

Background: A cytoplasmic PAS domain regulates Kv11.1 function, but its role in channel assembly is unclear.
Results: PAS domain deletion does not alter assembly, but removal of the N-Cap that immediately precedes the PAS domain severely disrupts channel trafficking.
Conclusion: The N-Cap is vital for PAS domain stability and channel trafficking.
Significance: Kv11.1 channel assembly defects underlie the pathogenesis of long QT syndrome.
Keywords: hERG, Potassium Channels, Site-directed Mutagenesis, Trafficking, Western Blotting, PAS Domain, Subunit-Subunit Interaction
Abstract
The N-terminal cytoplasmic region of the Kv11.1a potassium channel contains a Per-Arnt-Sim (PAS) domain that is essential for the unique slow deactivation gating kinetics of the channel. The PAS domain has also been implicated in the assembly and stabilization of the assembled tetrameric channel, with many clinical mutants in the PAS domain resulting in reduced stability of the domain and reduced trafficking. Here, we use quantitative Western blotting to show that the PAS domain is not required for normal channel trafficking nor for subunit-subunit interactions, and it is not necessary for stabilizing assembled channels. However, when the PAS domain is present, the N-Cap amphipathic helix must also be present for channels to traffic to the cell membrane. Serine scan mutagenesis of the N-Cap amphipathic helix identified Leu-15, Ile-18, and Ile-19 as residues critical for the stabilization of full-length proteins when the PAS domain is present. Furthermore, mutant cycle analysis experiments support recent crystallography studies, indicating that the hydrophobic face of the N-Cap amphipathic helix interacts with a surface-exposed hydrophobic patch on the core of the PAS domain to stabilize the structure of this critical gating domain. Our data demonstrate that the N-Cap amphipathic helix is critical for channel stability and trafficking.
Introduction
Kv11.1, often referred to as the human ether-a-go-go-related gene K+ channel, encodes the rapid component of the delayed rectifier potassium channel, IKr. There are at least three alternatively spliced isoforms of Kv11.1, denoted Kv11.1a, Kv11.1b, and Kv11.1–3.1 (1). Kv11.1 is an important repolarizing channel in a wide range of tissues but most notably in the heart where Kv11.1a and Kv11.1b are the predominant isoforms expressed (1). Mutations in KCNH2, which encodes the Kv11.1 protein, result in congenital long QT syndrome type 2 (LQT2)2 (2). LQT2 is associated with an increased risk of ventricular arrhythmia and sudden cardiac death often at a young age (3, 4). The majority of LQT2-associated missense mutations cause protein trafficking defects (5). There has therefore been considerable interest in understanding the mechanisms by which missense mutations result in reduced Kv11.1 trafficking, not just to gain insights into clinical genotype-phenotype relationships (6) but also to further understand the mechanisms of channel folding and assembly.
Like other voltage-gated K+ channels, Kv11.1 channels assemble as tetramers with each subunit containing cytoplasmic N- and C-terminal regions and a transmembrane region. The transmembrane region contains the voltage sensor (transmembrane helices 1–4) and pore (transmembrane helices 5–6, along with an intervening pore loop region) domains (Fig. 1A). In Kv11.1a channels, the N-terminal cytoplasmic region contains a PAS domain (residues 1–135), which has a very similar overall structure to PAS domains in other proteins (7). The Kv11.1a PAS domain contains an N-terminal Cap (residues 1–25), the PAS core (residues 26–75), a connector helix (residues 76–87), and a β-scaffold region (residues 88–135). There is very little known about the structure of the remainder of the N-terminal region (proximal N terminus, residues 136–405). The C-terminal region contains a cyclic nucleotide-binding homology domain (residues 748–865), which is connected to the transmembrane region via a structured domain usually referred to as the C-linker (residues 667–744). The cytoplasmic domains fine-tune channel gating (8, 9) and play a role in assembly and structural stability of the channels (10–12).
FIGURE 1.
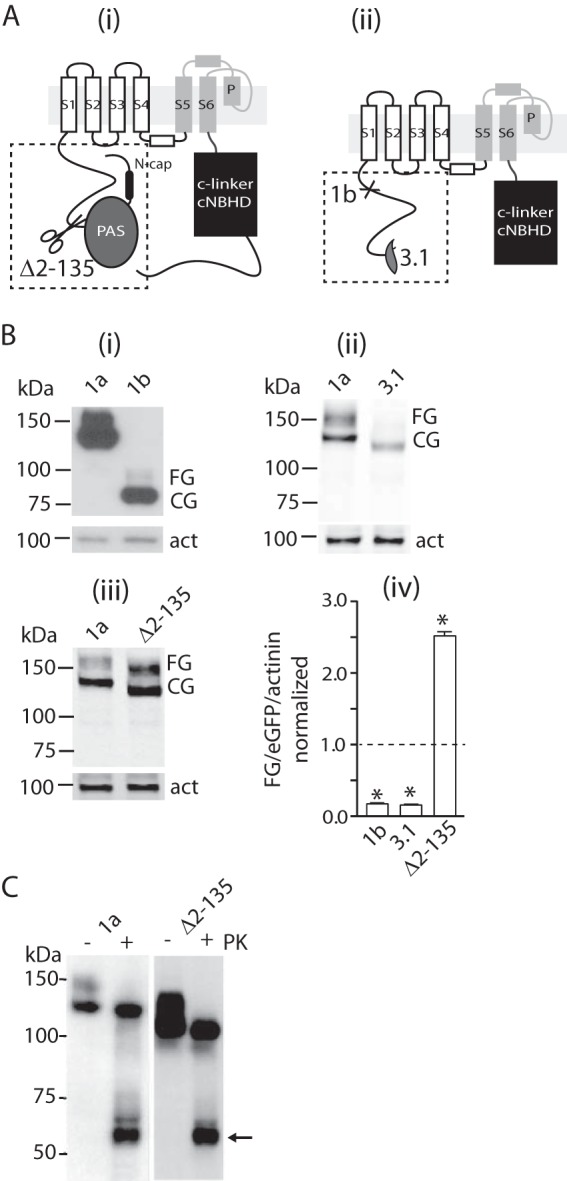
Effect of N-terminal deletions on trafficking of Kv11.1 channels. A, schematic diagrams of Kv11.1a monomers highlighting the N-terminal PAS domain (gray) and the C-linker and cyclic nucleotide binding homology domain (cNBHD, black). Scissors in panel i indicate site of deletion for the Δ2–135 construct. Panel ii illustrates alternatively spliced Kv11.1 isoforms. Kv11.1–3.1 starts midway through the PAS domain, and Kv11.1b is missing the first 373 amino acids, which are replaced by an alternative exon containing 33 amino acids. B, Western blots of whole cell lysates from HEK293 cells expressing Kv11.1a or -1b with C-terminal HA tag (panel i), Kv11.1a or Kv11.1–3.1 (panel ii), and Kv11.1a or Δ2–135 (panel iii) channels. Proteins were probed with anti-HA antibody and IRDye-conjugated secondary antibody for detection on a Li-Cor OdysseyTM system. α-Actinin (act) was probed simultaneously as an internal control for equal loading of samples. Molecular mass markers are indicated in kDa. One aliquot of cell lysates was scanned for expression of eGFP, which was used to normalize variations in transfection efficiency between experiments. Panel iv, summarizes the relative levels of expression of fully glycosylated proteins (FG/actinin/eGFP) normalized against the level for Kv11.1a (mean ± S.E., n = 3). There was a significant increase in expression of FG/actinin/eGFP for Δ2–135 compared with Kv11.1a but significant decreases in expression for Kv11.1b and Kv11.1–3.1 (* indicates p < 0.05). Dashed line indicates level of Kv11.1a expression. C, Western blot of lysates from intact cells transfected with Kv11.1a or Δ2–135 proteins treated with (+) or without (−) proteinase K (PK). The arrow indicates a C-terminal fragment of Kv11.1a generated by proteinase K digest. Molecular mass markers are indicated in kDa. Similar results were obtained for three independent experiments.
In the original x-ray crystal structure of the Kv11.1a PAS domain, the N-Cap was not sufficiently well ordered for its structure to be determined (9). Subsequent NMR studies from a number of laboratories have shown that the N-Cap contains an amphipathic helix (N-Cap helix, residues 13–23) and a flexible N-terminal tail (N-tail, residues 1–12) (13–15). More recently, using a construct in which the flexible N-terminal tail was deleted, Morais-Cabral and co-workers (26) showed that the N-Cap helix can fold back and interact with a surface-exposed hydrophobic patch on the remainder of the PAS domain. In numerous previous studies, it had been suggested that the hydrophobic patch on the surface of the PAS domain was important for domain-domain interactions (9, 16, 17), and it had also been suggested that it may interact with the cNBH domain in the cytoplasmic C terminus (18). Consistent with this hypothesis, Zagotta and co-workers (10) recently showed, using x-ray crystallography, that the interaction between the PAS and cNBH domains from the murine Kv10.1 channel protein (a closely related homologue of Kv11.1) involved the surface-exposed hydrophobic patches on the PAS and cNBH domains as well as the amphipathic N-Cap helix of the PAS domain.
The Kv11.1a PAS domain is a hot spot for clinical mutations that result in reduced stability of the isolated PAS domain and decreased trafficking of the full-length channel (17, 19). This is consistent with the hypothesis that the PAS domain plays an important role in channel assembly and/or stability of channels once they reach the plasma membrane. An alternatively spliced isoform, Kv11.1b, which lacks the PAS domain and a large proportion of the proximal N-terminal cytoplasmic region, also has impaired trafficking to the plasma membrane (20). More recently, another Kv11.1 isoform, denoted Kv11.1–3.1, that is missing the first 102 residues of the PAS domain (and therefore does not contain a folded PAS domain) has been described (see Fig. 1A) (21). As is the case for the Kv11.1b isoform (20), it is possible to record currents from Kv11.1–3.1 channels expressed alone in mammalian cells (21, 22), although the current densities are low. Thus, it is likely that the Kv11.1–3.1 has a reduced trafficking phenotype, but this has not been formally demonstrated. Nevertheless, the observation that currents can be recorded from Kv11.1–3.1 channels expressed alone at the very least suggests that an intact PAS domain may not be essential for stable assembly of Kv11.1 tetramers. The aim of this study was to test whether the PAS domain was critical for channel assembly and trafficking to the cell membrane. Here, we demonstrate that the PAS domain of Kv11.1a is not a prerequisite for the assembly or tetramerization of channels, nor is it required for trafficking of channels to the cell membrane. However, if the PAS domain is present, then the N-Cap is necessary for stabilizing the structure of the PAS domain and hence trafficking of Kv11.1a channels.
EXPERIMENTAL PROCEDURES
Molecular Biology
Expression and purification of recombinant Kv11.1 PAS domain proteins in bacteria were previously described in Ref. 13. A plasmid construct expressing full-length Kv11.1a with a C-terminal FLAG tag or HA tag in pIRES2eGFP (Clontech) has been previously described (23). Missense mutations in Kv11.1a were introduced by site-directed mutagenesis as described previously (17). Kv11.1b, Kv11.1–3.1, and N-terminally truncated Kv11.1a constructs were prepared by PCR cloning in pIRES2eGFP. Constructs were confirmed by automated Sanger sequencing.
Cell Culture and Transfections
Cell culture and transient transfection of the human embryonic kidney cell line (HEK293, European Cell Culture Collections) were previously described (17). In brief, liposome-based transfection reagent Lipofectamine 2000 (Invitrogen) was used to deliver plasmids into the cells. Cells were grown and transfected in 24-well plates and harvested at 48 h after transfection. To investigate the effect of lower temperature on Kv11.1 trafficking, transfected cells were incubated for 16–18 h at 27.5 °C before harvesting. In some experiments, cisapride (10 μm, a Kv11.1 channel pore-blocker) was added to the cell culture media 42 h before harvesting. Stock solutions of cisapride were prepared in DMSO. The final concentration of DMSO in the cell culture media was less than 0.1%. To determine the half-lives of cell surface Kv11.1a channels, cells were incubated in brefeldin A (17) to prevent forward trafficking from the Golgi apparatus, and the rate of degradation of protein at the cell surface was assayed by Western blotting as described below.
SDS-PAGE and Western Blot
Cell lysates of HEK293 cells transfected with Kv11.1a cNDAs were prepared for Western blot analysis as described previously (17). One aliquot of the cell lysates was scanned for eGFP activity (encoded by the pIRES2eGFP vector) on a PherastarTM multiwell plate reader (BMG LabTech, Melbourne, Victoria, Australia). Expression of eGFP in the cell lysates was used to normalize variations in transfection efficiencies of different Kv11.1 constructs. For quantitative Western blot analysis, the nitrocellulose membranes were probed simultaneously with a monoclonal anti-HA antibody (Covance, Princeton, NJ) and an anti-α-actinin or β-actin antibody (Cell Signaling Technology, Danvers, MA) followed by anti-mouse IRDye800 or IRDye680 (Li-Cor Biotechnology, Lincoln, NE) and scanned on a Li-Cor OdysseyTM system. The Odyssey application software (version 3) was used to quantify relevant protein bands with α-actinin or β-actin used as loading controls. For nonquantitative analysis, the membranes were probed with horseradish peroxidase (HRP)-conjugated anti-HA (HA-HRP, Sigma) or anti-FLAG antibody (FLAG-HRP, Sigma).
Double Mutant Cycle Analysis
To investigate whether pairs of residues were energetically coupled, we calculated whether the perturbation to trafficking caused by individual mutations (ΔTx and ΔTy) was additive when combined in the double mutant (ΔTxy). Additive effects in the double mutant (i.e. ΔTxy = ΔTx + ΔTy) would indicate a lack of energetic coupling, whereas nonadditive double mutants indicate an energetic interaction (24). To quantify the assembly of channels, the level of fully glycosylated protein was normalized for cell density (estimated from level of actinin expression) as well as transfection efficiency (estimated from the level of eGFP expression). As the difference in free energy between two end states is proportional to the logarithm of the equilibrium constant, we estimated the perturbation caused by a mutation as shown in Equation 1,
Proteinase K Digest of Cell Surface Kv11.1
Proteinase K digest of Kv11.1 proteins present at the surface of HEK293 cells has been described previously (17). In brief, transfected cells were incubated with 200 μg/ml proteinase K (Roche Applied Science) for 45 min at 37 °C and subsequently lysed for Western blot analysis after inactivating and removing proteinase K from the cells.
Coimmunoprecipitation
Kv11.1a or -1b constructs were tagged with a FLAG epitope (DYKDDDDK), and Kv11.1 constructs for immunoprecipitation were tagged with an HA epitope (YPYDVPDYA), and experiments performed as described previously (17).
Estimation of Molecular Mass
PAS domain proteins were expressed and purified as described previously (17). To determine the molecular mass of WT and mutant PAS domain constructs, we loaded the proteins onto a Superdex 75 10/300GL column (equilibrated with 10 mm HEPES, 150 mm NaCl, 3 mm tris(2-carboxyethyl)phosphine, and 5 mm n-octyl-β-d-glucopyranoside, pH 6.9) with a flow rate of 0.5 ml/min at room temperature. Light scattering, UV light, and refractive index measurements were made on the eluted proteins using either a Viscotek TDAmax (Malvern Instruments, Malvern, UK) or a Mini DAWN Treos (Wyatt Technology Corp., Santa Barbara, CA) system. Data were analyzed using either the OmniSEC (Malvern Instruments) or the Astra V (Wyatt Technology Corp.) software packages.
Thermal Shift Assay of Purified Human Ether-a-Go-Go Related Gene N-terminal Domain Proteins
Thermal shift assay to determine in vitro thermostability of recombinant Kv11.1 PAS proteins has been described previously (17). In brief, 4 μg of protein was mixed with SYPRO Orange (Molecular Probes Inc., Eugene, OR), and the samples were heated from 30 to 95 °C at a rate of 1 °C per min. Protein thermal unfolding curves were monitored by detection of changes in fluorescence of the SYPRO Orange. The melting temperature of WT and mutant PAS domain proteins (Tm) was determined by taking the temperature at which the relative fluorescence unit reached 50% of the maximum.
Statistical Analysis
All data were analyzed using GraphPad Prism version 6 (GraphPad Software Inc, San Diego). Data are presented as mean ± S.E. Statistical comparisons were carried out using a one-way analysis of variance followed by a Bonferroni unpaired t test. A p value of <0.05 was considered significant.
RESULTS
PAS Domain Is Not Required for Trafficking, Assembly, or Stability of Kv11.1 Channels
Transfection of HEK293 cells with full-length Kv11.1a gives rise to two bands on Western blot, one at ∼135 kDa, which represents the core-glycosylated protein, and another at ∼155 kDa, which represents the fully glycosylated (FG) protein (Fig. 1B). Incubation of intact Kv11.1a-expressing cells with proteinase K resulted in the loss of the higher molecular weight band, indicating that this band represents protein at the cell surface (Fig. 1C). In contrast to the full-length Kv11.1a protein, the entire N-terminal truncated isoform, Kv11.1b, or the partial PAS domain truncated isoform, Kv11.1–3.1, gave rise to a prominent lower band (at ∼95 and ∼125 kDa, respectively) associated with the core-glycosylated protein but only faint higher molecular weight bands (at ∼115 and ∼140 kDa, respectively, Fig. 1B) associated with the fully glycosylated forms, indicating that these truncated proteins do not make it to the membrane surface. Conversely, when cells were transfected with constructs lacking just the entire PAS domain (Δ2–135 channels), there was again two clear bands, at ∼120 and 140 kDa (Fig. 1B), the largest of which was removed by treatment with proteinase K (Fig. 1C). Thus, deletion of the PAS domain does not by itself reduce trafficking of the channel protein to the membrane surface.
Kv11.1a, Δ2–135-Kv11.1a, Kv11.1b, and Kv11.1–3.1 channels were all able to coimmunoprecipitate with coexpressed Kv11.1a or Kv11.1b in HEK293 cells (Fig. 2). Furthermore, Δ2–135 Kv11.1a was able to restore trafficking of the Kv11.1b subunits, similar to that seen when Kv11.1a was coexpressed with Kv11.1b (see highlighted box in Fig. 2B). Thus, the PAS domain is not required for subunit-subunit interactions of Kv11.1 channels.
FIGURE 2.
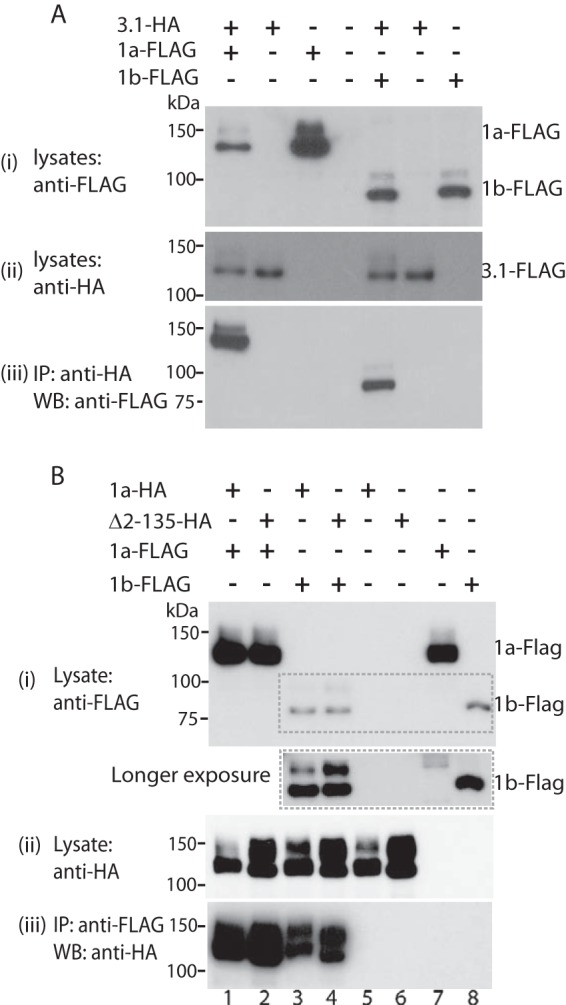
Effect of N-terminal deletions on subunit assembly of Kv11.1 channels. FLAG-tagged Kv11.1a or Kv11.1b was transiently cotransfected with HA tagged Kv11.1–3.1 (A), Kv11.1a or Δ2–135 (B) into HEK293 cells. Cell lysates were immunoprecipitated with anti-HA antibody and protein G beads. Cells transfected with Kv11.1a or Kv11.1b alone were used as negative controls. B, longer exposure of the region highlighted by the dashed box in the top panel (inset below panel) highlights the increased expression of FG protein for Kv11.1b in cells when coexpressed with Kv11.1a or Δ2–135 (compare lanes 3 and 4 with lane 8). Molecular mass markers are indicated in kDa. Similar results were obtained for three independent experiments. IP, immunoprecipitation. WB, Western blot.
To assess whether deletion of the PAS domain affected the stability of assembled channels, we used brefeldin-A (BFA) chase assays to compare the rate of degradation of the full-length and PAS-truncated (Δ2–135) Kv11.1a channels at the cell surface (see under “Experimental Procedures” for details). Typical examples of experiments for monitoring degradation of Kv11.1a and Δ2–135 channels are shown in Fig. 3A. The half-lives, calculated by fitting single exponentials to the time course data in Fig. 3B, were not statistically significantly different for Kv11.1a and Δ2–135 Kv11.1 proteins (12.5 ± 0.4, n = 3, and 13.6 ± 1.5, n = 3, respectively), suggesting that deletion of the PAS domain does not alter stability of the fully glycosylated form of the protein.
FIGURE 3.
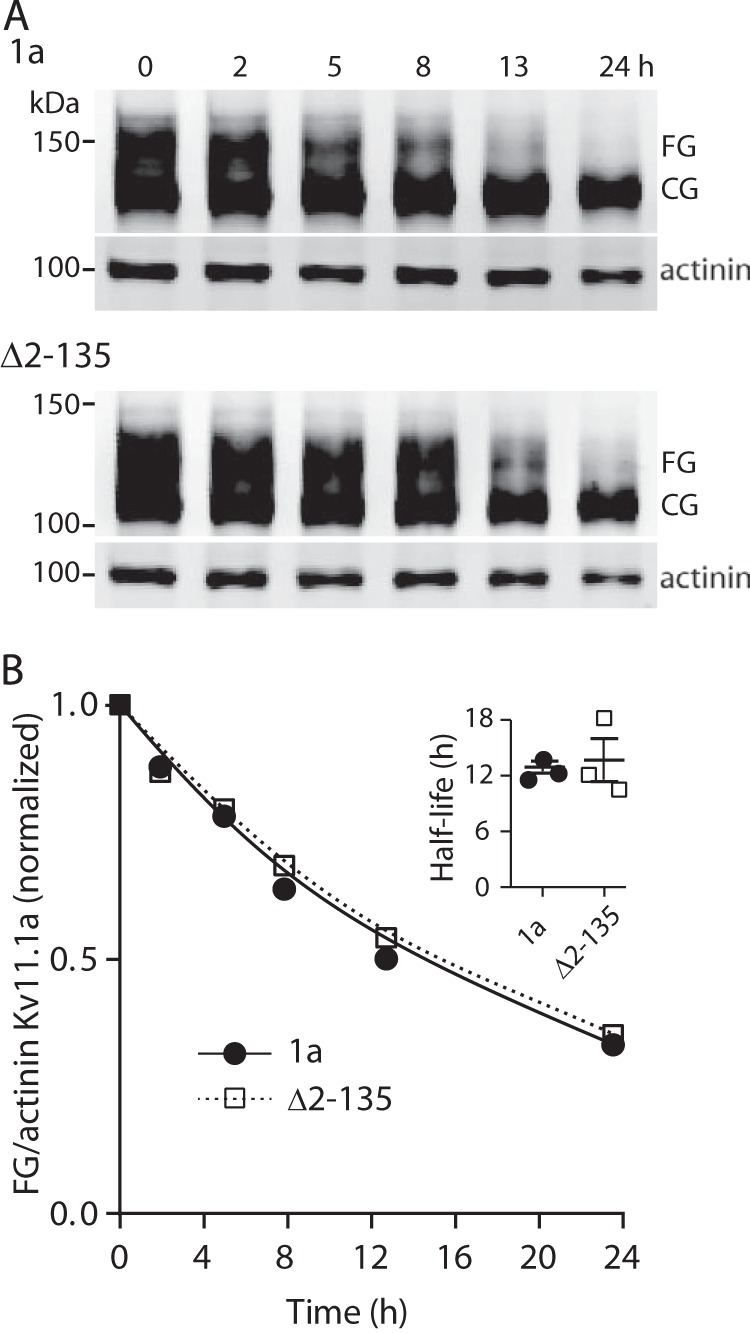
Effect of PAS domain deletion on stability of channels located at the plasma membrane. A, Western blot analysis of time-dependent degradation of FG protein of Δ2–135 and Kv11.1a. HEK293 cells expressing full-length Kv11.1a or Δ2–135 proteins were subject to overnight cell culture at 28 °C before BFA was added and returning to 37 °C. Cells were harvested at time points as indicated up to 24 h after BFA treatment. Molecular mass markers are indicated in kDa. B, intensities of the FG proteins (normalized by α-actinin) at each time point were normalized to the value at t = 0 and plotted against time. The data points were fitted with a single exponential to determine the rate of degradation. Inset shows summary of the values for the half-life (mean ± S.E., n = 3, p = not significant) of full-length and Δ2–135 Kv11.1a FG proteins.
N-Cap Is Required for Trafficking of Kv11.1a When the PAS Domain Is Present
The N-terminal PAS domain Cap (N-Cap, residues 1–25) of Kv11.1a contains an amphipathic helix (residues 13–23) and a flexible tail (residues 1–12). To investigate whether the N-Cap is important for protein trafficking, we expressed N-Cap truncated mutants, Δ2–9 and Δ2–25, as well as a GGS mutant in which residues 15–23 of the amphipathic helix were replaced with a flexible PDGGSGGSG linker (Fig. 4A). When the entire N-Cap was removed (Δ2–25), or the amphipathic helix disrupted (GGS), only a band at ∼135 kDa was seen on Western blots (Fig. 4B). Conversely, when just the flexible tail region of the N-Cap was removed (Δ2–9), the protein had a normal trafficking phenotype with both ∼135- and ∼155-kDa bands present (Fig. 4B). Proteinase K completely removed the ∼155-kDa FG form of the Δ2–9-construct (data not shown) similar to that seen for full-length Kv11.1a channels (see Fig. 1C). These results suggest that in channels containing a PAS domain, the amphipathic helix in the N-Cap must also be present for the channels to traffic normally. The N-Cap, however, is not required for assembly of the channels with either Kv11.1a or Kv11.1b subunits (Fig. 4, D and E).
FIGURE 4.
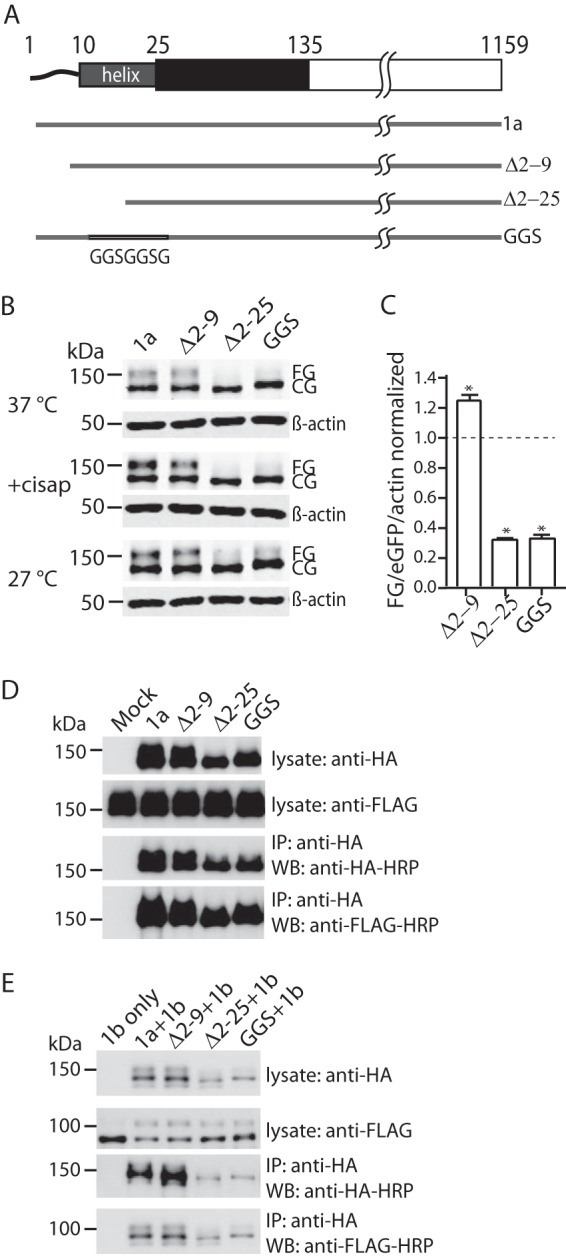
Effect of deleting the PAS domain N-Cap amphipathic helix on maturation of Kv11.1a. A, schematic drawings of the N-Cap mutant constructs used for trafficking assays. B, Western blot of Kv11.1a, Δ2–9, Δ2–25, and GGS mutants expressed in HEK293 cells. Cells were cultured at 37 °C (top panel), 27 °C for 18 h (bottom panel), or in the presence of cisapride (cisap) (10 μm) for 42 h (middle panel) before cell lysis. Note that images shown are from samples analyzed on the same SDS-PAGE and Western blot membrane. Molecular mass markers are indicated in kDa. C, summary of the relative expression of fully glycosylated proteins (FG/actin/eGFP) for Δ2–9, Δ2–25, and GGS normalized to the value for full-length Kv11.1a (mean ± S.E., n = 3). * indicates p < 0.05 for mutants compared with full-length Kv11.1a. D, HA-tagged Δ2–9, Δ2–25, and GGS mutants were transiently transfected into a HEK293 cell line stably expressing FLAG tagged Kv11.1a. Cell lysates were immunoprecipitated with anti-HA antibody and protein G beads. All mutants were able to coimmunoprecipitate Kv11.1a channels. Similar results were obtained for three independent experiments. IP, immunoprecipitation. WB, Western blot. E, HA-tagged Δ2–9, Δ2–25, and GGS mutants were transiently cotransfected with FLAG-tagged Kv11.1b into HEK293 cells. Cell lysates were immunoprecipitated with anti-HA antibody and protein G beads. All mutants were able to coimmunoprecipitate Kv11.1a channels. Similar results were obtained for three independent experiments. IP, immunoprecipitation. WB, Western blot.
Effect of the N-Cap on Stability of the Isolated PAS Domain and Full-length Channels
To investigate why deletion of the N-Cap reduces channel trafficking, we first investigated how removal of the N-Cap affected the in vitro stability of recombinant Kv11.1a PAS domains (Fig. 5). The first notable finding from these in vitro studies was that when the N-Cap was deleted (Δ2–25) or the N-Cap amphipathic helix disrupted (GGS), then the isolated proteins eluted from the gel filtration columns much earlier than the WT PAS domain (Fig. 5A). This suggested that the Δ2–25 and GGS mutants might be forming dimers, which we confirmed was the case using multiangle light scattering (Fig. 5B). Conversely, the WT and Δ2–9 constructs were stable as monomers. Thus, an intact N-Cap helix was required to prevent dimerization of the isolated PAS domain. The Δ2–25 and GGS mutant PAS domains also had significantly reduced thermostabilities (40.9 ± 0.9 and 45.8 ± 0.4 °C, respectively, compared with 55.5 ± 0.6 °C for the WT PAS domain). There was a slight increase in the thermostability for the Δ2–9 PAS domain (58.3 ± 1.2 °C), although this was not statistically significantly different from the Tm for the WT PAS domain.
FIGURE 5.
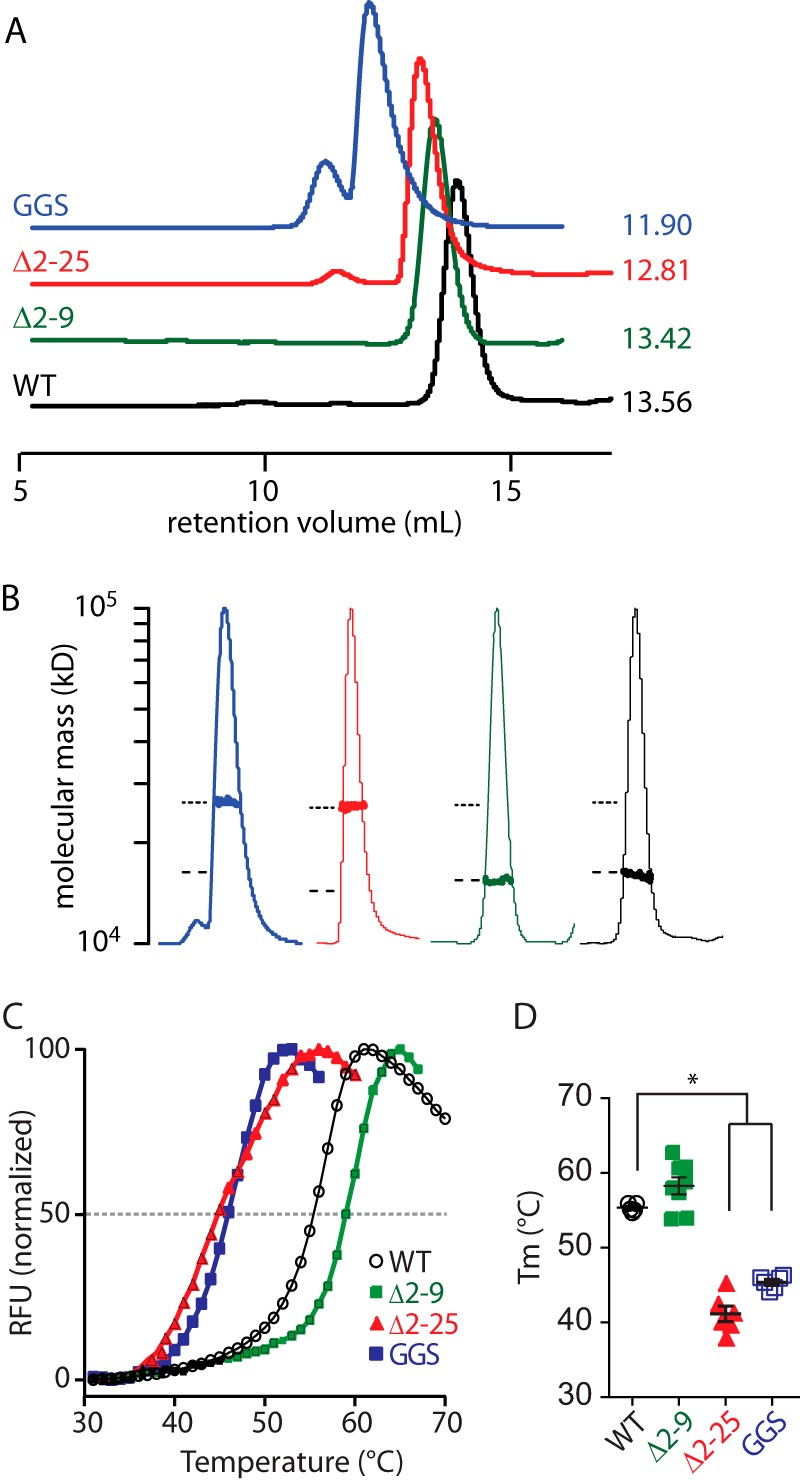
Role of the PAS domain N-Cap on the stability of the isolated Kv11.1a PAS domain. A, size exclusion chromatograms of WT (1–135, black) and mutant PAS (Δ2–9, green; GGS, blue; and Δ2–25, red) domains. Elutions were monitored by absorption at 280 nm. The retention volume for each peak is indicated next to the chromatogram. B, molecular weight of each PAS domain construct was measured using size exclusion chromatography-multiangle laser. The peak region of the normalized UV trace for each PAS domain is shown (thin lines, identical colors to A), and the measured molecular weight (bold lines) is plotted on a log scale for the peak region. The dashed and dotted lines indicate the expected molecular masses for a monomer and dimer. C, fluorescence traces of thermal stability assays for isolated Kv11.1a PAS domain proteins. The midpoint of the increasing fluorescence phase was determined as the melting temperature (Tm) of the protein, as indicated by dashed line. RFU, relative fluorescence unit. D, summary of Tm (mean ± S.E., n = 6–8) for mutant Kv11.1a PAS domains in comparison with WT Kv11.1a PAS domain. * indicates p < 0.05.
We analyzed the in vivo stability of full-length N-Cap mutant channels using a BFA-based chase assay as described above for Kv11.1a and Δ2–135 Kv11.1a proteins. We could not perform this assay on Δ2–25 and GGS mutant channels as these two mutants failed to express sufficient levels of FG proteins even after 16 h of cell culture at 28 °C. A typical example of Western blots obtained for Δ2–9 Kv11.1a proteins 1–24 h after incubation with BFA is shown in Fig. 6A, and the mean data from three independent experiments are shown in Fig. 6B. The gray line in Fig. 6B shows the data for full-length Kv11.1a proteins from Fig. 3B. The half-life for Δ2–9 Kv11.1a was 15.4 ± 0.6 h (n = 3), which is statistically significantly longer than that of full-length Kv11.1a (12.5 ± 0.4, n = 3, p = 0.04).
FIGURE 6.
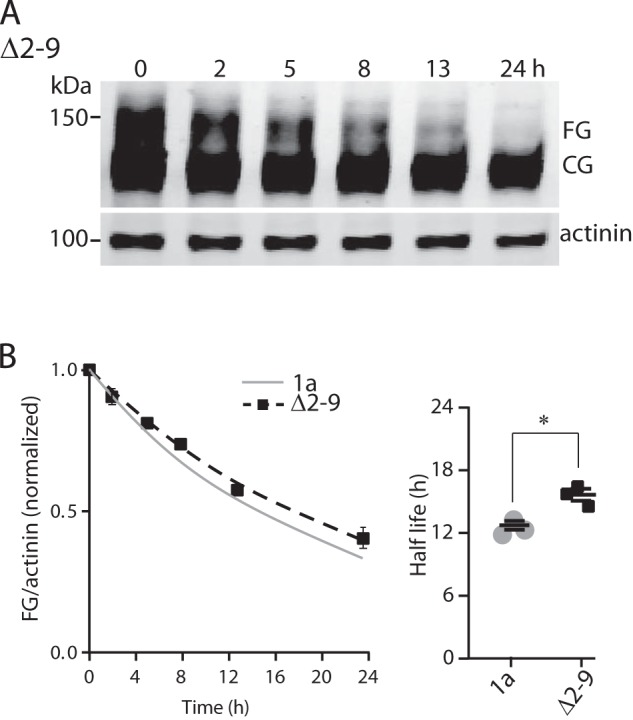
Role of the PAS domain N-Cap on the stability of the Kv11.1a proteins. A, Western blot analysis of time-dependent degradation of FG protein of Δ2–9 Kv11.1a. HEK293 cells expressing Δ2–9 Kv11.1a proteins were subject to overnight cell culture at 28 °C before BFA was added and returning to 37 °C. Cells were harvested at time points as indicated up to 24 h after BFA treatment. Molecular mass markers are indicated in kDa. B, intensities of the FG proteins (normalized by α-actinin) at each time point (mean ± S.E., n = 3) were normalized to the value at t = 0 and plotted again time. The data points were fitted with a single exponential to determine the rate of degradation. The gray line shows the fit for the Kv11.1a data from Fig. 3B. Inset shows summary of half-lives (mean ± S.E., n = 3) for Kv11.1a and Δ2–9 Kv11.1a FG proteins. * indicates p < 0.05
Role of Hydrophobic Face of the N-Cap Amphipathic Helix
Misfolding of proteins typically results in exposure of hydrophobic regions, which in turn causes the protein to be tagged for degradation before reaching the membrane surface (25). Recent crystallography studies suggest that the N-Cap amphipathic helix of Kv11.1a can bind to a surface-exposed hydrophobic patch on the PAS domain (26). Accordingly, we hypothesized that the reduced thermostability and severely impaired trafficking of the N-Cap truncated constructs was a result of the PAS domain hydrophobic patch no longer being covered by the N-Cap amphipathic helix. If this is the case, then we would also expect point mutations on the hydrophobic face of the N-Cap amphipathic helix to decrease the helix binding stability and result in decreased trafficking of mutant channels. To investigate this hypothesis, we performed mutagenesis scans of the N-Cap amphipathic helix to either a relatively hydrophobic alanine side chain or a polar serine side chain (Fig. 7). All individual alanine mutants resulted in channels with a relatively normal trafficking phenotype (Fig. 7A). Conversely, mutation of three bulky hydrophobic residues in the amphipathic helix (Leu-15, Ile-18, and Ile-19) to serine severely reduced trafficking (Fig. 7B). The other serine mutants had a normal trafficking phenotype (Fig. 7B). These results suggest that the hydrophobic face of the N-Cap amphipathic helix is important for normal trafficking of Kv11.1a channels.
FIGURE 7.
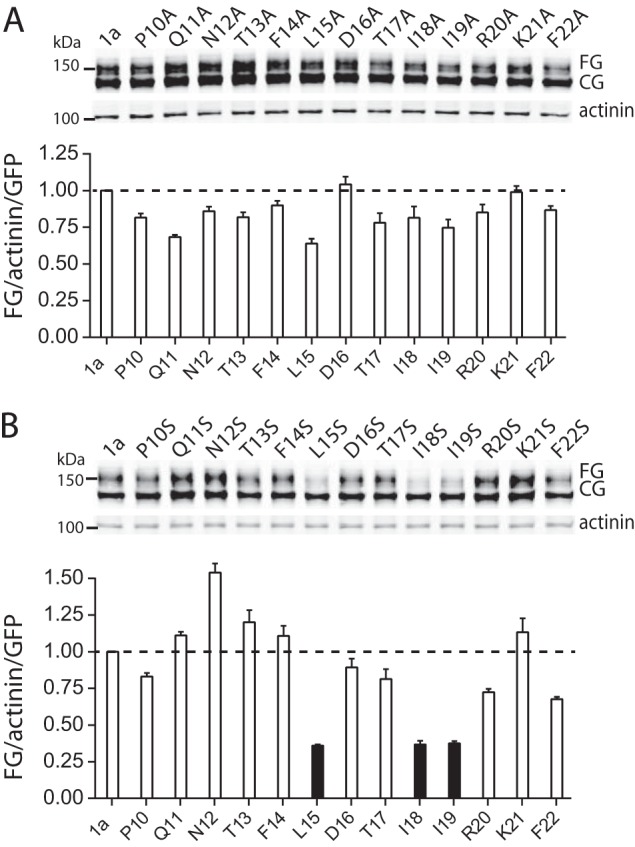
Alanine and serine scan mutagenesis of PAS domain N-Cap amphipathic helix. A, Western blot of alanine scan mutants expressed in HEK293 cells (top panel). Molecular mass markers are indicated in kDa. Summary (bottom panel) of the expression of fully glycosylated Kv11.1a proteins (FG/GFP/α-actinin) normalized to the value of WT Kv11.1a (bars indicate mean ± S.E., n = 3). B, Western blot of serine scan mutants expressed in HEK293 cells (top panel). Molecular mass markers are indicated in kDa. Summary (bottom panel) of the expression of fully glycosylated Kv11.1a proteins (FG/GFP/α-actinin) normalized to the value of WT Kv11.1a (bars indicate mean ± S.E., n = 3). * indicates p < 0.05.
In a previous study, we showed that mutations involving residues in the hydrophobic patch on the PAS domain resulted in either a severe reduction (F29L, I31S, I42N, and Y43C) or moderate reduction (M124R) in trafficking of the full-length protein. Structural models of the truncated PAS domain (residues 10–135), based on the crystal structure from Adaixo et al. (26), suggest that Leu-15, Ile-18, and Ile-19 would interact with this patch. We used a double mutant cycle approach to investigate whether L15S, I18S, and/or I19S had additive effects on the trafficking phenotype of the M124R mutant. We chose to use M124R as it had the mildest phenotype of the PAS domain hydrophobic patch mutants (17), and so it should be easier to see if there was an additive effect.
Example Western blots for L15S, I18S or I19S in either a WT or M124R background are shown in Fig. 8A. We estimated the effect each single and double mutant had on trafficking, ΔTx, from the logarithm of the ratio of the FG bands for a mutant relative to WT on the Western blot (after normalization for transfection efficiency and protein levels, Fig. 8A). The schematic double mutant cycle analysis for I18S and M124R, shown in Fig. 8B, illustrates that the combination of these two mutants would be expected to cause a perturbation of −1.74 log units if the perturbation caused by each mutant were independent of each other. The lower value for the observed perturbation, −1.25 log units, suggests that these two residues are energetically coupled. The perturbations caused by each single and double mutant combination for L15S/I18S/I19S with M124R are summarized in Fig. 8C. These data suggest that Leu-15, Ile-18, and Ile-19 are all energetically coupled to Met-124 and are consistent with the structural model that shows an interaction of the N-Cap amphipathic helix with the hydrophobic patch on the PAS domain (Fig. 8D). These data are also consistent with the structural model, in which the N-Cap amphipathic helix interacts with the hydrophobic patch on the PAS domain (Fig. 8D). Together, these results suggest that the hydrophobic face of the N-Cap amphipathic helix covers a hydrophobic patch on the PAS domain and that this interaction is critical for normal trafficking of the Kv11.1a channel, which would reduce early degradation of the channel protein.
FIGURE 8.
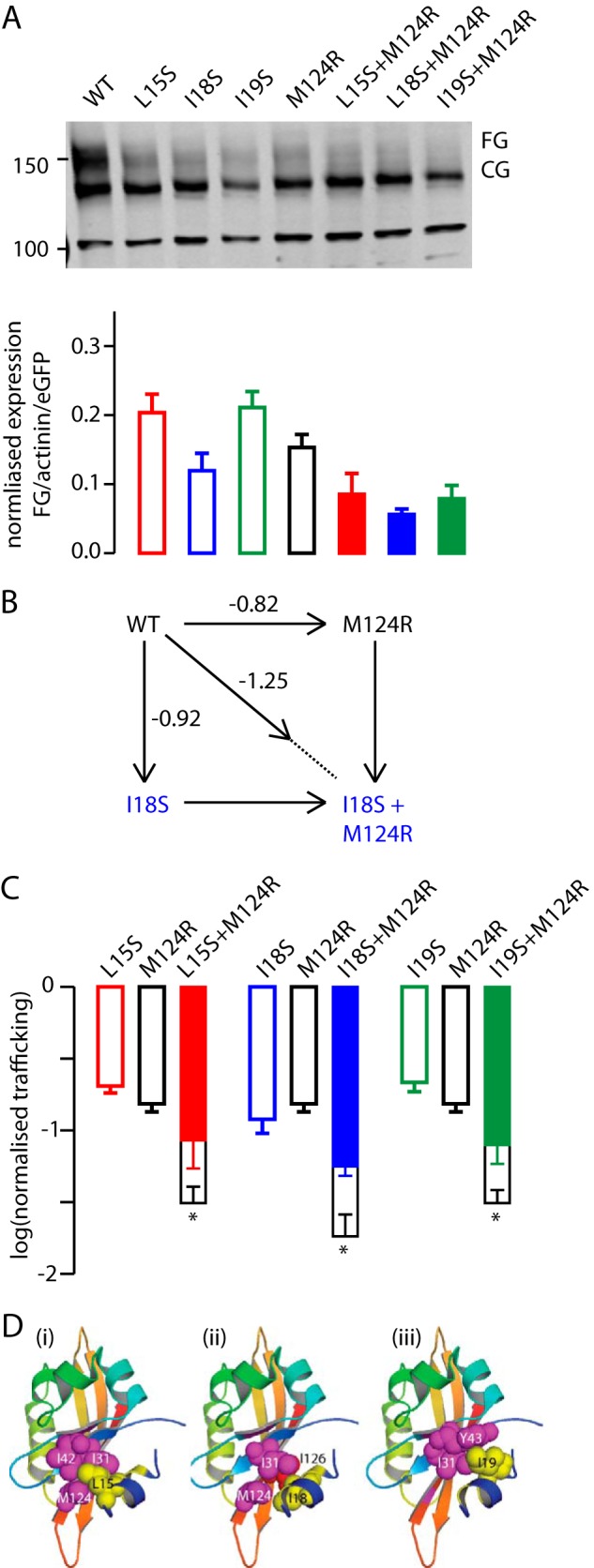
Double mutant cycle analysis of N-Cap-PAS domain interactions. A, Western blot of individual and double mutant Kv11.1a channels expressed in HEK293 cells (top panel). Molecular mass markers are indicated in kDa. Summary (bottom panel) of the expression of fully glycosylated Kv11.1a proteins (FG/GFP/α-actinin) normalized to the value of WT Kv11.1a (bars indicate mean ± S.E., n = 3). Individual mutants are shown as empty bars and double mutants as filled bars. B, double mutant cycle analysis for I18S/M124R. The individual mutants resulted in a Δlog(normalized trafficking) of −0.82 and −0.92 log units, respectively. If these mutants acted independently, then one would expect the double mutant to have perturbed the trafficking by −1.74 log units (i.e. the sum of the individual perturbations). The observed perturbation, however, was only −1.25 log units. C, summary of double mutant cycle analyses for L15S, I18S, and I19S with M124R. In each panel, the observed perturbation for the double mutant is shown as the filled bar and the predicted perturbation (i.e. sum of the individual mutations) is shown as the empty bar. The error bars represent S.E., and for the predicted value for the double mutants, the error bars show the sum of the S.E. values for the corresponding individual mutants. * indicates p < 0.05 (analysis of variance). D, structural models of the Kv11.1a PAS domain (Protein Data Bank code 4HP9) highlighted in yellow, Leu-15 (panel i), Ile-18 (panel ii), and Ile-19 (panel iii). Residues highlighted in magenta are hydrophobic residues that have clinical mutants known to affect the stability of the PAS domain and trafficking of the channels (17).
DISCUSSION
Previous work has demonstrated that the cytoplasmic N-terminal PAS domain of Kv10.1 channels can bind to the cNBH domain in the cytoplasmic C terminus (10). This might also be the case for Kv11.1 channels as these domains share significant sequence homology. Furthermore, it has been postulated that this interaction is critical for normal gating (18). There are numerous studies that have shown that Kv11.1 channels with deletion of large portions of the cytoplasmic N-terminal domain can give rise to robust currents when channels are expressed in Xenopus oocytes (8, 9, 27–29). However, there are only a couple of reports of currents recorded from N-terminal deletion constructs expressed in mammalian cells (22, 30), and in general the current densities are lower than those seen for full-length Kv11.1a channels (22). It is therefore commonly assumed that, although not essential, the presence of the N terminus helps to stabilize the assembled channels (10, 16, 17). Here, we show that deletion of just the PAS domain together with the N-Cap (Δ2–135) results in channels that traffic perfectly well (Fig. 1B). Furthermore, the half-life at the plasma membrane of Kv11.1 channels lacking a PAS domain is just as long as full-length Kv11.1a channels (Fig. 3). Thus, the Kv11.1a PAS domain is not required for the assembly or stability of assembled channels at the cell surface, although it is required for normal gating of the channels (16).
How then can we explain the numerous studies in the literature indicating that mutations in the PAS domain affect trafficking of full-length Kv11.1a channels (17, 19, 31) and in many instances affect domain-domain interactions involving the PAS domain (17, 18)? First, if the PAS domain is present, then it must be fully intact and properly folded. Thus, in Kv11.1–3.1, which has a partially truncated PAS domain, the trafficking is poor. Second, LQT2 mutants that alter stability of the PAS domain result in improper folding and cause impaired trafficking (17, 19). The data in this study clearly demonstrate that if the PAS domain is present, then the N-Cap amphipathic helix of the PAS domain must also be present (Fig. 4). In other PAS domains, the N-Cap can contribute to protein-protein interactions, e.g. in NifL, the hydrophobic face of the N-Cap helix forms a hydrophobic interaction with a neighboring PAS monomer in a domain swapping interaction (7). In the Kv11.1a PAS domain, the amphipathic helix can fold back to interact with the surface-exposed hydrophobic patch on the core of the PAS domain (which incorporates many clinical mutants, including F29L, I31S, I42N, Y43C, and M124R) (26). Here, we report that the interaction between the N-Cap amphipathic helix and the core of the PAS domain has a critical role in stabilizing the isolated PAS domain (Fig. 5). Hence, we suggest that in the intact channels, removal of the N-Cap amphipathic helix would result in exposure of the hydrophobic patch on the surface of the PAS domain, which would likely be recognized by the cellular machinery as being misfolded and therefore tagged for degradation (25). This would explain the severe trafficking defect observed in the N-Cap truncated protein (Fig. 4). Similarly, we suggest it is likely that clinical mutations in the hydrophobic patch on the core of the PAS domain disrupt this self-liganded structure, and hence they are tagged for degradation resulting in poor expression at the membrane, a factor that underlies their pathophysiology.
It is also worth noting that in both the NMR and crystal structures of the full-length Kv11.1a N-Cap/PAS domain, the amphipathic helix was not bound to the PAS domain core, although the amphipathic helix is bound in the recent crystal structure of the Δ2–9 construct (26). This suggests that in the Δ2–9 construct the interaction between the amphipathic helix and the PAS domain could be stronger than in the full-length domain, which is consistent with our findings that the trafficking and stability of Δ2–9 Kv11.1a proteins are improved compared with full-length Kv11.1a and consistent with a slightly higher thermostability of the Δ2–9 PAS domain compared with the full-length PAS domain.
The hydrophobic surface of the N-Cap amphipathic helix contains five bulky hydrophobic residues as follows: Phe-14, Leu-15, Ile-18, Ile-19, and Phe-22. However, it is only the middle three (Leu-15, Ile-18, and Ile-19) that when mutated to serine result in significant perturbation to the trafficking phenotype (Fig. 7). This suggests that it is only this central region that is important for binding to the core of the PAS domain. In the recent crystal structure of the N-truncated Kv11.1 PAS domain, the side chains of Phe-14 and Phe-22 were not discernible. This suggests that these side chains are more flexible and therefore less likely to contribute to binding of the N-Cap amphipathic α-helix, which is consistent with the data in our study.
There are at least three isoforms of Kv11.1, denoted Kv11.1a, Kv11.1b (32), and Kv11.1–3.1 (Fig. 1A) (21). Kv11.1b and Kv11.1–3.1 both lack a folded PAS domain, and Kv11.1b also lacks a large portion of the remainder of the N-terminal cytoplasmic domain. Although it is possible to record currents from homotetrameric Kv11.1b (20) and homotetrameric Kv11.1–3.1 channels (21, 22), they clearly traffic less efficiently (Fig. 1B). The N-terminal region of the Kv11.1b isoform contains an ER retention motif that is not present in Kv11.1a or Kv11.1–3.1, which has been shown to be responsible for the poor trafficking of the Kv11.1b isoform when it is expressed alone (20). Upon coexpression with Kv11.1a or the Δ2–135 construct (Fig. 2), Kv11.1b trafficking is dramatically improved, presumably due to masking of the ER retention motif (20). That this ER motif cannot be masked by coassembly with other Kv11.1b subunits, but can be when expressed with the Δ2–135 construct, suggests that it is the proximal N-terminal domain in Kv11.1a (or Δ2–135) that binds the ER retention motif in heterotetrameric channels. This is also consistent with the finding from Phartiyal et al. (33) who showed that the N-terminal regions of Kv11.1a and Kv11.1b are sufficient for interaction. In the case of the Kv11.1–3.1 isoform, the last 33 residues of the PAS domain are present (21 of these residues are hydrophobic), which we suggest are unlikely to fold into a compact structure and so are more likely to be recognized as unfolded and be tagged for degradation.
A recent x-ray crystallography study of the murine Kv10.1 channel showed a partially truncated PAS domain (residues 2–6 deleted) co-crystallized with the cNBH domain. In this crystal structure, the N-Cap amphipathic helix was intercalated between the PAS domain and the cNBH domain with the interaction surface, including the hydrophobic patch on the core of the PAS domain and a small hydrophobic pocket on the surface of the cNBH domain (10). The hydrophobic pocket on the cNBH domain is conserved between murine Kv10.1 and Kv11.1a (corresponding to residues 794VVVAIL799 in human Kv11.1a). Many mutations in this domain result in channels that have significantly altered gating (34) suggesting that this region also plays an important role in Kv11.1a function. If, as is likely, given the very high sequence homology between murine Kv10.1 and human Kv11.1a, the Kv11.1a PAS and cNBH domains form a similar structure to that reported for murine Kv10.1, then presumably in the Δ2–135 construct the hydrophobic patch on the cNBH domain would be left exposed. As such, one might expect the Δ2–135 channels to be degraded more rapidly than full-length Kv11.1a channels. That this is not the case (see Fig. 3) suggests two possibilities. First, the hydrophobic patch on the exposed cNBH domain is too small, or flanking acidic residues (Asp-793 and Asp-803) prevent this region from being recognized as unfolded. Second, in the absence of the PAS domain, another portion of the channel can bind to and cover the exposed cNBH domain hydrophobic patch. Distinguishing between these possibilities will require further investigation.
The final unexpected finding of this study is that the Δ2–135 channels not only traffic efficiently but they achieve significantly higher steady-state levels of protein expression at the plasma membrane compared with full-length Kv11.1a channels (see Fig. 1B), despite the fact that the rate of degradation of the Δ2–135 protein is comparable with that of full-length Kv11.1a proteins (Fig. 3). Accordingly, we suggest that the higher steady-state levels of Δ2–135 FG protein must be due to an increase in forward trafficking. The synthesis, assembly, and forward trafficking of proteins are complex multistep processes (35). From our results, it is not possible to determine which of the forward processes is altered by deletion of the PAS domain, but clearly this warrants further investigation.
SUMMARY
In this study, we demonstrated that the PAS domain is not necessary for assembly and stability of Kv11.1a channels. However, if the PAS domain is present, then the amphipathic helix in the N-Cap region of the PAS domain must also be present as it plays a critical role in the folding stability of the PAS domain. There is strong evidence to indicate that the PAS domain interacts with cNBH domain of Kv11.1a; however, rather than being an interaction that stabilizes folding of the channel, the PAS-cNBH domain interaction regulates the gating of Kv11.1a channels.
This work was supported by Grant-in-aid G11S 5829 from the Health Foundation of Australia and National Health and Medical Research Council Senior Research Fellowship 1019693 (to J. V.).
- LQT2
- long QT syndrome type 2
- BFA
- brefeldin A
- FG
- fully glycosylated
- PAS
- Per-Arnt-Sim
- eGFP
- enhanced GFP
- N-Cap
- N-terminal Cap
- ER
- endoplasmic reticulum.
REFERENCES
- 1. Vandenberg J. I., Perry M. D., Perrin M. J., Mann S. A., Ke Y., Hill A. P. (2012) hERG K(+) channels: structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478 [DOI] [PubMed] [Google Scholar]
- 2. Curran M. E., Splawski I., Timothy K. W., Vincent G. M., Green E. D., Keating M. T. (1995) A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803 [DOI] [PubMed] [Google Scholar]
- 3. Kaltman J. R., Thompson P. D., Lantos J., Berul C. I., Botkin J., Cohen J. T., Cook N. R., Corrado D., Drezner J., Frick K. D., Goldman S., Hlatky M., Kannankeril P. J., Leslie L., Priori S., Saul J. P., Shapiro-Mendoza C. K., Siscovick D., Vetter V. L., Boineau R., Burns K. M., Friedman R. A. (2011) Screening for sudden cardiac death in the young: report from a national heart, lung, and blood institute working group. Circulation 123, 1911–1918 [DOI] [PubMed] [Google Scholar]
- 4. Knollmann B. C., Roden D. M. (2008) A genetic framework for improving arrhythmia therapy. Nature 451, 929–936 [DOI] [PubMed] [Google Scholar]
- 5. Anderson C. L., Delisle B. P., Anson B. D., Kilby J. A., Will M. L., Tester D. J., Gong Q., Zhou Z., Ackerman M. J., January C. T. (2006) Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 113, 365–373 [DOI] [PubMed] [Google Scholar]
- 6. Moss A. J., Schwartz P. J. (2005) 25th Anniversary of the International Long-QT Syndrome Registry: an ongoing quest to uncover the secrets of long-QT syndrome. Circulation 111, 1199–1201 [DOI] [PubMed] [Google Scholar]
- 7. Key J., Hefti M., Purcell E. B., Moffat K. (2007) Structure of the redox sensor domain of Azotobacter vinelandii NifL at atomic resolution: signaling, dimerization, and mechanism. Biochemistry 46, 3614–3623 [DOI] [PubMed] [Google Scholar]
- 8. Wang J., Trudeau M. C., Zappia A. M., Robertson G. A. (1998) Regulation of deactivation by an amino terminal domain in human ether-a-go-go-related gene potassium channels. J. Gen. Physiol. 112, 637–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morais Cabral J. H., Lee A., Cohen S. L., Chait B. T., Li M., Mackinnon R. (1998) Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 95, 649–655 [DOI] [PubMed] [Google Scholar]
- 10. Haitin Y., Carlson A. E., Zagotta W. N. (2013) The structural mechanism of KCNH-channel regulation by the eag domain. Nature 501, 444–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akhavan A., Atanasiu R., Noguchi T., Han W., Holder N., Shrier A. (2005) Identification of the cyclic-nucleotide-binding domain as a conserved determinant of ion-channel cell-surface localization. J. Cell Sci. 118, 2803–2812 [DOI] [PubMed] [Google Scholar]
- 12. Akhavan A., Atanasiu R., Shrier A. (2003) Identification of a COOH-terminal segment involved in maturation and stability of human ether-a-go-go-related gene potassium channels. J. Biol. Chem. 278, 40105–40112 [DOI] [PubMed] [Google Scholar]
- 13. Ng C. A., Hunter M. J., Perry M. D., Mobli M., Ke Y., Kuchel P. W., King G. F., Stock D., Vandenberg J. I. (2011) The N-terminal tail of hERG contains an amphipathic α-helix that regulates channel deactivation. PLoS One 6, e16191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Q., Gayen S., Chen A. S., Huang Q., Raida M., Kang C. (2010) NMR solution structure of the N-terminal domain of hERG and its interaction with the S4-S5 linker. Biochem. Biophys. Res. Commun. 403, 126–132 [DOI] [PubMed] [Google Scholar]
- 15. Muskett F. W., Thouta S., Thomson S. J., Bowen A., Stansfeld P. J., Mitcheson J. S. (2011) Mechanistic insight into human ether-a-go-go-related gene (hERG) K+ channel deactivation gating from the solution structure of the EAG domain. J. Biol. Chem. 286, 6184–6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gustina A. S., Trudeau M. C. (2012) HERG potassium channel regulation by the N-terminal eag domain. Cell. Signal. 24, 1592–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ke Y., Ng C. A., Hunter M. J., Mann S. A., Heide J., Hill A. P., Vandenberg J. I. (2013) Trafficking defects in PAS domain mutant Kv11.1 channels: roles of reduced domain stability and altered domain-domain interactions. Biochem. J. 454, 69–77 [DOI] [PubMed] [Google Scholar]
- 18. Gustina A. S., Trudeau M. C. (2011) hERG potassium channel gating is mediated by N- and C-terminal region interactions. J. Gen. Physiol. 137, 315–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harley C. A., Jesus C. S., Carvalho R., Brito R. M., Morais-Cabral J. H. (2012) Changes in channel trafficking and protein stability caused by LQT2 mutations in the PAS domain of the HERG channel. PLoS One 7, e32654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Phartiyal P., Sale H., Jones E. M., Robertson G. A. (2008) Endoplasmic reticulum retention and rescue by heteromeric assembly regulate human ERG 1a/1b surface channel composition. J. Biol. Chem. 283, 3702–3707 [DOI] [PubMed] [Google Scholar]
- 21. Huffaker S. J., Chen J., Nicodemus K. K., Sambataro F., Yang F., Mattay V., Lipska B. K., Hyde T. M., Song J., Rujescu D., Giegling I., Mayilyan K., Proust M. J., Soghoyan A., Caforio G., Callicott J. H., Bertolino A., Meyer-Lindenberg A., Chang J., Ji Y., Egan M. F., Goldberg T. E., Kleinman J. E., Lu B., Weinberger D. R. (2009) A primate-specific, brain isoform of KCNH2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nat. Med. 15, 509–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heide J., Mann S. A., Vandenberg J. I. (2012) The schizophrenia-associated Kv11.1–3.1 isoform results in reduced current accumulation during repetitive brief depolarizations. PLoS One 7, e45624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao J. T., Hill A. P., Varghese A., Cooper A. A., Swan H., Laitinen-Forsblom P. J., Rees M. I., Skinner J. R., Campbell T. J., Vandenberg J. I. (2009) Not all hERG pore domain mutations have a severe phenotype: G584S has an inactivation gating defect with mild phenotype compared to G572S, which has a dominant negative trafficking defect and a severe phenotype. J. Cardiovasc. Electrophysiol. 20, 923–930 [DOI] [PubMed] [Google Scholar]
- 24. Perry M. D., Ng C. A., Vandenberg J. I. (2013) Pore helices play a dynamic role as integrators of domain motion during Kv11.1 channel inactivation gating. J. Biol. Chem. 288, 11482–11491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ellgaard L., Helenius A. (2003) Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191 [DOI] [PubMed] [Google Scholar]
- 26. Adaixo R., Harley C. A., Castro-Rodrigues A. F., Morais-Cabral J. H. (2013) Structural properties of PAS domains from the KCNH potassium channels. PLoS One 8, e59265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aydar E., Palmer C. (2001) Functional characterization of the C-terminus of the human ether-a-go-go-related gene K(+) channel (HERG). J. Physiol. 534, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schönherr R., Heinemann S. H. (1996) Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J. Physiol. 493, 635–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Viloria C. G., Barros F., Giráldez T., Gómez-Varela D., de la Peña P. (2000) Differential effects of amino-terminal distal and proximal domains in the regulation of human erg K(+) channel gating. Biophys. J. 79, 231–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fernández-Trillo J., Barros F., Machín A., Carretero L., Domínguez P., de la Peña P. (2011) Molecular determinants of interactions between the N-terminal domain and the transmembrane core that modulate hERG K+ channel gating. PLoS One 6, e24674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gianulis E. C., Trudeau M. C. (2011) Rescue of aberrant gating by a genetically encoded PAS (Per-Arnt-Sim) domain in several long QT syndrome mutant human ether-a-go-go-related gene potassium channels. J. Biol. Chem. 286, 22160–22169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones E. M., Roti Roti E. C., Wang J., Delfosse S. A., Robertson G. A. (2004) Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. J. Biol. Chem. 279, 44690–44694 [DOI] [PubMed] [Google Scholar]
- 33. Phartiyal P., Jones E. M., Robertson G. A. (2007) Heteromeric assembly of human ether-a-go-go-related gene (hERG) 1a/1b channels occurs cotranslationally via N-terminal interactions. J. Biol. Chem. 282, 9874–9882 [DOI] [PubMed] [Google Scholar]
- 34. Al-Owais M., Bracey K., Wray D. (2009) Role of intracellular domains in the function of the hERG potassium channel. Eur. Biophys. J. 38, 569–576 [DOI] [PubMed] [Google Scholar]
- 35. Delisle B. P., Anson B. D., Rajamani S., January C. T. (2004) Biology of cardiac arrhythmias: ion channel protein trafficking. Circ. Res. 94, 1418–1428 [DOI] [PubMed] [Google Scholar]