

Background: Mycobacterium tuberculosis PknB plays a critical role in modulating cell division and cell wall synthesis.
Results: We present a comprehensive evaluation of the importance of various domains of PknB in modulating cell survival.
Conclusion: The intracellular kinase domain and extracytoplasmic PASTA domains of PknB are essential for cell survival.
Significance: PknB is essential for both in vitro growth and survival of the pathogen in vivo.
Keywords: Bacterial Protein Kinases, Cell Growth, Cell Signaling, Mycobacteria, Protein Phosphorylation, PASTA Domain, PknB
Abstract
The Mycobacterium tuberculosis protein kinase B (PknB) comprises an intracellular kinase domain, connected through a transmembrane domain to an extracellular region that contains four PASTA domains. The present study describes the comprehensive analysis of different domains of PknB in the context of viability in avirulent and virulent mycobacteria. We find stringent regulation of PknB expression necessary for cell survival, with depletion or overexpression of PknB leading to cell death. Although PknB-mediated kinase activity is essential for cell survival, active kinase lacking the transmembrane or extracellular domain fails to complement conditional mutants not expressing PknB. By creating chimeric kinases, we find that the intracellular kinase domain has unique functions in the virulent strain, which cannot be substituted by other kinases. Interestingly, we find that although the presence of the C-terminal PASTA domain is dispensable in the avirulent M. smegmatis, all four PASTA domains are essential in M. tuberculosis. The differential behavior of PknB vis-à-vis the number of essential PASTA domains and the specificity of kinase domain functions suggest that PknB-mediated growth and signaling events differ in virulent compared with avirulent mycobacteria. Mouse infection studies performed to determine the role of PknB in mediating pathogen survival in the host demonstrate that PknB is not only critical for growth of the pathogen in vitro but is also essential for the survival of the pathogen in the host.
Introduction
Protein phosphorylation/dephosphorylation is a critical regulatory mechanism by which extracellular cues are transduced into cellular responses. Traditionally, two-component systems comprising a histidine kinase sensor and the associated response regulator were thought to be responsible for phosphorylation-mediated signaling in prokaryotes (1, 2). Two-component pathways are rare in eukaryotes, and phosphotransfer-based signaling predominantly involves phosphorylation/dephosphorylation on serine, threonine, or tyrosine residues, often in a cascade. The presence of a eukaryotic-like serine/threonine protein kinase (STPK)4 in a prokaryote was first reported in 1991 (pkn1 in Myxococcus xanthus) (3). These “eukaryotic-like” STPKs play important roles in bacterial cellular processes, including cell division, cell wall synthesis, cell metabolism, and dormancy exit (4–7). Analysis of the Mycobacterium tuberculosis genome sequence suggested the presence of 11 putative eukaryotic-like STPKs and three protein phosphatases (8). Except for PknG and PknK, all of these kinases were predicted to have a transmembrane domain (8, 9). All of the kinases possessed the protein kinase “signature” motifs, including 11 conserved subdomains as per Hanks' criteria, and amino acid sequence alignment of these STPK family members revealed that 15 catalytically important residues were conserved across all of them (9, 10). The M. tuberculosis STPKs affect key mycobacterial processes: signal transduction mediated by PknA and PknB plays an important role in determining cell shape, morphology, and possibly cell division (7); PknG and PknH influence M. tuberculosis virulence, adaptation, and growth within the host (11, 12); and PknF affects cell division, growth rate, morphology, and glucose transport (13). Recently, Ortega et al. (14) proposed a role for PknB as a replication switch in response to hypoxia. They demonstrated that PknB activity is necessary for reactivation of cells from the hypoxic state.
Protein kinases A and B, encoded by pknA and pknB, respectively, are part of the operon carrying cistrons coding for protein phosphatase pstP, rodA (involved in cell shape control), and pbpA (involved in peptidoglycan synthesis). This locus also includes two FHA (forkhead-associated) domain-containing genes and is found near the origin of replication throughout the Mycobacterium genus (15). As in most other kinases, PknA and PknB consist of a kinase domain, a juxtamembrane region, a short transmembrane domain, and an extracellular region (9, 16). Both kinases have been found to be essential based on transposon-insertion experiments (17), and the pknB gene can be disrupted by allelic replacement in M. tuberculosis and M. smegmatis only in the presence of a second functional copy of the gene (18). In 2009, Forti et al. (19) generated the M. tuberculosis H37Rv-pptr-pknB conditional mutant in which the genomic copy of pknB was converted to a pristinamycin-inducible copy and demonstrated its essentiality, reaffirming the previous findings.
PknB is among the highly characterized serine/threonine protein kinases of mycobacteria. The kinase domain of PknB (aa 1–279) by itself has been shown to be sufficient for its activity (20); however, the extent of its activity in comparison with full-length protein has not been determined. Crystal structure analysis of the PknB kinase domain found it to be a two-lobed structure (N- and C- terminal lobes) showing conservation of protein fold and catalytic machinery to eukaryotic STPK homologs (21, 22). The N-terminal lobe contains the ATP binding site, whereas the C-terminal lobe is involved in rendering an active state and in stabilizing interactions with the substrate (23). Based on the structures, PknB is proposed to form both back-to-back and front-to-front dimers (24). The dimerization of PknB was shown to be essential for autophosphorylation and activation of kinase through an allosteric mechanism (23). The extracellular domain of PknB is predicted to have four conserved PASTA (penicillin-binding protein and serine/threonine kinase-associated) domains (22, 25). This domain has been suggested to play a role in the recognition of d-alanyl-d-alanine dipeptides used to build up the peptidoglycan layers (25). Using nuclear magnetic resonance and small angle x-ray scattering, the four PASTA domains have been shown to be organized in a linear fashion, in contrast to penicillin-binding protein PBP2x (from Streptococcus pneumoniae), where the two PASTA domains fold over in a compact arrangement (26, 27). The extracellular domain of M. tuberculosis PknB has also been shown to bind muropeptides, and the specific amino acids in the stem peptide that impact the efficacy of the interaction have been identified (28).
Although the PknB structure and the mode of activation in vitro are now understood, structure-function relationships of the various domains have not been investigated in the context of mycobacterial growth and survival. The present study was undertaken to comprehensively evaluate the importance of the different domains in modulating PknB function in vivo. Using pknB conditional mutants in Mycobacterium smegmatis and M. tuberculosis strains, we observe that variable expression of PknB regulates cell growth and morphology, with both overexpression and depletion leading to cell death. Interestingly, we find that the minimum number of PASTA domains required in the extracytoplasmic region varies between avirulent and virulent mycobacteria. Infection studies with a mouse model reveal that depletion of PknB results in clearance of pathogen from the host tissues, indicating definitively that PknB is essential for survival of the pathogen within the host.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Reagents, and Radioisotopes
Bacterial strains used in the study are listed in Table 1. Cloning and expression vectors were purchased or obtained from their respective sources: pENTR-D-TOPO vector (Invitrogen), pMAL-c2x expression vector (New England Biolabs), pNit-1 vector (29), pST-HiT vector (30), and p2Nil suicide delivery vector (31). [γ-32P]ATP (6000 Ci/mmol) was purchased from PerkinElmer Life Sciences. Pristinamycin 1A was purchased from Molcan Corp. All medium components were purchased from BD Biosciences. Restriction/modification enzymes were purchased from New England Biolabs and MBI Fermentas. DNA oligonucleotides and analytical grade chemicals and reagents were purchased from Sigma-Aldrich or GE Healthcare.
TABLE 1.
Strains used in the study
| Strains | Description | Source |
|---|---|---|
| DH5α | E. coli strain used for cloning experiments | Invitrogen |
| BL21 (DE3) codon plus | E. coli strain used for protein expression | Stratagene |
| mc2155 | Wild type M. smegmatis strain | ATCC, 700084 |
| mc2HiT-PknB | mc2155 strain transformed with integrative vector containing ATc inducible PknB at L5 site; Hygr | This study |
| mc2::pNit | mc2155 strain transformed with pNit vector | This study |
| mc2::PknB | mc2155 strain transformed with pNit PknB | This study |
| mc2ΔpknB | M. smegmatis pknB conditional gene replacement mutant | This study |
| mc2ΔpknB::pNit | mc2ΔpknB transformed with pNit-1 vector | This study |
| mc2ΔpknB::PknB | mc2ΔpknB transformed with pNit-PknB construct | This study |
| mc2ΔpknB::PknB-K40M | mc2ΔpknB transformed with pNit-PknB-K40M construct | This study |
| mc2ΔpknB::PknB-KD | mc2ΔpknB transformed with pNit-PknB-KD (aa 1–279) construct | This study |
| mc2ΔpknB::PknB-JM | mc2ΔpknB transformed with pNit-PknB-JM (aa 1–331) construct | This study |
| mc2ΔpknB::PknB-TM | mc2ΔpknB transformed with pNit-PknB-TM (aa 1–352) construct | This study |
| mc2ΔpknB::PknB-(A-TM) | mc2ΔpknB transformed with pNit-PknB- (A-TM) construct | This study |
| mc2ΔpknB::PknB-(H-TM) | mc2ΔpknB transformed with pNit-PknB- (H-TM) construct | This study |
| mc2ΔpknB::PknB-ΔTM | mc2ΔpknB transformed with pNit-PknB-ΔTM construct | This study |
| mc2ΔpknB::PknB-PASTA1 | mc2ΔpknB transformed with pNit-PknB-PASTA1 (aa 1–422) construct | This study |
| mc2ΔpknB::PknB-PASTA2 | mc2ΔpknB transformed with pNit-PknB-PASTA2 (aa 1–490) construct | This study |
| mc2ΔpknB::PknB-PASTA3 | mc2ΔpknB strain transformed with pNit-PknB-PASTA3 (aa 1–557) construct | This study |
| mc2ΔpknB::PknA-B | mc2ΔpknB transformed with pNit PknA-B (aa 1–338 of PknA fused with aa 332–626 of PknB) construct | This study |
| mc2ΔpknB::PknH-B | mc2ΔpknB transformed with pNit PknH-B (aa 1–402 of PknH fused with aa 332–626 of PknB) construct | This study |
| H37Rv | Wild type M. tuberculosis strain | ATCC |
| H37Rv::pNit | H37Rv strain transformed with pNit-1 vector | This study |
| H37Rv::PknB | H37Rv strain transformed with pNit-PknB construct | This study |
| Rv-pptr-B | M. tuberculosis H37Rv pknB conditional expression strain under regulation of pptr promoter | Ref. 19 |
| Rv-pptr-B::pNit | Rv-pptr-B transformed with pNit-1 vector | This study |
| Rv-pptr-B::PknB | Rv-pptr-B transformed with pNit PknB construct | This study |
| Rv-pptr-B::PknB-K40M | Rv-pptr-B transformed with pNit-PknB-K40M construct | This study |
| Rv-pptr-B::PknB-KD | Rv-pptr-B transformed with pNit-PknB-KD construct | This study |
| Rv-pptr-B::PknB-JM | Rv-pptr-B transformed with pNit-PknB-JM construct | This study |
| Rv-pptr-B::PknB-TM | Rv-pptr-B transformed with pNit-PknB-TM construct | This study |
| Rv-pptr-B::PknB-(A-TM) | Rv-pptr-B transformed with pNit PknB- (A-TM) construct | This study |
| Rv-pptr-B::PknB-(H-TM) | Rv-pptr-B transformed with pNit PknB- (H-TM) construct | This study |
| Rv-pptr-B::PknB-ΔTM | Rv-pptr-B transformed with pNit PknB-ΔTM construct | This study |
| Rv-pptr-B::PknB-PASTA1 | Rv-pptr-B transformed with pNit PknB-PASTA1construct | This study |
| Rv-pptr-B::PknB-PASTA2 | Rv-pptr-B transformed with pNit PknB-PASTA2 construct | This study |
| Rv-pptr-B::PknB-PASTA3 | Rv-pptr-B transformed with pNit PknB-PASTA3 construct | This study |
| Rv-pptr-B::PknA-B | Rv-pptr-B transformed with pNit PknA-B construct | This study |
| Rv-pptr-B::PknH-B | Rv-pptr-B transformed with pNit PknH-B construct | This study |
Creation of pknB Conditional Gene Replacement Mutant in M. smegmatis (mc2ΔpknB)
The integration-proficient HiT-PknB construct expressing M. tuberculosis pknB under a tetracycline-inducible promoter was electroporated into M. smegmatis mc2155 to generate the merodiploid strain mc2HiT-pknB. Upstream and downstream flanks of pknB were amplified using specific primers, and the flanks were cloned into HpaI-HindIII sites in the p2Nil vector. This was followed by the cloning of a 6-kb PacI cassette from pGOAL17 into the corresponding site on p2Nil to generate the p2Nil-ΔpknB construct. The two-step recombination procedure described by Parish and Stoker was used to disrupt the pknB gene at its native locus in the genome (31). The mutant mc2ΔpknB (clone numbers 23 and 26) surviving only in the presence of the inducer anhydrotetracycline (ATc) was used for further studies (Fig. 1, A–C). The creation of the M. tuberculosis H37Rv pristinamycin-inducible pknB mutant pptr-pknB (Rv-pptr-B) used in this study was reported earlier (19).
FIGURE 1.

PknB depletion and PknB over expression affects growth of mycobacteria. A, schematic representation of mc2ΔpknB conditional mutant. The primers used for PCR-based confirmation are depicted as F1 and F2 (forward primers) and R1 and R2 (reverse primers). B, agarose gels showing PCR amplification using the respective genomic DNA of different M. smegmatis strains, with various primer sets as indicated. Expected sizes of PCR amplicons are as follows: for the F1-R1 primer pair: wild type, 0.95 kb; mutants, no amplicon; for the F1-R2 primer pair: wild type, 1.98 kb; mutants, 0.49 kb; for the F2-R2 primer pair: wild type, 2.9 kb; mutants, 1.42 kb. Leftmost lane, gene ruler 1-kb DNA ladder; lane 1, mc2155 (wild type); lane 2, mc2HiT-PknB (merodiploid PknB strain); lane 3, mc2ΔpknB-23 (conditional depletion strain); lane 4, mc2ΔpknB-26 (conditional depletion strain); lane 5, negative conditional depletion strain. C, growth of M. smegmatis mc2155, mc2::pNit-PknB, negative mutant clone, and mc2ΔpknB clones 23 and 26 on 7H10 agar plates containing either no antibiotic, hygromycin only, or hygromycin with ATc inducer. D, all of the cultures were seeded at an initial A600 of 0.2 in order to obtain sufficient cell pellets for whole cell lysate (WCL) preparations. WCLs from M. smegmatis mc2155 (mc2155), mc2155 transformed with pNit (mc2::pNit), or pNit-PknB (mc2::PknB) grown for 6 h in the absence or presence of 5 μm IVN inducer and PknB conditional mutant strain M. smegmatisΔpknB (mc2ΔpknB) grown for 6 h in the absence or presence of ATc were subjected to Western blotting with rabbit polyclonal α-PknB and α-GroEL1 antibodies. GroEL1 was used as a loading control to demonstrate equal loading of lysates. E, in vitro growth pattern analysis. All of the cultures were seeded at an initial A600 of 0.02. Growth of the different strains was monitored as described under “Experimental Procedures.” F, schematic representation of M. tuberculosis-pptr-pknB (Rv-pptr-B) mutant. G, M. tuberculosis H37Rv (H37Rv), H37Rv transformed with pNit (Rv::pNit), or pNit-PknB (Rv::PknB) grown to A600 ∼0.8 were used to seed fresh cultures. Cultures were seeded at an initial A600 of 0.2 in order to obtain sufficient cell pellets for WCL preparations. Cultures were grown in the absence or presence of inducer (5 μm IVN) for 4 days. In the case of Rv-pptr-B, freshly seeded cultures (A600 of 0.2) were grown in the presence or absence of pristinamycin for 4 days. WCLs were prepared and subjected to Western blotting with α-PknB, α-PknA, and α-GroEL1 (loading control) antibodies. H, in vitro growth pattern analysis of M. tuberculosis H37Rv cells that are either overexpressing PknB or depleted of PknB. All of the cultures were seeded at an initial A600 of 0.1. Error bars, S.E.
Generation of Plasmid Constructs
Full-length M. tuberculosis pknA, pknB, and pknH genes were amplified from BAC clones (8) using gene-specific primers and Phusion DNA polymerase (New England Biolabs), and the amplicons were cloned into pENTR-D-TOPO, pMAL-c2X, or pNit vector. HiT-PknB was created by subcloning the NdeI-HindIII fragment from pENTR-PknB into the corresponding sites in pST-HiT. The PknB-KD (aa 1–279), PknB-JM (aa 1–331), PknB-TM (aa 1–352), PknB-PASTA1 (aa 1–422), PknB-PASTA2 (aa 1–490), and PknB-PASTA3 (aa 1–557) fragments were amplified from pENTR-PknB, using PknB-specific forward and domain-specific reverse primers, and the amplicons were cloned into pMAL-c2X and pNit vectors. All point mutations were made by overlapping PCR using appropriate mutagenic primers, and the mutations were confirmed by DNA sequencing. To make the PknA-B and PknH-B chimeric proteins, the intracellular domains of PknA or PknH were fused to the transmembrane and extracellular domains of PknB using overlapping PCR.
Protein Expression, Purification, and in Vitro Kinase Assay
pMAL-c2X constructs expressing PknB or its mutants were transformed into Escherichia coli BL21 (DE3) Codon Plus cells (Stratagene), and the MBP-tagged proteins were purified as described (32). His-GarA was expressed and purified as described earlier (33). An in vitro kinase assay was performed as described previously using 2.5 pmol of PknB kinase or its mutants and using 100 pmol of GarA or MyBP as the substrate (33).
Growth Pattern Analysis
The strategy used for creation of the pknB conditional gene replacement mutant in M. smegmatis (mc2ΔpknB) is described above. Electrocompetent cells of mc2155 or H37Rv were transformed with pNit or pNit-PknB to generate mc2::pNit, mc2::PknB, Rv::pNit, and Rv::PknB. To analyze the growth patterns of M. smegmatis mc2155, mc2::pNit, mc2::PknB, and mc2ΔpknB, each strain was grown in LB medium containing 0.05% Tween 80, 0.2% glycerol, and appropriate antibiotic to A600 ∼0.8, either in the absence of inducer isovaleronitrile (IVN) in the case of mc2::pNit and mc2::PknB or in the presence of inducer ATc in the case of mc2ΔpknB mutant. M. tuberculosis transformants Rv::pNit and Rv::PknB as well as the mutant strain Rv-pptr-B were grown to A600 ∼0.8 in 7H9-ADC containing 0.05% Tween 80, 0.2% glycerol, and appropriate antibiotic in the presence of inducer pristinamycin 1A in the case of Rv-pptr-B or in the absence of inducer IVN in the case of H37Rv, Rv::pNit and Rv::PknB. The cultures were washed twice in PBS containing 0.05% Tween 80 and were used to seed fresh cultures at an initial A600 of 0.02 (in the case of M. smegmatis) or 0.1 (in the case of M. tuberculosis) in the presence or absence of inducer. To analyze growth in M. smegmatis strains, A600 was measured every 3–30 h after initiation. To analyze growth in M. tuberculosis strains, A600 was measured every 24 h to 8 days after initiation. Experiments were performed in triplicates, and the average A600 values were plotted as a function of time. S.E. values were calculated using SigmaPlot for each time point. For Western blot analysis, cultures of M. smegmatis strains were initiated at an A600 of 0.2 and were grown for 9 h in the absence or presence of ATc. Cultures of M. tuberculosis strains were likewise initiated and grown in the absence or presence of inducer for 6 days. In order to determine whether the absence of PknB has bacteriostatic or bacteriocidal effects, cultures were withdrawn on days 0, 2, and 5, and different dilutions were spotted on plates containing the inducer pristinamycin 1A. In growth analysis of complementation mutants, mc2ΔpknB or Rv-pptr-B strains were electroporated with pNit or pNit-PknB or pNit constructs containing various PknB mutants, including chimeric proteins, and growth patterns were analyzed as described above.
Scanning Electron Microscopy
In order to perform scanning electron microscopy, cells were incubated for 3 h in fixative (2.5% glutaraldehyde, 4% paraformaldehyde in 0.1 m sodium cacodylate buffer, pH 7.3). Fixed cells were then treated with increasing concentrations of ethanol (ranging from 25 to 100%) stepwise, followed by hexamethyldisilazane treatment. This was followed by coating with gold particles and visualizing under the microscope at ×10,000 magnification (Carl Zeiss, Evo LS scanning electron microscope).
Infection of Mice
M. tuberculosis H37Rv or Rv-pptr-B or Rv-pptr-B::PknB were grown in the presence of pristinamycin until midlogarithmic phase (A600 of 0.6). The bacilli were harvested by centrifugation at 3000 × g, washed twice with PBS, and resuspended in saline. To evenly disperse the bacteria, the suspension was passed through a 27.5-gauge needle 10 times. C57BL/6 mice (6–8 weeks old of either sex) were housed in individually ventilated cages at the Tuberculosis Aerosol Challenge Facility, International Centre for Genetic Engineering and Biotechnology (New Delhi, India). Mice (n = 6) were infected with 2 × 108 colony-forming units of either H37Rv, Rv-pptr-B, or Rv-pptr-B::PknB by the aerosol route using the Madison Aerosol Chamber (University of Wisconsin, Madison, WI) precalibrated to deliver ∼80–150 bacilli/mice. Two mice each were sacrificed at 24 h postexposure, and the lung homogenates were plated on nutrient 7H11 agar in triplicates for assessing the bacterial load. Bacterial loads for each set were again assessed 4 weeks and 8 weeks after aerosol exposure. This was achieved by homogenizing aseptically removed organs in 2 ml of sterile saline containing 0.05% Tween 80 (Sigma-Aldrich). Serial dilutions of homogenates thus obtained were plated on 7H11 plates supplemented with 10% OADC (BD Biosciences), and colonies were counted after 21 days.
Histopathology
Harvested organs were fixed in 10% neutral buffered formalin. The tissue was embedded in paraffin wax and cut into 5-μm sections using a microtome. Sections were then stained with hematoxylin and eosin (H&E) stain and subjected to histopathological analyses. The tissue samples were coded, and a pathologist having no prior knowledge of the experimental groups evaluated lung sections for granulomatous architecture, and the numbers of granulomas were counted. Each granuloma was graded by the following criteria: (a) granulomas with necrosis were given a score of 5; (b) granulomas without necrosis were given a score of 2.5; (c) granulomas with fibrous connective tissue were given a score of 1. Total granuloma score was calculated by multiplying the number of granulomas of each type by the score and then adding them up to obtain a total granuloma score for each sample.
RESULTS
Stringent Regulation of PknB Expression in the Cell Is Necessary for Cell Growth
In order to investigate the role of PknB in modulating physiological events in mycobacteria, we created a PknB conditional knock-out strain. We adopted the approach of integrating an inducible copy of the gene in the genome prior to deleting the pknB gene in M. smegmatis at its native locus. A comparison of the PCR amplicons obtained in potential deletion mutants, wild type strain, merodiploid strain, and negative clone revealed that the native pknB gene had been successfully disrupted (Fig. 1, A–C). In a previous study, M. tuberculosis H37Rv-pptr-PknB (Rv-pptr-B) mutant strain was created with the help of a suicide delivery plasmid, in which the pknB gene in the genome was replaced with a pristinamycin- inducible copy (19) (Fig. 1F). To characterize the functions of PknB, we used the M. smegmatis ΔpknB (mc2ΔpknB) strain created in this study and the Rv-pptr-B mutant from the previous study.
PknB is the last gene of the operon, which, in addition to others, carries the pknA gene, the only other essential serine/threonine protein kinase in M. tuberculosis. We therefore ascertained whether the expression of pknA, which lies immediately upstream of pknB, is affected in the mutants. We found that although the down-regulation of PknB expression in the absence of ATc was evident even after 6 h, it takes up to 9 h for substantial depletion (Figs. 1D and 2C). The expression of PknA was unaffected by the depletion of PknB (Fig. 2C). Conversely, PknB was considerably overexpressed upon induction with IVN in strain mc2::PknB (Fig. 1D). Analysis carried out with M. tuberculosis strains revealed a considerable diminution in the levels of PknB when Rv-pptr-B was grown in the absence of pristinamycin and substantial overexpression of PknB in Rv::PknB strain upon induction with IVN (Figs. 1G and 2A). Here too PknA expression was unaffected by depletion or overexpression of PknB (Fig. 1G). These results suggest that disrupting the pknB at its native location did not have any impact on the expression of other genes in the operon.
FIGURE 2.

Depletion of PknB results in cell lysis. A, in order to monitor the extent of PknB depletion over a period of 6 days, 100 ml of fresh Rv-pptr-B cultures were seeded at an initial A600 of 0.1 (in order to obtain sufficient quantities) and grown in the absence or presence of Pristinamycin 1A for 2, 4, and 6 days. WCLs were subjected to Western blotting with α-PknB and α-GroEL1 antibodies. B, H37Rv or Rv-pptr-B were grown to an A600 of ∼0.8 in presence of inducer were washed twice in PBS containing 0.05% Tween 80 and were used to seed fresh cultures at an initial A600 of 0.1. Cultures were grown in the presence or absence of pristinamycin for 0, 2 or 5 days. Cultures were serially diluted and spotted on 7H10 agar plates containing pristinamycin. Drastic reduction in the titer of Rv-pptr-B grown in the absence of inducer could be observed between days 2 and 5. C, Western blot analysis to check PknB expression in M. smegmatis (mc2155), merodiploid (mc2HiT-PknB), and conditional depletion (mc2ΔpknB) strains. Whole cell lysates were prepared and probed with α-PknB, α-PknA, and α-GroEL1 antibodies. D, morphology of mc2155, mc2ΔpknB, and mc2::PknB, observed by scanning electron microscopy. Cultures were grown in the absence of ATc in the case of mc2ΔpknB and in the presence of 5 μm IVN in the case of mc2::PknB for 9 or 12 h. Scale bar, 1 μm.
We found that the growth of mycobacterial strains, mc2::PknB and Rv::PknB, was not significantly different from that of wild type cells in the absence of inducer IVN (Fig. 1, E and H). Importantly, we found that the overexpression of PknB dramatically compromised growth in the case of both mc2::PknB and Rv::PknB strains (Fig. 1, E and H). Similarly, mc2ΔpknB and Rv-pptr-B growth patterns did not significantly diverge from those of the corresponding wild type strains in the presence of inducers. Depletion of PknB had strong effects on mycobacterial growth in both M. smegmatis and M. tuberculosis (Fig. 1, E and H). Although it was apparent that mycobacteria showed compromised growth upon PknB depletion, it was not clear if the absence of PknB was leading to a bacteriostatic or bactericidal phenotype. In order to differentiate between these possibilities, H37Rv and Rv-pptr-B cultures grown in the presence or absence of inducer for 0, 2, or 5 days were spotted at different dilutions on plates containing pristinamycin (Fig. 2B). Western blot analysis of lysates of Rv-pptr-B prepared from cultures grown with or without pristinamycin after 2, 4, or 6 days showed that efficient depletion of PknB could only be observed after 4–6 days of growth (Fig. 2A). In agreement with this observation, inducer depletion for 2 days had no obvious effect on cell growth (Fig. 2B). However, when cultures were spotted on plates after 5 days of growth in the absence of inducer, we clearly observed drastic reduction (∼3 log fold) in the titer (Fig. 2B). These results strongly suggest that depletion of PknB eventually leads to cell death.
The overexpression of PknB in M. smegmatis and Mycobacterium bovis BCG has been reported to alter the appearance of the cells, making them wider and bulging (7). Because efficient reduction of PknB expression could only be observed 9 h after the removal of inducer (Fig. 2C), scanning electron microscopy was performed after 9 and 12 h of overexpression or removal of inducer. PknB overexpression for 9 h results in cells of uneven width, with certain parts of the cells bulging out (Fig. 2D). Overexpression of PknB for 12 h resulted in the lysis of a majority of the cells, causing an accumulation of cell debris (Fig. 2D). Depletion of PknB over a period of 9 h had an extensive impact on the morphology of the cells. Cells appeared shrunken, and some of the cells seemed to be on the verge of lysis (Fig. 2D). When cells were visualized after depletion over a period of 12 h, extensive lysis was observed in a majority of the cells. Taken together, we conclude that PknB is absolutely essential in both avirulent and virulent strains of mycobacteria, and its depletion or overexpression alters cell morphology, eventually leading to cell death.
Kinase Activity of PknB Is Essential for Cell Survival
PknB is a 626-aa protein with four distinct domains: an N-terminal kinase domain spanning 1–279 aa, a juxtamembrane domain located between 279–331 aa, a 20-amino acid transmembrane domain (residues 332–353 aa), and an extracellular segment 273 amino acids in length, containing four PASTA domains (Fig. 3A). The invariant lysine residue in the kinase domain that interacts with the glutamate residue of αC helix and stabilizes ATP by making contacts with α- and β-phosphates is present at position 40. We expressed MBP-tagged PknB and PknB-K40M (putative kinase-dead mutant) in E. coli and purified the recombinant proteins for in vitro kinase assays using GarA (an FHA domain containing protein that has previously been identified as a substrate of PknG and PknB (34, 35)) as substrate. Results obtained (Fig. 3, B and C) unequivocally demonstrated the necessity of the invariant lysine for PknB kinase activity, in agreement with previous data (36, 37). To examine the importance of PknB kinase activity in mediating cell survival, Rv-pptr-B and mc2ΔpknB strains were transformed with pNit vector, pNit-PknB, or pNit-PknB-K40M, and growth patterns were analyzed. Western blot analysis of whole cell extracts prepared from mc2ΔpknB transformants in the absence of IVN and in the presence and absence of ATc demonstrated robust expression of PknB and PknB-K40M (Fig. 3D). Western blot analysis of Rv-pptr-B transformants also demonstrated robust expression of proteins (Fig. 3F), suggesting that leaky expression from the IVN-inducible promoter is adequate. Analysis of growth (Fig. 3, E and G) showed that conditional mutants of mycobacteria transformed with vector failed to survive in the absence of inducers. Rv-pptr-B and mc2ΔpknB transformed with pNit-PknB grew very well, demonstrating that wild type PknB expressed episomally was capable of functional complementation. On the other hand, mycobacterial strains transformed with plasmids expressing the kinase-dead PknB-K40M did not show growth recovery, thus underlining the importance of PknB kinase activity in mediating cell survival.
FIGURE 3.

Kinase activity of PknB is essential for its functionality. A, schematic representation of PknB depicting the various domains and critical residues. B, in vitro kinase assays were carried out using 80 nm MBP or PknB or PknB-K40M, 3.33 μm GarA, 10 μm ATP, and 10 μCi [γ-32P]ATP. The band corresponding to the phosphorylated GarA (p-GarA) is indicated. C, bands corresponding to phospho-GarA were excised from the gel and quantified by liquid scintillation counting in three independent experiments. The activity was calculated as cpm in phospho-GarA/min/μm enzyme. Activity of PknB was normalized to 100% in each experiment, and the percentage of activity in other samples was calculated with respect to (w.r.t.) PknB activity. The results were plotted with percentage of activity on the y axis and the samples on the x axis in the form of histograms. D, mc2ΔpknB strain was transformed with pNit vector, pNit-PknB, or pNit-PknB-K40M constructs. Cultures grown to A600 ∼0.8 in the presence of inducer (ATc) were washed twice, and fresh cultures were seeded at an initial A600 of 0.2. Cultures were grown in the presence or absence of ATc as indicated above for 6 h and WCLs were resolved and subjected to Western blotting with α-PknB and α-GroEL1 antibodies. E, growth pattern analysis of mc2ΔpknB transformants grown in the absence of ATc. F, Rv-pptr-B transformants grown to A600 ∼0.8 in the presence of inducer pristinamycin 1A were washed twice, and fresh cultures were seeded at an initial A600 of 0.2. Cultures were grown for 4 days in the presence or absence of pristinamycin as indicated above, and WCLs were resolved from transformant strains and subjected to Western blotting with α-PknB and α-GroEL1 antibodies. G, growth pattern analysis of Rv-pptr-B transformants in the absence of pristinamycin. All of the cultures were seeded at an initial A600 of 0.1. Error bars, S.E.
The Extracellular Domain of PknB Is Necessary for Survival
The kinase domain of PknB is capable of autophosphorylation as well as phosphorylation of substrates in vitro (38, 39). However, to date, the relative activities of different deletion constructs of PknB in comparison with the full-length PknB have not been determined. Accordingly, PknB-KD, PknB-JM, and PknB-TM deletion mutants (Fig. 4A) were purified, and their ability to phosphorylate substrates in vitro was determined at a limiting concentration of enzyme (Fig. 4, B and C). The activity of PknB deletion mutants ranged from 25 to 50% when compared with full-length PknB, with PknB-JM being the least active.
FIGURE 4.

Extracellular domain of PknB is essential for in vitro growth. A, schematic representation of PknB and deletion constructs. B, in vitro kinase assays performed using 80 nm MBP or PknB or PknB deletion mutants, 3.33 μm GarA, 10 μm ATP, and 10 μCi of [γ-32P]ATP. Samples were resolved on 12% SDS-PAGE and autoradiographed. C, bands corresponding to phosphorylated GarA (p-GarA) were excised from the gel and quantified by liquid scintillation counting in three independent experiments. Activity of PknB was normalized to 100% in each experiment, and the percentage of activity in other samples was calculated with respect to (w.r.t.) PknB activity. The results were plotted with percentage of activity on the y axis and the samples on the x axis in the form of histograms. D, cultures were seeded at an initial A600 of 0.2. Cultures were grown in the presence or absence of ATc as indicated above for 6 h. For mc2ΔpknB::pNit, mc2ΔpknB::PknB-KD, mc2ΔpknB::PknB-JM, and mc2ΔpknB::PknB-TM strains, 0.2 μm IVN inducer was added during depletion. WCLs prepared were subjected to Western blotting with α-PknB and α-GroEL1 antibodies. The arrows indicate bands corresponding to PknB deletion mutants. E, growth pattern analysis of mc2ΔpknB transformants grown in the absence of ATc. Cultures were seeded at an initial A600 of 0.02. mc2ΔpknB::PknB-KD, mc2ΔpknB::PknB-JM, and mc2ΔpknB::PknB-TM were grown in the absence of ATc and in the presence of 0.2 μm inducer IVN. F, the Rv-pptr-B transformant strains were streaked on 7H10-agar plates containing 2 μg/ml pristinamycin or 0.2 μm IVN. Error bars, S.E.
Next we sought to examine the possibility of various PknB deletion mutants rescuing growth of mycobacteria PknB conditional mutants. Western blot analysis of cell lysates from mc2ΔpknB transformants showed IVN-inducible expression of PknB fragments (Fig. 4D). Growth analysis of mc2ΔpknB transformants revealed that although the wild type PknB was able to rescue growth in cells depleted of endogenous PknB, all of the PknB deletion mutant proteins failed to do so (Fig. 4E). To determine the ability of PknB mutants to complement the Rv-pptr-B, transformants were streaked on solid media in the presence or absence of pristinamycin. Although all of the transformed strains grew robustly in the presence of inducer, full-length PknB alone was able to rescue growth in absence of inducer (Fig. 4F). Thus, whereas PknB kinase activity was essential for functional complementation (Fig. 3), the catalytic domain alone (PknB-KD or PknB-JM) or anchoring of the active kinase to the cell membrane (PknB-TM) was insufficient for rescuing the growth defects. These results suggest that in addition to PknB kinase activity, the extracellular segment is essential for cell survival.
The Localization of PknB to the Cell Membrane Is Essential for Cell Survival
The intracellular and extracellular domains of PknB are connected through a transmembrane (TM) domain of ∼20 amino acids. However, the precise role of the TM in targeting PknB to the cell membrane has not been elucidated. We reasoned that, if the only function of TM domain is to interact with the lipid bilayer, then replacing it with TM domains of other transmembrane kinases should not have any impact on PknB function. To test this hypothesis, we created plasmid constructs in which the PknB transmembrane domain was either deleted or swapped with that of PknB (control), PknA, or PknH (Fig. 5A). Western blot analysis of mc2ΔpknB cells transformed with the various constructs demonstrated proficient expression of the PknB-TM deletion mutant as well as the PknB proteins carrying TM domains of other kinases (Fig. 5B). Growth analysis of transformants showed that swapping the TM domain of PknB with that of PknA or PknH did not affect their ability to complement ΔpknB (Fig. 5C), although PknB-(A-TM) grew marginally slower. However, deletion of the TM domain was not tolerated. This was reflected in the inability of PknB-ΔTM to complement mc2ΔpknB (Fig. 5C), suggesting that localization of PknB to the membrane is essential for cell viability. Results obtained with Rv-pptr-B transformed with the various engineered PknB constructs reflected those obtained with M. smegmatis, thus indicating that the function of the TM domain is conserved between the avirulent and virulent strains (Fig. 5D). These data suggest that although the presence of the TM domain is essential, perhaps for targeting the protein to the cell membrane, specific sequences do not dictate the outcome.
FIGURE 5.

Anchoring of PknB to the cell membrane is essential for cell survival. A, schematic representation of PknB-TM deletion and swap mutations. B, mc2ΔpknB was transformed with pNit vector, pNit-PknB, pNit-PknB-(A-TM), pNit-PknB-(H-TM), or pNit-PknB-ΔTM constructs. Cultures were seeded at an initial A600 of 0.2 and grown in the presence or absence of ATc for 6 h. WCLs prepared were subjected to Western blotting with α-PknB and α-GroEL1 antibodies. C, in vitro growth analysis of mc2ΔpknB transformants performed in the absence of inducer ATc. Cultures were seeded at an initial A600 of 0.02. D, growth analysis of Rv-pptr-B transformants. Rv-pptr-B transformants were streaked on 7H10 agar plates in the presence or absence of 2 μg/ml pristinamycin 1A. Error bars, S.E.
All Four Extracellular PASTA Domains of PknB Are Essential in M. tuberculosis
PASTA domains have been shown to interact with penicillin and are believed to be involved in sensing the environment (25). Further PASTA domains along with the TM domain were shown to be necessary and sufficient for appropriate localization of PknB (28). The data presented in Fig. 4 demonstrate that the extracellular PASTA domains are essential for PknB function. PknB has four extracellular PASTA domains, which have been denoted here as PASTA1 to -4 (Fig. 6A). In order to determine the minimal number of PASTA domains required, we created three constructs, wherein PknB has one (aa 1–421), two (aa 1–490), or three (aa 1–557) PASTA domains (Fig. 6A). Whereas PknB-PASTA2 and PknB-PASTA3 expression was robust, we could not detect PknB-PASTA1 expression, suggesting that the protein may be unstable (Fig. 6, B and D). Growth analysis of mc2ΔpknB transformants unambiguously demonstrated that only PknB-PASTA3 could complement ΔpknB phenotype. PknB-PASTA1 and PknB-PASTA2 failed to rescue mc2ΔpknB growth defects in the absence of ATc (Fig. 6C). In order to determine whether the same holds true in M. tuberculosis, Rv-pptr-B cells were transformed with the various constructs, and the growth patterns of transformants in liquid culture were analyzed. To our surprise, in Rv-pptr-B, none of the PASTA deletion mutants could complement the conditional mutant phenotype (Fig. 6E). The data described above clearly demonstrate that whereas the presence of the PASTA4 domain is dispensable in M. smegmatis, in M. tuberculosis, all four PASTA domains are necessary for PknB function and cell viability.
FIGURE 6.

PknB extracellular PASTA domains are essential for cell survival. A, schematic representation of PknB PASTA domain deletion mutations. B, mc2ΔpknB was transformed with pNit, pNit-PknB, pNit-PknB-PASTA1, pNit-PknB-PASTA2, or pNit-PknB-PASTA3 and were seeded at an A600 of 0.2 for 6 h. WCLs from these strains grown in the presence/absence of ATc and in the presence of 0.2 μm IVN in the case of mc2ΔpknB harboring deletion constructs were probed with α-PknB and α-GroEL1 antibodies. Arrows, bands corresponding to PknB deletion mutants. C, growth pattern analysis of mc2ΔpknB transformant strains in the absence of ATc. 0.2 μm IVN was added to mc2ΔpknB strains harboring pNit vector or PknB-PASTA1, -2, or -3 constructs. Cultures were seeded at an initial A600 of 0.02. D, Rv-pptr-B transformants were seeded at A600 of 0.2. WCLs prepared from different Rv-pptr-B transformants grown in the absence or presence of 0.2 μm IVN were resolved and probed with α-PknB and α-GroEL1 antibodies. Arrows, bands corresponding to PknB deletion mutants. E, growth pattern analysis of the Rv-pptr-B transformants in the absence of pristinamycin. All of the cultures were seeded at an initial A600 of 0.1. Error bars, S.E.
The Intracellular Domain of M. tuberculosis PknB Has Unique Functions
Extracellular PASTA domains have been shown to interact with muropeptides in vitro (5, 28). Our data establish that PASTA domains are critical to PknB function in mycobacteria. Taken together, it is possible that the extracellular PASTA domains may work akin to a sensor, which, upon interaction with muropeptides, transmits signals to the intracellular kinase domain, leading to the phosphorylation of specific substrates. We sought to address the question of specificity in such a communication between the PASTA and the kinase domains. For this, we created chimeric kinases wherein the intracellular kinase and juxtamembrane domains of other kinases were fused to the transmembrane and extracellular PASTA domains of PknB (Fig. 7A). InhA, KasA, KasB, FabH, and EmbR are phosphorylated by PknA, PknB, and PknH (32, 40–43). Because the three kinases have overlapping substrates, we selected PknA and PknH for the domain swap experiments. Both kinases were selected because one (PknA) belongs to the same clade as PknB, whereas the other (PknH) belongs to a different clade (15).
FIGURE 7.

Intracellular domain of M. tuberculosis has a specific and distinct indispensable role. A, schematic representation of PknB chimeric proteins. B, MBP-tagged PknB, PknA-B, and PknH-B proteins were purified, and in vitro kinase assays were carried out using 25 nm MBP, PknB, PknA-B, or PknH-B; 6.25 μm MyBP; and 10 μCi of [γ-32P]ATP. Auto-p, autophosphorylation of respective kinases. p-MyBP, phosphorylated MyBP. Samples were resolved on 12% SDS-PAGE and autoradiographed. C, bands corresponding to phospho-MyBP were excised from the gel and quantified by liquid scintillation counting in three independent experiments. The activity was calculated as cpm of phospho-MyBP/min/μm enzyme. The activity of PknB was normalized to 100% in each experiment, and the percentage of activity in other samples was calculated with respect to (w.r.t.) PknB activity. The results were plotted with percentage of activity on the y axis and the samples on the x axis. D, mc2ΔpknB strain was transformed with pNit, pNit-PknB, pNit-PknA-B, or pNit-PknH-B constructs was seeded at A600 of 0. 2 and grown in the presence or absence of ATc. WCLs were resolved and probed with α-PknB and α-GroEL1 antibodies. E, growth analysis of mc2ΔpknB transformants seeded at A600 of 0.02 in the absence of inducer ATc. F, Rv-pptr-B transformed with pNit, pNit-PknB, pNit-PknA-B, or pNit-PknH-B was seeded at an A600 of 0.2. WCLs prepared in the presence or absence of pristinamycin (as indicated) were resolved and probed with α-PknB and α-GroEL1 antibodies. G, Rv-pptr-B transformants were streaked on 7H10 agar plates with or without pristinamycin. Error bars, S.E.
First, we sought to determine relative activities of chimeric proteins with respect to PknB. To accomplish this, MBP-tagged PknB, PknA-B, and PknH-B proteins were purified, and their ability to phosphorylate universal substrate myelin basic protein (MyBP) was determined in vitro at a limiting concentration of enzymes (Fig. 7, B and C). Although all three proteins were catalytically active, the quantitation of activity per μm enzyme showed that the activity of PknH-B was ∼34% as compared with PknB. In contrast, the PknA-B chimera was found to be almost 60% active in comparison with PknB (Fig. 7C). Western blot analysis of extracts prepared from mc2ΔpknB transformants showed that wild type PknB as well as the PknA-B and PknH-B chimeric proteins showed robust expression, PknH-B migrating more slowly as expected due to its larger size (Fig. 7D). Interestingly, mc2ΔpknB transformants expressing PknH-B displayed growth patterns similar to mc2ΔpknB transformants expressing full-length wild type PknB (Fig. 7E). This suggests that PknB PASTA/TM domains are able to communicate with the kinase/JM domains of PknH, and the chimeric kinase is able to phosphorylate PknB substrates, enabling cell survival in M. smegmatis (Fig. 7E). This result was quite interesting because the PknH-B chimera, which had substantially lower activity, showed successful complementation. We speculate that higher expression levels of PknH-B chimera probably compensated for its inefficient activity. On the other hand, the PknA-B chimeric protein, which was an efficient enzyme in vitro, failed to rescue mc2ΔpknB growth defects, indicating either lack of communication between the extracellular and intracellular domains of the chimeric protein or unique substrate recognition/specificity of the PknA intracellular/JM domains (Fig. 7E). This result was somewhat surprising because PknA and PknB both belong to the same clade, and both play a role in modulation of cell shape and cell division.
A similar analysis was carried out in the Rv-pptr-B mutant, wherein transformants were streaked on plates with or without pristinamycin. However, unlike in the mc2ΔpknB mutant, both PknH-B and PknA-B chimeric proteins failed to complement Rv-pptr-B (Fig. 7G). Further, we observed that even in the presence of pristinamycin (which induces expression of full-length PknB from the chromosomal copy), growth of Rv-pptr-B::PknA-B was compromised (Fig. 7G). Thus, even basal level of expression of PknA-B appears to be toxic to these cells. This was also supported by the observation that we obtained very few transformants, and they were smaller in size and grew much slower. These results reflect unique functions of intracellular domains of kinases in the virulent strain compared with the avirulent strain.
PknB Is Essential for Survival of Pathogen in the Host
In order to elucidate the importance of PknB in the growth and survival of M. tuberculosis inside the host, we have employed the murine model of experimental tuberculosis. Data in Fig. 2A demonstrated that in the absence of pristinamycin, PknB protein in Rv-pptr-B strain would be depleted in 4 days. Data in Fig. 3E suggested that PknB protein levels are restored even in the absence of IVN when the Rv-pptr-B strain was transformed with pNit-PknB (Rv-pptr-B::PknB). To investigate the role of PknB in vivo, C57BL/6 mice were infected with H37Rv, Rv-pptr-B, or Rv-pptr-B::PknB strains independently through the aerosol route. The animals were euthanized after day 1 or after 4 and 8 weeks of infection, and the impact of disruption of PknB on virulence was evaluated by bacterial enumeration in lungs and spleens (Fig. 8A). Aerosol infection in mice led to implantation of ∼100 bacilli in lungs at day 1 postinfection (Fig. 8B). Gross evaluation of lungs and spleen morphology 8 weeks postinfection revealed discrete tubercles distributed throughout the tissue in the lungs of mice infected with H37Rv and Rv-pptr-B::PknB. On the other hand, markedly reduced inflammation was observed in lungs of animals infected with Rv-pptr-B strain (Fig. 8A). Moreover, the spleens of H37Rv- and Rv-pptr-B::PknB-infected mice exhibited substantial enlargement (splenomegaly) in comparison with that of Rv-pptr-B-infected mice (Fig. 8A).
FIGURE 8.
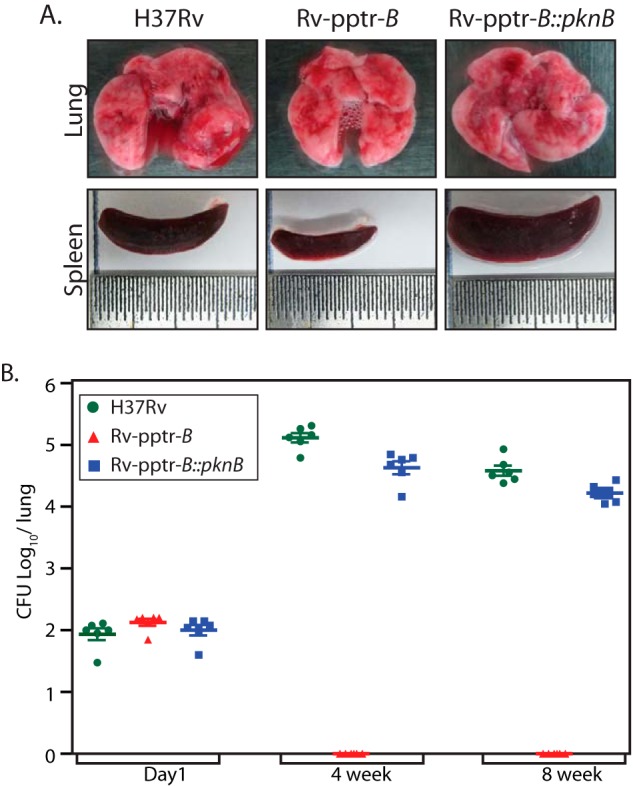
PknB is essential for in vivo survival. A, gross anatomy of lung and spleen from mice after 8 weeks of infection. B, C57Bl/6 mice were infected with H37Rv, Rv-pptr-B, or Rv-pptr-B::PknB as described under “Experimental Procedures.” Bacterial loads in the lungs of mice at different time points post infection were determined and represented as mean ± S.D. (error bars) for each group (n = 2 for day 1 and n = 6 for weeks 4 and 8). The results were plotted with cfu log10/lung on the y axis and the samples on the x axis in the form of a dot plot.
At 4 weeks postinfection, the bacillary load in the lungs of mice infected with H37Rv and Rv-pptr-B::PknB was 5.12 and 4.63 log10 cfu, respectively (Fig. 8B). Likewise, the bacillary load in the lungs of mice infected with H37Rv and Rv-pptr-B::PknB 8 weeks postinfection was 4.54 and 4.25 log10 cfu, respectively (Fig. 8B). However, no bacilli were recovered from the lungs of mice infected with Rv-pptr-B strain at both time points, even after incubation of the plates at 37 °C for a prolonged period. Correspondingly, at 4 weeks postinfection, the splenic bacillary load of H37Rv- and Rv-pptr-B::PknB-infected mice corresponded to 5.6 × 103 and 4.75 × 103 cfu, respectively (data not shown). In accordance with lung data, no bacilli were recovered from the spleen of mice infected with Rv-pptr-B strain (data not shown).
We assessed the impact of disruption of PknB on the pathogenic ability of M. tuberculosis to establish infection and cause disease progression to pathological the state. The gross pathological changes observed were in agreement with observed pulmonary and splenic bacillary loads. At 8 weeks postinfection, the lungs of the animals infected with either H37Rv or Rv-pptr-B::PknB displayed substantial infection with numerous large tubercles and necrosis, whereas the lungs of animals infected with Rv-pptr-B exhibited significantly reduced gross pathological changes (Fig. 9A, arrows). The gross pathological scores observed for the lungs isolated from the Rv-pptr-B-infected mice were considerably lower than those seen with H37Rv- and Rv-pptr-B::PknB- infected animals (Fig. 9B). In contrast, mice infected with Rv-pptr-B displayed negligible lesions compared with those infected with the parental and complemented strains (Fig. 9, A and B). In agreement with the gross pathological scores, animals infected with the H37Rv and Rv-pptr-B::PknB exhibited enhanced histopathological damage as compared with animals infected with Rv-pptr-B. However, in the case of infection with Rv-pptr-B, lung parenchyma exhibited normal microarchitecture, with infiltration of a few leukocytes (Fig. 9C). The absence of significant lesions in the tissues of the animals infected with Rv-pptr-B demonstrate that PknB is crucial for the pathogenesis of M. tuberculosis in the host tissues. Thus, the data obtained using the murine model of experimental tuberculosis clearly indicate that PknB is extremely crucial and plays an indispensable role in the pathogenesis of M. tuberculosis.
FIGURE 9.

PknB plays indispensable role in the pathogenesis. A, histopathological examination of six sections from each candidate mouse was carried out. The images shown are representative of typical lung sections in each experimental group. B, granuloma score of H&E-stained lung sections for each experimental group was performed as described. C, images depict H&E-stained lung sections of mice infected with H37Rv or Rv-pptr-B or Rv-pptr-B::PknB after 8 weeks. Images at ×100 are shown in the left panels. Images of boxed regions at ×400 are depicted in the corresponding right panels. G, granuloma; L, lymphocytes; FC, foamy histiocytic cells; AS, alveolar spaces; E, epithelioid cells.
DISCUSSION
In this report, we describe the comprehensive evaluation of the importance of various domains of PknB in modulating its function in avirulent and virulent mycobacteria. To accomplish this objective, first, we sought to evaluate the effect of PknB overexpression and depletion on cell growth, survival, and morphology. Results presented in Fig. 2 demonstrate that both overexpression and depletion of PknB resulted in cells with varied morphology with eventual cell death. This is in line with the previous finding, wherein overexpression of PknA and PknB in M. bovis BCG was shown to result in a deviation from normal cell morphology (7). Although the morphology observed was not exactly the same, we observed that overexpression led to the appearance of bulging cells and cells with uneven width (Fig. 2). Results obtained upon PknB depletion are also consistent with earlier observations, wherein allelic disruption could only be achieved in the presence of a functional copy of the gene or when pknB was converted into an inducible gene (18, 19). Cell morphology observed upon depletion was similar to that observed consequent to overexpression of PknB (Fig. 2). Interestingly, the effect of overexpression and depletion of PknB was identical in both avirulent and virulent strains, suggesting a central role for PknB in all mycobacterial species. The lethality of PknB overexpression or depletion is probably due to the critical roles played by the kinase in modulating a number of cellular processes via its ability to phosphorylate multiple cellular substrates. Phosphorylation events do not have similar effects on the modulation of substrate enzymatic activities. For example, the activities of KasA, MabA, FabH, and InhA are negatively regulated by phosphorylation (32, 40–42, 44), whereas the activity of KasB is enhanced upon phosphorylation by the kinase (41). These results suggest that stringent regulation of PknB expression and substrate phosphorylation by PknB is necessary for maintaining cellular homeostasis and for appropriate responses to the multitude of intracellular and extracellular cues in the bacteria.
We found that although none of the deletion mutants are as efficient as full-length PknB, these proteins are proficient in phosphorylating GarA, with comparative activity ranging from 25 to 50% (Fig. 4). However, none of the PknB deletion mutants could complement the mc2ΔpknB or Rv-pptr-B mutants (Fig. 4), revealing the significance of the extracytoplasmic domain in modulating PknB signaling. Recently, Mir et al. (28) have demonstrated that the extracytoplasmic domain along with the transmembrane domain of PknB is necessary and sufficient for targeting it to the poles and septum of the cell. Thus, the inability of the deletion mutants to complement mc2ΔpknB or Rv-pptr-B mutants might be due to their failure to localize to the septum and poles. The fact that PknBΔTM also failed to complement the conditional mutants supports this notion (Fig. 5). Although the TM domain per se is necessary for PknB function, the sequence of the TM domain does not seem to be vital because both PknB-TM swap mutants successfully complemented mutant strains (Fig. 5).
Most of the substrates identified in M. tuberculosis are actually phosphorylated by multiple STPKs (45, 46), and thus a key feature among all of these kinase-substrate interactions is functional redundancy. Assuming that the primary function of the PASTA domain is either accurate targeting of PknB to the septum/poles or being a sensor for the extracellular muropeptides and that the kinases have overlapping substrates, we speculated that replacing the PknB kinase domain with that of other kinases should not negatively affect the enzyme's activity. To test this hypothesis, we generated PknA-B and PknH-B chimeric kinases. Although the PknH-B chimera was able to completely complement the mc2ΔpknB mutant, the PknA-B chimera showed only partial functional complementation in mc2ΔpknB (Fig. 7). Interestingly, neither PknH-B nor PknA-B chimeras could complement the PknB mutant in Rv-pptr-B. These results suggest that the extracellular PASTA domains are able to communicate with the intracellular kinase domains in the chimeric kinases in M. smegmatis, thereby carrying out PknB functions. In contrast, in M. tuberculosis, the PknB kinase domain seems to have specific functions, which cannot be replicated by other kinases. Although M. smegmatis and M. tuberculosis belong to the same genus and PknB per se is important in both organisms for growth and survival, the PknB kinase domain seems to have critical and non-redundant functions in the slow growing and virulent M. tuberculosis.
Considering the different environments the pathogen encounters, it should have the capability to adapt to a variety of different available nutrients (47, 48). The PASTA domains contained in protein kinases in various organisms are implicated to mediate important roles in cell division, growth, and development (49). Interactions of the extracellular PASTA domains of PrkC (a PknB ortholog in Bacillus subtilis) with muropeptides have been shown to be essential for germination of spores (5). Findings by Mir et al. (28) suggested that exogenous muropeptides might not be capable of penetrating through the lipid-rich mycobacterial outer membrane. In the absence of exogenous muropeptides, the extracellular domain of PknB is likely to interact with either the peptidoglycan precursor muropeptides or peptidoglycan hydrolysis muropeptide products or with more complex muropeptide ligands generated in vivo (28). PknB has been shown to phosphorylate PbpA (penicillin-binding protein), MurD (a ligase involved in the process of peptidoglycan synthesis), GlmU (N-acetylglucosamine-1-phosphate uridyltransferase) (a key enzyme that synthesizes UDP-N-acetylglucosamine), and FhaA, an FHA domain-containing protein (46, 50–53). Recently, PknB-mediated phosphorylation of the pseudokinase Mt-MviN was shown to regulate the interaction of Mt-MviN with FhaA (54). The phospho-MviN-FhaA complex was interpreted to regulate late stage peptidoglycan biosynthesis, thus establishing a regulatory feedback loop that can control peptidoglycan biosynthesis (54). In addition, Mur synthetases have been shown to interact with PknA and PknB (50). Based on our findings and previous data, we think that in addition to dictating PknB localization, the PASTA domain-ligand interaction also regulates the activity levels of PknB (23, 26).
An intriguing finding of this study is that, although PknB-PASTA3 lacking the C-terminal PASTA4 domain could rescue mc2ΔpknB mutant growth defects, it failed to complement the Rv-pptr-B mutant. Our data also showed that, in contrast to M. smegmatis, the PknB kinase domain has non-redundant functions in M. tuberculosis (Fig. 7). It is possible that the binding of muropeptides is critical for PknB kinase domain function, and the signaling amplitude may be determined by the number of binding sites occupied at any point of time. Assuming that each PASTA domain contributes one binding site, the amplitude of signal generated by the binding of ligand to three PASTA domains may be sufficient to trigger a response in M. smegmatis. In M. tuberculosis, a response may be elicited only when all four PASTA domains are engaged. The notably distinct requirements of the number of PASTA domains and the non-redundant functions of the kinase domain in PknB-mediated signaling in M. tuberculosis reflect differential modulation of signaling events in virulent versus avirulent mycobacteria.
Our study has demonstrated that M. tuberculosis H37Rv or Rv-pptr-B::PknB exhibited normal growth and survival in the organs of infected mice as expected, although the growth of Rv-pptr-B::PknB was marginally lower than H37Rv at 4 and 8 weeks postinfection. On the contrary, no bacterial infection was apparent in organs harvested from the animals infected with Rv-pptr-B at both time points. These observations demonstrate for the first time that PknB is indispensable for the mycobacterium to initiate growth and to persist in the face of host innate and acquired immunity, a fact that is further corroborated by the observations that mice infected with Rv-pptr-B exhibited a significantly reduced gross pathological damage as compared with the animals infected with H37Rv or Rv-pptr-B::PknB. Thus, this study highlights the importance of PknB in the survival of M. tuberculosis both in vitro and in vivo and strongly suggests that PknB may be a potential target for therapeutic intervention against tuberculosis.
Acknowledgments
We acknowledge Dr. Rajmani for help with animal infection experiments. We thank the Tuberculosis Aerosol Challenge Facility, International Centre for Genetic Engineering and Biotechnology (ICGEB) (New Delhi, India) for providing the facility to perform animal infection experiments. We acknowledge Dr. Amit Singh and Dr. Dhiraj Kumar (ICGEB) for scientific discussions. We thank Dr. Christopher M. Sassetti for providing pNit-1, Dr. Tanya Parish for providing p2Nil and pGOAL17 vectors, and Dr. Cole for providing the M. tuberculosis BAC library. We thank Rekha Rani (National Institute of Immunology) for support in obtaining scanning electron microscopy images and Aditya Yadav for generating pMAL constructs.
This work was supported by grants from the National Institute of Immunology (New Delhi, India) and the Department of Biotechnology, India (to V. K. N.).
- STPK
- serine/threonine protein kinase
- JM
- juxtamembrane
- TM
- transmembrane
- PASTA domain
- penicillin-binding protein and serine threonine kinase-associated domain
- FHA domain
- forkhead-associated domain
- MyBP
- myelin-binding protein
- MBP
- maltose-binding protein
- aa
- amino acids
- ATc
- anhydrotetracycline
- IVN
- isovaleronitrile
- BAC
- bacterial artificial chromosome.
REFERENCES
- 1. Stock J. B., Ninfa A. J., Stock A. M. (1989) Protein phosphorylation and regulation of adaptive responses in bacteria. Microbiol. Rev. 53, 450–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Robinson V. L., Buckler D. R., Stock A. M. (2000) A tale of two components: a novel kinase and a regulatory switch. Nat. Struct. Biol. 7, 626–633 [DOI] [PubMed] [Google Scholar]
- 3. Muñoz-Dorado J., Inouye S., Inouye M. (1991) A gene encoding a protein serine/threonine kinase is required for normal development of M. xanthus, a Gram-negative bacterium. Cell 67, 995–1006 [DOI] [PubMed] [Google Scholar]
- 4. Echenique J., Kadioglu A., Romao S., Andrew P. W., Trombe M. C. (2004) Protein serine/threonine kinase StkP positively controls virulence and competence in Streptococcus pneumoniae. Infect. Immun. 72, 2434–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shah I. M., Laaberki M. H., Popham D. L., Dworkin J. (2008) A eukaryotic-like Ser/Thr kinase signals bacteria to exit dormancy in response to peptidoglycan fragments. Cell 135, 486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leiba J., Hartmann T., Cluzel M. E., Cohen-Gonsaud M., Delolme F., Bischoff M., Molle V. (2012) A novel mode of regulation of the Staphylococcus aureus catabolite control protein A (CcpA) mediated by Stk1 protein phosphorylation. J. Biol. Chem. 287, 43607–43619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kang C. M., Abbott D. W., Park S. T., Dascher C. C., Cantley L. C., Husson R. N. (2005) The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev. 19, 1692–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 9. Av-Gay Y., Everett M. (2000) The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 8, 238–244 [DOI] [PubMed] [Google Scholar]
- 10. Hanks S. K., Hunter T. (1995) Protein kinases 6: the eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9, 576–596 [PubMed] [Google Scholar]
- 11. Cowley S., Ko M., Pick N., Chow R., Downing K. J., Gordhan B. G., Betts J. C., Mizrahi V., Smith D. A., Stokes R. W., Av-Gay Y. (2004) The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol. Microbiol. 52, 1691–1702 [DOI] [PubMed] [Google Scholar]
- 12. Walburger A., Koul A., Ferrari G., Nguyen L., Prescianotto-Baschong C., Huygen K., Klebl B., Thompson C., Bacher G., Pieters J. (2004) Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304, 1800–1804 [DOI] [PubMed] [Google Scholar]
- 13. Deol P., Vohra R., Saini A. K., Singh A., Chandra H., Chopra P., Das T. K., Tyagi A. K., Singh Y. (2005) Role of Mycobacterium tuberculosis Ser/Thr kinase PknF: implications in glucose transport and cell division. J. Bacteriol. 187, 3415–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ortega C., Liao R., Anderson L. N., Rustad T., Ollodart A. R., Wright A. T., Sherman D. R., Grundner C. (2014) Mycobacterium tuberculosis Ser/Thr protein kinase B mediates an oxygen-dependent replication switch. PLoS Biol. 12, e1001746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Narayan A., Sachdeva P., Sharma K., Saini A. K., Tyagi A. K., Singh Y. (2007) Serine threonine protein kinases of mycobacterial genus: phylogeny to function. Physiol. Genomics 29, 66–75 [DOI] [PubMed] [Google Scholar]
- 16. Chakraborti P. K., Matange N., Nandicoori V. K., Singh Y., Tyagi J. S., Visweswariah S. S. (2011) Signalling mechanisms in Mycobacteria. Tuberculosis 91, 432–440 [DOI] [PubMed] [Google Scholar]
- 17. Sassetti C. M., Boyd D. H., Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 [DOI] [PubMed] [Google Scholar]
- 18. Fernandez P., Saint-Joanis B., Barilone N., Jackson M., Gicquel B., Cole S. T., Alzari P. M. (2006) The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J. Bacteriol. 188, 7778–7784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forti F., Crosta A., Ghisotti D. (2009) Pristinamycin-inducible gene regulation in mycobacteria. J. Biotechnol. 140, 270–277 [DOI] [PubMed] [Google Scholar]
- 20. Durán R., Villarino A., Bellinzoni M., Wehenkel A., Fernandez P., Boitel B., Cole S. T., Alzari P. M., Cerveñansky C. (2005) Conserved autophosphorylation pattern in activation loops and juxtamembrane regions of Mycobacterium tuberculosis Ser/Thr protein kinases. Biochem. Biophys. Res. Commun. 333, 858–867 [DOI] [PubMed] [Google Scholar]
- 21. Ortiz-Lombardía M., Pompeo F., Boitel B., Alzari P. M. (2003) Crystal structure of the catalytic domain of the PknB serine/threonine kinase from Mycobacterium tuberculosis. J. Biol. Chem. 278, 13094–13100 [DOI] [PubMed] [Google Scholar]
- 22. Young T. A., Delagoutte B., Endrizzi J. A., Falick A. M., Alber T. (2003) Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases. Nat. Struct. Biol. 10, 168–174 [DOI] [PubMed] [Google Scholar]
- 23. Lombana T. N., Echols N., Good M. C., Thomsen N. D., Ng H. L., Greenstein A. E., Falick A. M., King D. S., Alber T. (2010) Allosteric activation mechanism of the Mycobacterium tuberculosis receptor Ser/Thr protein kinase, PknB. Structure 18, 1667–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mieczkowski C., Iavarone A. T., Alber T. (2008) Auto-activation mechanism of the Mycobacterium tuberculosis PknB receptor Ser/Thr kinase. EMBO J. 27, 3186–3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yeats C., Finn R. D., Bateman A. (2002) The PASTA domain: a β-lactam-binding domain. Trends Biochem. Sci. 27, 438. [DOI] [PubMed] [Google Scholar]
- 26. Barthe P., Mukamolova G. V., Roumestand C., Cohen-Gonsaud M. (2010) The structure of PknB extracellular PASTA domain from mycobacterium tuberculosis suggests a ligand-dependent kinase activation. Structure 18, 606–615 [DOI] [PubMed] [Google Scholar]
- 27. Dessen A., Mouz N., Gordon E., Hopkins J., Dideberg O. (2001) Crystal structure of PBP2x from a highly penicillin-resistant Streptococcus pneumoniae clinical isolate: a mosaic framework containing 83 mutations. J. Biol. Chem. 276, 45106–45112 [DOI] [PubMed] [Google Scholar]
- 28. Mir M., Asong J., Li X., Cardot J., Boons G. J., Husson R. N. (2011) The extracytoplasmic domain of the Mycobacterium tuberculosis Ser/Thr kinase PknB binds specific muropeptides and is required for PknB localization. PLoS Pathog. 7, e1002182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pandey A. K., Raman S., Proff R., Joshi S., Kang C. M., Rubin E. J., Husson R. N., Sassetti C. M. (2009) Nitrile-inducible gene expression in mycobacteria. Tuberculosis 89, 12–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parikh A., Kumar D., Chawla Y., Kurthkoti K., Khan S., Varshney U., Nandicoori V. K. (2013) Development of a new generation of vectors for gene expression, gene replacement, and protein-protein interaction studies in mycobacteria. Appl. Environ. Microbiol. 79, 1718–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parish T., Stoker N. G. (2000) Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 146, 1969–1975 [DOI] [PubMed] [Google Scholar]
- 32. Khan S., Nagarajan S. N., Parikh A., Samantaray S., Singh A., Kumar D., Roy R. P., Bhatt A., Nandicoori V. K. (2010) Phosphorylation of enoyl-acyl carrier protein reductase InhA impacts mycobacterial growth and survival. J. Biol. Chem. 285, 37860–37871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tiwari D., Singh R. K., Goswami K., Verma S. K., Prakash B., Nandicoori V. K. (2009) Key residues in Mycobacterium tuberculosis protein kinase G play a role in regulating kinase activity and survival in the host. J. Biol. Chem. 284, 27467–27479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Villarino A., Duran R., Wehenkel A., Fernandez P., England P., Brodin P., Cole S. T., Zimny-Arndt U., Jungblut P. R., Cerveñansky C., Alzari P. M. (2005) Proteomic identification of M. tuberculosis protein kinase substrates: PknB recruits GarA, a FHA domain-containing protein, through activation loop-mediated interactions. J. Mol. Biol. 350, 953–963 [DOI] [PubMed] [Google Scholar]
- 35. O'Hare H. M., Durán R., Cerveñansky C., Bellinzoni M., Wehenkel A. M., Pritsch O., Obal G., Baumgartner J., Vialaret J., Johnsson K., Alzari P. M. (2008) Regulation of glutamate metabolism by protein kinases in mycobacteria. Mol. Microbiol. 70, 1408–1423 [DOI] [PubMed] [Google Scholar]
- 36. Carrera A. C., Alexandrov K., Roberts T. M. (1993) The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proc. Natl. Acad. Sci. U.S.A. 90, 442–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spitaler M., Villunger A., Grunicke H., Uberall F. (2000) Unique structural and functional properties of the ATP-binding domain of atypical protein kinase C-ι. J. Biol. Chem. 275, 33289–33296 [DOI] [PubMed] [Google Scholar]
- 38. Av-Gay Y., Jamil S., Drews S. J. (1999) Expression and characterization of the Mycobacterium tuberculosis serine/threonine protein kinase PknB. Infect. Immun. 67, 5676–5682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boitel B., Ortiz-Lombardía M., Durán R., Pompeo F., Cole S. T., Cerveñansky C., Alzari P. M. (2003) PknB kinase activity is regulated by phosphorylation in two Thr residues and dephosphorylation by PstP, the cognate phospho-Ser/Thr phosphatase, in Mycobacterium tuberculosis. Mol. Microbiol. 49, 1493–1508 [DOI] [PubMed] [Google Scholar]
- 40. Molle V., Gulten G., Vilchèze C., Veyron-Churlet R., Zanella-Cléon I., Sacchettini J. C., Jacobs W. R., Jr., Kremer L. (2010) Phosphorylation of InhA inhibits mycolic acid biosynthesis and growth of Mycobacterium tuberculosis. Mol. Microbiol. 78, 1591–1605 [DOI] [PubMed] [Google Scholar]
- 41. Molle V., Brown A. K., Besra G. S., Cozzone A. J., Kremer L. (2006) The condensing activities of the Mycobacterium tuberculosis type II fatty acid synthase are differentially regulated by phosphorylation. J. Biol. Chem. 281, 30094–30103 [DOI] [PubMed] [Google Scholar]
- 42. Veyron-Churlet R., Molle V., Taylor R. C., Brown A. K., Besra G. S., Zanella-Cléon I., Fütterer K., Kremer L. (2009) The Mycobacterium tuberculosis β-ketoacyl-acyl carrier protein synthase III activity is inhibited by phosphorylation on a single threonine residue. J. Biol. Chem. 284, 6414–6424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sharma K., Gupta M., Pathak M., Gupta N., Koul A., Sarangi S., Baweja R., Singh Y. (2006) Transcriptional control of the mycobacterial embCAB operon by PknH through a regulatory protein, EmbR, in vivo. J. Bacteriol. 188, 2936–2944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Veyron-Churlet R., Zanella-Cléon I., Cohen-Gonsaud M., Molle V., Kremer L. (2010) Phosphorylation of the Mycobacterium tuberculosis β-ketoacyl-acyl carrier protein reductase MabA regulates mycolic acid biosynthesis. J. Biol. Chem. 285, 12714–12725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prisic S., Dankwa S., Schwartz D., Chou M. F., Locasale J. W., Kang C. M., Bemis G., Church G. M., Steen H., Husson R. N. (2010) Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U.S.A. 107, 7521–7526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grundner C., Gay L. M., Alber T. (2005) Mycobacterium tuberculosis serine/threonine kinases PknB, PknD, PknE, and PknF phosphorylate multiple FHA domains. Protein Sci. 14, 1918–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jayachandran R., BoseDasgupta S., Pieters J. (2013) Surviving the macrophage: tools and tricks employed by Mycobacterium tuberculosis. Curr. Top Microbiol. Immunol. 374, 189–209 [DOI] [PubMed] [Google Scholar]
- 48. Hett E. C., Rubin E. J. (2008) Bacterial growth and cell division: a mycobacterial perspective. Microbiol. Mol. Biol. Rev. 72, 126–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ruggiero A., De Simone P., Smaldone G., Squeglia F., Berisio R. (2012) Bacterial cell division regulation by Ser/Thr kinases: a structural perspective. Curr. Protein Pept. Sci. 13, 756–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Munshi T., Gupta A., Evangelopoulos D., Guzman J. D., Gibbons S., Keep N. H., Bhakta S. (2013) Characterisation of ATP-dependent Mur ligases involved in the biogenesis of cell wall peptidoglycan in Mycobacterium tuberculosis. PLoS One 8, e60143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parikh A., Verma S. K., Khan S., Prakash B., Nandicoori V. K. (2009) PknB-mediated phosphorylation of a novel substrate, N-acetylglucosamine-1-phosphate uridyltransferase, modulates its acetyltransferase activity. J. Mol. Biol. 386, 451–464 [DOI] [PubMed] [Google Scholar]
- 52. Dasgupta A., Datta P., Kundu M., Basu J. (2006) The serine/threonine kinase PknB of Mycobacterium tuberculosis phosphorylates PBPA, a penicillin-binding protein required for cell division. Microbiology 152, 493–504 [DOI] [PubMed] [Google Scholar]
- 53. Roumestand C., Leiba J., Galophe N., Margeat E., Padilla A., Bessin Y., Barthe P., Molle V., Cohen-Gonsaud M. (2011) Structural insight into the Mycobacterium tuberculosis Rv0020c protein and its interaction with the PknB kinase. Structure 19, 1525–1534 [DOI] [PubMed] [Google Scholar]
- 54. Gee C. L., Papavinasasundaram K. G., Blair S. R., Baer C. E., Falick A. M., King D. S., Griffin J. E., Venghatakrishnan H., Zukauskas A., Wei J. R., Dhiman R. K., Crick D. C., Rubin E. J., Sassetti C. M., Alber T. (2012) A phosphorylated pseudokinase complex controls cell wall synthesis in mycobacteria. Sci. Signal. 5, ra7. [DOI] [PMC free article] [PubMed] [Google Scholar]