

Background: PP2A is a serine/threonine phosphatase playing a central role in the pathology of the autoimmune disease SLE.
Results: Ikaros binds to an intronic site in PP2A and modulates its expression.
Conclusion: Ikaros represses PP2A expression by recruiting histone deacetylase HDAC1.
Significance: This study proposes a novel pathway for regulation of PP2A, a critical molecule in SLE pathogenesis.
Keywords: Chromatin Modification, Gene Regulation, Protein Phosphatase, T Cell, Transcription Repressor
Abstract
Protein phosphatase 2A (PP2A) is a highly conserved and ubiquitous serine/threonine phosphatase. We have shown previously that PP2A expression is increased in T cells of systemic lupus erythematosus patients and that this increased expression and activity of PP2A plays a central role in the molecular pathogenesis of systemic lupus erythematosus. Although the control of PP2A expression has been the focus of many studies, many aspects of its regulation still remain poorly understood. In this study, we describe a novel mechanism of PP2A regulation. We propose that the transcription factor Ikaros binds to a variant site in the first intron of PP2A and modulates its expression. Exogenous expression of Ikaros leads to reduced levels of PP2Ac message as well as protein. Conversely, siRNA-enabled silencing of Ikaros enhances the expression of PP2A, suggesting that Ikaros acts as a suppressor of PP2A expression. A ChIP analysis further proved that Ikaros is recruited to this site in T cells. We also attempted to delineate the mechanism of Ikaros-mediated PP2Ac gene suppression. We show that Ikaros-mediated suppression of PP2A expression is at least partially dependent on the recruitment of the histone deacetylase HDAC1 to this intronic site. We conclude that the transcription factor Ikaros can regulate the expression of PP2A by binding to a site in the first intron and modulating chromatin modifications at this site via recruitment of HDAC1.
Introduction
Systemic lupus erythematosus (SLE)3 is a multifactorial autoimmune disease that primarily affects women in their reproductive years (1). It affects many different organs, like the skin, joints, brain, heart, and kidney. In addition to genetic, environmental, and hormonal factors, immune system irregularities, especially in T cells, is one of the contributing factors toward the pathology of this disease (2). In T cells isolated from SLE patients, aberrant signaling leads to atypical characteristics, such as enhanced tyrosine phosphorylation as well as increased calcium influx (3). One of the signaling defects is the reduced expression of the T cell receptor (TCR)-associated CD3ζ chain. In SLE T cells, the FcRγ structurally and functionally replaces the CD3ζ chain (4). FcRγ couples to spleen tyrosine kinase (syk) instead of ζ-associated protein 70 (ZAP70) kinase, thus resulting in defective signaling (5). Another hallmark of SLE T cells is a reduction in their ability to produce IL-2, an essential cytokine for T cell proliferation and effector functions (6).
One of the key components contributing to the signaling defects in SLE pathogenesis is the serine/threonine protein phosphatase 2A (PP2A). PP2A is a ubiquitously expressed, highly conserved serine/threonine phosphatase that plays a key role in a number of cellular processes like cell division, motility, cytoskeletal dynamics, etc. (7). PP2A has a tripartite structure consisting of the scaffold subunit A and the catalytic subunit C forming the core enzyme and one of the many regulatory subunits binding to the core enzyme to form a functional holoenzyme (8). We have shown previously that PP2A protein and mRNA levels as well as the enzymatic activity of the catalytic subunit are increased in T cells from SLE patients compared with healthy individuals (9). This enhanced expression of PP2A is one of the factors contributing to the molecular defects seen in SLE (10).
We have demonstrated previously that a cAMP response element in the PP2A promoter is hypomethylated in SLE T cells and that the binding of the transcription factors cAMP response element-binding protein and SP1 contribute to the expression levels of PP2A (11). However, additional mechanisms that affect the expression of PP2A are still not clear and warrant an extensive study. In 2011, a genome wide association study (GWAS) study identified an SNP in the first intron of PPP2CA that was associated with SLE (12). The risk allele of this SNP was associated with renal disease, anti-double-stranded DNA, and anti-ribonucleoprotein antibodies. Moreover, PP2A expression was higher in patients carrying this allele, suggesting that this SNP may play a role in regulating PP2A expression.
We hypothesized that a transcription factor binding at this site might be controlling PP2A expression. Here, we report that the transcription factor Ikaros binds to this variant site in the first intron of PP2A. Ikaros acts as a repressor, and its binding reduces the expression of PP2A. We further show that Ikaros exerts its repressive effect by the recruitment of the histone deacetylase HDAC1, which maintains the chromatin in a closed conformation, precluding the binding of the transcription machinery. Thus, we demonstrate, for the first time, that Ikaros acts as a repressor for the serine/threonine phosphatase PP2A and, thus, identify a novel means of control of PP2A, a critical molecule involved in lupus pathogenesis.
EXPERIMENTAL PROCEDURES
Purification of T cells
Peripheral blood was collected by venipuncture, and CD3+ T cells were purified using a rosette T cell purification kit (Stem Cell Technologies) as described before (11). T cells were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum and 1% penicillin-streptomycin (Sigma-Aldrich) and maintained in a humidified incubator (37 °C, 5% CO2). All studies were approved by the institutional review board (Committee on Clinical Investigations) at the Beth Israel Deaconess Medical Center, and informed consent was obtained from all participants.
Oligonucleotide Pulldown Assay
Custom-synthesized, biotin-labeled WT as well as control oligos were purchased from Integrated DNA Technologies. Sequences of the oligos were as follows: WT oligonucleotide, 5′-TTCCAGCCTCCGCCTCCCAAAGCACCAGGATTG-3′; control oligonucleotide, 5′-ACCTGACGCCTAAGACGCTTACTCGCCTCGCGC-3′. The pulldown assay was carried out using streptavidin magnetic beads (PureBiotech LLC, Middlesex, NJ) as described before (13). The eluates were run on a 4–20% BisTris NuPAGE precast gel (Invitrogen), transferred to a PVDF membrane, and blotted with anti-Ikaros antibody (Santa Cruz Biotechnology, Inc.).
Luciferase Assay
PP2A core promoters followed by 384 bases spanning the Ikaros binding site in the first intron were cloned upstream of the luciferase gene in the pGL3 reporter vector. Two million 293T cells were transfected with either the reporter plasmid by itself (250 ng) or in combination with different amounts of the Ikaros expression vector (50, 100, or 200 ng) using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. Twenty-four hours after transfection, cells were collected and lysed using passive lysis buffer (Promega), and luciferase activity was quantified by using the Dual-Luciferase assay system (Promega) according to the instructions of the manufacturer.
Site-directed Mutagenesis of the Reporter Plasmid
The luciferase reporter plasmid described above was mutated using Pfu Turbo polymerase (Stratagene). The mutagenesis primers were as follows: Del-1, 5′-CCTACAATCCTGGTGCCGGAGGCTGGAAGCTC-3′; Del-2, 5′-CCTACAATCCTGGTGCTTGGCGGAGGCTGGAAGCTC-3′; mutant, 5′-CCTACAATCCTGGTGCGTCTGAAGTCGGAGGCTGGAAG-3′. The reverse primer in each case was the respective reverse complement of the forward primer.
Reporter ChIP and ChIP Assays
293T cells were transfected with either only the wild-type or the mutant reporter (described above) or the reporter in combination with the Ikaros expression plasmid using Lipofectamine 2000. Twenty-four hours after transfection, cells were collected, and the ChIP assay was performed using the MAGnify ChIP kit (Invitrogen) according to the instructions of the manufacturer. Briefly, cells were fixed for 10 min with 1% formaldehyde to cross-link DNA-protein and protein-protein complexes. The cross-linking reaction was stopped using 1.25 m glycine for 5 min. The cells were lysed, sonicated to shear DNA, and sedimented, and then the diluted supernatants were incubated overnight with the respective antibodies. 10% of the diluted supernatants were saved as “inputs” for normalization. Several washing steps were followed by protein digestion using proteinase K. Reverse cross-linking was carried out at 65 °C. DNA was subsequently purified and amplified by quantitative PCR on a LightCycler 480 real-time PCR system (Roche) using specific primers flanking the intronic site and the luciferase gene (reverse primer). Threshold cycle (i.e. Ct) values were used to calculate relative mRNA expression by the ΔΔCt relative quantification method.
In the case of regular ChIP assays, 5 million freshly isolated primary T cells were used for each antibody/sample. There was no transfection, and endogenous proteins were used for immunoprecipitation of the chromatin. The ChIP assay was carried out as described above using the MAGnify ChIP kit. The primers used were as follows: 5′-CAGAATTGATGATACAGAAACTTGA-3′ (forward) and 5′TAGAAACGGTGGTCTCACTACAT-3′ (reverse). The antibodies used for the ChIP assays were anti-Ikaros (Santa Cruz Biotechnology, Inc.) and anti-HDAC1 (Millipore).
Transfections in T Cells
Plasmid or siRNA transfections in primary T cells were carried out using the Nucleofector system (Lonza). Five million freshly isolated T cells were resuspended in 100 μl of Nucleofector solution, and the respective amount of plasmid or siRNA was added. For most studies, 0.5, 1, or 2 μg of the plasmid/1 million cells was used. In the case of siRNA, either 5 or 10 nm final concentration was used. Cells were transfected using the U-014 program and rescued immediately in prewarmed RPMI medium supplemented with 10% fetal calf serum and 1% penicillin-streptomycin. For plasmid transfections, cells were harvested 36 h post-transfection for real-time and protein analysis. For silencing studies, 72 h after siRNA transfection, cells were harvested for further analysis.
Real-time PCR Analysis
After harvesting the cells, total RNA was isolated using the RNeasy Plus kit (Qiagen). 300 ng of the total RNA was reverse-transcribed into cDNA using RNA-to-cDNA premix (Clontech). Real-time PCR amplification was carried out with SYBR Green I using LightCycler 480 (Roche). Threshold cycles (Ct values) were used to calculate relative mRNA expression by the relative quantification method.
Coimmunoprecipitation Assay
293T cells were transfected with the various combinations of plasmids (0.5 μg each) using Lipofectamine 2000. Twenty-four hours after transfection, the cells were lysed in 1 ml of radioimmune precipitation assay buffer (Boston BioProducts) supplemented with EDTA-free complete protease inhibitor mixture (Roche). After spinning down the cells, the supernatants were incubated with 1 μg of Ikaros antibody (catalog no. H-100X, Santa Cruz Biotechnology, Inc.) for 2 h at 4 °C. 10% of the supernatant was saved as input to run on the gel along with the immunoprecipitated samples. After the preincubation with the antibody, 20 μl of agarose A/G beads (Santa Cruz Biotechnology, Inc.) were added to each sample and incubated overnight at 4 °C. The immunoprecipitates were washed four times with PBS containing 1% Triton X-100. The samples were then boiled with the reducing agent, run on a gel, transferred to a PVDF membrane, and blotted for the indicated proteins. The saved inputs were also run on the gel to confirm equal expression of proteins in all samples.
RESULTS
IKZF1 Binds to a Specific Site in the First Intron of PP2Ac
Elevated levels of PP2A have been observed in T cells isolated from lupus patients, and this increased expression is a contributing factor in a number of molecular abnormalities. In addition to the already known factors that affect the expression of PP2A, a GWAS study by Tan et al. (12) revealed a genetic variant in the first intron of PPP2CA that was not only positively associated with SLE but also affected the expression levels of PP2A in patients (Fig. 1A). Thus, we hypothesized that a transcription factor binding at this site might be responsible for the observed changes in PP2A levels. Using an in silico transcription factor binding search, we identified Ikaros (IKZF1) as a putative factor binding at this specific intronic site. To confirm the binding of Ikaros to this site, we used a custom-synthesized, biotin-conjugated DNA oligonucleotide spanning the particular site. A random oligonucleotide of similar length served as a control. The oligos were incubated with Jurkat cell nuclear extracts, and streptavidin magnetic beads were used to pull down the interacting proteins. The eluates were run on a gel, and the membrane was probed for Ikaros. As shown in Fig. 1B, Ikaros was pulled down specifically with the oligo corresponding to the PP2A intronic site. The control oligo was unable to bring down Ikaros protein, suggesting that the binding of Ikaros to this site is specific.
FIGURE 1.
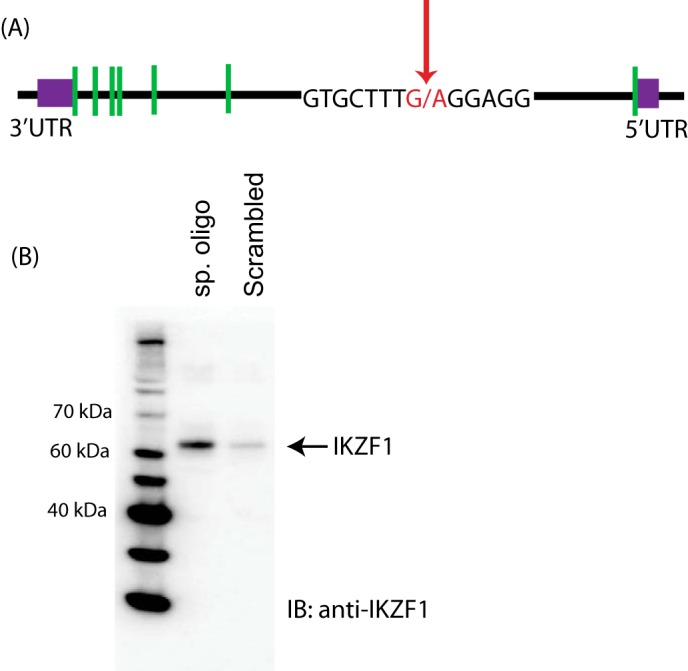
Ikaros binds to a specific site in the first intron of PP2A. A, schematic of the gene structure of IKZF1. The green boxes represent the introns. The sequence containing the variant site is depicted in red and an arrow. B, biotin-conjugated, intron-specific oligo (sp. oligo) or a random control oligo were used in pulldown assays with Jurkat cell nuclear extracts. The eluates were run on a gel and probed with anti-Ikaros antibody. This figure is representative of four independent experiments. IB, immunoblot.
Ikaros Binding Reduces PP2A promoter activity
To determine whether the binding of Ikaros to this site has any functional consequences, we employed a luciferase reporter system. The PP2A core promoter, followed by a 384-base pair region surrounding the intronic site, was cloned upstream of a gene encoding firefly luciferase (Fig. 2A). The expression of the luciferase gene is thus under direct control of the PP2A promoter as well as the intronic region. As compared with the empty vector, the PP2A reporter vector had significant activity (Fig. 2B). However, the exogenous expression of Ikaros, achieved by cotransfecting the expression plasmid along with the reporter vector, reduced the reporter activity to about half. Moreover, Ikaros had a dose-dependent effect, with increasing amounts of the expression plasmid leading to an even greater reduction in PP2A reporter activity.
FIGURE 2.
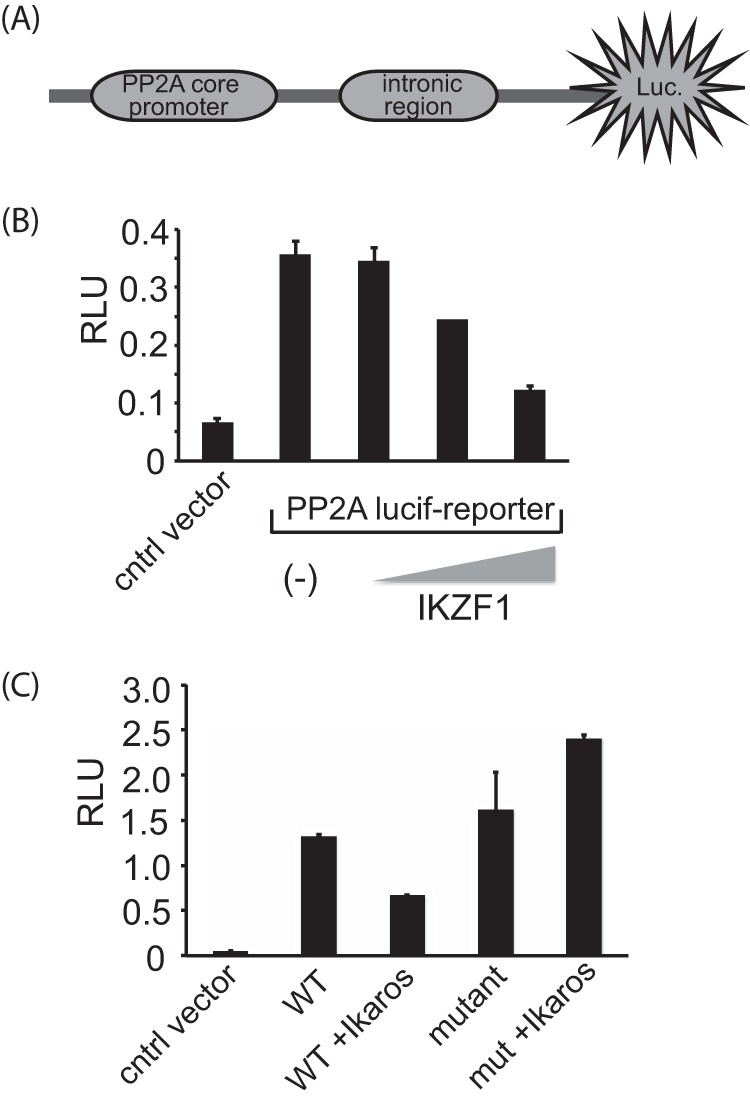
Ikaros binding reduces PP2A promoter activity. A, schematic of the luciferase reporter construct used in the assays. Luc, luciferase. B, 2 million 293T cells were transfected with either the reporter plasmid by itself (250 ng) or in combination with different amounts of the Ikaros expression vector (50, 100, or 200 ng) using Lipofectamine 2000. Twenty-four hours after transfection, cells were collected and lysed, and luciferase activity was quantified by using the Dual-Luciferase assay system. Renilla activity was used for normalization. The results represent the mean ± S.D. of three observations. RLU, relative light unit; cntrl, control. C, similar experiments were conducted with the mutant (mut) reporter construct in which the Ikaros binding site was deleted. The results represent mean ± S.D. of three observations.
To further prove that it is indeed the binding of Ikaros to the intronic region of PP2A that is responsible for the reduction in reporter activity, we generated a mutant version of the reporter plasmid, with the Ikaros binding site deleted. As compared with the wild-type reporter, the mutant reporter had enhanced reporter activity. Furthermore, in contrast to the wild-type construct, the mutant reporter activity was unaffected by the expression of Ikaros (Fig. 2C). This suggests that the binding of Ikaros to the specific site in the first intron of PP2A results in reduction of PP2A reporter activity.
Ikaros Represses the Expression of PP2Ac
The luciferase reporter assays with the transient expression of Ikaros suggest that Ikaros acts as a repressor of PP2A induction. Thus, we asked whether it can regulate the expression of PP2A. To this end, we overexpressed Ikaros in CD3+ T cells isolated from healthy individuals. After 36 h in culture, cells were collected and processed for immunoblotting. Forced expression of Ikaros led to a significant decrease in the protein levels of PP2A (Fig. 3A, right panel). Moreover, this decrease was dose-dependent. Increasing the amount of Ikaros transfected into the cells led to an even greater decrease in PP2A expression. To determine whether Ikaros represses PP2A expression at a transcriptional level, we assessed the mRNA levels of PP2A in cells expressing Ikaros. PP2A mRNA levels were similarly reduced in response to Ikaros overexpression (Fig. 3A, left panel). We next investigated the effect of Ikaros silencing on PP2A expression. siRNA-enabled silencing of Ikaros led to increased levels of PP2A message as well as protein (Fig. 3B, left and right panels, respectively). Thus, Ikaros acts as a repressor for PP2A expression.
FIGURE 3.
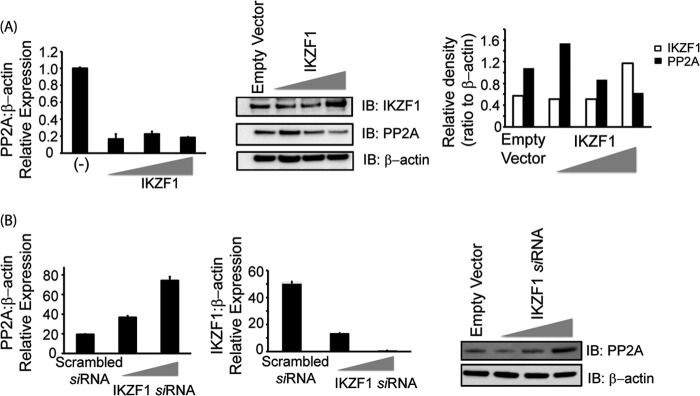
Ikaros suppresses the expression of PP2A. A, T cells isolated from healthy individuals were transfected with the empty vector or 0.5, 1, or 2 μg of the Ikaros expression plasmid/1 million cells using the nucleofector AMAXA. Five million cells were used per condition. 36 h post-transfection, cells were harvested to proceed with either real-time PCR (left panel) or protein analysis using Western blotting (center panel). For real-time PCR, the housekeeping gene β-actin was used for normalization. In the case of Western blot analyses, the membrane was probed with anti-Ikaros, stripped off the proteins, and reprobed with anti-PP2A. β-actin was used as a loading control. Band densities of IKZF1 and PP2A were quantified using Quantity One software and normalized to β-actin. Open bars, IKZF1; black bars, PP2A. IB, immunoblot. B, T cells were transfected with either scrambled siRNA or IKZF specific siRNA (5 or 10 nm final concentration) using AMAXA. 72 h after transfection, cells were harvested and subjected to either real-time PCR analysis or protein analysis using Western blotting. For real-time PCR, the housekeeping gene β-actin was used for normalization. In the case of Western blot analyses, the membrane was probed with anti-Ikaros, stripped off the proteins, and reprobed with anti-PP2A. β-actin was used as a loading control.
Ikaros Is Recruited to the First Intron in PP2A
We employed ChIP assays to confirm the recruitment of Ikaros to this particular site in a cellular setting. Reporter ChIP assays were performed in 293T cells. The cells were transfected with the PP2A promoter-intron reporter construct (described above) in combination with either the empty vector (pCMV) or an Ikaros expression vector. Twenty-four hours post-transfection, DNA-protein complexes were cross-linked and cells were lysed, sonicated, and immunoprecipitated with Ikaros antibody or a control IgG. Immunoprecipitated DNA was purified, and the presence of the specific intronic DNA was quantified using real-time PCR analysis. We observed a significantly increased recruitment of Ikaros to the intron of PP2A in the Ikaros-transfected samples compared with the pCMV-transfected cells, suggesting the recruitment of Ikaros to this site in the first intron of PP2A (Fig. 4B, left panel). We also mutated the wild-type reporter to generate three mutants (depicted in Fig. 4A) to test the specificity of this site. Two of these mutants were deletion mutants where either only the core Ikaros binding site (Del-2) or a few bases in addition to the core site were deleted (Del-1), and the third mutant had some of the bases changed, leaving the Ikaros binding site partially intact. We observed that both deletion mutants were unable to recruit Ikaros, suggesting that the absence of this site precludes the binding of Ikaros (Fig. 4B, right panel). With the mutant where some of the bases were changed, leaving the Ikaros binding site partially intact, we could see a reduction in recruitment compared with the wild-type reporter but not a complete abrogation. This suggests that an intact Ikaros binding site is necessary for the recruitment of Ikaros to the PP2A intron. We also wanted to verify the recruitment of Ikaros to the endogenous PPP2CA gene in T cells by ChIP assays in primary T cells. In this case, we immunoprecipitated the endogenous Ikaros protein using an Ikaros-specific antibody and analyzed its recruitment to the PP2A intron in a more physiologically relevant setting. We observed enhanced recruitment to this site in the case of Ikaros-specific antibody in comparison with control IgG, thus confirming that Ikaros is recruited to the intron of the endogenous PPP2CA gene.
FIGURE 4.
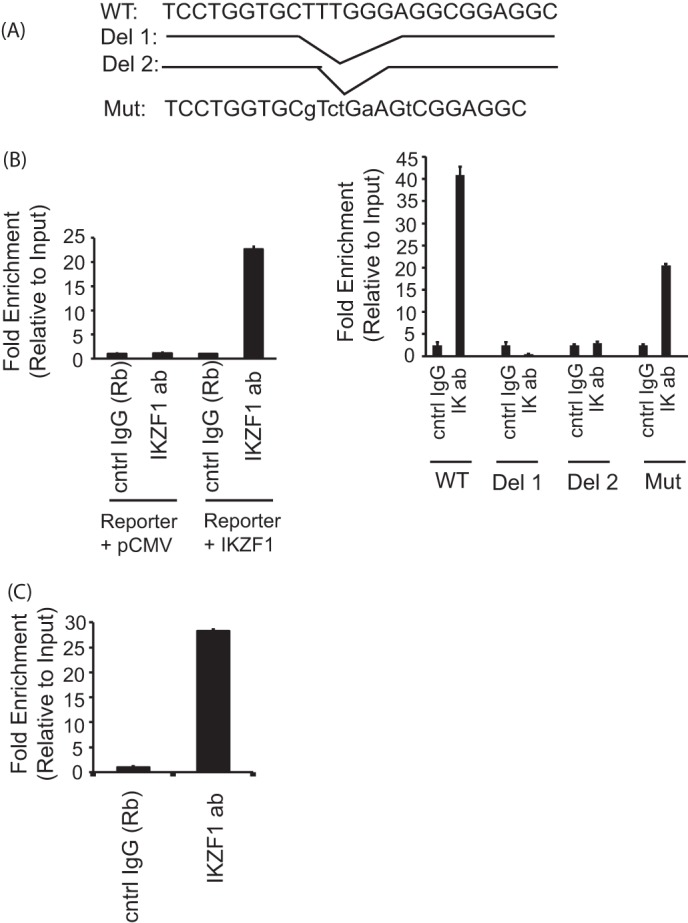
Ikaros is recruited to the specific site in the intron of PP2A. A, schematic of the mutant (Mut) reporters used in these experiments. B, reporter ChIP in 293T cells. 293T cells were transfected with either only the wild type (left panel) or the mutant reporter (right panel) or the reporter in combination with the Ikaros expression plasmid using Lipofectamine 2000. Twenty-four hours after transfection, cells were collected, and a ChIP assay was performed using the MAGnify ChIP kit. The region spanning the specific intron site was amplified by quantitative PCR and normalized to the values obtained from the input DNA. The graph shows mean ± S.D. of three observations. cntrl, control; ab, antibody; Rb, rabbit. C, ChIP assay in primary T cells. 5 million freshly isolated primary T cells were used for each antibody/sample. There was no transfection, and endogenous proteins were used for immunoprecipitation of the chromatin. A ChIP assay was carried out using the MAGnify ChIP kit. The region spanning the specific intron site was amplified by quantitative PCR and normalized to the values obtained from the input DNA. The graph shows mean ± S.D. of three observations.
Ikaros-mediated Repression of PP2A Is Dependent on the Recruitment of HDAC1
Ikaros is a zinc finger transcription factor that also harbors a dimerization domain at its carboxyl terminal through which it can recruit many different proteins and transcriptional regulators, including chromatin-modifying complexes like Sin3A, Sin3B, etc. (14). One of the proteins with which it is known to interact is histone deacetylase 1 or HDAC1. Thus, we asked whether the observed repressor activity of Ikaros in the case of PP2A could be due to the recruitment of HDAC1 to this particular intronic site. We first tested the interaction between Ikaros and HDAC1. Transient transfections in HEK 293T cells, followed by immunoprecipitation assays were carried out to address this question. As shown in Fig. 5A, HDAC1 could be coimmunoprecipitated with Ikaros, indicating that Ikaros and HDAC1 do indeed physically interact. Using biotinylated oligos, we also show that HDAC1 binds to the particular site in the first intron of PP2A (Fig. 5B). In both reporter ChIP as well as endogenous ChIP in primary T cells, we could see increased recruitment of HDAC1 to the intronic site compared with control IgG (Fig. 5C, left and right panels, respectively), thus confirming that HDAC1 does bind to this site in PP2A intron 1.
FIGURE 5.
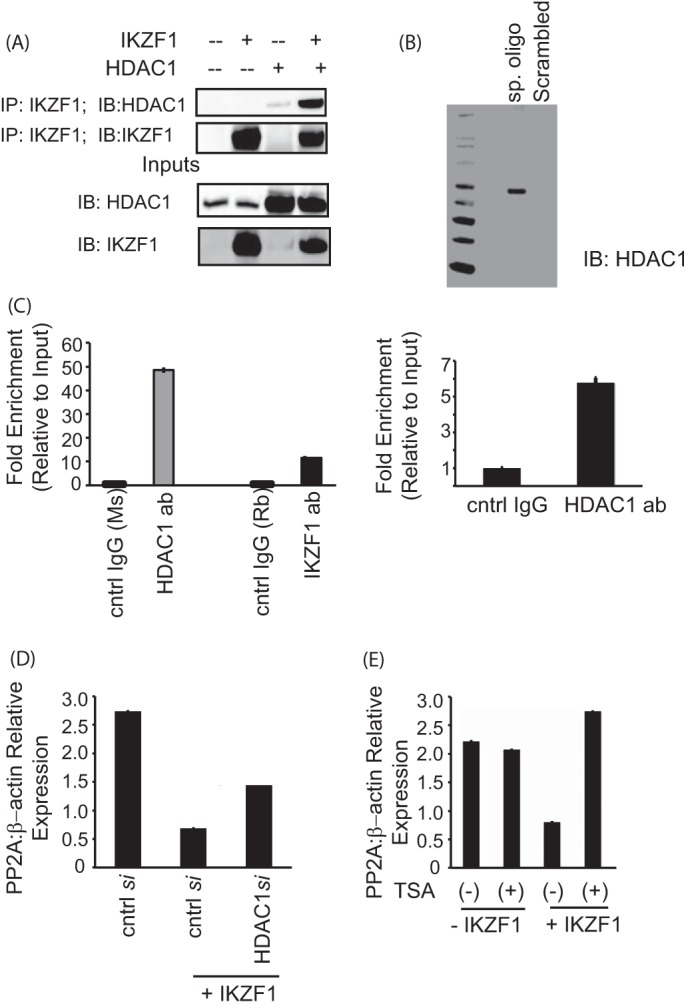
Ikaros-mediated repression of PP2A expression is dependent on HDAC1. A, coimmunoprecipitation (IP) assay showing the binding of Ikaros with HDAC1. 293T cells were transfected with the various combinations of plasmids using Lipofectamine 2000. Twenty-four hours after transfection, the cells were lysed, and the supernatants were incubated with Ikaros antibody for 2 h at 4 °C. After the preincubation with the antibody, agarose A/G beads were added to each sample and incubated overnight at 4 °C. The immunoprecipitates were subsequently run on a gel, transferred to a PVDF membrane, and blotted for the indicated proteins. The saved inputs were also run on the gel to confirm equal expression of proteins in all samples. IB, immunoblot. B, a biotin-conjugated, intron-specific oligo (sp. oligo) or a random control oligo were used in pulldown assays with Jurkat cell nuclear extracts. The eluates were run on a gel and probed with anti-HDAC1 antibody. C, reporter ChIP (left panel) and ChIP (right panel) in 293T and primary T cells, respectively. 293T cells (left panel) were transfected with the reporter in combination with the Ikaros and HDAC1 expression plasmids using Lipofectamine 2000. Twenty-four hours after transfection, cells were collected, and a ChIP assay was performed using the MAGnify ChIP kit. For ChIP with endogenous protein in primary T cells (right panel), 5 million freshly isolated primary T cells were used for each antibody (ab)/sample. The ChIP assay was carried out using the MAGnify ChIP kit. The region spanning the specific intron site was amplified by quantitative PCR and normalized to the values obtained from the input DNA. The graph shows mean ± S.D. of three observations. cntrl, control; Rb, rabbit; Ms, mouse. D, T cells were transfected with control siRNA or 10 nm HDAC1-specific siRNA with or without cotransfection of the Ikaros plasmid using AMAXA. 72 h after transfection, cells were harvested and subjected to real-time PCR analysis. The housekeeping gene β-actin was used for normalization. E, primary T cells were transfected with the Ikaros plasmid using AMAXA. Twenty-four hours after transfection, the cells were treated with 100 ng/ml trichostatin A (TSA) for 12 h. The same volume of dimethyl sulfoxide was used as in the vehicular control. The cells were harvested, and real-time PCR was carried out. The housekeeping gene β-actin was used for normalization.
Having established that HDAC1 interacts with Ikaros and is recruited to this site, we wanted to prove that the repression of PP2A expression that we see is due to HDAC1. To this effect, we used two different approaches. Using siRNA, we silenced HDAC1 in cells overexpressing Ikaros and assessed the mRNA levels of PP2Ac. As expected, the overexpression of Ikaros led to a reduction in PP2A mRNA levels. However, siRNA-mediated silencing of HDAC1 in these cells restored the PP2A levels to about 50% of the original, suggesting that, in the absence of HDAC1, Ikaros-mediated PP2A repression is not as competent (Fig. 5D). Secondly, we used an HDAC1 inhibitor, trichostatin A to confirm this. T cells were transfected with Ikaros, followed by a 12-hour treatment with trichostatin A. The treatment of cells with trichostatin A led to the complete restoration of PP2A expression (Fig. 5E). Thus, the repression of PP2A expression seen upon Ikaros overexpression is at least partly due to the recruitment of HDAC1 to the same site. HDAC1 recruitment to this site might influence the accessibility of transcription factors to the promoter, thus affecting gene expression of PP2A.
DISCUSSION
In this study we propose a novel mechanism for regulating the expression of PP2A, a serine/threonine phosphatase that is central to the molecular defects seen in the pathogenesis of SLE. We show, for the first time, that the expression of PP2A can be controlled by the binding of transcription factor Ikaros to a specific site in the first intron. Ikaros acts as a repressor for PP2A expression, and this can be explained by the ability of Ikaros to bind and recruit the histone deacetylase HDAC1 to this particular site. Thus, we provide evidence for a previously unknown regulatory mechanism of one of the most important factors involved in SLE.
T cells of SLE patients have higher levels of PP2A. This enhanced expression as well as activity translates to defects in the T cell signaling machinery, such as reduced expression of CD3ζ as well as decreased IL-2 production. We have also demonstrated recently that higher levels of PP2A might be one of the factors contributing to hypomethylation of DNA in patients, another hallmark of SLE pathogenesis (15). Although PP2A expression is clearly a significant aspect controlling the outcome of T cell signaling in SLE, not many studies have focused on the mechanisms affecting the expression of PP2A. We not only bring to the forefront the transcriptional control of PP2A, but also delineate a novel pathway for its regulation.
Conventionally, transcriptional regulation involves various factors binding to the promoter or the initiation site of the protein coding sequence. Nevertheless, there are countless studies exemplifying the instances where transcription factor binding to the introns can influence the expression of the gene, and intron-mediated enhancement of genes is a well studied branch of gene expression (16). A recent study by Hoffmann et al. (17) describes an intronic enhancer that promotes the expression of the UCP3 gene by the binding of SP1/SP3 transcription factors. Another report demonstrates the regulation of fibroblast growth factor receptor 3 by an intronic element (18). The intron-mediated control of genes is not limited to transcriptional enhancement. In fact, Ikaros itself has been shown to bind to an intronic site in the κ opioid receptor gene and cause its repression (19). How exactly an intronic element affects gene expression is not very clear. Perhaps the looping and folding of DNA brings these distal enhancer/repressor elements closer to the regulatory sequences of the promoter of the respective genes.
Ikaros (IKZF1) belongs to a family of zinc finger transcription factors (20). It binds to the regulatory elements of its target genes in a sequence-dependent manner. Ikaros can act as an activator or repressor of transcription, depending on which proteins it binds and recruits via its C terminus (21). Its repressive activity can be classified into three main categories: chromatin modification, corepressor recruitment, and competition with transcriptional activators (22). In the case of PP2A, we find that Ikaros recruits HDAC1 to this specific site in the first intron of PP2A. The repressive activity is dependent on the presence of HDAC1 because its silencing or the chemical inhibition of its activity restores the expression of PP2A. However, we believe that HDAC1 is not the sole factor responsible for Ikaros-mediated repression of PP2A because we only see a partial restoration of PP2A levels. Additional proteins/factors that might be involved in the regulation of PP2A levels remain to be investigated.
Because our study demonstrates that the binding of Ikaros to an intronic element reduces the levels of PP2A, it is conceivable that, in SLE patients, binding of Ikaros is compromised, thus giving rise to enhanced PP2A levels. However, the question remains why and how Ikaros differentiates between patients and healthy individuals. Is it the expression of Ikaros itself that controls its binding and, hence, the differential expression of PP2A? This would suggest that, in SLE patients, the expression of Ikaros would be decreased compared with normal controls. Indeed, a report by Hu et al. (24) does imply that in peripheral blood mononuclear cells from SLE patients, Ikaros mRNA levels are reduced (23). Whether this difference in expression of Ikaros translates to the regulation of PP2A remains to be seen.
In conclusion, we have shown that Ikaros is a modulator of PP2A expression. It binds to a site in the first intron of PPP2CA and reduces the expression levels of PP2A. The repressive ability of Ikaros depends, at least in part, on the recruitment of the histone deacetylase HDAC1, thus affecting the accessibility to the chromatin. Thus, we define a novel mechanism for the regulation of PP2A, a key component of SLE pathogenesis.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 AI068787 (to G. C. T.).
- SLE
- systemic lupus erythematosus
- PP2A
- protein phosphatase 2A
- HDAC
- histone deacetylase.
REFERENCES
- 1. Tsokos G. C. (2011) Systemic lupus erythematosus. N. Engl. J. Med. 365, 2110–2121 [DOI] [PubMed] [Google Scholar]
- 2. Grammatikos A. P., Tsokos G. C. (2012) Immunodeficiency and autoimmunity: lessons from systemic lupus erythematosus. Trends Mol. Med. 18, 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moulton V. R., Tsokos G. C. (2011) Abnormalities of T cell signaling in systemic lupus erythematosus. Arthritis Res. Ther. 13, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krishnan S., Warke V. G., Nambiar M. P., Tsokos G. C., Farber D. L. (2003) The FcR γ subunit and Syk kinase replace the CD3 ζ-chain and ZAP-70 kinase in the TCR signaling complex of human effector CD4 T cells. J. Immunol. 170, 4189–4195 [DOI] [PubMed] [Google Scholar]
- 5. Krishnan S., Farber D. L., Tsokos G. C. (2003) T cell rewiring in differentiation and disease. J. Immunol. 171, 3325–3331 [DOI] [PubMed] [Google Scholar]
- 6. Solomou E. E., Juang Y. T., Gourley M. F., Kammer G. M., Tsokos G. C. (2001) Molecular basis of deficient IL-2 production in T cells from patients with systemic lupus erythematosus. J. Immunol. 166, 4216–4222 [DOI] [PubMed] [Google Scholar]
- 7. Cohen P. T., Brewis N. D., Hughes V., Mann D. J. (1990) Protein serine/threonine phosphatases: an expanding family. FEBS Lett. 268, 355–359 [DOI] [PubMed] [Google Scholar]
- 8. Mumby M. C., Walter G. (1993) Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol. Rev. 73, 673–699 [DOI] [PubMed] [Google Scholar]
- 9. Katsiari C. G., Kyttaris V. C., Juang Y. T., Tsokos G. C. (2005) Protein phosphatase 2A is a negative regulator of IL-2 production in patients with systemic lupus erythematosus. J. Clin. Invest. 115, 3193–3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Juang Y. T., Wang Y., Jiang G., Peng H. B., Ergin S., Finnell M., Magilavy A., Kyttaris V. C., Tsokos G. C. (2008) PP2A dephosphorylates Elf-1 and determines the expression of CD3ζ and FcRγ in human systemic lupus erythematosus T cells. J. Immunol. 181, 3658–3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sunahori K., Juang Y. T., Tsokos G. C. (2009) Methylation status of CpG islands flanking a cAMP response element motif on the protein phosphatase 2Ac α promoter determines CREB binding and activity. J. Immunol. 182, 1500–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tan W., Sunahori K., Zhao J., Deng Y., Kaufman K. M., Kelly J. A., Langefeld C. D., Williams A. H., Comeau M. E., Ziegler J. T., Marion M. C., Bae S. C., Lee J. H., Lee J. S., Chang D. M., Song Y. W., Yu C. Y., Kimberly R. P., Edberg J. C., Brown E. E., Petri M. A., Ramsey-Goldman R., Vilá L. M., Reveille J. D., Alarcón-Riquelme M. E., Harley J. B., Boackle S. A., Stevens A. M., Scofield R. H., Merrill J. T., Freedman B. I., Anaya J. M., Criswell L. A., Jacob C. O., Vyse T. J., Niewold T. B., Gaffney P. M., Moser K. L., Gilkeson G. S., Kamen D. L., James J. A., Grossman J. M., Hahn B. H., Tsokos G. C., Tsao B. P., Alarcón G. S., BIOLUPUS Network, and GENLES Network (2011) Association of PPP2CA polymorphisms with systemic lupus erythematosus susceptibility in multiple ethnic groups. Arthritis Rheum. 63, 2755–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moulton V. R., Kyttaris V. C., Juang Y. T., Chowdhury B., Tsokos G. C. (2008) The RNA-stabilizing protein HuR regulates the expression of ζ chain of the human T cell receptor-associated CD3 complex. J. Biol. Chem. 283, 20037–20044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Popescu M., Gurel Z., Ronni T., Song C., Hung K. Y., Payne K. J., Dovat S. (2009) Ikaros stability and pericentromeric localization are regulated by protein phosphatase 1. J. Biol. Chem. 284, 13869–13880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Richardson B. (2003) DNA methylation and autoimmune disease. Clin. Immunol. 109, 72–79 [DOI] [PubMed] [Google Scholar]
- 16. Moabbi A. M., Agarwal N., El Kaderi B., Ansari A. (2012) Role for gene looping in intron-mediated enhancement of transcription. Proc. Natl. Acad. Sci. U.S.A. 109, 8505–8510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoffmann C., Zimmermann A., Hinney A., Volckmar A. L., Jarrett H. W., Fromme T., Klingenspor M. (2013) A novel SP1/SP3 dependent intronic enhancer governing transcription of the UCP3 gene in brown adipocytes. PloS ONE 8, e83426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McEwen D. G., Ornitz D. M. (1998) Regulation of the fibroblast growth factor receptor 3 promoter and intron I enhancer by Sp1 family transcription factors. J. Biol. Chem. 273, 5349–5357 [DOI] [PubMed] [Google Scholar]
- 19. Hu X., Bi J., Loh H. H., Wei L. N. (2001) An intronic Ikaros-binding element mediates retinoic acid suppression of the κ opioid receptor gene, accompanied by histone deacetylation on the promoters. J. Biol. Chem. 276, 4597–4603 [DOI] [PubMed] [Google Scholar]
- 20. Georgopoulos K., Winandy S., Avitahl N. (1997) The role of the Ikaros gene in lymphocyte development and homeostasis. Annu. Rev. Immunol. 15, 155–176 [DOI] [PubMed] [Google Scholar]
- 21. Sun L., Liu A., Georgopoulos K. (1996) Zinc finger-mediated protein interactions modulate Ikaros activity, a molecular control of lymphocyte development. EMBO J. 15, 5358–5369 [PMC free article] [PubMed] [Google Scholar]
- 22. Sellars M., Kastner P., Chan S. (2011) Ikaros in B cell development and function. World J. Biol. Chem. 2, 132–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He C. F., Liu Y. S., Cheng Y. L., Gao J. P., Pan T. M., Han J. W., Quan C., Sun L. D., Zheng H. F., Zuo X. B., Xu S. X., Sheng Y. J., Yao S., Hu W. L., Li Y., Yu Z. Y., Yin X. Y., Zhang X. J., Cui Y., Yang S. (2010) TNIP1, SLC15A4, ETS1, RasGRP3 and IKZF1 are associated with clinical features of systemic lupus erythematosus in a Chinese Han population. Lupus 19, 1181–1186 [DOI] [PubMed] [Google Scholar]
- 24. Hu W., Sun L., Gao J., Li Y., Wang P., Cheng Y., Pan T., Han J., Liu Y., Lu W., Zuo X., Sheng Y., Yao S., He C., Yu Z., Yin X., Cui Y., Yang S., Zhang X. (2011) Down-regulated expression of IKZF1 mRNA in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Rheumatol. Int. 31, 819–822 [DOI] [PubMed] [Google Scholar]