A mutational study reported that Ler activates gene expression exclusively by preventing H-NS-mediated repression in LEE2, LEE3, LEE4, and espG, which supports the H-NS-antagonizing mechanism for anti-silencing (26). In our in vitro transcription assays, we did not see stimulation of LEE5 by Ler in the absence of H-NS.5 This suggests that Ler relieves the H-NS-mediated gene silencing likely by inhibiting H-NS binding to DNA. Nevertheless, the molecular mechanism of the interplay between H-NS and Ler is largely unexplored. In addition, the mechanism by which Ler binds DNA has not been extensively studied. A better understanding of the mechanism of Ler's anti-silencing activity in bacteria demands further investigation of these questions.
In this work, we show that Ler binds DNA nonspecifically, which depends on factors such as force and the concentration of Ler. At lower concentrations of Ler, we find that Ler binding can result in DNA folding, consistent with the previously reported DNA wrapping by Ler. At higher concentrations, Ler binding no longer folds DNA, instead it increases the DNA bending rigidity as measured by single-DNA stretching experiments. In contrast to DNA stiffening by H-NS, which is caused by formation of nucleoprotein filaments, we found that the DNA stiffening induced by Ler proteins is not caused by a cooperative polymerization process. We also investigated the influence of environmental factors such as KCl concentration, MgCl2 concentration, pH, and temperature on Ler's DNA binding activity. Importantly, we provide direct evidence that Ler robustly replaces H-NS nucleoprotein filaments under various buffer conditions. We believe that the DNA binding properties of Ler and its ability to displace H-NS from DNA as revealed in this study provide new perspectives on the molecular basis of regulation of the LEE pathogenicity islands as well as other genes controlled by these proteins.
EXPERIMENTAL PROCEDURES
Proteins
Ler protein was purified by the protein expression core facility of the Mechanobiology Institute, Singapore, as described previously (19).
Transverse Magnetic Tweezers
The magnetic tweezers used in this study are similar to the ones used in our previous studies (12, 14, 15). One end of the DNA was tethered to a coverslip edge that had been chemically coated with streptavidin, and the other end was attached to a streptavidin-coated paramagnetic bead. A pair of magnets was used to apply a force to the bead, and the DNA is stretched along the focal plane. Because of the shadow of the edge, a minimal distance of the bead from the edge of >2 μm is required for accurate determination of the bead position (14). Therefore, in the experiments λ-DNA (48,502 bp) with a long contour length (∼16 μm) was used. The tether was inside a glass channel fabricated in-house, which allows the automated exchange of buffer using a syringe pump to ensure consistency. The magnet's position was controlled by an XYZ manipulator to control the amount of force applied on the magnetic bead, which was calculated based on the bead's thermal motion shown in Equation 1,
 |
where kB is the Boltzmann constant; T is the temperature; z is the measured DNA extension, and δ is the variance of bead fluctuation perpendicular to the force applied on the magnetic bead. The data acquisition was done in real time using a home-written LabVIEW program.
In our magnetic tweezers experiments, we stretched a single DNA and used two different procedures to generate force-extension curves, a force-scanning procedure and a force-jumping procedure. Together, these two techniques allow us to draw a more complete picture of DNA binding properties of the protein studied.
The force-scanning procedure was done as follows. We initially stretched a single DNA molecule at high force, ∼10 pN, to prevent possible DNA folding during buffer exchange. The force was then reduced successively to ∼0.1 pN, after which the force was increased back through the same set of force values. The DNA was held at each force value for ∼30 s, and the average extension during this period of time was recorded. The two resulting force-extension curves are referred to as the force-decrease and force-increase curves, respectively. Whether the protein binding to DNA reaches a steady state can be inferred from the hysteresis (as evidenced by nonoverlapping values) between the force-decrease and the force-increase curves.
The force-jumping procedure was carried out as follows. A single DNA was initially held at a high force (∼10 pN), before jumping to a lower force for around 7–10 s, during which time the DNA extension was measured. Next, the force was jumped back to ∼10 pN to ensure that the DNA folding is negligible (<300 nm below the naked DNA at ∼10 pN). Repeating this procedure for a series of lower forces, a force-extension curve was obtained without accumulating DNA folding at low forces. The DNA spends significantly less time at low forces under this force-jumping procedure compared with the force-scanning procedure. Using the force-scanning procedure, the DNA is incubated for 6.5 min at forces <1 pN. In contrast, the incubation time at <1 pN for each force with the force-jumping procedure is only 7–10 s. Any folding during the time is not accumulated, because the DNA is unfolded at high force before jumping to the next low force step.
Atomic Force Microscopy
The AFM imaging experiments were done on glutaraldehyde-coated mica surfaces. Compared with a 3-aminopropyltriethoxysilane-mica surface, the glutaraldehyde mica surface is less charged, and therefore, the perturbation to the protein·DNA complex is significantly reduced (27, 28). To make the glutaraldehyde mica surface, a fresh layer of mica was obtained by peeling with double-sided tape, followed by incubation in a 0.1% solution of 3-aminopropyltriethoxysilane for 15 min. The mica was then thoroughly cleaned with deionized water before a 1% solution of glutaraldehyde was put on top of the mica to form glutaraldehyde-coated mica. Linearized ΦX174 DNA (5,386 bp, New England Biolabs) was incubated with various concentrations of protein for 15 min in a test tube in 50 mm KCl, 10 mm Tris, pH 7.5, at room temperature. Next, the samples containing DNA or protein·DNA complexes were incubated on the glutaraldehyde-coated mica for 15 min to allow for attachment, gently washed with deionized water, and imaged. The AFM imaging was done using a Molecular Imaging 5500 AFM (Molecular Imaging, Agilent Technologies) with AC mode. Image acquisitions were typically done with a resolution of 2048 × 2048 pixels at 1 line/s scan speed. The images were processed with Gwyddion software.
RESULTS
Ler Binds Extended DNA and Increases DNA Bending Rigidity
It has been suggested that Ler antagonizes H-NS-mediated gene silencing based on its DNA binding activity (20, 21, 26). Previously, DNA binding by Ler has been studied using traditional biochemical methods such as the electrophoretic mobility shift assay (EMSA) and DNase footprinting, and more recently by EM and AFM (20, 29). The dissociation constant (Kd) of Ler and DNA has been reported to be in the range of 200 nm based on binding to proU and LEE5 operons, which is lower than the Kd of H-NS binding to the same operons (20). In addition, Ler was able to induce DNA wrapping into toroid-like structures at Ler to DNA-binding site ratios lower than 1:1 (20, 29).
As the nucleoid is attached to the cell wall (30), one can expect tension of a few piconewtons on DNA in vivo due to DNA packaging by nucleoid-associated proteins. This assumes that the interaction energy between the DNA and associated proteins is in the range of kBT (the typical energy level associated with reversible binding and unbinding driven by thermal fluctuations),and that the interaction distance is on the scale of nanometers. In addition, forces up to 25 pN can be exerted on DNA by RNA polymerase (31). In this study, we stretched a single 48,502-bp-long molecule of λ-DNA in the focal plane (∼16 μm) by exerting a few piconewtons of force using magnetic tweezers. We examined the binding properties of Ler by its effect on the force response of the DNA. Because of the possibility that Ler's binding mechanism is magnesium-dependent, as is often observed for H-NS and H-NS-like proteins (12, 14, 15), we first examined the DNA binding properties of Ler in the absence of magnesium to isolate the potential effect of magnesium. The effects of magnesium will be investigated below. We used a long DNA tether of ∼16 μm, which assumes a random coiled conformation at low force, to make the DNA distortions generated by protein binding, such as stiffening, more distinguishable.
We first examined the DNA-binding mode of Ler using a force-scanning procedure, as detailed under “Experimental Procedures.” At a Ler concentration of 6 nm, the force-decrease curve resembles that of naked DNA, but the force-increase curve is below the naked DNA curve (Fig. 1A, magenta data). The hysteresis indicated by the nonoverlapping curves suggests that the binding does not reach a steady state. In addition, the fact that the force-increase curve falls below the naked DNA curve indicates DNA folding when DNA is held at low forces. The Ler-bound DNA is still ∼1 μm shorter than the naked DNA when the force is increased back to ∼10 pN. A higher force (∼15 pN) must be applied to nearly recover the original DNA extension before the experiment is repeated with a 100 nm Ler concentration. Because the reaction does not reach a steady state over the experimental time scale during this force-scanning procedure, the level of hysteresis between the force-decrease and force-increase curves varies from one experiment to another in the same solution conditions under the same force-scanning procedure.
FIGURE 1.

Single molecule stretching and imaging experiments on Ler·DNA complexes. A, force-extension curves generated from single DNA stretching experiments on Ler·DNA complexes at various protein concentrations using the force-scanning procedure detailed under “Experimental Procedures.” Reduction in DNA extension accompanied with hysteresis between the force-decrease (FD) and force-increase (FI) curve was observed at low protein concentration. At higher protein concentrations, Ler stiffened the DNA, indicated by higher extension compared with naked DNA. B, force-extension curves obtained by the force-jumping procedure detailed under “Experimental Procedures,” which prevents DNA folding, show increased DNA extension that commensurate with Ler concentration. The error bars represent the standard deviation obtained from three successive force-jumping data from the same DNA molecule. C–F, representative AFM images of Ler·DNA complexes at various protein concentrations in 50 mm KCl: naked DNA (C), DNA complexed with 30 nm Ler (D), 300 nm Ler (E), and 3 μm Ler (F). Ler binding on DNA results in more compact nucleoprotein structures. At 3 μm Ler, co-existing extended nucleoprotein complexes were also observed. G, line profiles of naked DNA (magenta), DNA complexed with 30 nm Ler (green), DNA complexed with 300 nm Ler (blue), and DNA complexed with 3 μm Ler (orange) as indicated by the lines in C–F. All the AFM images are 0.7 × 0.7 μm in size.
At a Ler concentration of 100 nm, the force-decrease curve falls above the naked DNA curve, indicating DNA stiffening (Fig. 1A, green data) (32). At this concentration, hysteresis is still observed, and the force-increase curve falls below the naked DNA curve, indicating DNA folding. Force scanning experiments at 300 and 600 nm Ler showed DNA stiffening but significantly reduced amount of hysteresis (Fig. 1A, blue and orange data, respectively). In addition, at ∼10 pN, the measured extension of Ler-bound DNA is slightly (∼500 nm, 3% of DNA contour length) below that of naked DNA. This may be caused by a small fraction of Ler·DNA complexes that remain in the folded conformation. The general trend of Ler concentration-dependent DNA force-extension curves is reproducible in multiple independent experiments, although the level of hysteresis may vary from one experiment to another (data not shown).
Our experiments reveal that Ler can bind DNA and cause DNA stiffening. Interestingly, DNA stiffening has been observed in general for H-NS and H-NS-like proteins, often in association with the formation of nucleoprotein filaments through cooperative polymerization (12, 14, 15, 18). This is posited to be a possible mechanism of gene silencing. To further determine the characteristics of the observed DNA stiffening, we performed a force-jumping procedure to measure the force-extension curve of DNA while minimizing interference from DNA folding during the measurement, as detailed under “Experimental Procedures.” This procedure revealed that Ler binding to DNA does not necessarily introduce distortion, such as bending or wrapping, of the DNA, which was not reported previously (Fig. 1B). Under such conditions, Ler stiffens DNA to a degree commensurate with the concentration of Ler until saturation at about 300 nm (Fig. 1B).
To relate our single-molecule stretching experiments with the protein·DNA complex conformations formed by Ler, we performed AFM imaging experiments by incubating 0.2 ng/μl ΦX174 DNA (∼300 nm DNA base pairs) in 50 mm KCl at various Ler concentrations to give appropriate density on mica surface for imaging. The DNA exhibited random-coiled conformations in the absence of protein (Fig. 1C). This is consistent with predictions for DNA lengths much longer than the DNA bending persistence length of ∼50 nm, in the absence of force (33, 34).
Unlike our previous single-molecule stretching experiments, the DNA was not under force during the imaging experiments, and therefore the protein-DNA interaction may be different. As 30 nm Ler was introduced (corresponding to one protein monomer per 10 bp), the DNA folds into more compact structures (Fig. 1D). As we increased the Ler concentration to 300 nm (corresponding to 1 protein monomer for every base pair), the DNA becomes further compacted (Fig. 1E). Further increasing the Ler concentration to 3 μm results in a mixture of compact DNA structures and extended protein·DNA complexes (Fig. 1F). These extended DNA complexes are consistent with Ler-coated DNA, because their width and height are significantly greater than those of naked DNA (Fig. 1G). These observations of DNA compaction at a lower concentration and extended DNA complexes at a higher concentration are consistent with our single-molecule stretching experiments, where DNA folding occurs at lower Ler concentrations, and DNA stiffening starts to dominate at higher Ler concentrations and correspondingly higher protein to DNA stoichiometry.
Ler Binds Extended DNA through a Noncooperative Binding Process
We have shown that when DNA is extended by force, Ler can bind the DNA without the need to fold it. This is different from the previously reported mode of Ler·DNA binding in which Ler wraps the DNA (20, 29). To better understand the properties of this new mode of Ler binding to extended DNA, we fit the force-extension data of Ler·DNA complexes at various concentrations measured using the force-jumping procedure, which prevents DNA wrapping or folding, using the Marko-Siggia formula that describes DNA force-extension curves (33, 34) as shown in Equation 2,
 |
where z is the measured DNA extension; L is the contour length of the DNA used; kB is the Boltzmann constant; T is the temperature, and F is the stretching force. A is the persistence length of the DNA that describes the bending stiffness of DNA. For naked DNA, the measured bending stiffness is in the range of 44–53 nm (34, 35), which weakly depends on the solution conditions.
Two-parameter fitting gives us the apparent persistence length and contour length of the Ler nucleoprotein complex (Fig. 2A), where the error bars represent the mean ± S.E. obtained from at least four independent experiments using different DNA. The apparent persistence length of Ler·DNA complexes is 120 ± 9 nm at the highest Ler concentration tested under these buffer conditions. The fully coated Ler·DNA complex has an apparent contour length very close to that of naked DNA (a <500 nm reduction corresponding to a <3% relative contour length reduction). As either DNA folding/wrapping or disruptions to the DNA duplex structure should change the DNA contour length, our results indicate that under the force-jumping protocol, DNA in the Ler nucleoprotein complex assumes a nearly extended duplex conformation.
FIGURE 2.

Single molecule characterization of Ler and H-NS binding to DNA. A, Ler·DNA complex's bending persistence length and contour length were quantified using the data generated from force-jumping procedure by fitting with the Marko-Siggia formula in panel. B, average occupancy from multiple independent experiments at each concentration was fitted with the Hill equation. The values of the Hill coefficient and dissociation constant of Ler binding to DNA were obtained from the average values of each individual experiment. C, H-NS·DNA complex's bending persistence length and contour length in stiffening binding mode were quantified using the same method as A. D, values of the Hill coefficient and dissociation constant of H-NS binding to DNA were quantified using the same method as in B. The error bars for each panel represent the mean ± S.E., and the error bars in B and D were calculated based on the error propagation from the standard error of persistence length measurements in A and C, respectively, at each protein's concentration obtained from multiple independent experiments.
Because the force-extension curve obtained by force-jumping experiments is largely unaffected by DNA folding, the nucleoprotein complex persistence length allows us to quantify the occupation fraction of the protein on the DNA by Equation 3,
 |
as derived previously (14, 16), where Aapparent is the measured persistence length; Asaturated is the persistence length of the fully coated DNA, and Anaked is the persistence length of the naked DNA.
To obtain the cooperativity of protein binding to DNA, we can either fit the occupancy with the McGhee-von Hippel equation or with the Hill equation. Because of the lack of information about the size of the Ler-binding site, we fit our data for Ler occupancy of DNA over various Ler concentrations with the Hill equation as shown in Equation 4 (36),
 |
The Hill equation represents the cooperativity and the dissociation constant of Ler binding to the extended DNA, where α is the occupation fraction; Kd is the apparent dissociation constant; [C] is the protein concentration, and n is the Hill coefficient, which describes the cooperativity.
We plotted the average occupancy using force-jumping data from multiple independent experiments (Fig. 2B). From these data, we obtain an average Hill coefficient value of 1.08 ± 0.19 and a Kd value of 23.4 ± 5.4 nm for Ler (Fig. 2B). A Hill coefficient value of 1 signifies noncooperative or independent protein binding process, whereas a value >1 indicates a cooperative binding process.
The average value of 1.08 that we obtain from our experiments indicates that Ler binding to extended DNA is essentially noncooperative. We emphasize that the values of Kd and the Hill coefficient measured in our experiments represent Ler binding to extended DNA, which may be different from ensemble experiments where DNA molecules are not subjected to force and are therefore able to interact with each other. It has been reported that Ler could form higher order oligomers in solution at sufficiently high concentrations (20). Our results suggest that on extended DNA, regardless of its oligomer state in solution, the binding units of Ler bind DNA noncooperatively. This is also consistent with previous AFM imaging experiments that have reported random binding of Ler oligomers along the DNA, although in those experiments DNA was not subjected to force (20, 29).
To better understand the properties of Ler binding to extended DNA, we compared them to the H-NS·DNA binding properties using a similar force-jumping procedure and data analysis. We derived the persistence length and contour length of H-NS nucleoprotein filament formed in various H-NS concentrations (Fig. 2C). We also derived the average occupancy fraction of H-NS on DNA as a function of H-NS concentration from these data (Fig. 2D). At binding saturation, the persistence length of the H-NS nucleoprotein filament is 227 ± 26 nm. From these measurements, we obtained Hill coefficient values for H-NS of 1.73 ± 0.12 and a Kd value of 46.3 ± 5.6 nm. The positive cooperativity of H-NS binding to DNA is consistent with the segregated islands of H-NS nucleoprotein filaments reported in our previous studies (12).
Comparing the properties we obtained for Ler and H-NS, we find the following: 1) the persistence length of the Ler nucleoprotein complexes on extended DNA is about half that of the H-NS nucleoprotein complexes; 2) Ler binds extended DNA with higher affinity than H-NS as indicated by the 2-fold lower dissociation constant; and 3) Ler has a markedly lower cooperativity in DNA binding than H-NS. The last point is of particular interest, because low cooperativity may indicate that Ler does not bind DNA through a cooperative polymerization process, in contrast to H-NS and other H-NS family proteins (12, 14–16).
Ler Responses to Environmental Factors
Virulence gene expression in pathogens is often regulated by the surrounding environment. Ler is known for its anti-silencing of the LEE pathogenicity islands, where it can activate genes that are repressed by H-NS. In enterohemorrhagic E. coli and enteropathogenic E. coli, temperature has been found to affect the virulence gene expression in the H-NS/Ler system (26, 37). H-NS nucleoprotein filaments have also been found to respond sensitively to environmental factors such as KCl concentration, pH, magnesium concentration, and temperature (12). To the best of our knowledge, the effects of these factors on Ler·DNA binding have not been investigated. To examine the possibility that responses to these factors are related to Ler's anti-silencing activity, we examined the changes in the Ler-DNA interaction to these environmental factors.
It has been reported that the monovalent salt concentration in bacteria ranges from 50 mm to a few hundred millimolars under physiological conditions (38). Our force-scanning data show that Ler binding to DNA leads to longer extension of DNA (i.e. DNA stiffening), with some small hysteresis between force-decrease and force-increase curves in the 50–200 mm range (Fig. 3A). The DNA extension is significantly reduced at higher KCl concentrations of 400 mm, indicating reduced Ler DNA-binding affinity. This observation can be explained by the electrostatic nature of Ler·DNA interactions; higher KCl concentration gives rise to electrostatic screening that may reduce Ler's DNA-binding affinity.
FIGURE 3.

Ler·DNA complex response to environmental factors. A, Ler·DNA complex response to KCl concentration. Ler binding to DNA was reduced as KCl concentration was increased, as apparent in the reduction of nucleoprotein stiffness at 400 mm KCl. The data points at 50 mm KCl (magenta) overlap the data points at 100 mm KCl (green) and 200 mm KCl (blue) and are not visible in the panel. B, Ler·DNA complexes were relatively insensitive to variations in magnesium concentration. The force-decrease curves were largely unaffected as the magnesium concentration is varied from 2 to 10 mm MgCl2, whereas an increased hysteresis was observed between the force-decrease and force-increase curves. C, Ler·DNA complexes were relatively insensitive to pH. The force-decrease curves were largely unaffected as pH is varied from 8.8 to 6.8, although an increased hysteresis was observed between the force-decrease and force-increase curves. D, Ler·DNA complex binding was reduced when the temperature was increased from 23 to 37 °C, indicated by the decrease in DNA extension and increased level of hysteresis at higher temperatures.
Another important environmental factor is magnesium, which has been shown to alter the binding modes of H-NS and StpA (12, 15) and to increase the DNA folding of MvaT (14). The physiological concentration of magnesium has been reported to be in the range of a few millimolars (39, 40). We examined the effect of MgCl2 concentrations up to 10 mm (Fig. 3B). The curves obtained in the force-decrease scan are significantly longer than the naked DNA extension, indicating DNA stiffening. In addition, these force-decrease curves nearly overlap each other, indicating that Ler's DNA-binding affinity is insensitive over the MgCl2 concentration range. Increasing amount of hysteresis between the force-decrease and force-increase curves is observed as MgCl2 concentration is increased, suggesting that MgCl2 may have a role in promoting Ler-dependent DNA folding at low forces (Fig. 3B). The folding may not be due to a decrease in Ler's DNA-binding affinity, because the force-decrease curves are largely unaffected by changes in the MgCl2 concentration.
Similar experiments are performed to probe the effects of pH on Ler binding (Fig. 3C). Decreasing pH increases the apparent hysteresis, which indicates an increased level of DNA folding. Similar to the effects of increasing MgCl2 concentration, the force-decrease curve is largely unchanged by alterations to the pH value. Therefore the increased DNA folding cannot be explained by a reduction in Ler binding.
The effect of temperature is important because temperature is often reported to affect the virulence gene expression (26, 37). We observed a significant decrease in DNA stiffening at temperatures greater than 30 °C (Fig. 3D). These results indicate reduced Ler·DNA binding at higher temperatures. At 37 °C, besides significantly reduced DNA stiffening, a large hysteresis is observed in the force-increase curve. The force-extension curves obtained at 37 °C are similar to those recorded at low Ler concentrations and those recorded at high KCl concentration, consistent with decreased Ler's DNA-binding affinity at higher temperatures (Figs. 1A and 3, A and D).
Among the four important environmental factors tested, only KCl concentration and temperature were able to regulate Ler binding to DNA over physiological ranges. Their physiological implications will be discussed under “Discussion.”
Ler Replaces Prebound H-NS on DNA
After elucidating the binding properties of Ler on extended DNA, we then focused on understanding its function as an anti-silencer of H-NS-silenced LEE genes. Regulation of these operons is hypothesized to be controlled by competitive binding by H-NS and Ler on DNA (20, 21). Previously, this hypothesis has been examined by the EMSA experiments, which showed that Ler could change the migration speed of DNA fragments prebound with H-NS in 5 mm NaCl, 50 mm KCl, pH 7.4, at room temperature (20). This suggests that Ler can compete H-NS off the DNA. Here, we provide more direct evidence on the impacts of Ler on preformed H-NS nucleoprotein complexes at the single-molecule level.
In the experiments, DNA was pre-coated with H-NS by incubating the DNA tether in 600 nm H-NS at 50 mm KCl at 24 °C for 30 min. Under these conditions, a fully coated rigid H-NS nucleoprotein filament forms (12). The resulting force-extension curve obtained by the force-scanning procedure exhibits negligible hysteresis (Fig. 4A, magenta data). It lies above the naked DNA curve, which is expected from the increased bending rigidity. The bending persistence length of the H-NS nucleoprotein filament formed under this condition is determined by the force-jumping procedure to be 227 ± 26 nm (Fig. 2C). Based on this measured persistence length, we plotted the predicted force-extension curve of fully coated H-NS·DNA complexes as reference, where the dark gray region indicates 1 S.D. in persistence length around the mean (Fig. 4A). The experimental data lies close to the predicted values, indicating that the DNA is fully coated with H-NS.
FIGURE 4.
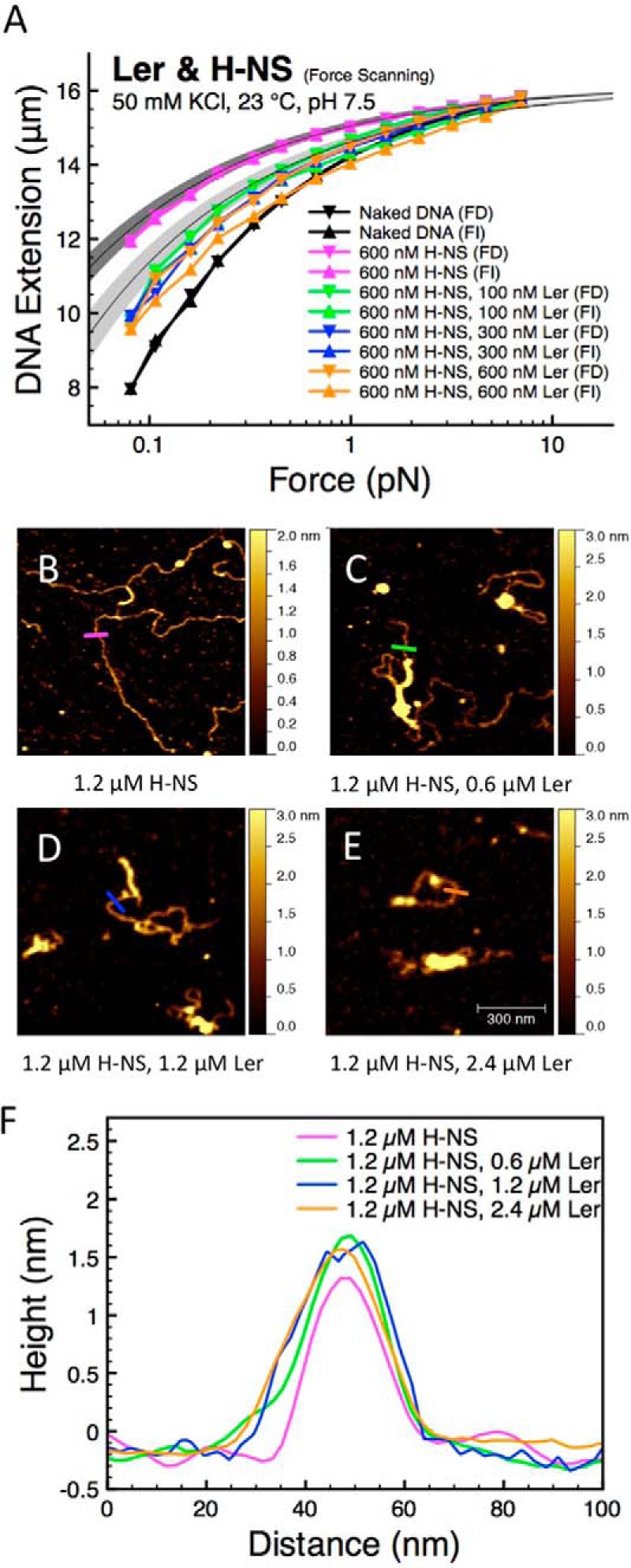
Ler replaces H-NS in stiffening mode to form a stable Ler·DNA complex. A, at 50 mm KCl, H-NS forms rigid nucleoprotein filaments, which can be replaced by Ler when the concentration of Ler was progressively increased in the presence of 600 nm H-NS. This is indicated by the decrease in nucleoprotein stiffness to the level of Ler nucleoprotein complex (light gray region) when Ler concentration in solution exceeds 100 nm. B, DNA complexed with 1.2 μm H-NS resulted in rigid nucleoprotein filaments. C–E, DNA was first incubated in 1.2 μm H-NS for 30 min before various concentrations of Ler were introduced in the solution for another 20 min (0.6 μm Ler for C, 1.2 μm Ler for D, and 2.4 μm Ler for E). Some parts of the H-NS filaments were compacted by Ler proteins, indicating that Ler can replace H-NS filaments. F, line profiles of DNA complexed with 1.2 μm H-NS (magenta), H-NS pre-coated DNA with 0.6 μm Ler (green), H-NS pre-coated DNA with 1.2 μm Ler (blue), and H-NS pre-coated DNA with 2.4 μm Ler (orange) as indicated by the lines in B–E. All the AFM images are 0.7 × 0.7 μm in size.
Next, on the pre-coated H-NS nucleoprotein filament and in the same buffer conditions, Ler concentration was progressively increased from 100 to 600 nm, maintaining a concentration of 600 nm H-NS. A significant downward shift of the force-extension curve was observed when 100 nm Ler was introduced, with a somewhat increased level of hysteresis (Fig. 4A, green data). No further shift was observed when the Ler concentration is increased to 300 and 600 nm, indicating saturation (Fig. 4A, blue and orange data, respectively). The force-extension curves obtained in the presence of 100, 300, and 600 nm Ler lie just slightly below the curve predicted based on the persistence length of Ler·DNA complex determined earlier (Fig. 2A). The light gray region indicates 1 S.D. in persistence length around the mean (Fig. 4A). These results indicate that with a 600 nm H-NS concentration, the H-NS molecules bound on DNA are nearly completely replaced by Ler in our experimental time scale when the concentration of Ler exceeds 100 nm.
The conformations of DNA formed when Ler was introduced into pre-coated H-NS nucleoprotein filaments were visualized by AFM. In the single-DNA stretching experiments, the ratio between the numbers of proteins in solution to the available binding sites on DNA is vast. In such experiments, we only need to control the concentration of the protein in solution. However, in AFM imaging experiments, as in any bulk experiments, there are numerous DNA molecules providing numerous binding sites to proteins. To ensure saturated binding, higher concentrations of H-NS and Ler compared with the single-DNA stretching experiments are used. First we incubated the DNA with 1.2 μm H-NS for 30 min before adding various concentrations of Ler. At 50 mm KCl, H-NS is known to form rigid nucleoprotein filaments (11, 12). The majority of H-NS·DNA complexes in 50 mm KCl at 1.2 μm H-NS are rigid H-NS nucleoprotein filaments (Fig. 4B). After such H-NS filaments were formed, we introduced 0.6 μm Ler, resulting in a mixture of 0.6 μm Ler and 1.2 μm H-NS. After 20 min of incubation, AFM imaging revealed more compacted DNA·protein complexes than before Ler was introduced (Fig. 4C). Repeating the same procedure with 1.2 and 2.4 μm Ler while maintaining the same concentration of 1.2 μm H-NS in solution revealed similar Ler-dependent DNA condensations (Fig. 4, D and E). Overall, these results are consistent with the single-DNA stretching experiments in which Ler was found to be able to replace H-NS on DNA. Observation of DNA conformations in the presence of Ler and H-NS revealed that, besides the condensed DNA regions, there are also extended filamentous structures (Fig. 4, C–E). These filaments could either be the remaining H-NS·DNA filaments or the extended Ler·DNA complexes, which are indistinguishable by AFM as the line scanning profiles of H-NS·DNA filaments and Ler·DNA filaments are similar (Fig. 4F). However, because we have shown in Fig. 4A that Ler can efficiently replace H-NS on DNA at concentrations comparable with those in these experiments, we reason that in these experiments where Ler has equal or higher concentrations than H-NS, the majority of the DNA will be associated with Ler (Fig. 4, D and E).
Effect of KCl and MgCl2 Concentration in H-NS and Ler Competition
Transcriptional gene regulation by H-NS and Ler may correspond directly to their DNA binding activity, which is sensitive to changes in environmental factors such as KCl and MgCl2 concentration. Hence, we would like to examine the competition of H-NS and Ler under different buffer conditions. We thus varied the KCl concentration from 50 to 200 mm KCl, at which concentration H-NS does not give rise to DNA stiffening (12), possibly indicating a low binding affinity of H-NS at 200 mm KCl or simply an inability to form a rigid H-NS nucleoprotein filament under these circumstances. Consistent with previous data, we found that the introduction of 600 nm H-NS in 200 mm KCl only generates a slight upward shift in the force-extension curve of DNA (Fig. 5A, magenta data), as expected for the reduced DNA stiffening effect in higher KCl concentrations (11, 12).
FIGURE 5.
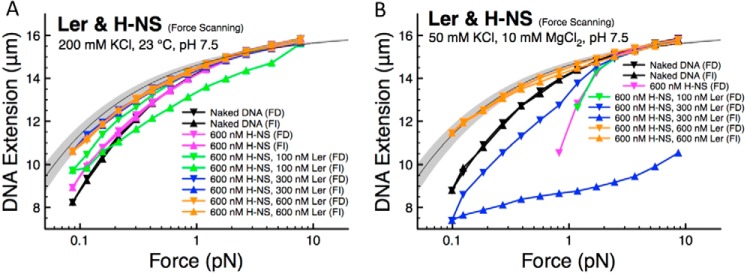
Effects of variations in KCl and MgCl2 concentrations on Ler and H-NS competition for DNA binding. A, at 200 mm KCl and 600 nm H-NS, the measured extension was close to that of naked DNA. Increasing Ler concentrations while maintaining 600 nm H-NS gave progressive increases in DNA extension. The resulting force-extension curve approaches the predicted curve for saturated Ler-bound DNA when Ler concentration exceeds 300 nm. B, at 50 mm KCl, 10 mm MgCl2, where H-NS predominantly induces DNA bridging with negligible DNA stiffening, Ler replaced H-NS when the concentration of Ler was progressively increased while maintaining 600 nm H-NS. This is indicated by reduced DNA folding and an increase in the extension of protein·DNA complexes. At 600 nm Ler, the curves are close to the predicted curve for Ler nucleoprotein complex (light gray region).
When 100 nm of Ler was introduced while maintaining the H-NS concentration of 600 nm, a further small upward shift was observed, indicating Ler binding. However, the experimental data still lie below the predicted curve for fully coated Ler·DNA complexes (Fig. 5A, light gray region), suggesting that, under these conditions, there is a mixture of H-NS and Ler on the DNA. We also observe a hysteresis between the force-decrease and force-increase curves, a signature of unsaturated Ler binding that is generally observed in the presence of Ler at lower concentrations or high KCl concentrations or high temperature (Figs. 1A and 3, A and D). When the Ler concentration was further increased up to 600 nm, increased DNA stiffening and less hysteresis were observed. The curves obtained at 300 and 600 nm Ler in the presence of 600 nm H-NS were close to the predicted curves for fully coated Ler·DNA complexes (Fig. 5A, light gray region), suggesting that at these conditions the majority of H-NS has been replaced with Ler.
The conformation of the nucleoprotein complexes formed by H-NS is sensitive to the magnesium concentration in the range of 0–10 mm (12). At low magnesium concentration (< 2 mm) and 50 mm KCl, the nucleoprotein complex exhibits a rigid extended filamentous conformation at 50 mm KCl (DNA stiffening). At high magnesium concentrations (> 5 mm) and 50 mm KCl, the level of DNA stiffening is reduced, and the resulting nucleoprotein complexes mainly adopt hairpin-like conformations (DNA bridging).
To understand Ler and H-NS competition for DNA binding when H-NS predominantly binds DNA in the bridging mode (12), we examined the Ler/H-NS competition in the presence of 10 mm MgCl2 and 50 mm KCl (Fig. 5B). The hysteresis for naked DNA is negligible under these conditions, as expected because no DNA folding should occur in the absence of protein. At 600 nm H-NS in the absence of Ler, the force-decrease curve lies below the naked DNA curve at forces below 2 pN, indicating DNA folding (Fig. 5B, magenta data). The data below 0.8 pN are not recorded due to quick DNA folding at these low forces (and therefore the force-increase scan cannot be recorded either), as our instrument cannot measure DNA extension shorter than 2 μm. A similar phenomenon was observed when 100 nm Ler was introduced in the presence of 600 nm H-NS, indicating that H-NS still dominates the DNA binding. As the Ler concentration was increased to 300 nm, both the force-decrease and the force-increase scans were below that of naked DNA, but the level of DNA folding was significantly reduced. Therefore, the data in the force-increase scan can be recorded without the DNA extension falling below the shortest measurable extension. A further increase of Ler concentration to 600 nm led to force-extension curves lying above the naked DNA curve and exhibited negligible hysteresis (Fig. 5B, orange data). These curves were in the range of the force-extension curves for pure Ler·DNA nucleoprotein complexes (Fig. 5B, light gray region), as predicted based on the DNA bending persistence length (Fig. 2A). This suggests that Ler has nearly completely replaced prebound H-NS on DNA.
The competitions observed between H-NS and Ler in Fig. 5 are reproduced in multiple independent experiments. In summary, taking results from this and the previous sections, we conclude that Ler can replace prebound H-NS on DNA over wide concentration ranges of KCl (50–200 mm) and MgCl2 (0–10 mm).
DISCUSSION
Binding Modes of Ler to DNA
Ler-mediated anti-silencing of H-NS-controlled genes silenced by H-NS has been proposed to occur through competitive binding of DNA (20, 21). A thorough understanding of the Ler-DNA interaction is critical to evaluate this hypothesis. Despite a wealth of important knowledge from previous biochemical and imaging studies, the dependences of the Ler-DNA interaction on environmental factors, such as KCl concentration, temperature, pH, magnesium concentration, and force have been largely unexplored. Our work demonstrates that Ler binding to DNA is sensitive to variations in temperature and KCl concentration over physiological ranges. These results were obtained using long DNA molecules with nonspecific sequences. We did not stretch specific promoter sequences because short DNA fragments are intrinsically rigid, meaning that there would be a negligible amount of DNA stiffening to measure. However, we expect that similar DNA-binding modes and environment-induced effects might occur in specific promoter sequences at lower Ler concentration because of higher affinity for these specific sites.
Our results obtained from the force-scanning procedure demonstrate the existence of DNA folding at low Ler concentrations under low force regime and DNA stiffening at high Ler concentrations over the range of forces studied (Fig. 1, A and B). Based on previous imaging studies, which reported particle-like Ler nucleoprotein complexes randomly distributed along DNA (20, 29), our results suggest that DNA folding is caused by the formation of Ler nucleoprotein complexes, which wraps a certain length of DNA. At high concentrations of Ler and a high protein to DNA stoichiometry, we observed DNA stiffening, which can be explained by the unwrapping of DNA from the nucleoprotein unit to accommodate more Ler oligomers. This is consistent with the DNA stiffening we observed, due to steric interactions between the closely packed Ler oligomers. This result implies that the association of unwrapped Ler with DNA is an intermediate state of the Ler·DNA complex preceding DNA wrapping at low Ler·DNA stoichiometry and low tension. In vitro, this mode of DNA binding may be stabilized at DNA regions that are subjected to force. Such a scenario is observed in DNA wrapping by the E. coli integration host factor and heat-unstable proteins on dsDNA and single-stranded DNA-binding proteins on single-stranded DNA (41–43).
The previously reported random distribution of the Ler nucleoprotein complex along DNA suggested that in the wrapping mode Ler binds DNA in a noncooperative process. Our single molecule studies using the force-jumping procedure revealed that Ler binds extended DNA with low cooperativity in the unwrapped binding mode. Together, this information suggests that Ler binds DNA through a largely noncooperative process in both the wrapped and unwrapped modes. The model of Ler binding to DNA is schematized in Fig. 6.
FIGURE 6.
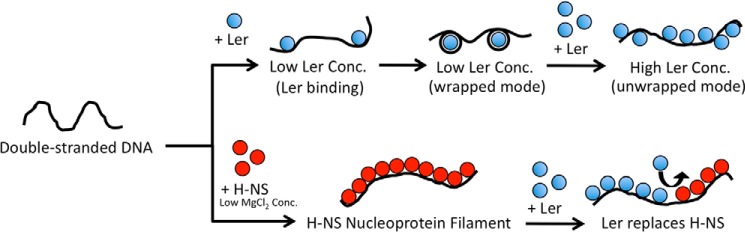
Models of Ler binding to DNA and its role in gene regulation. At nonsaturated condition or low tension, Ler can bind DNA in the wrapped mode. At a higher concentration (Conc.) of Ler or high tension, Ler binds DNA in an unwrapped mode in a largely noncooperative process to form Ler·DNA nucleoprotein complex array. H-NS, however, forms rigid nucleoprotein filament at low MgCl2 concentrations (<5 mm) through a cooperative binding process. The formation of nucleoprotein filament may silence genes by blocking RNA polymerases from accessing the promoters or blocking translocation of RNA polymerases. Ler can replace H-NS nucleoprotein filaments in some environmental conditions (such as low KCl concentration). This replacement of H-NS by Ler, together with Ler's lack of cooperativity, may serve as the basis to understand Ler's anti-silencing activity.
Our results indicating low cooperativity of Ler binding to extended DNA contrast with H-NS and other H-NS-like proteins, all of which bind DNA cooperatively via a polymerization process that results in the formation of rigid nucleoprotein filaments. Although Ler exhibits 24% sequence identity to Salmonella enterica H-NS (19), its N terminus is markedly different from the N terminus of H-NS. Because the N-terminal domain of H-NS is responsible for its oligomerization activity and thereby the cooperativity of its binding to DNA, we reason that this difference is the basis for Ler's lack of cooperativity.
Implications of Ler Responses to Environmental Factors
It has been shown that Ler's anti-silencing activity is regulated by environmental factors such as temperature (26, 37). Previous studies did not reveal whether this occurs through regulation of Ler·DNA binding or through regulation of H-NS·DNA binding. Our previous results demonstrated that E. coli and Salmonella enterica H-NS·DNA binding is sensitive to many environmental factors such as KCl concentration, temperature, pH, and magnesium concentration (12). In this work, we show that Ler·DNA binding is largely insensitive to changes of magnesium concentration and pH, and it exhibits moderate sensitivity to temperature and KCl concentration. The insensitivity of Ler·DNA binding to KCl concentration compared with H-NS·DNA binding may suggest that Ler has less electrostatic interactions with DNA, making the KCl dependence of its observed association constant for DNA binding less severe than that for H-NS (44). These results, in conjunction with our previous data for H-NS, provide the basis for understanding the competition for DNA binding between Ler and H-NS under various solution conditions. As this competition is directly related to the transcriptional regulatory activities of these two proteins, this is vital to understanding how they function in vivo.
Implications on Ler-mediated Anti-silencing Activity
Although the anti-silencing of Ler at genes repressed by H-NS is well characterized (2–4), the mechanism of this process is still unclear. In this work, we shed light on the issue by resolving the binding modes of Ler to DNA. Ler can bind DNA in a wrapped or unwrapped mode, depending on protein concentration and tension, possibly as well as other as yet unidentified factors. We provide further evidence that Ler can compete H-NS off the DNA with a 50 mm KCl condition, which supports the hypothesis that Ler acts as anti-silencer by directly competing with H-NS for DNA binding. We propose that Ler antagonizes H-NS filaments for DNA binding based on the following mechanism. Thermal fluctuations at the ends of H-NS filaments cause H-NS unbinding and rebinding. In the presence of Ler, the transiently vacated DNA-binding sites are occupied by Ler. This prevents rebinding of H-NS and results in a progressive replacement of H-NS along the DNA.
An important question that has not yet been answered is how the replacement of H-NS by Ler results in transcriptional activation of genes previously silenced by H-NS. In our previous work, we posited two possible mechanisms for H-NS-mediated gene silencing as follows: the restriction of promoter accessibility to RNA polymerase or the blocking of the translocation of RNA polymerase by the formation of H-NS filaments downstream of respective promoters (17). Although RNA polymerase may have a higher affinity for promoter sequence compared with H-NS, the formation of continuous filaments that results from H-NS polymerization can effectively block access to DNA, as we expect a slow exchange rate between H-NS in the middle of the filament with RNA polymerase in solution, because this requires the multiple H-NS molecules to vacate the DNA simultaneously. The antagonistic function of Ler follows naturally from this model and our new data. We found that Ler binds DNA with a noncooperative manner, which may permit easier dissociation from the middle of the nucleoprotein complex. As a result, although Ler has a higher binding affinity than H-NS, Ler bound on the DNA may have faster exchange rate with RNA polymerase. This would lead to increased transcription. We plan to investigate this possibility with single RNA polymerase activity assays.
In this study, we also examine the susceptibility of Ler binding to DNA to various environmental factors. Ler binding to DNA is insensitive to changes in pH and magnesium concentration, whereas higher temperature and higher KCl concentration can reduce the DNA binding. Furthermore, we examine the competitive binding of H-NS and Ler on the DNA under different buffer conditions. We find that Ler binding predominates over wide ranges of KCl concentration (50–200 mm) and MgCl2 concentration (0–10 mm). Given that Ler likely antagonizes H-NS gene-silencing function based on competitive DNA binding, our results suggest that Ler anti-silencing activities are robust against changes in these environmental factors.
Finally, we want to emphasize that there exist many anti-silencing proteins for H-NS-silenced genes. It is not clear how diverse these proteins are with respect to their anti-silencing mechanisms (45). Generally speaking, anti-silencing can be achieved by competing with H-NS for DNA binding, competing with DNA for H-NS binding, or interfering with the H-NS N-terminal oligomerization domain. The Ler protein studied in this work is a DNA-binding protein, which most likely belongs to the first category. However, we do not exclude the possibility that Ler may also antagonize H-NS-mediated gene silencing by one or more additional mechanisms.
Acknowledgments
We thank Linda Kenney (University of Illinois at Chicago and the Mechanobiology Institute Singapore) for stimulating discussions. We also thank Dr. Adam Yuan and the protein expression core facility of the Mechanobiology Institute for protein purification.
This work was supported by the Ministry of Education of Singapore, National Research Foundation of Singapore Grant MOE 2013-T2-1-154, and by the Mechanobiology Institute Singapore (to J. Y.).
J. L. Mellies, unpublished data.
- H-NS
- histone-like nucleoid-structuring protein
- AFM
- atomic force microscopy
- pN
- piconewton.
REFERENCES
- 1. Lucchini S., Rowley G., Goldberg M. D., Hurd D., Harrison M., Hinton J. C. (2006) H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog. 2, e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dorman C. (2004) H-NS: a universal regulator for a dynamic genome. Nat. Rev. Microbiol. 2, 391–400 [DOI] [PubMed] [Google Scholar]
- 3. Fang F. C., Rimsky S. (2008) New insights into transcriptional regulation by H-NS. Curr. Opin. Microbiol. 11, 113–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ali S. S., Xia B., Liu J., Navarre W. W. (2012) Silencing of foreign DNA in bacteria. Curr. Opin. Microbiol. 15, 175–181 [DOI] [PubMed] [Google Scholar]
- 5. Torres A. G., López-Sánchez G. N., Milflores-Flores L., Patel S. D., Rojas-López M., Martínez de la Peña C. F., Arenas-Hernández M. M., Martínez-Laguna Y. (2007) Ler and H-NS, regulators controlling expression of the long polar fimbriae of Escherichia coli O157:H7. J. Bacteriol. 189, 5916–5928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berdichevsky T., Friedberg D., Nadler C., Rokney A., Oppenheim A., Rosenshine I. (2005) Ler is a negative autoregulator of the LEE1 operon in enteropathogenic Escherichia coli. J. Bacteriol. 187, 349–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smyth C. P., Lundbäck T., Renzoni D., Siligardi G., Beavil R., Layton M., Sidebotham J. M., Hinton J. C., Driscoll P. C., Higgins C. F., Ladbury J. E. (2000) Oligomerization of the chromatin-structuring protein H-NS. Mol. Microbiol. 36, 962–972 [DOI] [PubMed] [Google Scholar]
- 8. Esposito D., Petrovic A., Harris R., Ono S., Eccleston J. F., Mbabaali A., Haq I., Higgins C. F., Hinton J. C., Driscoll P. C., Ladbury J. E. (2002) H-NS oligomerization domain structure reveals the mechanism for high order self-association of the intact protein. J. Mol. Biol. 324, 841–850 [DOI] [PubMed] [Google Scholar]
- 9. Ceschini S., Lupidi G., Coletta M., Pon C. L., Fioretti E., Angeletti M. (2000) Multimeric self-assembly equilibria involving the histone-like protein H-NS. A thermodynamic study. J. Biol. Chem. 275, 729–734 [DOI] [PubMed] [Google Scholar]
- 10. Ueguchi C., Seto C., Suzuki T., Mizuno T. (1997) Clarification of the dimerization domain and its functional significance for the Escherichia coli nucleoid protein H-NS. J. Mol. Biol. 274, 145–151 [DOI] [PubMed] [Google Scholar]
- 11. Amit R., Oppenheim A. B., Stavans J. (2003) Increased bending rigidity of single DNA molecules by H-NS, a temperature and osmolarity sensor. Biophys. J. 84, 2467–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y., Chen H., Kenney L. J., Yan J. (2010) A divalent switch drives H-NS/DNA-binding conformations between stiffening and bridging modes. Genes Dev. 24, 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dame R. T., Wyman C., Goosen N. (2000) H-NS mediated compaction of DNA visualised by atomic force microscopy. Nucleic Acids Res. 28, 3504–3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Winardhi R. S., Fu W., Castang S., Li Y., Dove S. L., Yan J. (2012) Higher order oligomerization is required for H-NS family member MvaT to form gene-silencing nucleoprotein filament. Nucleic Acids Res. 40, 8942–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lim C. J., Whang Y. R., Kenney L. J., Yan J. (2012) Gene silencing H-NS paralogue StpA forms a rigid protein filament along DNA that blocks DNA accessibility. Nucleic Acids Res. 40, 3316–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qu Y., Lim C. J., Whang Y. R., Liu J., Yan J. (2013) Mechanism of DNA organization by Mycobacterium tuberculosis protein Lsr2. Nucleic Acids Res. 41, 5263–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lim C. J., Lee S. Y., Kenney L. J., Yan J. (2012) Nucleoprotein filament formation is the structural basis for bacterial protein H-NS gene silencing. Sci. Rep. 2, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walthers D., Li Y., Liu Y., Anand G., Yan J., Kenney L. J. (2011) Salmonella enterica response regulator SsrB relieves H-NS silencing by displacing H-NS bound in polymerization mode and directly activates transcription. J. Biol. Chem. 286, 1895–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sperandio V., Mellies J. L., Delahay R. M., Frankel G., Crawford J. A., Nguyen W., Kaper J. B. (2000) Activation of enteropathogenic Escherichia coli (EPEC) LEE2 and LEE3 operons by Ler. Mol. Microbiol. 38, 781–793 [DOI] [PubMed] [Google Scholar]
- 20. Mellies J. L., Benison G., McNitt W., Mavor D., Boniface C., Larabee F. J. (2011) Ler of pathogenic Escherichia coli forms toroidal protein-DNA complexes. Microbiology 157, 1123–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bustamante V. H., Santana F. J., Calva E., Puente J. L. (2001) Transcriptional regulation of type III secretion genes in enteropathogenic Escherichia coli: Ler antagonizes H-NS-dependent repression. Mol. Microbiol. 39, 664–678 [DOI] [PubMed] [Google Scholar]
- 22. Dorman C. J., Hinton J. C., Free A. (1999) Domain organization and oligomerization among H-NS-like nucleoid-associated proteins in bacteria. Trends Microbiol. 7, 124–128 [DOI] [PubMed] [Google Scholar]
- 23. Cordeiro T. N., Schmidt H., Madrid C., Juárez A., Bernadó P., Griesinger C., García J., Pons M. (2011) Indirect DNA readout by an H-NS related protein: structure of the DNA complex of the C-terminal domain of Ler. PLoS Pathog. 7, e1002380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elliott S. J., Sperandio V., Girón J. A., Shin S., Mellies J. L., Wainwright L., Hutcheson S. W., McDaniel T. K., Kaper J. B. (2000) The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 68, 6115–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yerushalmi G., Nadler C., Berdichevski T., Rosenshine I. (2008) Mutational analysis of the locus of enterocyte effacement-encoded regulator (Ler) of enteropathogenic Escherichia coli. J. Bacteriol. 190, 7808–7818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Umanski T., Rosenshine I., Friedberg D. (2002) Thermoregulated expression of virulence genes in enteropathogenic Escherichia coli. Microbiology 148, 2735–2744 [DOI] [PubMed] [Google Scholar]
- 27. Wang H., Bash R., Yodh J. G., Hager G. L., Lohr D., Lindsay S. M. (2002) Glutaraldehyde modified mica: a new surface for atomic force microscopy of chromatin. Biophys. J. 83, 3619–3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fu H., Freedman B. S., Lim C. T., Heald R., Yan J. (2011) Atomic force microscope imaging of chromatin assembled in Xenopus laevis egg extract. Chromosoma 120, 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. García J., Cordeiro T. N., Prieto M. J., Pons M. (2012) Oligomerization and DNA binding of Ler, a master regulator of pathogenicity of enterohemorrhagic and enteropathogenic Escherichia coli. Nucleic Acids Res. 40, 10254–10262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Toro E., Shapiro L. (2010) Bacterial chromosome organization and segregation. Cold Spring Harbor Perspect. Biol. 2, a000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang M. D., Schnitzer M. J., Yin H., Landick R., Gelles J., Block S. M. (1998) Force and velocity measured for single molecules of RNA polymerase. Science 282, 902–907 [DOI] [PubMed] [Google Scholar]
- 32. Yan J., Marko J. (2003) Effects of DNA-distorting proteins on DNA elastic response. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 68, 011905 [DOI] [PubMed] [Google Scholar]
- 33. Bustamante C., Marko J. F., Siggia E. D., Smith S. (1994) Entropic elasticity of λ-phage DNA. Science 265, 1599–1600 [DOI] [PubMed] [Google Scholar]
- 34. Marko J., Siggia E. (1995) Stretching DNA. Macromolecules 28, 8759–8770 [Google Scholar]
- 35. Smith S. B., Finzi L., Bustamante C. (1992) Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science 258, 1122–1126 [DOI] [PubMed] [Google Scholar]
- 36. Hill A. V. (1910) The possible effects of the aggregation of the molecules of hæmoglobin on its dissociation curves. J. Physiol. 40, iv–vii [Google Scholar]
- 37. Steinmann R., Dersch P. (2013) Thermosensing to adjust bacterial virulence in a fluctuating environment. Future Microbiol. 8, 85–105 [DOI] [PubMed] [Google Scholar]
- 38. Christian J. H., Waltho J. A. (1961) The sodium and potassium content of non-halophilic bacteria in relation to salt tolerance. J. Gen. Microbiol. 25, 97–102 [DOI] [PubMed] [Google Scholar]
- 39. Lusk J. E., Williams R. J., Kennedy E. P. (1968) Magnesium and the growth of Escherichia coli. J. Biol. Chem. 243, 2618–2624 [PubMed] [Google Scholar]
- 40. Martin-Orozco N., Touret N., Zaharik M. L., Park E., Kopelman R., Miller S., Finlay B. B., Gros P., Grinstein S. (2006) Visualization of vacuolar acidification-induced transcription of genes of pathogens inside macrophages. Mol. Biol. Cell 17, 498–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Holbrook J. A., Tsodikov O. V., Saecker R. M., Record M. T. (2001) Specific and non-specific interactions of integration host factor with DNA: thermodynamic evidence for disruption of multiple IHF surface salt-bridges coupled to DNA binding. J. Mol. Biol. 310, 379–401 [DOI] [PubMed] [Google Scholar]
- 42. Lin J., Chen H., Dröge P., Yan J. (2012) Physical organization of DNA by multiple non-specific DNA-binding modes of integration host factor (IHF). PLoS One 7, e49885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fu H., Le S., Chen H., Muniyappa K., Yan J. (2013) Force and ATP hydrolysis dependent regulation of RecA nucleoprotein filament by single-stranded DNA binding protein. Nucleic Acids Res. 41, 924–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Record M. T., Jr., Lohman M. L., De Haseth P. (1976) Ion effects on ligand-nucleic acid interactions. J. Mol. Biol. 107, 145–158 [DOI] [PubMed] [Google Scholar]
- 45. Stoebel D. M., Free A., Dorman C. J. (2008) Anti-silencing: overcoming H-NS-mediated repression of transcription in Gram-negative enteric bacteria. Microbiology 154, 2533–2545 [DOI] [PubMed] [Google Scholar]