

Background: Mammalian Rif1 is a regulator of DNA replication timing and repair.
Results: We characterized DNA binding by Rif1 C terminus and identified the critical residues.
Conclusion: Rif1 shows a highly selective binding of DNA cruciform structures.
Significance: This is a step forward in the understanding of Rif1 functions.
Keywords: Cell Cycle, Directed Evolution, DNA Replication, Intrinsically Disordered Proteins, NMR
Abstract
Mammalian Rif1 is a key regulator of DNA replication timing, double-stranded DNA break repair, and replication fork restart. Dissecting the molecular functions of Rif1 is essential to understand how it regulates such diverse processes. However, Rif1 is a large protein that lacks well defined functional domains and is predicted to be largely intrinsically disordered; these features have hampered recombinant expression of Rif1 and subsequent functional characterization. Here we applied ESPRIT (expression of soluble proteins by random incremental truncation), an in vitro evolution-like approach, to identify high yielding soluble fragments encompassing conserved regions I and II (CRI and CRII) at the C-terminal region of murine Rif1. NMR analysis showed CRI to be intrinsically disordered, whereas CRII is partially folded. CRII binds cruciform DNA with high selectivity and micromolar affinity and thus represents a functional DNA binding domain. Mutational analysis revealed an α-helical region of CRII to be important for cruciform DNA binding and identified critical residues. Thus, we present the first structural study of the mammalian Rif1, identifying a domain that directly links its function to DNA binding. The high specificity of Rif1 for cruciform structures is significant given the role of this key protein in regulating origin firing and DNA repair.
Introduction
Rif1 was initially identified as a telomeric protein in budding yeast, where it regulates telomerase access to the shortest telomeres (1–4). In Saccharomyces cerevisiae, Rif1 telomeric binding depends upon its interaction with the sequence-specific DNA-binding protein Rap1. In contrast to the yeast orthologue, mammalian Rif1 is not involved in telomere maintenance but has diverse roles in DNA repair and replication. Mammalian Rif1 was reported to accumulate at DNA double-stranded breaks (DSBs)4 (5, 6) and stalled replication forks (7). Recently, it was further shown that mammalian Rif1 is important for DSB repair pathway choice. In G1 phase of the cell cycle, Rif1 is recruited to DSB by P53BP1 and promotes nonhomologous end joining. In S phase, when sister chromatids are present, BRCA1 antagonizes Rif1 accumulation, ensuring a switch to the less error-prone homologous recombination pathway (8–12). We and others have demonstrated that global regulation of replication timing is a key evolutionary conserved function of Rif1. In fission yeast, human, and mouse cells, the absence of Rif1 causes extensive deregulation of the temporal order of activation of replication origins (13–15). All these studies clearly indicate that this protein plays an important function in multiple aspects of DNA metabolism. Furthermore, in mammalian cells, Rif1 is found in the insoluble nuclear matrix fraction (14, 15), where DNA replication factories and repair foci are organized (16, 17). Because of this association with the insoluble nuclear fraction, it is technically challenging to address in vivo the questions of whether and how Rif1 binds DNA; similarly, it is also not possible to purify significant quantities of active material from the native source necessitating a recombinant approach.
There are two conserved regions (CR) in the murine Rif1 (2418 amino acids): an N-terminal ∼1000-amino acid region and a C-terminal ∼350-amino acid region (5, 18). These mediate protein-protein and protein-nucleic acid interactions, although functional mapping of domains or regions is limited. It was reported that Rif1 requires its N terminus to accumulate at DSB sites (11), whereas the C terminus is required for interaction with BLM helicase (18). The N terminus of Rif1 is predicted to form 21 HEAT-type α-helical repeats (5, 18), whereas the C-terminal region comprises three smaller parts: CRI, CRII, and CRIII (see Fig. 1A). CRI contains the canonical phosphoprotein phosphatase 1 (PP1) docking motif RVXF (RVSF in Rif1) together with a conserved SILK motif that has been identified as an additional binding motif necessary for a subset of phosphoprotein phosphatase 1-binding proteins (19–21). CRII is assumed to be a DNA binding domain due to its weak similarity with the C-terminal domain of RNA polymerase α-subunit (18). The C terminus of human Rif1, when expressed in Escherichia coli as a maltose-binding protein (MBP) fusion protein, bound DNA with some preference for branched structures but showed a tendency for self-association or aggregation (18). Although the maltose-binding protein fusion of Rif1 C terminus was useful in EMSA, this protein material is less appropriate for analytical and structural methods requiring large quantities of monodisperse purified protein. Recently, a crystal structure of the S. cerevisiae Rif1 C terminus (4) revealed a tetramerization function; however, it is unclear whether this is found in mammalian homologues.
FIGURE 1.
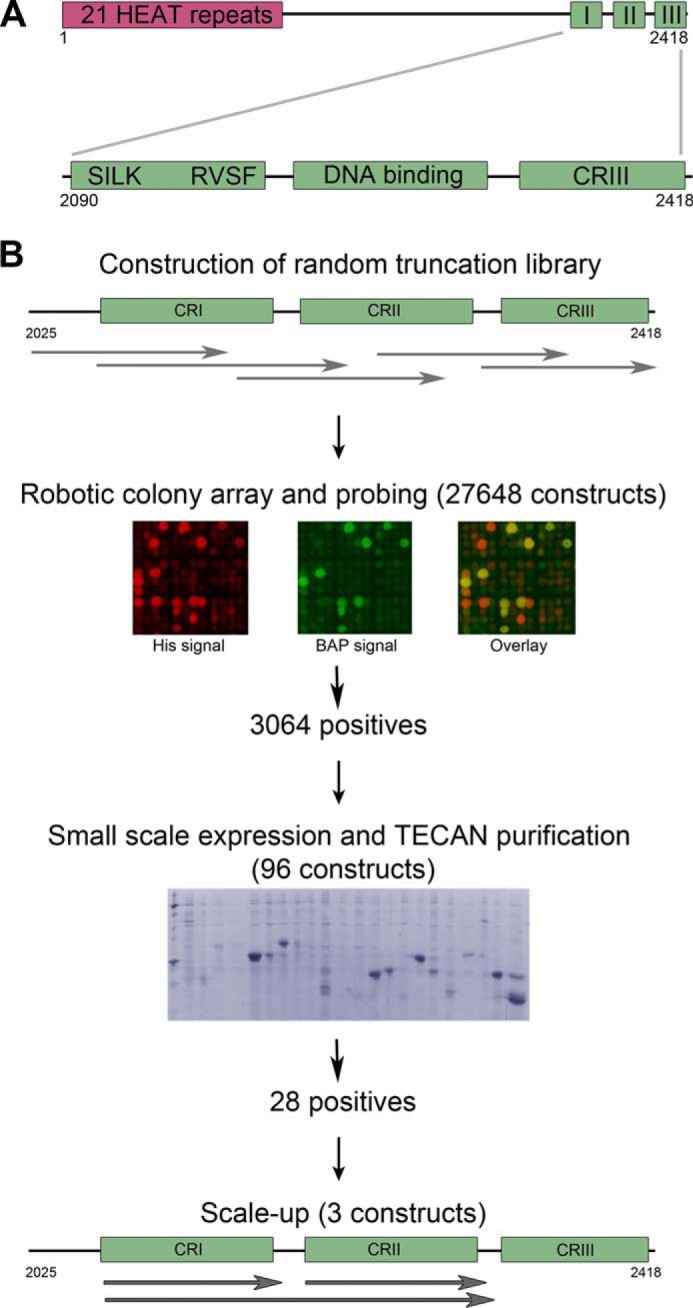
A, conserved regions in mammalian Rif1. The conserved N terminus is predicted to form 21 HEAT-type repeats, and there are three conserved regions at the C terminus: CRI, which contains phosphoprotein phosphatase 1-binding motifs, CRII, which could be a DNA binding domain, and CRIII. B, high throughput library screening for soluble Rif1 fragments. The region coding for residues 2025–2418 was subjected to exonuclease III truncation from both termini to generate a library for expression screening in colony blot format. The clones showing the highest signals from both biotin acceptor peptide (BAP) and hexahistidine tag (His) were selected for small scale expression followed by Ni2+-affinity chromatography.
To understand better the distinct functionalities of the mammalian Rif1 C-terminal region, we sought well behaving constructs spanning these conserved regions that could be produced at the multi-milligram quantities needed for biophysical and structural analyses including NMR. However, the low level of annotation, predicted absence of conserved globular domains, and high level of intrinsic disorder prevent construct design using conventional bioinformatics tools. We circumvented this problem by using the high throughput ESPRIT technology (22, 23), an in vitro evolution-type approach, to screen ∼28,000 discrete truncated variants of the ∼350-amino acid C-terminal region. In this way, we identified mammalian Rif1 protein fragments that could be labeled isotopically and used in the elucidation of structure-specific DNA binding by NMR. These well behaving constructs showed none of the aggregation problems observed previously (18) and revealed a strong selective binding to cruciform structures, but not to the closely related branched DNA. Although the in vivo functions of cruciform DNA are poorly understood, they have been associated with replication origin firing, promoter melting, and recombination intermediates (24). Thus, our data represent an important step forward in the understanding of the contribution of Rif1 DNA binding to the general Rif1 function.
EXPERIMENTAL PROCEDURES
High Throughput Screening for Solubility
The region coding for mouse Rif1 (GenBankTM: AAR87833.2) residues 2025–2418 was codon-optimized and synthesized by DNA2.0. The ESPRIT process was performed as described (22, 25) following subcloning of this fragment into pESPRIT002, a pET9a (Novagen)-derived vector that provides two restriction site pairs for directional exonuclease III truncation, an upstream tobacco etch virus protease-cleavable hexahistidine tag and a downstream biotin acceptor peptide. The first unidirectional truncation of the Rif1 gene was performed from the 5′ terminus as described (22, 25). Linearized plasmids were recircularized by ligation and recovered by transformation of E. coli Mach1 ultracompetent cells (Life Technologies). Plasmid DNA was then extracted from about 10,000 pooled colonies and subjected to a second unidirectional truncation from the 3′ terminus. Following this second step, plasmids containing inserts between 300 and 1200 nucleotides (potentially encoding protein inserts of 100–400 amino acids) were excised from agarose gel and recovered by transformation of Mach1. Plasmids from ∼60,000 pooled colonies were purified, yielding a library of random but size-focused inserts. For analysis of expressed protein products, competent E. coli BL21 AI cells containing the RIL plasmid (Stratagene) were electroporated with the plasmid library. Approximately 28,000 clones were picked robotically and arrayed on nitrocellulose membranes. Protein expression was induced by transferring membranes to fresh agar plates containing 0.2% w/v arabinose and then colony filters prepared by NaOH lysis (22, 25). The colony blot was probed for the presence of expressed proteins bearing both N-terminal and C-terminal tags, thereby confirming correct frame, absence of proteolysis, and putative solubility of expressed proteins from individual colonies, the latter correlating with the efficiency of in vivo biotinylation of the biotin acceptor peptide. Detection was performed using a primary antibody against the N-terminal hexahistidine tag with corresponding Alexa Fluor 532 secondary antibody (Life Technologies) and Alexa Fluor 488 streptavidin (Life Technologies) against the C-terminal in vivo biotinylated biotin acceptor peptide. Membranes were imaged using a Typhoon imaging system (GE Healthcare) with colony fluorescence intensities measured with VisualGrid software (GPC Biotech). Clones with detectable hexahistidine tags were ranked according to their biotinylation signal; the first 96 most intense clones were expressed in small scale 4-ml cultures followed by Ni2+-affinity chromatography and SDS-PAGE (26). Clones exhibiting a clearly visible band of purified protein after Coomassie Blue staining of gels were selected for DNA sequencing to determine construct boundaries and for subsequent scale-up expression and purification following deletion of the biotin acceptor peptide.
Purification of Rif1 Fragments
The Rif1 protein fragments CRI (2093–2190), CRII (2226–2340), CRI+II (2090–2316), and CRII+III (2226–2418) were expressed in E. coli BL21 AI (RIL). Cells grown in TB medium until A595 ∼0.6 were induced by the addition of 0.2% w/v arabinose for 20 h at 25 °C. The pellets were lysed by sonication in 50 mm sodium phosphate (pH 7.4) containing 300 mm NaCl and protease inhibitor cocktail (Roche Applied Science), and the cleared extracts were loaded on TALON resin (Clontech Laboratories). The eluted proteins were incubated at 4 °C with tobacco etch virus protease overnight to remove the hexahistidine tag. The fragments CRII, CRI+II, and CRII+III were dialyzed against a buffer A (20 mm Tris-HCl, 200 mm NaCl and 1 mm DTT, pH 7.5), and loaded on a heparin-Sepharose column (GE Healthcare). The CRI construct was further purified by gel filtration on a Superdex 75 column (GE Healthcare) in buffer A. Mutations were introduced using the QuikChange kit (Stratagene).
Gel Mobility Shift Assay
DNA substrates were prepared by annealing the oligonucleotides (Table 1) described previously (18, 27). Oligonucleotide H1 was synthesized with a 5′-Alexa Fluor 488 dye label. The proteins were incubated with 1 μm labeled DNA in binding buffer (25 mm Tris-HCl, 4 mm MgCl2, 50 mm KCl, 1 mm DTT, 0.1 mg/ml BSA, 5% glycerol, pH 7.5) for 10 min. The indicated amounts of unlabeled DNA were added for the competition assay. The mixtures were resolved on 6% polyacrylamide gels in 0.5× Tris/Borate/EDTA buffer (TBE).
TABLE 1.
Oligonucleotides used to form DNA substrates by annealing
(H1+H5), double-stranded DNA; (H1+H4), Y-shaped DNA; (H1+H3.5+H4), flap; (H1+H2.5+H3.5+H4), fork; (H1+H2+H3+H4), cruciform.
| Name | Sequence |
|---|---|
| H1 | GTGACCGTCTCCGGGAGCTGGAAACGCGCGAGACGAAAGG |
| H5 | CCTTTCGTCTCGCGCGTTTCCAGCTCCCGGAGACGGTCAC |
| H4 | CGAGTTGCTCTTGCCCGGCGCAGCTCCCGGAGACGGTCAC |
| H3.5 | CGCCGGGCAAGAGCAACTCG |
| H2.5 | CCTTTCGTCTCGCGCGTTTC |
| H2 | CCTTTCGTCTCGCGCGTTTCGCCAGCCCCGACACCCGCCA |
| H3 | TGGCGGGTGTCGGGGCTGGCCGCCGGGCAAGAGCAACTCG |
For determination of the dissociation constant (Kd), mixtures were prepared containing protein (from 0.3 to 120 μm) and cruciform DNA (0.3 or 1 μm). The samples were analyzed by fluorescence imaging with quantification of the lanes using ImageQuant software (GE Healthcare). The Kd values were calculated by fitting data to the equation (28)
Here, y is the concentration of protein-DNA complex (μm) at the total protein concentration x, and s0 is the total DNA concentration in the binding mixture. Each Kd was determined twice, and the average value was calculated.
NMR
Rif1 fragments were labeled in M9 minimal medium containing [15N]ammonium chloride with or without [13C6]glucose and purified as above. The samples were concentrated to 100–330 μm in NMR buffer (50 mm sodium phosphate, 200 mm NaCl, 1 mm DTT, pH 6.8). NMR measurements were performed at 10 °C for CRI and 25 °C for CRII on Varian spectrometers operating at a 1H frequency of 600 or 800 MHz. The interactions of CRII and DNA were studied by the addition of DNA to the 15N- or 15N,13C-labeled protein. The spectra were recorded for a 1:1.5 molar excess of cruciform DNA and 1:1 molar ratio of dsDNA. The 1H-15N heteronuclear single quantum coherence (HSQC) spectrum of the complex between CRII and cruciform DNA was assigned using a set of triple resonance experiments: HNCO, intraresidue HN(CA)CO, HN(CO)CA, intraresidue HNCA, HN(COCA)CB, and intraresidue HNCACB (29).
RESULTS
Identification of Soluble Rif1-C Fragments
We applied the in vitro evolution-like ESPRIT technology (22) to identify soluble Rif1 C-terminal fragments (Fig. 1B). DNA encoding the C terminus of Rif1 (residues 2025–2418) was randomly truncated at both ends to generate a diverse construct library. About 28,000 clones were picked and arrayed onto a nitrocellulose membrane and probed for signals from the N-terminal hexahistidine tag and the C-terminal biotin acceptor peptide. The highest ranking 96 positive clones were expressed in liquid culture and analyzed using small scale affinity purification. Twenty-eight expressed soluble purifiable proteins were clearly visible by Coomassie Blue-stained SDS-PAGE. DNA sequencing identified the construct boundaries with all positive clones showing the correct native reading frame despite only one in nine constructs of the naive library being in-frame with the flanking tags (polypeptides with >100 amino acids arising from alternative reading frames are virtually nonexistent due to high stop codon frequency). We selected fragments for study that corresponded to the Rif1 C-terminal conserved regions (Fig. 1B): CRI, CRII, and a fragment spanning through both these regions (CRI+II). No soluble constructs containing CRIII were obtained from the screen, although once subcloned as a fusion to the identified CRII fragment (CRII+III), small amounts could be purified.
Characterization of Rif1-C Fragments
According to the DISPROT (30), IUPred (31, 32), and PrDos (33) disorder predictors, more than half of Rif1 is intrinsically disordered. Folded regions are predicted at the N terminus (HEAT repeat region) and at the very C terminus of Rif1 (CRII and CRIII) (Fig. 2A) where there are also short interaction motifs (2116–2122, 2149–2172, and 2243–2248) predicted via ANCHOR (34). Thus, CRI is predicted to be unfolded, whereas CRII and CRIII may be partially folded.
FIGURE 2.

A, disorder prediction for murine Rif1 by DISPROT (30) (blue), IUPred (31, 32) (red), and PrDos (33) (green) servers. A probability value higher than 0.5 indicates a tendency to be unfolded. The approximate boundaries of the conserved regions are shown below. B, resistance of Rif1 fragments to aggregation after heating. Rif1 fragments at 0.5 mg/ml were incubated at 90 °C for 10 or 30 min and spun down afterward. The supernatants were analyzed by SDS-PAGE. C, analytical gel filtration of Rif1 fragments. Chromatography was performed on a Superdex 75 column in buffer containing 20 mm Tris-HCl (pH = 7.5 at 25 °C), 200 mm NaCl, and 1 mm DTT. The apparent Stokes radii (Rs) values were calculated by interpolation from the standard curve obtained using a set of Stokes radii standards. Kav: measured partition coefficient; Kav = (Ve − Vo)/(Vt − Vo) where Ve is elution volume, Vo is void volume, and Vt is total volume. D, two-dimensional 1H-15N HSQC NMR spectra of 15N-labeled CRI and CRII. The dispersion of resonances in the 1HN dimension shows that CRI is intrinsically disordered (indicated by blue arrow), whereas CRII contains both a folded region as well as disordered parts. The spectra of CRI and CRII were acquired at 10 and 25 °C, respectively.
The recombinant Rif1-C fragments showed behaviors typical of intrinsically disordered proteins, with higher molecular masses observed by SDS-PAGE than calculated from their sequences; intrinsically disordered proteins are typically sized 1.2–1.8-fold larger (35). CRI (10.8 kDa) migrated as a ∼15-kDa protein (Fig. 2B), whereas CRII (12.9 kDa) and CRI+II (25.0 kDa) also migrated slowly. Secondly, the apparent Stokes radii calculated from gel-filtration elution volumes for CRI, CRII, and CRI+II were increased as compared with spherical standard proteins of similar molecular mass (Fig. 2C and Table 2); this likely resulted from unstructured regions (36). Finally, intrinsically disordered proteins often possess high resistance toward heat denaturation and aggregation (35). CRI remained in solution after 30 min at 90 °C, whereas CRII and CRI+II were less heat-resistant (Fig. 2B), in agreement with the prediction of some degree of CRII structure.
TABLE 2.
Apparent Stokes radii (Rs) and molecular masses of Rif C-terminal fragments determined by analytical size exclusion chromatography
Aprotinin, ribonuclease A, carbonic anhydrase, ovalbumin, and conalbumin (GE Healthcare) were used as Stokes radii standards.
| Protein | Molecular massa | Rs | Apparent Rs | Apparent molecular mass |
|---|---|---|---|---|
| kDa | Å | Å | kDa | |
| CRI | 10.8 | 23 | 28 | |
| CRII | 12.9 | 21 | 24 | |
| CRI+II | 25.0 | 29 | 48 | |
| Aprotinin | 6.50 | 13 | 11 | 6.1 |
| Ribonuclease A | 13.7 | 16 | 17 | 14 |
| Carbonic anhydrase | 29.0 | 20 | 23 | 28 |
| Ovalbumin | 44.0 | 30 | 28 | 45 |
| Conalbumin | 75.0 | 36 | 36 | 72 |
a Calculated from amino acid sequence.
To characterize the structural disorder of Rif1 fragments in more detail, we recorded two-dimensional 1H-15N HSQC NMR spectra of 15N-labeled CRI and CRII (Fig. 2D). The resonances in the CRI spectrum showed very little chemical shift dispersion in the 1HN dimension, confirming that CRI is intrinsically disordered. The larger dispersion for CRII chemical shifts in the 1HN dimension indicates that the CRII fragment contains both folded and unstructured regions.
Structure-specific DNA Binding
The C terminus of human Rif1 (residues 2250–2466) was previously reported to bind branched DNA and Holliday junction-type structures, with a low affinity for single-stranded DNA (18). Using the constructs CRI, CRII, CRI+CRII, and CRII+III, we used EMSA to perform a detailed DNA binding study of the mouse Rif1 C terminus. Employing EMSA allowed us to discriminate cruciform structure-specific and nonspecific DNA binding by competition with unlabeled oligonucleotides, and additionally provided approximate and relative binding affinities. The CRII fragment formed a micromolar affinity (Kd = 2.0 ± 0.23 μm) complex with cruciform DNA (Fig. 3, A and B). Almost no binding of CRII to single-stranded, double-stranded DNA, splayed arm, 3′ flap, or fork structures was observed under the same conditions (Fig. 3A). A titration experiment revealed that CRII remains bound to labeled cruciform in the presence of 25-fold excess of unlabeled single-stranded, double-stranded, or other branched DNA (Fig. 4). These results show that CRII is a structure-specific DNA binding domain with a higher specificity toward cruciform DNA than previously observed. The CRI fragment, which is as positively charged as CRII (pI = 9.14 and 9.21, respectively), showed almost no binding to DNA (Fig. 3A). However, the CRI+II fragment bound cruciform DNA with the same specificity (Figs. 3A and Fig. 4) and four times higher affinity (Kd = 0.5 ± 0.05 μm) than CRII (Fig. 3B), suggesting that CRI may further stabilize the complex. CRII+III showed slightly lower affinity to cruciform DNA (Kd = 3.0 ± 1.1 μm, Fig. 3B) than CRII alone (Kd = 2.0 ± 0.23 μm). Also, although CRII and CRI+II formed a single complex with cruciform DNA, several complexes were observed with CRII+III. Similar behavior was also reported for human CRII+III (18). In conclusion, our data indicate that the Rif1 C-terminal CRII region mediates DNA binding with a remarkable preference for cruciform structures, and this may be further enhanced by CRI.
FIGURE 3.

DNA binding by Rif1-C fragments. A, fluorescently labeled DNA fragments at a concentration of 1 μm were mixed with 1; 1.5; 2; 3; 5; and 8 μm Rif1-C fragments. The mixtures were resolved in native gels as described under “Experimental Procedures.” B, calculation of the dissociation constant (Kd) values for Rif1 fragments by fitting data from gel mobility shift assay as described under “Experimental Procedures.” Each Kd was determined twice and the average value calculated.
FIGURE 4.

Competition gel-shift assay. The proteins (5 μm CRII or 3 μm CRI+II) were incubated with 1 μm labeled Holliday junction DNA in presence of 0; 0.2; 1; 5; and 25 μm unlabeled single-stranded, double-stranded, Holliday junction, splayed arm, 3′ flap, or fork DNA (indicated below the relevant lanes). The mixtures were resolved as above.
Structural Analysis of the CRII/DNA Interaction Region
The preference for cruciform DNA observed in this study offers new insights into Rif1 function and the potential for selectively inhibiting this interaction, either by mutation or via an interfering molecule. To this end, it was of interest to determine which residues within Rif1 CRII directly interact with DNA. Thus, we recorded a two-dimensional 1H-15N HSQC spectrum of 15N-labeled CRII in the presence of cruciform DNA. Comparison with the HSQC spectrum of CRII recorded without DNA revealed that a significant fraction of the resonances disappear from the spectrum in the presence of cruciform DNA, whereas the remaining resonances were unperturbed and retained their chemical shift values (Fig. 5A). The former correspond to the residues directly involved in the CRII interaction with cruciform DNA, and from the dispersion of the NMR signals that disappear, it is clear that the DNA interacts with a folded region within CRII. A second experiment was performed with CRII and double-stranded DNA; here no major differences between the HSQC spectra were observed (Fig. 5B), confirming that CRII binding is cruciform-specific.
FIGURE 5.

Analysis of CRII binding to cruciform DNA by NMR. A, superposition of the 1H-15N HSQC spectra of 15N-labeled CRII (red) and 15N-labeled CRII in the presence of cruciform DNA (blue). The spectra were acquired at 25 °C. B, superposition of the 1H-15N HSQC NMR spectra of 15N-labeled CRII (red) and 15N-labeled CRII in the presence of 12-bp DNA. The spectra were acquired at 25 °C. C, DNA binding region in Rif1 CRII. Interacting residues identified by NMR analysis are highlighted in red, and residues subjected to mutations are shown in bold.
The NMR assignment of the spectrum of CRII in complex with DNA revealed that the DNA binding region of CRII is located at residues 2246–2315 in mouse Rif1 (Fig. 5C). Interestingly, this region aligns closely with that identified by Xu et al. (18) on the basis of sequence alignment of Rif1 C terminus with the αCTD domain of bacterial RNA polymerase α (2256–2315). In this previous study, based upon sequence alignments, clusters of residues were simultaneously mutated, confirming the role of the entire region in DNA binding. We took a contrasting approach and systematically substituted every lysine and arginine by alanine within the NMR-identified binding region of CRII (Fig. 5C) to identify and/or confirm the contribution of individual residues in DNA binding. All mutant proteins harboring single amino acid changes were still able to form a complex with cruciform DNA; however, the affinity decreased at least two times for each of them (Figs. 6 and Fig. 7). However, when the two mutations with the most pronounced effect, R2294A and K2303A, were combined, the double-mutated protein lost its ability to bind cruciform DNA (Fig. 6). In summary, these results not only identify and characterize structurally the region 2246–2315 in mouse Rif1, revealing it to be a structure-specific DNA binding domain, but also pinpoint the residues crucial to this function. Intriguingly, this region shows weak sequence similarity with the recently crystallized C-terminal tetramerization domain from budding yeast Rif1 (4). We did not observe any CRII oligomers in solution (Table 2), so it is unclear whether this is significant in the murine homologue.
FIGURE 6.
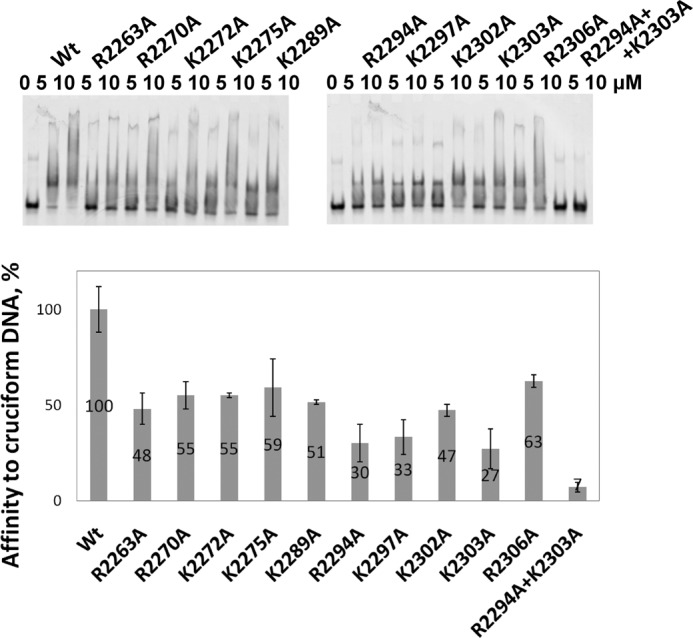
Effects of mutations on DNA binding by Rif1 CRII. The wild type (Wt) and mutant proteins were mixed with 1 μm labeled cruciform DNA and loaded on native gels. The Kd values were determined by gel mobility assay as described under “Experimental Procedures.” The affinity of mutant proteins to cruciform DNA is shown as the percentage of wild type. Error bars indicate mean ± S.D.
FIGURE 7.

Calculation of the dissociation constant (Kd) values for Rif1 CRII mutants by fitting data from gel mobility shift assay as described under “Experimental Procedures.” Each Kd was determined twice, and the average value was calculated.
DISCUSSION
Mammalian Rif1 is an essential nuclear protein involved in DNA repair and replication. Recent studies revealed that Rif1 is a regulator of both DNA replication timing (14, 15) and pathway choice during DSB repair (8–12). However, the identification of Rif1 molecular function has been limited by challenges encountered while producing recombinant material for in vitro studies. Mammalian Rif1 is a large protein (≥2400 residues) that does not contain any well defined functional domains. Moreover, more than half of Rif1 is predicted to be intrinsically disordered. Sequence analysis indicates that the N terminus of Rif1 forms a scaffold of HEAT-type α-helical repeats, whereas the middle region and much of the C terminus remain unfolded (Fig. 2A). To address problems of designing constructs in unstructured proteins, we applied the ESPRIT technology, previously used only on folded targets (22, 23), to identify well behaving fragments in the conserved, but largely unstructured C-terminal region of mouse Rif1 (Fig. 1). As such, this study provides the first example of application of an in vitro evolution-type method to generate functional protein constructs from large intrinsically disordered proteins for structural, biophysical, and biochemical characterization. The screening revealed two regions (CRI and CRII) that can be expressed solubly at quantities and qualities suitable for structural and functional studies in vitro. Biophysical characterization of these protein fragments revealed that, as predicted, parts of the Rif1 C terminus are intrinsically disordered. CRI is completely unfolded, whereas CRII contains both folded and unfolded regions.
The C terminus of human Rif1 was previously reported to bind various branched DNA structures in vitro (18); therefore we performed a detailed study of DNA binding using the new Rif1-C fragments to localize precisely the functional regions and critical residues therein. EMSA assays with various DNA substrates showed the CRII region to bind cruciform DNA with micromolar affinity and with a higher degree of specificity than previously reported (18). It is not known yet whether there is any sequence preference in cruciform binding. In addition, although not directly involved in contacting the DNA, the CRI region contributes to the affinity of DNA binding by CRII. NMR analysis showed that CRII residues 2246–2315 are involved in the interaction with cruciform DNA and guided alanine mutagenesis experiments that identified Arg-2294 and Lys-2303 as key residues in DNA-protein interactions.
These results, when taken together, show that the conserved region CRII of mammalian Rif1 contains a selective structure-specific DNA binding domain. Its specificity for cruciform structures suggests that structure-specific DNA binding plays a conserved and important role in Rif1 function. Although the biological significance of this finding is still unclear, these data will assist the design of cellular and animal-based experiments that may help uncover why and when Rif1 binds DNA during cellular function. Cruciform DNA arises naturally from inverted sequence repeats that can be further stabilized by supercoiling. In addition to well characterized junction-resolving enzymes, a number of proteins have been reported to interact with cruciform DNA and are involved in regulation of transcription, DNA replication, and repair. These include PARP1, BRCA1, HMG family proteins, P53, 14-3-3, and many other proteins (reviewed in Ref. 24). Few affinity data are available; however, the DNA binding domains of BRCA1 (37, 38) and HMGB1 (39, 40) bind cruciform structures with a similar affinity to Rif1 CRII.
Over-representation of inverted repeats in the vicinity of breakpoint junctions, promoter regions, and at sites of replication initiation suggests that cruciform structures may be important in many biological processes, notably DNA repair and initiation of DNA replication (24, 41). These data support a model in which Rif1 mediates the association of other proteins or complexes, perhaps via its N-terminal HEAT repeat region, with these DNA structural elements using a C-terminal DNA binding domain.
Acknowledgments
The ESPRIT platform is supported by EU FP7 contracts P-CUBE (227764) and BioStruct-X (283570). This work used the platforms of the Grenoble Instruct centre (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). Technical assistance was provided by G. El Masri.
This work was supported by an EMBL Interdisciplinary Postdoc (EIPOD) under Marie Curie Actions COFUND (229597) (to R. S.). This work was also supported by the French Agence Nationale de la Recherche (ANR) through ANR JCJC Protein Disorder (to M. R. J.); ANR MALZ TAUSTRUCT (to M. B); and TGIR-RMN-THC FR3050 CNRS. D. J. H. is an inventor on an EMBL patent for the ESPRIT technology used to express Rif1 fragments.
- DSB
- DNA double-stranded break
- ESPRIT
- expression of soluble proteins by random incremental truncation
- CR
- conserved region
- HSQC
- heteronuclear single quantum coherence.
REFERENCES
- 1. Hardy C. F., Sussel L., Shore D. (1992) A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 6, 801–814 [DOI] [PubMed] [Google Scholar]
- 2. Teixeira M. T., Arneric M., Sperisen P., Lingner J. (2004) Telomere length homeostasis is achieved via a switch between telomerase-extendible and -nonextendible states. Cell 117, 323–335 [DOI] [PubMed] [Google Scholar]
- 3. Gallardo F., Laterreur N., Cusanelli E., Ouenzar F., Querido E., Wellinger R. J., Chartrand P. (2011) Live cell imaging of telomerase RNA dynamics reveals cell cycle-dependent clustering of telomerase at elongating telomeres. Mol. Cell 44, 819–827 [DOI] [PubMed] [Google Scholar]
- 4. Shi T., Bunker R. D., Mattarocci S., Ribeyre C., Faty M., Gut H., Scrima A., Rass U., Rubin S. M., Shore D., Thomä N. H. (2013) Rif1 and Rif2 shape telomere function and architecture through multivalent Rap1 interactions. Cell 153, 1340–1353 [DOI] [PubMed] [Google Scholar]
- 5. Silverman J., Takai H., Buonomo S. B., Eisenhaber F., de Lange T. (2004) Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev. 18, 2108–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xu L., Blackburn E. H. (2004) Human Rif1 protein binds aberrant telomeres and aligns along anaphase midzone microtubules. J. Cell Biol. 167, 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buonomo S. B., Wu Y., Ferguson D., de Lange T. (2009) Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J. Cell Biol. 187, 385–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chapman J. R., Barral P., Vannier J. B., Borel V., Steger M., Tomas-Loba A., Sartori A. A., Adams I. R., Batista F. D., Boulton S. J. (2013) RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 49, 858–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zimmermann M., Lottersberger F., Buonomo S. B., Sfeir A., de Lange T. (2013) 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339, 700–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Di Virgilio M., Callen E., Yamane A., Zhang W., Jankovic M., Gitlin A. D., Feldhahn N., Resch W., Oliveira T. Y., Chait B. T., Nussenzweig A., Casellas R., Robbiani D. F., Nussenzweig M. C. (2013) Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339, 711–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Escribano-Díaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J. T., Tkáč J., Cook M. A., Rosebrock A. P., Munro M., Canny M. D., Xu D., Durocher D. (2013) A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883 [DOI] [PubMed] [Google Scholar]
- 12. Feng L., Fong K. W., Wang J., Wang W., Chen J. (2013) RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem. 288, 11135–11143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hayano M., Kanoh Y., Matsumoto S., Renard-Guillet C., Shirahige K., Masai H. (2012) Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev. 26, 137–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cornacchia D., Dileep V., Quivy J. P., Foti R., Tili F., Santarella-Mellwig R., Antony C., Almouzni G., Gilbert D. M., Buonomo S. B. (2012) Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 31, 3678–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamazaki S., Ishii A., Kanoh Y., Oda M., Nishito Y., Masai H. (2012) Rif1 regulates the replication timing domains on the human genome. EMBO J. 31, 3667–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anachkova B., Djeliova V., Russev G. (2005) Nuclear matrix support of DNA replication. J. Cell. Biochem. 96, 951–961 [DOI] [PubMed] [Google Scholar]
- 17. Rivera-Mulia J. C., Hernández-Muñoz R., Martínez F., Aranda-Anzaldo A. (2011) DNA moves sequentially towards the nuclear matrix during DNA replication in vivo. BMC Cell Biol. 12, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu D., Muniandy P., Leo E., Yin J., Thangavel S., Shen X., Ii M., Agama K., Guo R., Fox D., 3rd, Meetei A. R., Wilson L., Nguyen H., Weng N. P., Brill S. J., Li L., Vindigni A., Pommier Y., Seidman M., Wang W. (2010) Rif1 provides a new DNA-binding interface for the Bloom syndrome complex to maintain normal replication. EMBO J. 29, 3140–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bollen M., Peti W., Ragusa M. J., Beullens M. (2010) The extended PP1 toolkit: designed to create specificity. Trends Biochem. Sci. 35, 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hendrickx A., Beullens M., Ceulemans H., Den Abt T., Van Eynde A., Nicolaescu E., Lesage B., Bollen M. (2009) Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem. Biol. 16, 365–371 [DOI] [PubMed] [Google Scholar]
- 21. Sreesankar E., Senthilkumar R., Bharathi V., Mishra R. K., Mishra K. (2012) Functional diversification of yeast telomere associated protein, Rif1, in higher eukaryotes. BMC Genomics 13, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yumerefendi H., Tarendeau F., Mas P. J., Hart D. J. (2010) ESPRIT: an automated, library-based method for mapping and soluble expression of protein domains from challenging targets. J. Struct. Biol. 172, 66–74 [DOI] [PubMed] [Google Scholar]
- 23. Hart D. J., Waldo G. S. (2013) Library methods for structural biology of challenging proteins and their complexes. Curr. Opin. Struct. Biol. 23, 403–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brázda V., Laister R. C., Jagelská E. B., Arrowsmith C. (2011) Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 12, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tarendeau F., Boudet J., Guilligay D., Mas P. J., Bougault C. M., Boulo S., Baudin F., Ruigrok R. W., Daigle N., Ellenberg J., Cusack S., Simorre J. P., Hart D. J. (2007) Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat. Struct. Mol. Biol. 14, 229–233 [DOI] [PubMed] [Google Scholar]
- 26. Scheich C., Sievert V., Büssow K. (2003) An automated method for high-throughput protein purification applied to a comparison of His-tag and GST-tag affinity chromatography. BMC Biotechnol. 3, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xue Y., Li Y., Guo R., Ling C., Wang W. (2008) FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Hum. Mol. Genet. 17, 1641–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rippe K. (1997) Analysis of protein-DNA binding at equilibrium. B. I. F. Futura 12, 20–26 [Google Scholar]
- 29. Lescop E., Schanda P., Brutscher B. (2007) A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J. Magn. Reson. 187, 163–169 [DOI] [PubMed] [Google Scholar]
- 30. Obradovic Z., Peng K., Vucetic S., Radivojac P., Brown C. J., Dunker A. K. (2003) Predicting intrinsic disorder from amino acid sequence. Proteins 53, Suppl. 6, 566–572 [DOI] [PubMed] [Google Scholar]
- 31. Dosztányi Z., Csizmok V., Tompa P., Simon I. (2005) IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21, 3433–3434 [DOI] [PubMed] [Google Scholar]
- 32. Dosztányi Z., Csizmók V., Tompa P., Simon I. (2005) The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 347, 827–839 [DOI] [PubMed] [Google Scholar]
- 33. Ishida T., Kinoshita K. (2007) PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 35, W460–W464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dosztányi Z., Mészáros B., Simon I. (2009) ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics 25, 2745–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Uversky V. N. (2010) The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 568068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Uversky V. N., Dunker A. K. (2010) Understanding protein non-folding. Biochim. Biophys. Acta 1804, 1231–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sturdy A., Naseem R., Webb M. (2004) Purification and characterisation of a soluble N-terminal fragment of the breast cancer susceptibility protein BRCA1. J. Mol. Biol. 340, 469–475 [DOI] [PubMed] [Google Scholar]
- 38. Naseem R., Sturdy A., Finch D., Jowitt T., Webb M. (2006) Mapping and conformational characterization of the DNA-binding region of the breast cancer susceptibility protein BRCA1. Biochem. J. 395, 529–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grasser K. D., Teo S. H., Lee K. B., Broadhurst R. W., Rees C., Hardman C. H., Thomas J. O. (1998) DNA-binding properties of the tandem HMG boxes of high-mobility-group protein 1 (HMG1). Eur. J. Biochem. 253, 787–795 [DOI] [PubMed] [Google Scholar]
- 40. Assenberg R., Webb M., Connolly E., Stott K., Watson M., Hobbs J., Thomas J. O. (2008) A critical role in structure-specific DNA binding for the acetylatable lysine residues in HMGB1. Biochem. J. 411, 553–561 [DOI] [PubMed] [Google Scholar]
- 41. Pearson C. E., Zorbas H., Price G. B., Zannis-Hadjopoulos M. (1996) Inverted repeats, stem-loops, and cruciforms: significance for initiation of DNA replication. J. Cell. Biochem. 63, 1–22 [DOI] [PubMed] [Google Scholar]