

Background: TSPO has been proposed to be a critical regulator of the permeability transition pore (PTP).
Results: TSPO-null mitochondria and cardiac tissue show no difference from controls in pore function, response to ligands, or response to ischemia/reperfusion injury.
Conclusion: Regulation of the PTP by the outer membrane must rely on unknown proteins.
Significance: Our results call into question studies implicating TSPO in pathological processes through the PTP.
Keywords: Drug Action, Gene Knockout, Mitochondria, Mitochondrial Permeability Transition, Porphyrin, Photodynamic Effect
Abstract
Translocator protein of 18 kDa (TSPO) is a highly conserved, ubiquitous protein localized in the outer mitochondrial membrane, where it is thought to play a key role in the mitochondrial transport of cholesterol, a key step in the generation of steroid hormones. However, it was first characterized as the peripheral benzodiazepine receptor because it appears to be responsible for high affinity binding of a number of benzodiazepines to non-neuronal tissues. Ensuing studies have employed natural and synthetic ligands to assess the role of TSPO function in a number of natural and pathological circumstances. Largely through the use of these compounds and biochemical associations, TSPO has been proposed to play a role in the mitochondrial permeability transition pore (PTP), which has been associated with cell death in many human pathological conditions. Here, we critically assess the role of TSPO in the function of the PTP through the generation of mice in which the Tspo gene has been conditionally eliminated. Our results show that 1) TSPO plays no role in the regulation or structure of the PTP, 2) endogenous and synthetic ligands of TSPO do not regulate PTP activity through TSPO, 3) outer mitochondrial membrane regulation of PTP activity occurs though a mechanism that does not require TSPO, and 4) hearts lacking TSPO are as sensitive to ischemia-reperfusion injury as hearts from control mice. These results call into question a wide variety of studies implicating TSPO in a number of pathological processes through its actions on the PTP.
Introduction
Translocator protein of 18 kDa (TSPO3; previously called peripheral benzodiazepine receptor) is a well conserved, ubiquitous protein primarily localized in the outer mitochondrial membrane (OMM). Structural analysis has suggested that it consists of five transmembrane α helices forming a channel-like structure that may accommodate the import of lipophilic molecules into mitochondria (1). The C-terminal, cytosolic domain of TSPO contains a high affinity cholesterol recognition sequence, and TSPO has been suggested to play an important role in facilitating the transport of cholesterol from the OMM to the inner mitochondrial membrane (IMM) (2, 3). Mitochondrial transport of cholesterol is necessary for its metabolization to pregnenolone (the precursor of all steroid hormones and the rate-limiting step in steroidogenesis) via the activity of cytochrome P450 cholesterol side chain cleavage enzyme (4). TSPO is particularly abundant in cells and tissues where active steroidogenesis takes place (adrenals, gonads, and brain), and its role in steroidogenesis has been extensively investigated (2, 4). TSPO gene disruption in Leydig cells suppressed intramitochondrial cholesterol transport and steroid production, whereas overexpression of exogenous TSPO in cells devoid of the protein induced cholesterol uptake and transport (5). A second isoform of TSPO (TSPO2) has also been identified that is exclusively localized in the endoplasmic reticulum (6). The role of TSPO in steroidogenesis has been unexpectedly challenged by conditional elimination of TSPO in Leydig cells in mice, which was not followed by the expected effects on synthesis of steroid hormones (7).
The identification of TSPO was through its ability to bind benzodiazepines, leading to its initial characterization as the peripheral benzodiazepine receptor (8) located to mitochondria (9). Natural and synthetic ligands of TSPO have been identified and developed (10), and these compounds have been used extensively to characterize TSPO function both in vitro and in vivo. Endogenous ligands include cholesterol and porphyrins (protoporphyrin (PP) IX, mesoporphyrin IX, deuteroporphyrin (DP) IX, and hemin), whereas synthetic ligands classically include 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)isoquinoline-3-carboxamide (PK11195) and 4-chlorodiazepam (Ro5-4864) (8, 10, 11). Both endogenous and synthetic ligands exhibit nanomolar affinity for TSPO when tested in vitro, but the former bind exclusively TSPO across all species and show high levels of stereoselectivity, whereas binding of the latter differs between species (12). The distinctive properties of the two sets of ligands suggest that their binding sites are not identical. For example, although cholesterol clearly binds to the C terminus of TSPO (13, 14), all other ligands (and presumably porphyrins) are thought to bind near the N terminus (15).
TSPO has been proposed to play a role in the mitochondrial permeability transition (PT) (16–21), a sudden permeability increase in ions and solutes up to roughly 1500 Da of the otherwise impermeable IMM (22). The prevailing view is that the PT is due to opening of a regulated, high conductance IMM channel (23–25), the mitochondrial PT pore (PTP) (26, 27). A great deal is now understood about the regulation of the PTP, whose opening requires the presence of matrix Ca2+, which is an essential permissive factor, and of additional agents or conditions that are collectively termed “inducers” (28). Under the conditions used in most in vitro studies, opening of the PTP is accompanied by depolarization, Ca2+ efflux, depletion of matrix pyridine nucleotides, matrix swelling, cristae unfolding, OMM rupture, and release of intermembrane space (IMS) proteins, including cytochrome c, Smac/Diablo, and AIF (27). Consequently, persistent opening of the PTP has been associated with the cell death associated with many human pathological conditions (27). It has also been demonstrated that transient PTP openings without detrimental consequences on cell survival can occur both in isolated mitochondria and in situ in response to physiological signals (29–35) (see Ref. 36 for a recent review).
Although convincing insights into the molecular composition of the PTP have been obtained only recently (37, 38), previous biochemical attempts to define core constituents of the pore resulted in the persistent hypothesis that key molecular constituents include the voltage-dependent anion channel (VDAC) in the OMM, the adenine nucleotide translocase (ANT) in the IMM, and the regulatory protein cyclophilin D (CyPD) in the matrix (39). TSPO has been included in such models of the PTP because it can be co-purified with the putative pore components, VDAC and ANT (40). Involvement of TSPO in the “PTP pore complex” was also tested following treatment of mitochondria and cells with both endogenous and synthetic TSPO ligands. These studies suggested that TSPO ligands, such as PK11195, Ro5-4864, and PP IX, affect PTP opening, although the potency depended on the experimental setting and on the concentration used (varying from nanomolar to micromolar and in some cases switching from inhibition to activation) (16, 18, 19, 41). In addition, PK11195 was originally classified as an antagonist of TSPO, and Ro5-4864 was classified as an agonist (8), yet both activate or inhibit PTP in the same cell type, depending on the ligand concentration (17, 18, 42). The role of TSPO in PTP function was further muddled by the demonstration that the same compounds could inhibit PTP and/or protect cells from apoptosis and that individual TSPO ligands can have the opposite effect on the same cell. For example, Ro5-4864 and PK11195 can either stimulate or inhibit apoptosis, respectively, with the addition of Ro5-4864 overcoming the antiapoptotic effect of PK11195 (43, 44).
Genetic analysis has demonstrated conclusively that the PTP still functions in the absence of all genes encoding ANT (45) or VDAC (46, 47), discounting these proteins as mediating structural roles in the formation of the PTP. Furthermore, these studies have also clearly demonstrated that CyPD is a key regulator of the PTP and the target of the PTP-inhibitory action of cyclosporin A (CsA) but not an essential component of the pore (48–51). Similar genetic studies directed at testing the role of TSPO in PTP formation have not been possible to date because non-conditional elimination of TSPO expression results in embryonic lethality (52). Here, we report on the generation of mice in which the Tspo gene has been conditionally eliminated in the liver and heart, allowing us to test whether TSPO participates in PTP formation and whether it plays a role in the regulatory effects of the OMM on the PTP (53–57).
EXPERIMENTAL PROCEDURES
Reagents
PP IX, DP IX, and hematoporphyrin (HP) IX were obtained from Frontier Scientific (Logan, UT), and stock solutions were prepared in dimethyl sulfoxide. Cationic monobromobimane (MBM+) was from Calbiochem. CsA, PK11195, Ro5-4864, N-Ethylmaleimide, digitonin, phenylarsine oxide (PhAsO), arachidonic acid oligonucleotides and all generic reagents were from Sigma-Aldrich. Copper-o-phenanthroline (Cu(OP)2) was prepared immediately before use by mixing CuSO4 with o-phenanthroline at a molar ratio of 1:2 in bidistilled water. Calcium Green-5N was from Invitrogen. [3H]PK11195 (85.7 Ci/mmol) and Ultima Gold scintillation mixture were from PerkinElmer Life Sciences. Genomic mouse DNA was isolated with the DNeasy blood and tissue kit (Qiagen). 2× Phusion HF master mix (Thermo Scientific) was used for PCR. Nitrocellulose membrane was from GE Healthcare, and the enhanced chemiluminescence kit was from Millipore.
Generation of TspoloxP Mice
Mice in which the endogenous gene encoding TSPO was replaced with a gene containing loxP sites is outlined under “Results” and in Fig. 1A. Five candidate ES cell lines carrying the TspoloxPneo construct were identified, injected into C57Bl/6J (C57) MF-1 blastocysts (an albino variant of black C57), and evaluated for the ability to generate chimeric offspring. Of the five ES cell lines identified, two were able to successfully generate chimeric offspring (≥95% black). Male chimeras were subsequently mated with C57 female mice and all offspring evaluated for germ line transmission. Several of the males from two targeted ES cell lines were able to pass the modified Tspo gene to progeny. F1 heterozygotes carrying the targeted allele were mated to mice expressing the FLP1 recombinase from yeast driven by the Gt(ROSA)26Sor promoter (Jackson Laboratory stock number 009086). To generate TSPO-null livers, Cre recombinase was driven by the hepatocyte-specific albumin (Alb) promoter (Jackson Laboratory stock number 003574). To generate TSPO-null hearts, TspoloxP mice were crossed to mice in which the cardiac-specific α myosin heavy chain promoter directs expression of a tamoxifen-inducible Cre recombinase in juvenile and adult cardiac myocytes (Myh6Cre/Esr1; Jackson Laboratory stock number 005657) (58). Genotyping was performed by PCR using the set of primers shown in Fig. 1A, primers targeting the Cre recombinase, or, when necessary, standard Southern blotting. TspoloxP mice described here are now available from Jackson Laboratory as stock number 024976.
FIGURE 1.

Generation and characterization of liver-specific TSPO-null mitochondria. A, outline of the creation of TspoloxP mice and outline of final genetic structure, with the location of primers used for genotyping and analysis indicated. See “Experimental Procedures” for details. B, Western blot analysis of mitochondria isolated from Tspo+/+, TspoloxP, and AlbCre;TspoloxP mouse livers. Ndufs1 antibody was used as a loading control. C, [3H]PK11195 binding to TspoloxP (black symbols) and AlbCre;TspoloxP liver mitochondria (red symbols) was assessed as described under “Experimental Procedures.” D, Scatchard plots of experiments described in C. Error bars, mean ± S.D. of three independent experiments performed in duplicate. B refers to bound, and F refers to free.
Isolation of Mitochondria and Preparation of Mitoplasts
TSPO-null and control mouse liver mitochondria were prepared from mice between 2 and 6 months old by standard differential centrifugation. Livers were removed and placed in a glass beaker containing ice-cold 0.25 m sucrose, 10 mm Tris-HCl, 0.1 mm EGTA-Tris, pH 7.4, buffer (isolation buffer; IB) supplemented with bovine serum albumin. Livers were then cut into small pieces with scissors, rinsed with ice-cold IB, and passed through a prechilled Potter homogenizer with Teflon pestle. The homogenate (∼60 ml/liver) was transferred to centrifuge tubes, and unbroken cells and nuclei were removed by centrifugation at 685 × g for 10 min at 4 °C. The supernatant containing mitochondria and other organelles was transferred to new tubes and centrifuged at 6010 × g for 10 min at 4 °C. The resulting supernatant was discarded, and mitochondrial pellet was carefully suspended in ice-cold IB buffer and spun at 9390 × g for 5 min at 4 °C. The mitochondrial pellet was suspended in IB to give a protein concentration of about 60–80 mg/ml and stored on ice. Mitoplasts were prepared essentially as described previously (56). Briefly, mitochondria were suspended in IB at 20 mg/ml, supplemented with 1.5 mm digitonin, and incubated under gentle stirring at 4 °C for 20 min. The suspension was centrifuged at 10,000 × g for 5 min and washed once with IB, and the resulting pellet was resuspended in the same buffer. Protein concentration was determined by the Biuret method.
Western Blotting
Mitochondrial proteins were solubilized in SDS-PAGE loading buffer (2% SDS, 50 mm Tris-HCl, pH 6.8, 10% glycerol, 2.5% β-mercaptoethanol, 0.002% bromphenol blue), boiled for 5 min, and separated by 15% SDS-PAGE. The proteins were then electrophoretically transferred to nitrocellulose membranes using a Mini Trans-Blot system (Bio-Rad). To minimize the nonspecific binding, the membranes were blocked with 10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.01% Tween 20 (TBST) buffer containing 5% nonfat dry milk at room temperature for 1 h, followed by an overnight incubation with an anti-TSPO (a generous gift of Dr. V. Papadopoulos), monoclonal anti-ANT (MitoSciences), anti-VDAC (a generous gift of F. Thinnes), anti-CypD (Calbiochem), anti-ATPB (ATPase subunit β; Abcam), anti-Ndufs1 (Complex I; Abcam), and polyclonal anti-SLC25A3 (phosphate carrier, PiC; Abcam) antibodies. The membrane was then washed in TBST and subjected to 1 h of incubation with appropriate secondary antibody conjugated to horseradish peroxidase. The membrane was washed again in TBST buffer, and immunoreactive bands were detected by enhanced chemiluminescence.
Radioligand Binding Assay
The presence of the TSPO in mitochondrial membranes was assayed by the use of [3H]PK11195 (85.7 Ci/mmol). Liver mitochondria were isolated as outlined above, and mitochondrial protein was adjusted to 0.1 mg/ml in Tris buffer (50 mm Tris-HCl, pH 7.4, 4 °C) to a final assay volume of 200 μl. The suspensions were incubated with 0.05–9.6 nm radioligand or 10 μm PK11195 (for background correction) for 60 min at 4 °C. Assays were terminated by rapid vacuum filtration though Whatman GF/C glass microfiber filters. The filters were immediately washed first with 1 ml of 10 μm PK11195 in Tris buffer and then with an additional 15 ml of ice-cold 50 mm Tris-HCl, pH 7.4. Subsequently, the membranes were dried, placed in 5 ml of Ultima gold liquid scintillation counting mixture, vortexed, and assayed for 3H using a Packard Tri-Carb 1500 liquid scintillation analyzer.
Measurement of Mitochondrial Respiration
Mitochondrial oxygen consumption was assessed with the Seahorse Extracellular Flux Analyzer XF24 essentially as described previously (59). Briefly, mitochondrial assay solution contained 220 mm mannitol, 70 mm sucrose, 25 mm MOPS-Tris, 10 mm Pi-Tris, 5 mm MgCl2, 1 mm EGTA-Tris, 0.2% fatty acid-free BSA, and 5 mm glutamate plus 2.5 mm malate or 5 mm succinate and 2 μm rotenone, pH 7.4, as specified in the legend to Fig. 2. 10 μg of mitochondria (suspended in 50 μl of mitochondrial assay solution) per well were added to an XF24 cell culture microplate, centrifuged at 2000 × g for 20 min, and supplemented with 450 μl of mitochondrial assay solution to initiate the experiments.
FIGURE 2.

Morphological and functional features of TSPO-null liver mitochondria. A, transmission electron microscopy of TspoloxP (left) and AlbCre;TspoloxP mouse livers (right); bar, 500 nm. B, oxygen consumption rate (OCR) of mitochondria of the indicated genotypes incubated with succinate plus rotenone (squares) or glutamate plus malate (circles). Where indicated, 4 mm ADP, 2.5 μg/ml oligomycin, 4 μm FCCP, and 2 μm antimycin plus 2 μm rotenone (circles only) were added. Error bars, mean ± S.D. C, typical recording traces of Rh123 fluorescence changes in TspoloxP (black trace) and AlbCre;TspoloxP (red trace) liver mitochondria. Where indicated, 0.5 mg/ml mitochondria, 5 mm succinate, and 0.5 μm FCCP were added. a.u., absorbance units.
Mitochondrial Membrane Potential, Porphyrin Accumulation, and Photosensitization
Mitochondrial inner membrane potential (Δψ) was assessed with the potentiometric fluorescent probe rhodamine 123 (Rh123), a lipophilic cation that readily equilibrates across the IMM and accumulates in the matrix of energized mitochondria in response to the inside negative Δψ. The probe concentrates in the matrix, forming aggregates that cause a process of fluorescence quenching. 300 nm Rh123 and 1 μm rotenone were added to 2 ml of assay buffer (AB) containing 250 mm sucrose, 10 mm MOPS-Tris, 10 μm EGTA-Tris, and 1 mm Pi-Tris, pH 7.4, followed by the addition of 0.5 mg/ml mitochondria, 5 mm succinate, and 0.5 μm FCCP. Rh123 fluorescence intensity changes (λex = 503 nm, λem = 523 nm) were monitored with a PerkinElmer Life Sciences LS50 spectrofluorimeter. To measure porphyrin accumulation, mitochondria (0.5 mg/ml) were suspended in AB medium supplemented with 5 mm succinate plus 1 μm rotenone, and PP IX, DP IX, and HP IX at different concentrations were added under gentle stirring at room temperature. After 5 min, the suspensions were centrifuged at 10,000 × g for 1 min, and porphyrin concentrations were determined fluorimetrically by calibration plots after extraction of supernatants and pellets with 2% SDS (λex = 505 nm, λem = 632 nm; λex = 498 nm, λem = 618 nm; and λex = 500 nm, λem = 620 nm for PP IX, DP IX, and HP IX, respectively).
Porphyrin-mediated photosensitization of mitochondria was achieved as described previously (55). Briefly, preparations were incubated for 1–2 min in the dark with the desired concentration of porphyrin in AB medium supplemented with 5 mm succinate plus 1 μm rotenone and then irradiated at 365 nm in a thermostated glass reaction vessel with a Philips HPW 125-watt lamp (Philips, Eindhoven, Netherlands). The fluence rate at the level of the preparations (56 watts/m2) was measured with a calibrated quantum photo-radiometer (Delta OHM HD 9021). All irradiations were performed at 25 °C under magnetic stirring. Proper controls were carried out to verify that neither incubation with the photosensitizer in the dark nor illumination in the absence of porphyrin produced any appreciable changes in the parameters under study. Statistical analysis of the results shown in Fig. 6 was based on relative z scores as outlined by Pocock (60).
FIGURE 6.
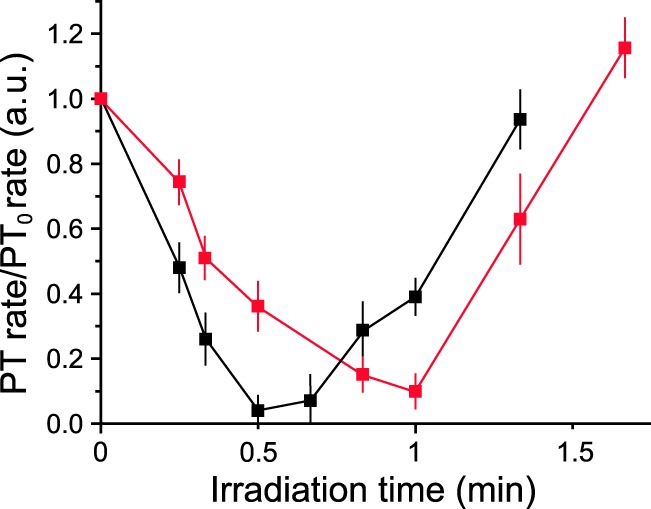
Effect of irradiation on the rate of permeabilization of TspoloxP or AlbCre;TspoloxP mitochondria. TspoloxP (black symbols) or AlbCre;TspoloxP (red symbols) liver mitochondria (0.5 mg/ml) were treated with 0.5 μm PP IX and kept in the dark or irradiated for the indicated time prior to the addition of 220 μm Ca2+, and mitochondrial volume changes were followed. Permeabilization rates were normalized to those of non-irradiated mitochondria. Error bars, S.D. of three independent experiments. a.u., absorbance units.
Assessment of PTP Activity
PTP activity in mitochondrial samples was evaluated by two methods. First, the Ca2+ retention capacity (CRC) was determined using Calcium Green-5N (λex = 505 nm, λem = 535 nm), a low affinity membrane-impermeant probe that increases its fluorescence emission upon Ca2+ binding. Mitochondria (0.5 mg/ml) were suspended in AB that also contained 0.5 μm Calcium Green-5N and respiratory substrate as specified in the figure legends; the suspension was subjected to trains of Ca2+ pulses, and fluorescence was measured either with the PerkinElmer LS50 spectrofluorimeter (final volume 2 ml) or (for the experiments of Fig. 4) with a Fluoroskan Ascent FL (Thermo Electron) plate reader. Second, the mitochondrial volume changes were followed as the decrease in light scattered at 540 nm with 0.5 mg/ml mitochondria suspended in AB supplemented with 5 mm succinate plus 1 μm rotenone. Permeabilization rates were calculated as the rate of change of light scattering immediately after the addition of Ca2+. All experiments were performed at 25 °C under magnetic stirring.
FIGURE 4.

Mitochondrial Ca2+ retention capacity of TspoloxP and AlbCre;TspoloxP mitochondria; effect of Pi, arachidonic acid, PhAsO, and Cu(OP)2. A–D, liver mitochondria from TspoloxP (black symbols) or AlbCre;TspoloxP mice (red symbols) were energized with 5 mm glutamate plus 2.5 mm malate in the presence of the indicated concentrations of Pi, arachidonic acid, PhAsO, or Cu(OP)2. In B–D, the Pi concentration was 1 mm. Values are the mean ± S.D. (error bars) of at least three independent experiments performed in duplicate.
Assessment of Cardiac Injury Due to Postischemic Reperfusion
Hearts were isolated from 1) untreated TspoloxP, 2) untreated Myh6Cre/Esr1;TspoloxP, 3) TspoloxP, and 4) Myh6Cre/Esr1;TspoloxP treated with tamoxifen (daily intraperitoneal injection of 40 mg/kg for 5 days). Mice were sacrificed 3 days after the last tamoxifen injection, and the ischemia/reperfusion (I/R) protocol and viability assay were performed as described (61). Briefly, isolated hearts were perfused under conditions of no-flow ischemia for 40 min, followed by the restoration of coronary flow (postischemic reperfusion) for 15 min. Loss of tissue viability was assessed as the release of lactic dehydrogenase (LDH) in the coronary effluent collected at 1-min intervals during the 15 min of reperfusion as described (61). At the end of reperfusion, hearts were collected and homogenized for assessing the residual activity of LDH in the whole tissue. Because all values were normalized to heart weight, the amount of LDH released was expressed as a percentage of the total (i.e. effluent + homogenate) to rule out possible changes due to variations in heart size (62). Results are presented as means ± S.E. of six experiments. Statistical analysis was performed utilizing the unpaired Student's t test.
RESULTS
Generation of TspoloxP Mice and TSPO-null Livers
TSPO is encoded by a mouse nuclear gene (Tspo) located on chromosome 15 that spans four exons (exon 1 is a non-translated exon) encompassing 10.6 kb of genomic DNA. A construct was generated that allowed conditional elimination of TSPO using the Cre/loxP system (generation of constructs outlined in Fig. 1A). Bacterial artificial chromosome clones containing the Tspo gene from C57 mice were obtained, and subclones containing Tspo protein-coding exons were modified in two ways. First, loxP sites were inserted into intronic sequence flanking exons 2 and 3, resulting in the elimination of residues encoding the translational start site through amino acid 124 following Cre-mediated recombination. Second, the neo gene, rendering cells resistant to the antibiotic G418, was inserted between loxP sites and flanked by flippase recognition target (FRT) sites recognized by the FRT recombinase (FLP) from yeast. To generate mice containing this construct, C57 ES cells were cultured using standard conditions and transfected by electroporation with the targeting construct described in Fig. 1A, transfectants were selected by growth in the presence of G418, and surviving ES cells were screened for replacement of the endogenous Tspo gene by PCR analysis and Southern blotting. Male chimeras were subsequently mated with C57 female mice, and all offspring were evaluated for germ line transmission. Several of the males from two targeted ES cell lines were able to pass the modified Tspo gene to progeny. F1 heterozygotes carrying the targeted allele were mated to mice expressing the FLP1 recombinase from yeast driven by a ubiquitous promoter. In this “deleter strain,” FLP1 recombinase is constitutively expressed from preimplantation onward, resulting in progeny in which the neo gene has been eliminated. The progeny were tested for the elimination of the neo gene by PCR analysis of genomic DNA (final gene shown in Fig. 1A), and homozygous lines for Tspo containing loxP sites (TspoloxP) were established. Finally, these mice were mated to mice heterozygous for Cre recombinase driven by a hepatocyte-specific Alb promoter. The offspring positive for Cre and homozygous TspoloxP generate experimental mice with livers lacking Tspo expression (AlbCre;TspoloxP) and littermate controls (TspoloxP).
We assessed TSPO expression in mitochondria prepared from livers lacking TSPO and control animals by two different methods. First, TSPO protein levels were determined by SDS-PAGE and Western blotting. TSPO expression in control liver mitochondria was not altered by the insertion of loxP sites when compared with C57 liver mitochondria, whereas TSPO was undetectable in AlbCre;TspoloxP mitochondria; expression of ANT, VDAC, CyPD, PiC, and ATPase subunit β was comparable in the three samples (Fig. 1B). Second, binding profiles of the synthetic TSPO ligand, [3H]PK11195, were assessed in control and liver mitochondria lacking TSPO (Fig. 1, C and D). [3H]PK11195 bound control mitochondria with an affinity similar to that previously reported (Kd = 1.2 nm), but no high affinity binding was apparent in mitochondria lacking TSPO. Given the absence of appreciable TSPO protein, as assessed by Western blotting and ligand binding, we have successfully created mitochondria in which the effects of TSPO on mitochondrial function and on PTP structure and regulation can be effectively examined.
Basic Mitochondrial Properties Are Not Altered in the Absence of TSPO
Mice lacking liver expression of TSPO were phenotypically unremarkable when compared with control mice, with apparently similar viability (mice lacking liver TSPO survive for over 18 months). At a gross level, the morphology of livers lacking TSPO was also not significantly different from controls; nor were the overall characteristics of hepatocytes (data not shown). Transmission electron microscopy analysis revealed no obvious ultrastructural differences in liver mitochondria obtained from control and TSPO-null donors; both possessed a well defined OMM, fingerlike cristae unfoldings, and an electron-dense matrix (Fig. 2A). Because TSPO ligands were demonstrated to increase state 4 and decrease state 3 respiration rates, resulting in a decreased respiratory control ratio (63), oxygen consumption rates were assessed in TspoloxP and AlbCre;TspoloxP liver mitochondria. Mitochondria lacking TSPO displayed the same rates of basal and ADP- and uncoupler-stimulated respiration as control mitochondria (Fig. 2B). Mitochondrial membrane potential, assessed on the changes in fluorescence quenching of Rh123, was evaluated in both mitochondrial preparations. Each readily developed a Δψ of about −180 mV upon the addition of succinate, and Δψ could be abolished in each by the uncoupler FCCP (Fig. 2C).
TSPO Does Not Play a Role in the Regulation of PT
The ability of TSPO-null mitochondria to undergo PT was tested with the sensitive CRC assay. Control mitochondria readily took up a train of Ca2+ pulses (Fig. 3A, trace a) in a process that was sensitive to CsA, as expected (Fig. 3A, trace b). Mitochondria lacking TSPO exhibited identical responses (Fig. 3B, traces a and b), the Ca2+ load required to trigger PTP opening, as well as the desensitizing effect of CsA, being indistinguishable in mitochondria from mice with the two genotypes. We next challenged mitochondria with a variety of endogenous and synthetic TSPO-specific ligands and tested their effect on the CRC. PK11195 (100 μm), Ro5-4864 (100 μm), and PP IX (8 μm) lowered the CRC of control mitochondria (Fig. 3C and Table 1), and exactly the same response was observed in TSPO-null mitochondria (Fig. 3D and Table 1). With either genotype, the sensitizing effect could be prevented by 1 μm CsA (data not shown). We also tested the well characterized PTP inducers Pi, arachidonic acid, PhAsO, and Cu(OP)2 and obtained superimposable effects irrespective of whether liver mitochondria from TspoloxP or AlbCre;TspoloxP mice were used (Fig. 4, A–D).
FIGURE 3.

Mitochondrial Ca2+ retention capacity of TspoloxP and AlbCre;TspoloxP mitochondria; effect of TSPO ligands. TspoloxP (A and C) and AlbCre;TspoloxP (B and D) mouse liver mitochondria (0.5 mg/ml) were suspended in AB supplemented with 5 mm succinate plus 1 μm rotenone and 0.5 μm Calcium Green-5N and loaded with a train of Ca2+ pulses. A and B, in traces b, the medium was supplemented with 1 μm CsA. C and D, in traces b, c, and d, the mitochondrial suspensions were supplemented with 100 μm PK11195, 100 μm Ro5-4864, and 8 μm PP IX, respectively, immediately prior to Ca2+ additions. Representative traces of at least three independent experiments are shown. a.u., absorbance units.
TABLE 1.
Effect of treatment with PK11195, Ro5-4864 and PP IX on the CRC/CRC0 of TspoloxP and AlbCre;TspoloxP mouse liver mitochondria
Mitochondria (1 mg in 2 ml of assay buffer supplemented with 5 mm succinate plus 1 μm rotenone) were incubated as described under “Experimental Procedures” in the presence of the indicated concentrations of TSPO ligands. The CRC was determined using Calcium Green-5N, and data refer to the mean ± S.D. of 4–6 experiments.
| Ligand | CRC/CRC0 |
|
|---|---|---|
| TspoloxP | AlbCre;TspoloxP | |
| PK11195 (100 μm) | 0.55 ± 0.08 | 0.53 ± 0.10 |
| Ro5-4864 (100 μm) | 0.71 ± 0.05 | 0.68 ± 0.08 |
| PP IX (8 μm) | 0.68 ± 0.10 | 0.71 ± 0.08 |
The OMM, but Not TSPO, Is Required for PTP Induction by N-Ethylmaleimide
The OMM is required for PTP induction by substituted maleimides in rat liver mitochondria (54). We confirmed that mouse liver mitochondria readily undergo Ca2+-dependent swelling after treatment with 0.5 mm N-ethylmaleimide (NEM) and that mitoplasts are completely protected from the PT-inducing effects of NEM (Fig. 5A). The novel finding is that the NEM-reactive sites are not contributed by TSPO because PT opening could be observed in both TspoloxP and AlbCre;TspoloxP mitochondria (Fig. 5B). We also tested whether TSPO contributes to the inducing effects of Cu(OP)2, which acts at IMM site(s) facing the OMM, and PhAsO, which acts at a matrix site (57). It can be seen that both inducers caused mitochondrial permeabilization irrespective of the presence (Fig. 5, C and E) or absence of TSPO (Fig. 5, D and F). The inducing effect of Cu(OP)2 was prevented by MBM+ (Fig. 5, C and D), which is not transported inside the matrix (55), and that of PhAsO was expectedly inhibited by CsA (64) (Fig. 5, E and F).
FIGURE 5.

Effect of NEM, Cu(OP)2, and PhAsO on mitochondria and mitoplasts. 0.5 mg/ml Tspo+/+ liver mitochondria (trace a) or mitoplasts (trace b) (A) and TspoloxP (trace a) or AlbCre;TspoloxP (trace b) liver mitochondria (B) were suspended in assay buffer supplemented with 5 mm succinate plus 1 μm rotenone; where indicated, 150 μm Ca2+, 0.5 mm NEM, and 5 μm alamethicin were added. C–F, 0.5 mg/ml TspoloxP (C and E) or AlbCre;TspoloxP mouse liver mitochondria (D and F) in the same buffer as in A and B were supplemented with 15 μm Ca2+ and 5 μm Cu(OP)2 (C and D) or 15 μm PhAsO (E and F) (traces a). In traces b (C and D) 0.2 mm MBM+ was added prior to Cu(OP)2. E and F, medium was supplemented with 1 μm CsA. Representative traces of three independent experiments are shown.
TSPO Is Not Required for Porphyrin-mediated Photodynamic Effects on the PTP
We have previously reported that in isolated mitochondria, the PT can be modulated by photooxidative stress following UV-visible light irradiation of PP IX-like dicarboxylic porphyrins associated with mitochondria, resulting in the production of reactive oxygen species (primarily singlet oxygen, 1O2) (65). Earlier studies have demonstrated that such photooxidative stress promotes modification of vicinal targets at two critical sites. At one site, low doses of light inactivate the PTP through modification of critical histidines, which in turn causes a drop in the reactivity of matrix-exposed cysteines, probably on the core components of the PTP, stabilizing the pore in a closed conformation (65). In contrast, at the second site, higher light doses coincide with modification of IMS-exposed cysteines that promote PTP activation (55, 57). This activation requires the presence of the OMM, given that mitoplasts are refractory to PT activation even after prolonged irradiation (56). Because porphyrins were reported to be ligands of TSPO (10), we have proposed that PT photoactivation occurs through TSPO (56). To test this hypothesis, we assessed occurrence of the PT (measured as a decrease of light scattered at 540 nm) in TspoloxP and AlbCre;TspoloxP mouse liver mitochondria upon photooxidative stress (Fig. 5). Mitochondria of both genotypes were incubated with 0.5 μm PP IX (a concentration that per se does not affect the PT (56)) and then loaded with 220 μm Ca2+. As reported previously for both rat and mouse liver mitochondria (57, 65), irradiation for short times inhibited, whereas irradiation for longer times activated, the PTP. Both photodependent pore inactivation and activation occur with identical kinetics (z scores (60) comparing slopes of inactivation, p = 0.94; activation, p = 0.96) in mice of both genotypes, but activation was shifted to slightly higher irradiation times in the absence of TSPO (i.e. AlbCre;TspoloxP mitochondria) (Fig. 6). These results are at striking variance from mitoplasts (56) in which porphyrin-mediated photoactivation of the PTP was eliminated. Because TSPO has been implicated in porphyrin transport from OMM to IMM (9, 15, 66), we suspected that the difference in irradiation time between the two genotypes might be explained by different amounts of porphyrin accumulated by mitochondria. This was not the case because identical amounts of porphyrin were accumulated at similar rates and levels in both sets of liver mitochondria (Table 2). These results indicate that under the present experimental conditions (i.e. with micromolar concentrations of PP IX), TSPO is not required for mitochondrial porphyrin uptake; nor is it required for porphyrin-mediated photomodification of critical matrix/IMS cysteines that, when modified, alter PTP activity.
TABLE 2.
Accumulation of porphyrins by TspoloxP and AlbCre;TspoloxP mouse liver mitochondria
Mitochondria (1 mg in 2 ml of assay buffer supplemented with 5 mm succinate plus 1 μm rotenone) were incubated as described under “Experimental Procedures” in the presence of the indicated amounts of porphyrins. Porphyrin accumulation was determined spectrofluorimetrically, and data refer to the mean ± S.D. of three independent experiments performed in duplicate.
| Porphyrin added | Porphyrin accumulated |
|
|---|---|---|
| TspoloxP | AlbCre;TspoloxP | |
| nmol × mg−1 | nmol × mg−1 | |
| PP IX | ||
| 1.00 | 0.72 ± 0.04 | 0.74 ± 0.05 |
| 2.00 | 1.44 ± 0.03 | 1.44 ± 0.05 |
| 3.00 | 2.15 ± 0.02 | 1.79 ± 0.26 |
| DP IX | ||
| 1.00 | 0.56 ± 0.02 | 0.51 ± 0.05 |
| 2.00 | 1.10 ± 0.12 | 1.03 ± 0.13 |
| 3.00 | 1.66 ± 0.18 | 1.53 ± 0.21 |
| HP IX | ||
| 1.00 | 0.30 ± 0.02 | 0.31 ± 0.07 |
| 2.00 | 0.60 ± 0.03 | 0.53 ± 0.01 |
| 3.00 | 0.92 ± 0.11 | 0.83 ± 0.05 |
TSPO Deletion Does Not Affect Postischemic Reperfusion in Isolated Hearts
We used a classical protocol of I/R injury in isolated, perfused hearts from mice treated with tamoxifen (Fig. 7A), which caused the expected disappearance of TSPO only from heart mitochondria of Myh6Cre/Esr1;TspoloxP mice (Fig. 7B, top). Deletion of cardiac TSPO did not significantly affect the extent of tissue necrosis (reflected by LDH release in the coronary effluent) in control animals not treated with tamoxifen and those treated with tamoxifen (Fig. 7B, bar graph). Therefore, in intact hearts, TSPO is not involved in I/R-induced tissue necrosis resulting from PTP activation.
FIGURE 7.

Effect of ischemia/reperfusion on LDH release in hearts from TspoloxP and Myh6Cre/Esr1;TspoloxP mice. A, tamoxifen was administered to mice as indicated, followed by sacrifice and heart isolation 3 days after the last tamoxifen injection. B, top, Western blot analysis of heart homogenates prepared from Myh6Cre/Esr1;TspoloxP mice treated with vehicle (lanes 1 and 2) or tamoxifen (lanes 3 and 4); each lane refers to one mouse. Bottom, LDH activity released in the coronary effluent during postischemic reperfusion (percentage of total LDH). Values are mean ± S.D. (error bars) of six independent experiments.
DISCUSSION
In this report, we outline the creation and initial characterization of mice in which the Cre/loxP system has been used for the tissue-specific elimination of TSPO, thereby bypassing the embryonic lethality noted when non-conditional elimination was initially attempted (52). Our goals in these studies were 1) to rigorously test the suggestion that TSPO is a component or a regulator of the mitochondrial PTP (16, 18, 19, 40, 41); 2) to assess whether the regulatory properties conferred to the PTP by the OMM (sensitivity to substituted maleimides and photoactivation by porphyrins) (53–56) are mediated by TSPO; and 3) to investigate whether TSPO is involved in heart I/R injury, a condition where the PTP is causally involved.
The idea that TSPO is involved in PTP formation originally stemmed from studies in which the TSPO could be purified in association with VDAC and the ANT (40), which were long considered to be core components of the PTP (38). This biochemical association evolved into a proposed functional role for TSPO in PTP formation through a variety of studies reporting that both endogenous (e.g. porphyrins) and synthetic TSPO ligands (e.g. PK11195 and Ro5-4864) altered the activity of the PTP (16–21). Here, we demonstrate that liver mitochondria prepared from AlbCre;TspoloxP mice do not express TSPO and totally lack high affinity binding sites for PK11195. When compared with TspoloxP (control) mice, transmission electron microscopy analysis demonstrated no ultrastructural alterations, and no significant differences in basal or ADP- and uncoupler-stimulated respiration were noted, as were no differences in mitochondrial membrane potential. On the basis of these studies, we conclude that liver mitochondria lacking TSPO function essentially as do control mitochondria. Whether proposed functions of TSPO in cholesterol and heme transport and in steroidogenesis (1–6) are compensated by mechanisms that we have not addressed here remains to be established. Indeed, recent similar genetic studies have called into question the precise role of TSPO in steroid hormone biogenesis (7). However, our study of PTP function in TSPO-null mitochondria allowed us to reach specific conclusions concerning the proposed role of TSPO as a component or regulator of the PTP.
When the ability of TSPO-null liver mitochondria to undergo the PT was tested by the sensitive CRC assay, the Ca2+ load required to trigger PTP opening, as well as the desensitizing effect of CsA, was indistinguishable from controls. Ro5-4864 and PK11195, archetypical synthetic ligands of TSPO whose affinity for TSPO is in the low nanomolar range, were also tested using this assay. Although each synthetic compound lowered the CRC of control mitochondria, the same response was observed in TSPO-null mitochondria, which totally lacked high affinity PK11195 binding sites. Thus, PTP sensitization to Ca2+ by “TSPO ligands” cannot be mediated by TSPO. This conclusion is significant because TSPO has been implicated in many pathological conditions, including uncontrolled cellular proliferation as occurs on oncogenic transformation, inflammation, and neurodegeneration (among others), primarily through modulation of each process in the presence of high concentrations (micromolar) of “TSPO-specific” ligands (20). This also applies to myocardial ischemia, where involvement of TSPO has been investigated only through pharmacological approaches without demonstrating the absolute specificity of the compounds tested (67). The PT is causally involved in I/R injury of the heart (61, 68), as is also shown by the cardioprotection afforded by CyPD deletion or inhibition not only in experimental models (48, 50, 61, 68) but also in a clinical setting (69). The lack of effect of TSPO ablation on heart I/R injury further calls into question the normal role of TSPO in disease processes as defined by the use of “TSPO-selective” drugs.
It appears reasonable to conclude that effects mediated by “TSPO ligands” may rather be due to interactions with other targets (20), and a prominent candidate is the mitochondrial F0F1-ATP synthase (37, 66, 70–72). Indeed, the ensemble of our current results indicates that the PTP is formed by a unique Ca2+-dependent conformation of dimers of the ATP synthase (37). For example, these studies have identified the site of Bz-423 binding on the OSCP subunit of this complex. Bz-423 both promotes PTP opening (37) and inhibits the F0F1-ATP synthase (66, 72). Moreover, many “TSPO ligands” also bind to and inhibit the mitochondrial FOF1-ATP synthase (66, 70–72), and this interaction probably best explains their effects in cell death and autophagy (73) through an inducing effect on PTP formation by ATP synthase dimers (37). Thus, our current picture calls for a reassessment of the mechanisms through which “TSPO ligands,” including PP IX (16), affect cell function and viability, which may be due to an effect on ATP synthase both through inhibition of ATP synthesis (70, 72) and through its transition to the PTP conformation (37, 38).
TSPO was originally identified by its ability to bind benzodiazepines with a distinct pharmacological profile from the benzodiazepine target in the central nervous system, GABA receptors (8, 9). In the search for endogenous ligands for this receptor, porphyrins were identified as being able to displace PK11195 from TSPO at nanomolar concentrations (10, 74). Based on these studies, naturally occurring dicarboxylic porphyrins, such as PP IX, other heme precursors, and heme itself have been considered endogenous ligands for TSPO (10). Our results, however, indicate that TSPO-null mitochondria accumulate porphyrins to similar extents as control mitochondria, suggesting either that porphyrin diffusion across the OMM does not require a specific transport protein or that such diffusion requires a protein that could coincide with Abcb6, an ATP-binding cassette transporter of the OMM (75). These results call into question our suggestion that PTP photoactivation mediated by porphyrins, which strictly requires an intact OMM, is mediated by TSPO (55–57), as discussed below.
The PT is an inner membrane event because mitoplasts (i.e. mitochondria stripped of the OMM) can readily undergo Ca2+-dependent PTP opening (56), yet the OMM is required for PTP induction in at least two paradigms (i.e. treatment with relatively high concentrations of substituted maleimides (54) and photosensitization (at high light doses) in the presence of porphyrins (55–57)). The sensitizing effect of the OMM is striking because mitoplasts become refractory to NEM (this work) and to other substituted maleimides (54) as well as to PTP opening by porphyrins plus visible light (56, 57), which activate the PTP through the OMM (55, 76). Photoactivation is not substantially affected by the absence of TSPO (this work), and the reactivity of the critical sites responsible for PTP inactivation and activation is only slightly modified in the absence of TSPO. Thus, any differences might be attributed to subtle structural differences in relevant proteins in the absence of TSPO that might modify the reactivity of critical residues. We must conclude, however, that the PTP-inducing effects of both porphyrin-dependent photoirradiation and NEM treatment depend on the OMM but not on TSPO and that the detailed mechanisms through which the OMM affects induction of the PTP remain to be established. In addition, we cannot exclude the possibility that any differences between those reported here and earlier studies might be due to species differences because most earlier work on the porphyrin-mediated photodynamic effect on the PTP was performed on rat liver mitochondria, whereas the current studies have used mouse mitochondria.
The vast majority of FOF1-ATP synthase dimers, which mediate PTP formation (37), are located in long rows of oligomers deep inside cristae (77–82), where contact with the OMM cannot occur. In principle, the PTP could form in a small population of dimers facing the IMS, where direct contact with the OMM is theoretically possible. We think that this possibility is not likely because the interaction should occur with FO, the membrane-embedded rotor of ATP synthase. We currently favor the idea that the OMM controls the diffusion of PTP-regulating species, including porphyrins, metabolites, and ions, including Ca2+ itself (83, 84), in a process that could be greatly favored by cristae remodeling (85). Alternatively, OMM effects on PTP activation may in part be due to mitochondrial structural reorganizations required for PTP formation that have yet to be appreciated and defined.
In summary, our results demonstrate that any effects on the activity of the PTP attributed to TSPO and to endogenous or synthetic ligands of TSPO must be due to their action at a yet to be defined site(s). In addition, porphyrin-mediated photoregulation of PTP activity must also be due to a currently unknown site, and TSPO cannot be responsible for mitochondrial accumulation of porphyrins. Most significantly, the results presented here invalidate any model of the PTP invoking a complex between VDAC, the ANT, and TSPO (39), including the most recent iterations (86). Indeed, the biochemical associations that led to these models have been conclusively demonstrated to be associations that may exist in mitochondria but play no structural role in the PTP complex because genetic analysis in each case has demonstrated that PTP activity is still present in mitochondria prepared from animals in which each protein has been eliminated (38). Combining our results with other recently published studies (7), we suggest that the role of TSPO-dependent mitochondrial processes remains to be established.
Acknowledgment
We thank Sarah Johnson for suggestions regarding the statistical analysis of the data in Fig. 6.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R01GM069883. This work was also supported by Telethon, Associazione Italiana per la Ricerca sul Cancro, Ministero dell'Istruzione, dell'Università e della Ricerca, and the University of Padova Progetto Strategico di Ateneo “Models of Mitochondrial Diseases.”
- TSPO
- translocator protein of 18 kDa
- ANT
- adenine nucleotide translocase
- AB
- assay buffer
- CRC
- Ca2+ retention capacity
- Cu(OP)2
- copper-o-phenanthroline
- CyPD
- cyclophilin D
- CsA
- cyclosporin A
- DP
- deuteroporphyrin
- Δψ
- mitochondrial inner membrane potential difference
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenyl hydrazone
- HP
- hematoporphyrin
- IB
- isolation buffer
- IMM
- inner mitochondrial membrane
- IMS
- intermembrane space
- I/R
- ischemia/reperfusion
- LDH
- lactic dehydrogenase
- OMM
- outer mitochondrial membrane
- PhAsO
- phenylarsine oxide
- PiC
- phosphate carrier
- PK11195
- 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)isoquinoline-3-carboxamide
- PP
- protoporphyrin
- PT
- permeability transition
- PTP
- permeability transition pore
- Rh123
- rhodamine 123
- Ro5-4864
- 4-chlorodiazepam
- VDAC
- voltage-dependent anion channel
- ES cell
- embryonic stem cell.
REFERENCES
- 1. Fan J., Lindemann P., Feuilloley M. G., Papadopoulos V. (2012) Structural and functional evolution of the translocator protein (18 kDa). Curr. Mol. Med. 12, 369–386 [DOI] [PubMed] [Google Scholar]
- 2. Papadopoulos V., Miller W. L. (2012) Role of mitochondria in steroidogenesis. Best Pract. Res. Clin. Endocrinol. Metab. 26, 771–790 [DOI] [PubMed] [Google Scholar]
- 3. Miller W. L. (2013) Steroid hormone synthesis in mitochondria. Mol. Cell. Endocrinol. 379, 62–73 [DOI] [PubMed] [Google Scholar]
- 4. Miller W. L., Auchus R. J. (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Papadopoulos V., Amri H., Li H., Boujrad N., Vidic B., Garnier M. (1997) Targeted disruption of the peripheral-type benzodiazepine receptor gene inhibits steroidogenesis in the R2C Leydig tumor cell line. J. Biol. Chem. 272, 32129–32135 [DOI] [PubMed] [Google Scholar]
- 6. Fan J., Rone M. B., Papadopoulos V. (2009) Translocator protein 2 is involved in cholesterol redistribution during erythropoiesis. J. Biol. Chem. 284, 30484–30497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morohaku K., Pelton S. H., Daugherty D. J., Butler W. R., Deng W., Selvaraj V. (2014) Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology 155, 89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Le Fur G., Vaucher N., Perrier M. L., Flamier A., Benavides J., Renault C., Dubroeucq M. C., Guérémy C., Uzan A. (1983) Differentiation between two ligands for peripheral benzodiazepine binding sites, [3H]RO5-4864 and [3H]PK 11195, by thermodynamic studies. Life Sci. 33, 449–457 [DOI] [PubMed] [Google Scholar]
- 9. Anholt R. R., Pedersen P. L., De Souza E. B., Snyder S. H. (1986) The peripheral-type benzodiazepine receptor: localization to the mitochondrial outer membrane. J. Biol. Chem. 261, 576–583 [PubMed] [Google Scholar]
- 10. Verma A., Nye J. S., Snyder S. H. (1987) Porphyrins are endogenous ligands for the mitochondrial (peripheral-type) benzodiazepine receptor. Proc. Natl. Acad. Sci. U.S.A. 84, 2256–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Romeo E., Auta J., Kozikowski A. P., Ma D., Papadopoulos V., Puia G., Costa E., Guidotti A. (1992) 2-Aryl-3-indoleacetamides (FGIN-1): a new class of potent and specific ligands for the mitochondrial DBI receptor (MDR) J. Pharmacol. Exp. Ther. 262, 971–978 [PubMed] [Google Scholar]
- 12. Scarf A. M., Luus C., Da Pozzo E., Selleri S., Guarino C., Martini C., Ittner L. M., Kassiou M. (2012) Evidence for complex binding profiles and species differences at the translocator protein (TSPO) (18 kDa) Curr. Mol. Med. 12, 488–493 [DOI] [PubMed] [Google Scholar]
- 13. Li H., Papadopoulos V. (1998) Peripheral-type benzodiazepine receptor function in cholesterol transport: identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 139, 4991–4997 [DOI] [PubMed] [Google Scholar]
- 14. Li H., Yao Z., Degenhardt B., Teper G., Papadopoulos V. (2001) Cholesterol binding at the cholesterol recognition/interaction amino acid consensus (CRAC) of the peripheral-type benzodiazepine receptor and inhibition of steroidogenesis by an HIV TAT-CRAC peptide. Proc. Natl. Acad. Sci. U.S.A. 98, 1267–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Farges R., Joseph-Liauzun E., Shire D., Caput D., Le Fur G., Ferrara P. (1994) Site-directed mutagenesis of the peripheral benzodiazepine receptor: identification of amino acids implicated in the binding site of Ro5-4864. Mol. Pharmacol. 46, 1160–1167 [PubMed] [Google Scholar]
- 16. Pastorino J. G., Simbula G., Gilfor E., Hoek J. B., Farber J. L. (1994) Protoporphyrin IX, an endogenous ligand of the peripheral benzodiazepine receptor, potentiates induction of the mitochondrial permeability transition and the killing of cultured hepatocytes by rotenone. J. Biol. Chem. 269, 31041–31046 [PubMed] [Google Scholar]
- 17. Berson A., Descatoire V., Sutton A., Fau D., Maulny B., Vadrot N., Feldmann G., Berthon B., Tordjmann T., Pessayre D. (2001) Toxicity of alpidem, a peripheral benzodiazepine receptor ligand, but not zolpidem, in rat hepatocytes: role of mitochondrial permeability transition and metabolic activation. J. Pharmacol. Exp. Ther. 299, 793–800 [PubMed] [Google Scholar]
- 18. Chelli B., Falleni A., Salvetti F., Gremigni V., Lucacchini A., Martini C. (2001) Peripheral-type benzodiazepine receptor ligands: mitochondrial permeability transition induction in rat cardiac tissue. Biochem. Pharmacol. 61, 695–705 [DOI] [PubMed] [Google Scholar]
- 19. Li J., Wang J., Zeng Y. (2007) Peripheral benzodiazepine receptor ligand, PK11195 induces mitochondria cytochrome c release and dissipation of mitochondria potential via induction of mitochondria permeability transition. Eur. J. Pharmacol. 560, 117–122 [DOI] [PubMed] [Google Scholar]
- 20. Ravagnan L., Marzo I., Costantini P., Susin S. A., Zamzami N., Petit P. X., Hirsch F., Goulbern M., Poupon M. F., Miccoli L., Xie Z., Reed J. C., Kroemer G. (1999) Lonidamine triggers apoptosis via a direct, Bcl-2-inhibited effect on the mitochondrial permeability transition pore. Oncogene 18, 2537–2546 [DOI] [PubMed] [Google Scholar]
- 21. Azarashvili T., Grachev D., Krestinina O., Evtodienko Y., Yurkov I., Papadopoulos V., Reiser G. (2007) The peripheral-type benzodiazepine receptor is involved in control of Ca2+-induced permeability transition pore opening in rat brain mitochondria. Cell Calcium 42, 27–39 [DOI] [PubMed] [Google Scholar]
- 22. Hunter D. R., Haworth R. A., Southard J. H. (1976) Relationship between configuration, function, and permeability in calcium-treated mitochondria. J. Biol. Chem. 251, 5069–5077 [PubMed] [Google Scholar]
- 23. Kinnally K. W., Campo M. L., Tedeschi H. (1989) Mitochondrial channel activity studied by patch-clamping mitoplasts. J. Bioenerg. Biomembr. 21, 497–506 [DOI] [PubMed] [Google Scholar]
- 24. Petronilli V., Szabò I., Zoratti M. (1989) The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett. 259, 137–143 [DOI] [PubMed] [Google Scholar]
- 25. Szabó I., Zoratti M. (1991) The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J. Biol. Chem. 266, 3376–3379 [PubMed] [Google Scholar]
- 26. Zoratti M., Szabò I., De Marchi U. (2005) Mitochondrial permeability transitions: how many doors to the house? Biochim. Biophys. Acta 1706, 40–52 [DOI] [PubMed] [Google Scholar]
- 27. Bernardi P., Krauskopf A., Basso E., Petronilli V., Blachly-Dyson E., Di Lisa F., Forte M. A. (2006) The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099 [DOI] [PubMed] [Google Scholar]
- 28. Gunter T. E., Pfeiffer D. R. (1990) Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 258, C755–C786 [DOI] [PubMed] [Google Scholar]
- 29. Ichas F., Jouaville L. S., Mazat J. P. (1997) Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89, 1145–1153 [DOI] [PubMed] [Google Scholar]
- 30. Hüser J., Blatter L. A. (1999) Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J. 343, 311–317 [PMC free article] [PubMed] [Google Scholar]
- 31. Petronilli V., Miotto G., Canton M., Brini M., Colonna R., Bernardi P., Di Lisa F. (1999) Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 76, 725–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Petronilli V., Penzo D., Scorrano L., Bernardi P., Di Lisa F. (2001) The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 276, 12030–12034 [DOI] [PubMed] [Google Scholar]
- 33. Wang W., Fang H., Groom L., Cheng A., Zhang W., Liu J., Wang X., Li K., Han P., Zheng M., Yin J., Wang W., Mattson M. P., Kao J. P., Lakatta E. G., Sheu S. S., Ouyang K., Chen J., Dirksen R. T., Cheng H. (2008) Superoxide flashes in single mitochondria. Cell 134, 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Elrod J. W., Wong R., Mishra S., Vagnozzi R. J., Sakthievel B., Goonasekera S. A., Karch J., Gabel S., Farber J., Force T., Brown J. H., Murphy E., Molkentin J. D. (2010) Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120, 3680–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barsukova A., Komarov A., Hajnóczky G., Bernardi P., Bourdette D., Forte M. (2011) Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur. J. Neurosci. 33, 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bernardi P., von Stockum S. (2012) The permeability transition pore as a Ca2+ release channel: new answers to an old question. Cell Calcium 52, 22–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Giorgio V., von Stockum S., Antoniel M., Fabbro A., Fogolari F., Forte M., Glick G. D., Petronilli V., Zoratti M., Szabó I., Lippe G., Bernardi P. (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 110, 5887–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bernardi P. (2013) The mitochondrial permeability transition pore: A mystery solved? Front. Physiol. 4, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zamzami N., Kroemer G. (2001) The mitochondrion in apoptosis: how Pandora's box opens. Nat. Rev. Mol. Cell Biol. 2, 67–71 [DOI] [PubMed] [Google Scholar]
- 40. McEnery M. W., Snowman A. M., Trifiletti R. R., Snyder S. H. (1992) Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. U.S.A. 89, 3170–3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kinnally K. W., Zorov D. B., Antonenko Y. N., Snyder S. H., McEnery M. W., Tedeschi H. (1993) Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc. Natl. Acad. Sci. U.S.A. 90, 1374–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Parker M. A., Bazan H. E., Marcheselli V., Rodriguez de Turco E. B., Bazan N. G. (2002) Platelet-activating factor induces permeability transition and cytochrome c release in isolated brain mitochondria. J. Neurosci. Res. 69, 39–50 [DOI] [PubMed] [Google Scholar]
- 43. Bono F., Lamarche I., Prabonnaud V., Le Fur G., Herbert J. M. (1999) Peripheral benzodiazepine receptor agonists exhibit potent antiapoptotic activities. Biochem. Biophys. Res. Commun. 265, 457–461 [DOI] [PubMed] [Google Scholar]
- 44. Bernardi P., Forte M. (2007) The mitochondrial permeability transition pore. Novartis Found. Symp. 287, 157–164; discussion 164–169 [PubMed] [Google Scholar]
- 45. Kokoszka J. E., Waymire K. G., Levy S. E., Sligh J. E., Cai J., Jones D. P., MacGregor G. R., Wallace D. C. (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krauskopf A., Eriksson O., Craigen W. J., Forte M. A., Bernardi P. (2006) Properties of the permeability transition in VDAC1−/− mitochondria. Biochim. Biophys. Acta 1757, 590–595 [DOI] [PubMed] [Google Scholar]
- 47. Baines C. P., Kaiser R. A., Sheiko T., Craigen W. J., Molkentin J. D. (2007) Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baines C. P., Kaiser R. A., Purcell N. H., Blair N. S., Osinska H., Hambleton M. A., Brunskill E. W., Sayen M. R., Gottlieb R. A., Dorn G. W., Robbins J., Molkentin J. D. (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 [DOI] [PubMed] [Google Scholar]
- 49. Basso E., Fante L., Fowlkes J., Petronilli V., Forte M. A., Bernardi P. (2005) Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 280, 18558–18561 [DOI] [PubMed] [Google Scholar]
- 50. Nakagawa T., Shimizu S., Watanabe T., Yamaguchi O., Otsu K., Yamagata H., Inohara H., Kubo T., Tsujimoto Y. (2005) Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658 [DOI] [PubMed] [Google Scholar]
- 51. Schinzel A. C., Takeuchi O., Huang Z., Fisher J. K., Zhou Z., Rubens J., Hetz C., Danial N. N., Moskowitz M. A., Korsmeyer S. J. (2005) Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 102, 12005–12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lacapère J. J., Papadopoulos V. (2003) Peripheral-type benzodiazepine receptor: structure and function of a cholesterol-binding protein in steroid and bile acid biosynthesis. Steroids 68, 569–585 [DOI] [PubMed] [Google Scholar]
- 53. Lê-Quôc K., Lê-Quôc D. (1982) Control of the mitochondrial inner membrane permeability by sulfhydryl groups. Arch. Biochem. Biophys. 216, 639–651 [DOI] [PubMed] [Google Scholar]
- 54. Lê-Quôc K., Lê-Quôc D. (1985) Crucial role of sulfhydryl groups in the mitochondrial inner membrane structure. J. Biol. Chem. 260, 7422–7428 [PubMed] [Google Scholar]
- 55. Petronilli V., Sileikyte J., Zulian A., Dabbeni-Sala F., Jori G., Gobbo S., Tognon G., Nikolov P., Bernardi P., Ricchelli F. (2009) Switch from inhibition to activation of the mitochondrial permeability transition during hematoporphyrin-mediated photooxidative stress. Unmasking pore-regulating external thiols. Biochim. Biophys. Acta 1787, 897–904 [DOI] [PubMed] [Google Scholar]
- 56. Sileikyte J., Petronilli V., Zulian A., Dabbeni-Sala F., Tognon G., Nikolov P., Bernardi P., Ricchelli F. (2011) Regulation of the inner membrane mitochondrial permeability transition by the outer membrane translocator protein (peripheral benzodiazepine receptor) J. Biol. Chem. 286, 1046–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ricchelli F., Sileikytė J., Bernardi P. (2011) Shedding light on the mitochondrial permeability transition. Biochim. Biophys. Acta 1807, 482–490 [DOI] [PubMed] [Google Scholar]
- 58. Sohal D. S., Nghiem M., Crackower M. A., Witt S. A., Kimball T. R., Tymitz K. M., Penninger J. M., Molkentin J. D. (2001) Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 89, 20–25 [DOI] [PubMed] [Google Scholar]
- 59. Rogers G. W., Brand M. D., Petrosyan S., Ashok D., Elorza A. A., Ferrick D. A., Murphy A. N. (2011) High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One 6, e21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pocock S. J. (2006) The simplest statistical test: how to check for a difference between treatments. Brit. Med. J. 332, 1256–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Di Lisa F., Menabò R., Canton M., Barile M., Bernardi P. (2001) Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 276, 2571–2575 [DOI] [PubMed] [Google Scholar]
- 62. Schlüter K. D., Schwartz P., Siegmund B., Piper H. M. (1991) Prevention of the oxygen paradox in hypoxic-reoxygenated hearts. Am. J. Physiol. 261, H416–H423 [DOI] [PubMed] [Google Scholar]
- 63. Hirsch J. D., Beyer C. F., Malkowitz L., Beer B., Blume A. J. (1989) Mitochondrial benzodiazepine receptors mediate inhibition of mitochondrial respiratory control. Mol. Pharmacol. 35, 157–163 [PubMed] [Google Scholar]
- 64. Lenartowicz E., Bernardi P., Azzone G. F. (1991) Phenylarsine oxide induces the cyclosporin A-sensitive membrane permeability transition in rat liver mitochondria. J. Bioenerg. Biomembr. 23, 679–688 [DOI] [PubMed] [Google Scholar]
- 65. Salet C., Moreno G., Ricchelli F., Bernardi P. (1997) Singlet oxygen produced by photodynamic action causes inactivation of the mitochondrial permeability transition pore. J. Biol. Chem. 272, 21938–21943 [DOI] [PubMed] [Google Scholar]
- 66. Stelzer A. C., Frazee R. W., Van Huis C., Cleary J., Opipari A. W., Jr., Glick G. D., Al-Hashimi H. M. (2010) NMR studies of an immunomodulatory benzodiazepine binding to its molecular target on the mitochondrial F1F0-ATPase. Biopolymers 93, 85–92 [DOI] [PubMed] [Google Scholar]
- 67. Surinkaew S., Chattipakorn S., Chattipakorn N. (2011) Roles of mitochondrial benzodiazepine receptor in the heart. Can. J. Cardiol. 27, 262.e3–13 [DOI] [PubMed] [Google Scholar]
- 68. Griffiths E. J., Halestrap A. P. (1995) Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 307, 93–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Piot C., Croisille P., Staat P., Thibault H., Rioufol G., Mewton N., Elbelghiti R., Cung T. T., Bonnefoy E., Angoulvant D., Macia C., Raczka F., Sportouch C., Gahide G., Finet G., André-Fouët X., Revel D., Kirkorian G., Monassier J. P., Derumeaux G., Ovize M. (2008) Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 359, 473–481 [DOI] [PubMed] [Google Scholar]
- 70. Cleary J., Johnson K. M., Opipari A. W., Jr., Glick G. D. (2007) Inhibition of the mitochondrial F1F0-ATPase by ligands of the peripheral benzodiazepine receptor. Bioorg. Med. Chem. Lett. 17, 1667–1670 [DOI] [PubMed] [Google Scholar]
- 71. Seneviratne M. S., Faccenda D., De Biase V., Campanella M. (2012) PK11195 inhibits mitophagy targeting the F1F0-ATP synthase in Bcl-2 knock-down cells. Curr. Mol. Med. 12, 476–482 [DOI] [PubMed] [Google Scholar]
- 72. Johnson K. M., Chen X., Boitano A., Swenson L., Opipari A. W., Jr., Glick G. D. (2005) Identification and validation of the mitochondrial F1F0-ATPase as the molecular target of the immunomodulatory benzodiazepine Bz-423. Chem. Biol. 12, 485–496 [DOI] [PubMed] [Google Scholar]
- 73. Gastaldello A., Callaghan H., Gami P., Campanella M. (2010) Ca2+-dependent autophagy is enhanced by the pharmacological agent PK11195. Autophagy 6, 607–613 [DOI] [PubMed] [Google Scholar]
- 74. Verma A., Snyder S. H. (1988) Characterization of porphyrin interactions with peripheral type benzodiazepine receptors. Mol. Pharmacol. 34, 800–805 [PubMed] [Google Scholar]
- 75. Krishnamurthy P., Xie T., Schuetz J. D. (2007) The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol. Ther. 114, 345–358 [DOI] [PubMed] [Google Scholar]
- 76. Moreno G., Poussin K., Ricchelli F., Salet C. (2001) The effects of singlet oxygen produced by photodynamic action on the mitochondrial permeability transition differ in accordance with the localization of the sensitizer. Arch. Biochem. Biophys. 386, 243–250 [DOI] [PubMed] [Google Scholar]
- 77. Allen R. D., Schroeder C. C., Fok A. K. (1989) An investigation of mitochondrial inner membranes by rapid-freeze deep-etch techniques. J. Cell Biol. 108, 2233–2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Paumard P., Vaillier J., Coulary B., Schaeffer J., Soubannier V., Mueller D. M., Brèthes D., di Rago J.-P., Velours J. (2002) The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 21, 221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dudkina N. V., Sunderhaus S., Braun H. P., Boekema E. J. (2006) Characterization of dimeric ATP synthase and cristae membrane ultrastructure from Saccharomyces and Polytomella mitochondria. FEBS Lett. 580, 3427–3432 [DOI] [PubMed] [Google Scholar]
- 80. Strauss M., Hofhaus G., Schröder R. R., Kühlbrandt W. (2008) Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 27, 1154–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Davies K. M., Strauss M., Daum B., Kief J. H., Osiewacz H. D., Rycovska A., Zickermann V., Kühlbrandt W. (2011) Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci. U.S.A. 108, 14121–14126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Davies K. M., Anselmi C., Wittig I., Faraldo-Gómez J. D., Kühlbrandt W. (2012) Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. U.S.A. 109, 13602–13607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Madesh M., Hajnóczky G. (2001) VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 155, 1003–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rapizzi E., Pinton P., Szabadkai G., Wieckowski M. R., Vandecasteele G., Baird G., Tuft R. A., Fogarty K. E., Rizzuto R. (2002) Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Scorrano L., Ashiya M., Buttle K., Weiler S., Oakes S. A., Mannella C. A., Korsmeyer S. J. (2002) A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2, 55–67 [DOI] [PubMed] [Google Scholar]
- 86. Pinton P., Kroemer G. (2014) Cancer therapy: altering mitochondrial properties. Nat. Chem. Biol. 10, 89–90 [DOI] [PubMed] [Google Scholar]