

Background: Aquaporins are channels permeable to water, and they are essential for immune cell migration.
Results: We demonstrate that aquaporin-mediated water fluxes are necessary for the NLRP3 inflammasome-dependent release of mature IL-1β in vitro and in vivo.
Conclusion: Aquaporins are implicated in the mechanisms of proinflammatory cytokine secretion during inflammation.
Significance: The discovery of a new function for AQPs opens new diagnostic and therapeutic opportunities in inflammatory disorders.
Keywords: Aquaporin, Caspase, Inflammation, Interleukin, Water Channel, RVD, Cell Swelling, Inflammasome
Abstract
Rapid changes in cell volume characterize macrophage activation, but the role of water channels in inflammation remains unclear. We show here that, in vitro, aquaporin (AQP) blockade or deficiency results in reduced IL-1β release by macrophages activated with a variety of NLRP3 activators. Inhibition of AQP specifically during the regulatory volume decrease process is sufficient to limit IL-1β release by macrophages through the NLRP3 inflammasome axis. The immune-related activity of AQP was confirmed in vivo in a model of acute lung inflammation induced by crystals. AQP1 deficiency is associated with a marked reduction of both lung IL-1β release and neutrophilic inflammation. We conclude that AQP-mediated water transport in macrophages constitutes a general danger signal required for NLRP3-related inflammation. Our findings reveal a new function of AQP in the inflammatory process and suggest a novel therapeutic target for anti-inflammatory therapy.
Introduction
Because of their ability to trigger or dampen inflammatory processes, macrophages are key effectors of innate immunity. They possess a wide range of specific receptors recognizing endogenous and foreign patterns indispensable to organize adequate clearance and immune responses. Upon detection of threat signals, macrophages elicit immune reactions, trying to eliminate danger and to inform other cells by releasing cytokines and chemokines. In particular, the macrophage-derived cytokine IL-1β drives inflammation by enhancing granulocyte differentiation, migration, and accumulation to inflammatory sites. This cytokine has been implicated in lung inflammatory reactions in response to bacteria, viruses, inorganic particles, and drugs (1–4).
The release of mature IL-1β by macrophages requires the assembly of a multiproteic inflammasome complex, of which the caspase 1 component is the final effector. The NLRP3 (NOD-like receptor protein 3) inflammasome is the most fully characterized inflammasome. It can be activated by a wide array of stimuli, including extracellular ATP, bacterial toxins such as nigericin, monosodium urate crystals, and environmental and industrial particles such as silica (5). How those structurally and chemically very different stimuli can activate this inflammasome in macrophages is still under investigation, but common cellular stress signals are likely involved. It has been established that ionic movements represent a common event required for inflammasome activation by diverse stimuli. A drop in cytosolic K+ is necessary and sufficient for caspase 1 activation and IL-1β release by macrophages (5). Cell distension and subsequent regulatory volume decrease (RVD)2 mediated by activation of volume sensor systems such as transient receptor potential (TRP) channels have also been associated with inflammasome activation in macrophages (6–8). However, the potential importance of channel-mediated water transport as a general pathway of inflammasome activation remains unknown.
Aquaporins (AQPs) are a family of channels that facilitates water transport across cell membranes in response to osmotic gradients. Aquaporin 1 (AQP1), the archetype of all aquaporins, is selectively permeable to water and involved in rapid cellular water fluxes and cell volume regulations (9). Although AQPs are primarily expressed in epithelial and endothelial cells, these channels are also expressed in erythrocytes and immune cells such as macrophages, dendritic cells, and thymocytes (10–15). AQPs are implicated in the phagocytic functions, activation, and migration of immune cells (10, 12, 14).
Here we explore the involvement of AQPs in mature IL-1β release by macrophages. We show that AQP-related cell water transport triggered by diverse soluble and particulate NLRP3 activators is sensed as a danger signal by macrophages and that AQPs are implicated in NLRP3 inflammasome activation in vitro and in vivo.
EXPERIMENTAL PROCEDURES
Animal Model
Female C57BL/6 mice were obtained from Janvier (Le Genest-Saint-Isle, France). Wild-type (AQP1+/+) and knockout (AQP1−/−) mice (16) were obtained from A. S. Verkman (University of California, San Francisco, CA) and developed as a specific pathogen-free colony at the Université Catholique de Louvain Medical School (Brussels, Belgium). Studies were performed on gender-matched littermates aged 8–12 weeks. The animals were housed in positive pressure, air-conditioned units (25 °C, 50% relative humidity) on a 12-h light/dark cycle and had access to standard diet and tap water ad libitum. The experiments were conducted in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals and approved by the institutional ethics committee. A suspension of crystalline silica particles (DQ12, d50 = 2.2 μm, a gift from Dr. Ambruster, Essen, Germany) in sterile 0.9% saline (Braun Medical, Diegem, Belgium) was injected directly into the lung by pharyngeal aspiration at a dose of 2.5 mg/mouse. To sterilize and inactivate any trace of endotoxins, particles were heated at 200°c for 2 h immediately before suspension and administration. Bleomycin (Aventis, Brussels, Belgium) was suspended in sterile 0.9% NaCl, and 1.25 units/kg body weight was injected directly into the lung by pharyngeal aspiration. The experimental protocol was approved by the local ethical committee for animal research at the Université Catholique de Louvain (2010/UCL/MD/034) and conformed to the Belgian and European Community regulations (LA1230312 and CEE no. 86/609).
Bronchoalveolar Lavage (BAL) and Alveolar Neutrophil Number
Mice were sacrificed after 6 h with an intraperitoneal injection of sodium pentobarbital (20 mg/mice), and bronchoalveolar lavage was performed by cannulating the trachea and lavaging the lung four times with 1 ml of NaCl 0.9%. The BAL fluid was centrifuged at 281 × g for 10 min at 4 °C (centrifuge 5804R, Eppendorf, Hamburg, Germany). Cells recovered in BAL were fixed in 1.25% paraformaldehyde and analyzed with FACSCalibur (BD Biosciences) using FlowJo software. Neutrophils were identified by labeling with anti-GR1-PE (clone RB6-8C5, BD Biosciences), and their number was calculated in a function of total BAL cells counted with a Burker cell chamber. Fc receptors were blocked with anti-CD16/CD32 (clone 2.4G2, BD Biosciences) to reduce nonspecific binding.
Lung Histopathology
Lungs were lavaged and perfused with 0.9% NaCl, and the superior left lung lobe was fixed in 3.6% formaldehyde solution for one night. Then, paraffin-embedded, 5-μm sections were stained with hematoxylin and eosin. Images were acquired with a microscope (Zeiss, Jena, Germany, model Jenalumar) equipped with an Apochromat ×12.5/0.35 for pictures in a large panel and a Planachromat fl ×50/0.95 for pictures in the insets (Carl Zeiss, Munich, Germany). The microscope was connected to a Coolpix 4500 camera (Nikon, Kingston upon Thames, UK).
RNA Extraction and Quantification
Total RNA extraction and quantification by quantitative PCR were performed as described previously (17). Sequences of interest were amplified by PCR using the following forward primers (Invitrogen): CGG CTA CCA CAT CCA AGC AA (mouse 18 S RNA), GAC GGA CCC CAA AAG ATG AAG (mouse IL-1β), CCA GCC AGA GTG GAA CTT TCG (mouse NLRP3), GGA GCT CAC AAT GAC TGT GCT TA (mouse ASC), GGA CTT CTC AAG ATC ATG GCT ACT T (mouse CXCR2), GAC TGC GAC TTC CTG GAG GAT (mouse ngp) and reverse primers: ATA CGC TAT TGG AGC TGG ATT ACC (mouse 18s RNA), CTC TTC GTT GAT GTG CTG CTG TG (mouse IL-1β), TAC AAA TGG AGA TGC GGG AGA (mouse NLRP3), CTG CCA CAG CTC CAG ACT CTT(mouse ASC), TAG TAG AGG TGT TTG CTG AAG ACG A (mouse CXCR2), and GTA TCC TCT CGA CTG CAA TCC CTG (mouse ngp).
ELISA
ELISA kits were used to measure IL-1β and IL-6 (R&D Systems, Wiesbaden-Nordenstadt, Germany). Assays were run according to the protocols of the manufacturer with a detection limit of 15 pg/ml. The IL-1β assay selectively measured the mature form of IL-1β (17 kDa) (18).
Cell Purification
Mice were sacrificed by intramuscular injection of 60 mg sodium pentobarbital. For peritoneal macrophages, the peritoneal cavity was lavaged with 10 ml of sterile NaCl 0.9% and centrifuged (280 × g, 10 min, 4 °C). Macrophages were purified from peritoneal lavage on the basis of their F4/80 expression by using magnetic cell separation (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the protocol of the manufacturer. For lung macrophages, lungs were perfused via the right heart ventricle with sterile NaCl 0.9%, and then 1 ml of enzyme mix containing 20 mg of Pronase (Sigma-Aldrich, Bornem, Belgium) and 1 mg of DNase (Worthington Biochemical Corp.) in Hanks' balanced salt solution (Invitrogen) with 1% of antibiotic antimycotic (Fungizone (25 μg/ml), penicillin-streptomycin (10,000 units and 10,000 μg/ml); Invitrogen) was infused in the cannulated trachea. After 20 min, lungs were excised and placed into a tube chilled on ice with FBS (Invitrogen). Collected lungs were then crushed by repeated aspiration and expulsion in a 20-ml syringe and passed on a 70-μm filter. The filtered fraction was grown in a 75-cm2 tissue culture flask in DMEM (Invitrogen) supplemented with 10% FBS and 1% of antibiotic antimycotic and then detached using trypsin (Invitrogen). Lung macrophages were purified on the basis of their CD45 expression by using magnetic cell separation (Miltenyi Biotec) according to the protocol of the manufacturer. Macrophages were primed for 18 h by exposure to 0.1 μg/ml of purified LPS (Enzo Life Sciences, Antwerpen, Belgium) in complete medium.
Inflammasome Activation
LPS-primed cells (200,000 cells/well in a 96-well plate, 7 × 106 cells/well in a 6-well plate) were exposed to an inflammasome activator (200 μl) for 30 min for ATP (5 mm), 1 h for nigericin (20 μm), Stöber silica nanoparticles (SNP) (100 μg/ml) and hypotonic shock, and 3 h for DQ12 (400 μg/ml). Supernatant was collected for mature IL-1β measurement, and the cell pellet was used to quantify active caspase 1. The Stöber-based synthesis process and characterization of SNP are described elsewhere (19). SNP diameter of 12.2 nm or 100 nm were obtained by transmission electron microscopy or by dynamic light scattering in DMEM, respectively. The total surface area evaluated by the BET (Brunauer, Emmet, and Teller) model was 400 m2/g. Isotonic and hypotonic media were prepared on the basis of Perregaux (6). The isotonic solution contained 0.9 m CaCl2, 0.5 mm MgCl2, 1.5 mm KH2PO4, 2.7 mm KCl, 5 mm glucose, 20 mm Hepes, and 137 mm NaCl. In hypotonic solutions, NaCl was absent (47 mosmol/kg H2O) or at a concentration reduced to 3.75 mm (53 mosmol/kg H2O), 7.5 mm (60 mosmol/kg H2O), 15 mm (66 mosmol/kg H2O), or 30 mm (100 mosmol/kg H2O). All solutions were prepared in LPS-free water (Baxter, Braine-l'Alleud, Belgium) and filtered on a 0.22-μm filter. Hypertonic solutions were obtained by adding 100 mm NaCl (415 mosmol/kg H2O) or 200 mm mannitol to DMEM. When a pretreatment was required, cells were first incubated with HgCl2 30 min prior to the activator. HgCl2 was maintained in the medium until the end of the experiment. For mercurial AQP inhibition after hypotonic shock, HgCl2 was added at different time points following hypotonic shock and maintained till the end of the experiment (60 min after the shock). For TRP inhibition, Ruthenium Red (Sigma-Aldrich) was added when hypotonic shock was applied and maintained until the end of the experiment (60 min after the shock). For the cytotoxicity assay, cells were incubated in DMEM with 2% WST1 (Roche Applied Science, Vilvoorde, Belgium) for 15 min.
Active Caspase 1 Measurement
After treatment with an inflammasome activator, cells were incubated with green fluorescent peptide, 5-carboxyfluorescein-Tyr-Val-Ala-Asp-fluoromethyl ketone, according to the recommendations of the manufacturer (Immunochemistry Technologies). Fluorescence was measured by using a SPECTRAmax GEMINI XS microplate spectrofluorometer (Molecular Devices). For the flow cytometry assay, 106 cells were exposed to hypotonic medium during 30 or 60 min in microtubes and then incubated with the probe according to the recommendation of the manufacturer and analyzed by using a FACSCalibur (BD Biosciences).
Size Modification Assessment
For microscopical analysis of size modification, LPS-primed cells (300,000 cells/well in a 48-well plate) were exposed to isotonic or hypotonic medium, and pictures were taken after the required time of incubation under optimal culture conditions. Images were acquired with a Zeiss AxioObserver.z1 equipped with a T° and pO2/pCO2 regulation chamber and an EC “Plan-Neofluar” ×10/0.30 objective (Carl Zeiss). The microscope was connected to an AxioCam camera using AxioVision software (Carl Zeiss). For flow cytometry assessment of cell volume modification, LPS-primed cells were incubated in microtubes in isotonic or hypotonic medium during the desired time with or without mercury treatment. Cells were then fixed by addition of paraformaldehyde directly to the medium and analyzed with FACSCalibur.
Western Blot Analysis
After treatments, supernatants of LPS-primed macrophages (7 106 cells/well in 6-well plates) were collected, and cell pellets were lysed on ice in Triton lysis buffer (1% Triton, 25 mm Tris (pH 7.4), 150 mm NaCl, and antiprotease tablets (Roche Applied Science) at 2 mg/ml). The supernatant was concentrated by methanol-chloroform precipitation and then suspended in 30 μl of Laemmli buffer (Bio-Rad). The supernatant and cell samples were then sonicated for 5 min in a bath (Bransonic-12, Geneva, Switzerland), heated at 99 °C for 5 min, and centrifuged for 10 min at 14,000 rpm. Samples were loaded onto a 4–20% polyacrylamide gel (Mini- Protean TGX, Bio-Rad) and transferred to a PVDF membrane (Millipore) by electroblotting. Membranes were incubated overnight at 4 °C with primary antibody (0.2 μg/ml, polyclonal goat anti-IL-1β antibody, catalog no. AF-401-NA, R&D Systems; 2 μg/ml, polyclonal rabbit anti-caspase 1 p10 antibody, Santa Cruz Biotechnology, Santa Cruz, CA or 1 μg/ml, monoclonal mouse anti-β-actin antibody, catalog no. A1978, Sigma-Aldrich). After washing, membranes were incubated for 1 h with RDye infrared secondary antibodies (1:15,000; donkey IgG anti-goat, donkey IgG anti-rabbit, or donkey IgG anti-mouse, Westburg, Leusden, The Netherlands). Detection was performed with an Odyssey CLx Western blot detection system (Westburg).
Statistics
Results were analyzed by Student Newman-Keuls test. Statistical significance was considered at p < 0.05.
RESULTS
AQP-facilitated Water Transport Induces NLRP3 Inflammasome-related IL-1β Activation and Release in Macrophages
Because macrophage swelling is commonly recorded following exposure to different NLRP3 inflammasome activators (8), we analyzed the importance of water transport in IL-1β release in vitro. We first contained cell swelling in ATP-activated lung macrophages by exposure to increased extracellular tonicity (20). Under extracellular hyperosmolar conditions, ATP-induced IL-1β release by LPS-primed lung macrophages was reduced significantly (Fig. 1a). The absence of cytotoxicity because of increased extracellular osmolarity was verified by measuring cellular mitochondrial activity (Fig. 1d). To determine whether this effect was specific to inflammasome activation, we also measured the levels of the proinflammatory cytokine IL-6, which does not directly require inflammasome activation for its secretion. Unlike IL-1β, the release of IL-6 was not modified by extracellular hyperosmolar conditions (Fig. 1e), suggesting that swelling is not required for the release of other proinflammatory cytokines that do not directly depend on inflammasomes for their release. The inhibitory effect of an extracellular hyperosmolarity on ATP-induced inflammasome activation was confirmed in peritoneal macrophages on the basis of the same protocol (Fig. 1b). IL-1β release by peritoneal macrophages was also reduced when osmolarity was increased by mannitol addition (Fig. 1c). These results show that rapid cell swelling is necessary for IL-1β release in macrophages.
FIGURE 1.

Water entry is necessary for IL-1β release in macrophages. Release of IL-1β by lung macrophages (LM) (a) or peritoneal macrophages (PM) (b) primed with LPS (0.1 μg/ml, overnight) and subsequently exposed or not exposed to ATP (5 mm) with addition or no addition of NaCl to the medium (415 mosmol/kg H2O). n = 4 replicates/condition. c, release of IL-1β by peritoneal macrophages exposed or not exposed to ATP (5 mm) with addition or no addition of mannitol to the medium (200 mm). n = 3 or 4. d, mitochondrial activity assessed by optical density (OD) of reduced WST-1 and release of IL-6 by lung macrophages exposed or not exposed to ATP (5 mm) with addition or no addition of NaCl to the medium (e). n = 3 or 4. Data are means ± S.E. ***, p < 0.001 upon exposure to hypotonic ATP with or without NaCl or mannitol addition; ns, no significant difference. p values were calculated by Student Newman-Keuls test.
To study water channel implication, a model of osmotic swelling-induced NLRP3 inflammasome activation and IL-1β release was used (7). IL-1β release was assessed in lung macrophages exposed for 60 min to hypotonic solutions with decreasing osmolarity. Reducing medium osmolarity to 100 and 66 mosmol/kg H2O had no significant impact on mature IL-1β detected in cell supernatant, but more drastic hypotonic conditions (from 60 to 47 mosmol/kg H2O) progressively induced significant IL-1β release (Fig. 2a). To test the involvement of water channels in that process, we treated cells 30 min before the hypotonic shock with mercury, which pharmacologically inhibits most AQPs by interacting with an extracellular cysteine residue (21). The implication of AQPs in osmotic swelling was studied 2 min after exposure of lung macrophages to 60 mosmol/kg H2O. At this time point, the size of cells exposed to hypotonic medium was increased compared with isotonic medium. This swelling was reduced in mercury-treated cells (Fig. 2b). AQP inhibition decreased the release of IL-1β by lung macrophages submitted to hypotonic shock (Fig. 2c). We also explored the response of peritoneal macrophages treated according to the same protocol. The reduction of IL-1β release was confirmed when AQPs were inhibited in peritoneal macrophages submitted to hypotonic shock (Fig. 2d). This effect was supported by Western blot data (Fig. 2e). The release of mature IL-1β (IL-1β p17) by peritoneal macrophages in response to hypotonic shock was reduced upon mercury exposure. In parallel, the decrease of the intracellular pool of the IL-1β proform (IL-1β p37) consecutive to hypotonic shock was moderated by AQP blockade (Fig. 2e). Mercurial AQP inhibition also resulted in reduced caspase 1 activation consecutive to hypotonic shock in lung macrophages (Fig. 2f). Together, these results demonstrated that rapid cell swelling via AQPs leads to NLRP3 inflammasome-related release of mature IL-1β by macrophages.
FIGURE 2.

AQP-facilitated water transport induces NLRP3 inflammasome-related caspase-1 activation and mature IL-1β release. a, IL-1β release by lung macrophages (LM) primed with LPS (0.1 μg/ml) exposed to isotonic (320 mosmol/kg H2O) or hypotonic medium (100 mosmol/kg H2O to 47 mosmol/kg H2O). n = 3 replicates/condition. b, flow cytometry forward scatter plots of lung macrophages subjected or not subjected to hypotonic shock (60 mosmol/kg H2O) in the absence or presence of HgCl2 (10 μm). Values indicated in the plots are differences between median forward scatter of cells exposed or not exposed to hypotonic shock. Levels of released IL-1β by lung (c) or peritoneal macrophages (PM) (d) exposed or not exposed to hypotonic shock (in the absence or presence of HgCl2 (10 μm). n = 3. e, Western blot analysis of mature IL-1β (IL-1β p17) in the supernatant (SN) and pro-IL-1β (IL-1β p37) in cell lysate of peritoneal macrophages exposed or not exposed to hypotonic shock in the absence or presence of HgCl2. f, fluorimetric measurement of cellular active caspase 1 in lung macrophages exposed or not exposed to hypotonic shock in the absence or presence of HgCl2 (10 μm). n = 3 or 4. Release of IL-1β by lung (g) or peritoneal macrophages (h) exposed or not exposed to silica (400 μg/ml) in the absence or presence of HgCl2 (10 μm)). n = 3 or 4. IL-1β release by lung (i) or peritoneal macrophages (j) exposed or not exposed to ATP (5 mm) in the absence or presence of HgCl2. n = 3 or 4. k, Western blot analysis of mature IL-1β and active caspase 1 (Casp1 p10) in the supernatant and pro-IL-1β in cell lysate of peritoneal macrophages exposed or not exposed to ATP in the absence or presence of HgCl2. l, mitochondrial activity assessed by optical density (OD) of reduced WST-1 and release of IL-6 by lung macrophages exposed or not exposed to ATP (5 mm) in the absence or presence of HgCl2 (10 μm) (m). n = 3 or 4. Data are means ± S.E. **, p < 0.01; ***, p < 0.001 upon exposure to hypotonic medium or ATP or no exposure, with HgCl2 or no exposure; ns, no significant difference. p values were calculated by Student Newman-Keuls test.
The requirement of AQPs was extended to other soluble and insoluble NLRP3 inflammasome activators. The concentration of IL-1β detected in the supernatant of lung macrophages after silica (DQ12) and ATP was significantly lower when AQPs were inhibited (Fig. 2, g and i). Similarly, mercury inhibition of AQP led to decreased silica-induced (Fig. 2h) and ATP-induced (Fig. 2j) IL-1β release in peritoneal macrophages. The implication of AQP in caspase 1 activation (active caspase 1, casp1 p10) and mature IL-1β release was confirmed by Western blotting in ATP-treated peritoneal macrophages (Fig. 2k). AQP blockade also decreased the ATP-induced reduction of the intracellular IL-1β proform. The absence of cytotoxicity (Fig. 2l) and IL-6 release modifications (Fig. 2m) in the presence of mercury was verified in lung macrophages exposed to ATP. Together, these data indicate that rapid water transport via AQPs leads to IL-1β activation and release by lung and peritoneal macrophages after exposure to soluble and particulate NLPR3 stimuli.
AQP-facilitated RVD Is Sufficient to Induce the NLRP3 Inflammasome-related Release of Mature IL-1β in Macrophages
We next investigated which phase of the AQP-dependent volume changes (initial swelling or subsequent volume restoration) mediates inflammasome activation in macrophages. First, the kinetics of volume modifications, caspase 1 activation, and IL-1β release after hypotonic shock in lung macrophages were monitored. Cell swelling was obvious 2 min after exposure to hypotonic medium and sustained until 10 min. The subsequent size reduction was already perceptible 20 min after hypotonic shock, and the return to the initial size was achieved after 45 and 60 min (Fig. 3, a–c). IL-1β excretion was detected 30 min after hypotonic shock when the cell RVD process started (Fig. 3c). Similarly, the active caspase 1 content in macrophages was detected 30 min after hypotonic shock and increased up to 1 h (Fig. 3d). The implication of cell volume sensors in IL-1β excretion was assessed by using a nonspecific TRP inhibitor, Ruthenium Red. TRP inhibition resulted in decreased IL-1β release by lung macrophages in response to hypotonic shock (Fig. 3e). These observations suggested that IL-1β release by macrophages arise from the RVD process consecutive to the initial cell swelling.
FIGURE 3.

AQPs mediate IL-1β activation during the cell volume reduction process in macrophages. Shown are optical microscopy (Scale bar = 4 μm) (a) and flow cytometry forward scatter plots (b) of lung macrophages primed with LPS (0.1 μg/ml) at different time points after hypotonic shock (60 mosmol/kg H2O). Values indicated in the flow cytometry plots are differences between median forward scatter of cells exposed or not exposed to hypotonic shock. c, IL-1β release (left y axis) and flow cytometry forward scatter (FCS) (right y axis) of lung macrophages at different time points after hypotonic shock. n = 4 replicates/condition. d, flow cytometry plot of intracellular active caspase 1 content in lung macrophages 30 or 60 min after hypotonic (Hypo) shock (Iso, isotonic). Shown is the IL-1β release by lung macrophages 1 h after hypotonic shock in the absence or presence of Ruthenium Red (RR, 30 μm) (e) or HgCl2 (10 μm, added 10, 20, 30, 40, or 50 min after hypotonic shock) (f). n = 3 or 4. g, Western blot analysis of mature IL-1β (IL-1β p17) in the supernatant (SN) and pro-IL-1β (IL-1β p37) in cell lysate of lung macrophages exposed or not exposed to hypotonic shock in the absence or presence of HgCl2 (10 μm, added 10, 20, or 50 min after hypotonic shock). h, IL-6 release by lung macrophages 1 h after hypotonic shock in the absence or presence of HgCl2 (10 μm, added 10, 20, 30, 40, or 50 min after hypotonic shock). n = 3 or 4. i, flow cytometry forward scatter plots of lung macrophages 1 h after hypotonic shock in the presence or absence of HgCl2 (10 μm, added 10 min after hypotonic shock). The values indicated in the plots are differences between median forward scatter of cells exposed or not exposed to hypotonic shock. Data are means ± S.E. *, p < 0.05; ***, p < 0.001 upon exposure to hypotonic medium with Ruthenium Red or HgCl2 or no exposure; ns, no significant difference. p values were calculated by Student Newman-Keuls test.
To clarify the role of AQPs in the RVD-related shrinkage process, HgCl2 was added after cell swelling (i.e. at 10, 20, 30, 40, or 50 min) upon hypotonic stress. Inhibition of AQPs after cell swelling and when RVD was initiated was sufficient to reduce mature IL-1β release (Fig. 3, f and g) without affecting the intracellular IL-1β proform pool. This effect was not related to mercurial cytotoxicity (data not shown) nor IL-6 release modifications (Fig. 3h). To confirm the inhibitory effect of mercury on RVD, cell size was assessed 1 h after the shock when AQPs were inhibited 10 min after the shock (Fig. 3i). These data indicate that volume restoration following cell swelling is implicated in NLRP3 inflammasome-related IL-1β release and that this process involves AQPs.
AQP1 Mediates NLRP3-related Caspase 1 Activation and Mature IL-1β Release in Macrophages
We further investigated the implication of AQPs in NLRP3 inflammasome activation by targeting AQP1, a water-specific channel expressed by immune cells (11, 12, 15). Cell swelling consecutive to hypotonic shock was reduced in AQP1-deficient cells (Fig. 4a). AQP1 deficiency also reduced the release of mature IL-1β by lung (Fig. 4, b and c) and peritoneal (Fig. 4d) macrophages submitted to hypotonic shock. Similarly, caspase 1 activation because of hypotonic shock was impaired in AQP1-deficient lung macrophages (Fig. 4e). These results demonstrate that AQP1 leads to a rapid cell volume modification necessary for NLRP3 inflammasome activation and IL-1β release in response to osmotic shock.
FIGURE 4.

AQP1 contributes to swelling, caspase 1 activation, and mature IL-1β release in macrophages. a, flow cytometry forward scatter plots of WT or AQP1 KO lung macrophages (LM) primed with LPS (0.1 μg/ml) and subsequently subjected or not subjected to hypotonic shock (60 mosmol/kg H2O). The values indicated in plots are differences between median forward scatter of cells exposed or not exposed to hypotonic shock. b, IL-1β released by WT and AQP1 KO lung macrophages exposed or not exposed to hypotonic medium for 1 h. n = 3 replicates/condition. c, Western blot analysis of mature IL-1β (IL-1β p17) in the supernatant (SN) of WT and AQP1 KO lung macrophages exposed or not exposed to hypotonic medium. d, IL-1β released by WT and AQP1 KO peritoneal macrophages (PM) exposed or not exposed to hypotonic medium (100 mosmol/kg H2O) for 1 h. n = 3. e, fluorimetric measurement of cellular active caspase 1 in WT and AQP1 KO lung macrophages exposed or not exposed to hypotonic medium (60 mosmol/kg H2O) for 1 h. n = 3. f, IL-1β release by WT and AQP1 KO lung macrophages primed with LPS (0.1 μg/ml) and subsequently exposed or not exposed to ATP (5 mm). n = 3 or 4. g, Western blot analysis of mature IL-1β and active caspase 1 (Casp1 p10) in the supernatant of WT and AQP1 KO lung macrophages exposed or not exposed to ATP. Shown is the IL-1β release by WT and AQP1 KO lung macrophages primed with LPS (0.1 μg/ml) and subsequently exposed or not exposed to nigericin (Ng, 20 μm) (h), silica (DQ12, 400 μg/ml) (i), or SNP (100 μg/ml) (j). n = 3 or 4. k, Western blot analysis of intracellular pro-IL-1β (IL-1β p37), procaspase 1 (Casp1 p45), and β-actin in LPS-primed WT and AQP1-deficient lung macrophages. l, intracellular levels of pro-IL-1β in WT and AQP1 KO lung macrophages, quantified by ELISA. n = 3 or 4. Expression of NLRP3 (m) and ASC transcripts (n) quantified by quantitative RT-PCR in WT and AQP1 KO lung. n = 3 or 4. Data are means ± S.E. ***, p < 0.001 in WT or AQP1 KO cells; ns, no significant difference. p values were calculated by Student Newman-Keuls test.
The implication of AQP1 in NLRP3 inflammasome activation was extended to soluble and non-soluble inflammasome activators. AQP1 deletion decreased mature IL-1β release and caspase 1 activation by lung macrophages exposed to ATP (Fig. 4, f and g). The concentration of IL-1β detected in the supernatant of AQP1-deficient lung macrophages after nigericin, silica, and SNP was also significantly lower in comparison to cells from WT mice (Fig. 4, h–j). We then verified whether AQP1 deletion could affect the expression of NLRP3 inflammasome components (Fig. 4, k–n). AQP1 deficiency did not modify the pool of the IL-1β proform after LPS stimulation (Fig. 4, k and l). Similarly, no decrease in the expression of procaspase 1 (Fig. 4k, casp1 p45), NLRP3 (Fig. 4m), or ASC (Fig. 4n) was found in AQP1-deficient cells. Thus, these data indicate that AQP1 contributes to IL-1β activation and release by macrophages after exposure to NLRP3 soluble and particulate stimuli.
AQP1 Mediates Crystal-induced NLRP3 Inflammasome-dependent Lung Inflammation in Mice
To verify whether aquaporin-mediated inflammasome activation has relevance in inflammation, we assessed the acute lung response of mice deficient in AQP1 to silica particles, a danger signal inducing NLRP3 inflammasome assembly and mature IL-1β release in vivo (22). Silica-induced lung inflammation is mainly characterized by an early accumulation of neutrophils and an increase of proinflammatory mediators, both believed to be mediated by IL-1β (1, 23). DQ12 was administered in the lung of AQP1+/+ and AQP1−/− mice, and acute lung inflammatory parameters were compared. Early neutrophilic inflammation was quantified by counting alveolar neutrophils and assessing the lung expression of specific markers such as ngp and CXCR2 (24). The cellular response to silica was evidenced by an increased number of GR1+ cells in BAL fluid (Fig. 5a) and expression of neutrophilic markers in WT mice (Fig. 5, b and c), whereas lower levels of neutrophils as well as ngp and CXCR2 transcripts were detected in the lung of AQP1−/− mice. Silica-induced neutrophilic inflammation was also investigated histologically (Fig. 5, e–h). In hematoxylin and eosin-stained lung sections obtained from untreated mice, neutrophils were absent in WT and AQP1−/− samples (Fig. 5, e and f). When treated with silica, WT mice presented a more pronounced neutrophilic infiltration than AQP1 deficient mice (Fig. 5, g and h). Inflammasome activation was evaluated by the measurement of mature IL-1β in the alveolar space. The BAL fluid concentration of this cytokine was increased significantly after silica in WT mice and, to a lower extent, in AQP1-deficient animals (Fig. 5d). In contrast to silica particles, lung inflammation induced by the antibiotic bleomycin (not considered a direct NLRP3 activator) was not reduced in AQP1-deficient mice (data not shown). In agreement with in vitro observations, we concluded that AQP1 contributes to IL-1β release and neutrophilic inflammation in response to silica particles in vivo.
FIGURE 5.
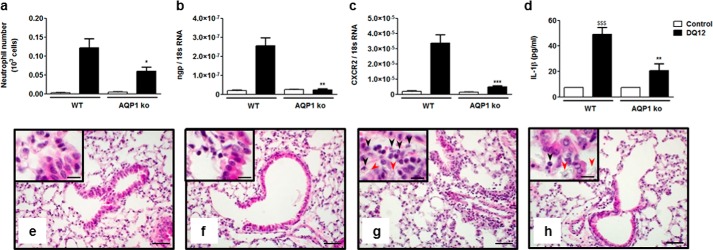
Acute lung inflammatory response to silica particles is reduced in AQP1-deficient mice. a, number of alveolar neutrophils (GR1+ cells) assessed by flow cytometry and expression of the pulmonary neutrophilic markers ngp (b) and CXCR2 (c) quantified by quantitative RT-PCR in WT and AQP1 KO mice 6 h after instillation of silica (DQ12, 2.5 mg) or no instillation (control). d, levels of mature IL-1β in BAL fluid collected 6 h after instillation of silica or no instillation. Data are means ± S.E. n = 3 to 6 animals/condition. e–h, hematoxylin and eosin-stained lung sections obtained from WT (e and g) or AQP1 KO mice (f and h), collected 6 h after instillation of silica (g and h) or no instillation (e and f). Black arrows identify neutrophils, and red arrows identify silica deposition. Scale bars = 50 μm (large panels) and 20 μm (insets). *, p < 0.05; **, p < 0.01; ***, p < 0.001 in silica-treated WT and AQP1 KO mice. p values were calculated by Student Newman-Keuls test.
DISCUSSION
Innate immune cells possess diverse sensors recognizing molecular patterns related to pathogenic or endogenous danger indicators, with most sensors recognizing a limited number of patterns. Inflammasomes can activate caspase 1 in response to a great variety of stimuli, including ATP, bacterial toxins, viral components, crystals, and particles (2, 15, 20, 22). The identification of a common element among these diverse stimuli that initiates inflammasome assembly may have a wide applicability in a range of inflammatory diseases (5).
Cell swelling is a hallmark of inflammatory conditions (25, 26). In particular, swelling has been linked to macrophage activation and to its ability to induce inflammation by cytokine release (27–29). Interestingly, macrophage swelling and following RVD are recorded in association with NLRP3 inflammasome activation and IL-1β release in response to different stimuli (6–8, 15, 30). In this study, we show the importance of AQP in cell volume changes, mainly RVD, for mature IL-1β release in vitro and in vivo. In particular, we demonstrate the implication of AQP1 in the development of neutrophilic inflammation. Rapid AQP-mediated water transport is thus sensed as a danger signal by the cell to trigger caspase 1 activation and mature IL-1β release in response to a variety of stimuli. The implication of AQPs in cell RVD and inflammasome activation is, however, not limited to AQP1. Mercury blocks most AQPs. Thus, its inhibitory effect on inflammasome activation could be related to different AQPs. Indeed, AQP9 is known to be expressed in leukocytes, and activation of the mononuclear cells following an intravenous endotoxin challenge results in modified expression of this water channel (13). Also, AQP9 inhibition has been linked to reduced inflammasome activation in response to monosodium urate crystals (15).
Several interactions of AQP with orchestrators of inflammasome activation can be proposed. Cells possess different volume sensor systems, such as the calcium-permeable channel protein TRP, driving RVD in macrophages. The activation of these channels and the associated ionic changes activate TAK1, a MAPKK kinase, necessary for hypotonicity-, alum-, ATP- and nigericin-induced mature IL-1β release and inflammation (7, 8). In this study, we confirmed the implication of TRP in IL-1β release by macrophages in response to hypotonic shock. Interestingly, it has been observed that hypotonicity in salivary gland cells increased the association of TRPV4 and AQP5. In this case, TRP activation required AQP (31). The RVD process implies an efflux of intracellular K+ that has been shown to be necessary for inflammasome activation in response to various stimuli, including hypotonic shock (5, 7, 20). In this study, we found that intracellular potassium decrease was not reduced by AQP1 deficiency in lung macrophages exposed to hypotonic shock (data not shown). Thus, the implication of AQP1 in IL-1β activation is not related to potassium efflux. Finally, it has been observed that inhibition of cell swelling and water flux by using mercury and phloretin reduced the capacity of macrophages to release mature IL-1β after monosodium urate crystals exposure (15). The authors concluded that water entry reduced intracellular potassium concentration to critical levels necessary for inflammasome assembly in this model. During the release process of mature IL-1β, active caspase 1/ASC aggregates abutted the external surfaces of actin filaments (32). Thus, it is also possible that AQP-mediated water efflux regulates caspase 1 processing and mature IL-1β release by modulating actin cytoskeletal rearrangements (33).
A possible implication of AQPs in inflammatory diseases has been proposed because of their role in cell migration and edema formation (34). At the alveolar level, AQP1 and AQP5 are the most abundant, with an endothelial or epithelial distribution, respectively (35, 36). It has been shown that AQP5 knockout mice develop less inflammation in a model of house dust mite-induced asthma (37). We observed that AQP1 deficiency reduces neutrophilic inflammation as well as cytokine release in silica-induced lung inflammation. Besides the respiratory tract, AQPs have been associated with inflammation in other organs (38, 39). In a model of peritoneal dialysis, reduced accumulation of leukocytes was observed in mice genetically deficient in AQP1 (40). AQP4-deficient mice developed less inflammation in a model of autoimmune encephalomyelitis (41). In light of our results, we propose that, in addition to their possible role in cell migration and edema formation, AQP-mediated water fluxes in macrophages, subsequent RVD, and, finally, mature IL-1β release contribute to the inflammatory responses in these experimental models.
In humans, AQP alterations have also been correlated with several inflammatory diseases. Overexpression of AQP1 has been evidenced in autoimmune and alcoholic pancreatitis (42), in Alzheimer disease (43), as well as in rheumatoid and psoriatic arthritis cases (44). AQP4 expression is enhanced in the brain of Alzheimer disease patients and cerebral amyloid angiopathy patients (43, 45). AQP5 polymorphisms have been associated with chronic obstructive pulmonary disease and chronic inflammation in smoking patients and survival rate in severe sepsis (46, 47). These data further support a role of AQPs in human inflammatory diseases. Hemorrhagic shock and sepsis result in multiple organ failure, in particular acute lung injury, which may lead to death. IL-1β and NLRP3 activation have been implicated in this process (48, 49). Interestingly, hypertonic solution has been shown to reduce inflammation and to increase the vital prognosis of these patients (50). The anti-inflammatory mechanism of this hypertonic resuscitation procedure is still unclear but, in light of our data, we postulate that it reduces AQP-mediated water transport and, consecutively, restricts inflammasome activation. Hence, AQPs should be considered as a new target in inflammation-associated disorders.
In conclusion, we demonstrated that rapid AQP-mediated cell swelling and mainly subsequent volume restoration are sensed as danger signals by macrophages and trigger NLRP3 inflammasome activation. AQP-mediated water movements appear as the common element unifying the variety of NLRP3 inflammasome activators. In vivo, AQPs contribute to inflammation by mediating mature IL-1β release.
This work was supported by the Programme d'excellence Marshall Diane convention, by the Fonds de la Recherche Scientifique Médicale (FRSM), by Actions de Recherche Concertées, Communauté Française de Belgique, Direction de la Recherche Scientifique grant ARC 09/14-021), by the Fondation Contre le Cancer, by Fonds de la Recherche Scientifique (FNRS) Project PDR T.0119.13 14633768, by the European Commission under FP7-HEALTH-F4-2008 contract no. 202047, and by Agence Nationale de Sécurité Sanitaire de L'Alimentation, de l'Environnement, et du Travail (ANSES, France). This work was also supported in part by the Fondation Saint-Luc at Université Catholique de Louvain, by a Baxter extramural grant, by the Inter-University Attraction Pole (IUAP, Belgium Federal Government), by the NCCR Kidney.CH program (Swiss National Science Foundation), and by Swiss National Science Foundation grant 310030-146490 (to O. D.).
- RVD
- regulatory volume decrease
- TRP
- transient receptor potential
- AQP
- aquaporin
- BAL
- bronchoalveolar lavage
- ASC
- apoptosis-associated speck-like protein containing a CARD
- ngp
- neutrophilic granule protein
- DQ12
- crystalline silica (α-quartz)
- SNP
- silica nanoparticles.
REFERENCES
- 1. Cassel S. L., Eisenbarth S. C., Iyer S. S., Sadler J. J., Colegio O. R., Tephly L. A., Carter A. B., Rothman P. B., Flavell R. A., Sutterwala F. S. (2008) The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. U.S.A. 105, 9035–9040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomas P. G., Dash P., Aldridge J. R., Jr., Ellebedy A. H., Reynolds C., Funk A. J., Martin W. J., Lamkanfi M., Webby R. J., Boyd K. L., Doherty P. C., Kanneganti T. D. (2009) The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30, 566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoshino T., Okamoto M., Sakazaki Y., Kato S., Young H. A., Aizawa H. (2009) Role of proinflammatory cytokines IL-18 and IL-1β in bleomycin-induced lung injury in humans and mice. Am. J. Respir. Cell Mol. Biol. 41, 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cai S., Batra S., Wakamatsu N., Pacher P., Jeyaseelan S. (2012) NLRC4 inflammasome-mediated production of IL-1β modulates mucosal immunity in the lung against Gram-negative bacterial infection. J. Immunol. 188, 5623–5635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muñoz-Planillo R., Kuffa P., Martínez-Colón G., Smith B. L., Rajendiran T. M., Núñez G. (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Perregaux D. G., Laliberte R. E., Gabel C. A. (1996) Human monocyte interleukin-1β posttranslational processing. Evidence of a volume-regulated response. J. Biol. Chem. 271, 29830–29838 [DOI] [PubMed] [Google Scholar]
- 7. Compan V., Baroja-Mazo A., López-Castejón G., Gomez A. I., Martínez C. M., Angosto D., Montero M. T., Herranz A. S., Bazán E., Reimers D., Mulero V., Pelegrín P. (2012) Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37, 487–500 [DOI] [PubMed] [Google Scholar]
- 8. Gross O., Yazdi A. S., Thomas C. J., Masin M., Heinz L. X., Guarda G., Quadroni M., Drexler S. K., Tschopp J. (2012) Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36, 388–400 [DOI] [PubMed] [Google Scholar]
- 9. Agre P., Sasaki S., Chrispeels M. J. (1993) Aquaporins: a family of water channel proteins. Am. J. Physiol. 265, F461. [DOI] [PubMed] [Google Scholar]
- 10. de Baey A., Lanzavecchia A. (2000) The role of aquaporins in dendritic cell macropinocytosis. J. Exp. Med. 191, 743–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moon C., Rousseau R., Soria J. C., Hoque M. O., Lee J., Jang S. J., Trink B., Sidransky D., Mao L. (2004) Aquaporin expression in human lymphocytes and dendritic cells. Am. J. Hematol. 75, 128–133 [DOI] [PubMed] [Google Scholar]
- 12. Jablonski E. M., Webb A. N., McConnell N. A., Riley M. C., Hughes F. M., Jr. (2004) Plasma membrane aquaporin activity can affect the rate of apoptosis but is inhibited after apoptotic volume decrease. Am. J. Physiol. Cell Physiol. 286, C975–985 [DOI] [PubMed] [Google Scholar]
- 13. Talwar S., Munson P. J., Barb J., Fiuza C., Cintron A. P., Logun C., Tropea M., Khan S., Reda D., Shelhamer J. H., Danner R. L., Suffredini A. F. (2006) Gene expression profiles of peripheral blood leukocytes after endotoxin challenge in humans. Physiol. Genomics 25, 203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu N., Feng X., He C., Gao H., Yang L., Ma Q., Guo L., Qiao Y., Yang H., Ma T. (2011) Defective macrophage function in aquaporin-3 deficiency. FASEB J. 25, 4233–4239 [DOI] [PubMed] [Google Scholar]
- 15. Schorn C., Frey B., Lauber K., Janko C., Strysio M., Keppeler H., Gaipl U. S., Voll R. E., Springer E., Munoz L. E., Schett G., Herrmann M. (2011) Sodium overload and water influx activate the NALP3 inflammasome. J. Biol. Chem. 286, 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma T., Yang B., Gillespie A., Carlson E. J., Epstein C. J., Verkman A. S. (1998) Severely impaired urinary concentrating ability in transgenic mice lacking aquaporin-1 water channels. J. Biol. Chem. 273, 4296–4299 [DOI] [PubMed] [Google Scholar]
- 17. Rabolli V., Lo Re S., Uwambayinema F., Yakoub Y., Lison D., Huaux F. (2011) Lung fibrosis induced by crystalline silica particles is uncoupled from lung inflammation in NMRI mice. Toxicol. Lett. 203, 127–134 [DOI] [PubMed] [Google Scholar]
- 18. Herzyk D. J., Berger A. E., Allen J. N., Wewers M. D. (1992) Sandwich ELISA formats designed to detect 17 kDa IL-1 β significantly underestimate 35 kDa IL-1 β. J. Immunol. Methods 148, 243–254 [DOI] [PubMed] [Google Scholar]
- 19. Thomassen L. C., Aerts A., Rabolli V., Lison D., Gonzalez L., Kirsch-Volders M., Napierska D., Hoet P. H., Kirschhock C. E., Martens J. A. (2010) Synthesis and characterization of stable monodisperse silica nanoparticle sols for in vitro cytotoxicity testing. Langmuir 26, 328–335 [DOI] [PubMed] [Google Scholar]
- 20. Perregaux D., Gabel C. A. (1994) Interleukin-1 β maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 269, 15195–15203 [PubMed] [Google Scholar]
- 21. Savage D. F., Stroud R. M. (2007) Structural basis of aquaporin inhibition by mercury. J. Mol. Biol. 368, 607–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hornung V., Bauernfeind F., Halle A., Samstad E. O., Kono H., Rock K. L., Fitzgerald K. A., Latz E. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dostert C., Pétrilli V., Van Bruggen R., Steele C., Mossman B. T., Tschopp J. (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Van Belle A. B., de Heusch M., Lemaire M. M., Hendrickx E., Warnier G., Dunussi-Joannopoulos K., Fouser L. A., Renauld J. C., Dumoutier L. (2012) IL-22 is required for imiquimod-induced psoriasiform skin inflammation in mice. J. Immunol. 188, 462–469 [DOI] [PubMed] [Google Scholar]
- 25. Solez K., Axelsen R. A., Benediktsson H., Burdick J. F., Cohen A. H., Colvin R. B., Croker B. P., Droz D., Dunnill M. S., Halloran P. F. (1993) International standardization of criteria for the histologic diagnosis of renal allograft rejection: the Banff working classification of kidney transplant pathology. Kidney Int. 44, 411–422 [DOI] [PubMed] [Google Scholar]
- 26. Bonventre J. V., Weinberg J. M. (2003) Recent advances in the pathophysiology of ischemic acute renal failure. J. Am. Soc. Nephrol. 14, 2199–2210 [DOI] [PubMed] [Google Scholar]
- 27. Sokol R. J., Hudson G., James N. T., Frost I. J., Wales J. (1987) Human macrophage development: a morphometric study. J. Anat. 151, 27–35 [PMC free article] [PubMed] [Google Scholar]
- 28. Pankow W., Neumann K., Rüschoff J., von Wichert P. (1995) Human alveolar macrophages: comparison of cell size, autofluorescence, and HLA-DR antigen expression in smokers and nonsmokers. Cancer Detect. Prev. 19, 268–273 [PubMed] [Google Scholar]
- 29. Daigneault M., Preston J. A., Marriott H. M., Whyte M. K., Dockrell D. H. (2010) The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PloS ONE 5, e8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Palomäki J., Välimäki E., Sund J., Vippola M., Clausen P. A., Jensen K. A., Savolainen K., Matikainen S., Alenius H. (2011) Long, needle-like carbon nanotubes and asbestos activate the NLRP3 inflammasome through a similar mechanism. ACS Nano 5, 6861–6870 [DOI] [PubMed] [Google Scholar]
- 31. Liu X., Bandyopadhyay B. C., Bandyopadhyay B., Nakamoto T., Singh B., Liedtke W., Melvin J. E., Ambudkar I. (2006) A role for AQP5 in activation of TRPV4 by hypotonicity: concerted involvement of AQP5 and TRPV4 in regulation of cell volume recovery. J. Biol. Chem. 281, 15485–15495 [DOI] [PubMed] [Google Scholar]
- 32. Pelegrin P., Surprenant A. (2009) Dynamics of macrophage polarization reveal new mechanism to inhibit IL-1β release through pyrophosphates. EMBO J. 28, 2114–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Papadopoulos M. C., Saadoun S., Verkman A. S. (2008) Aquaporins and cell migration. Pflugers Arch. 456, 693–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Verkman A. S. (2012) Aquaporins in clinical medicine. Annu. Rev. Med. 63, 303–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Towne J. E., Harrod K. S., Krane C. M., Menon A. G. (2000) Decreased expression of aquaporin (AQP)1 and AQP5 in mouse lung after acute viral infection. Am. J. Resp. Cell Mol. Biol. 22, 34–44 [DOI] [PubMed] [Google Scholar]
- 36. Jang A. S., Lee J. U., Choi I. S., Park K. O., Lee J. H., Park S. W., Park C. S. (2004) Expression of nitric oxide synthase, aquaporin 1 and aquaporin 5 in rat after bleomycin inhalation. Intensive Care Med. 30, 489–495 [DOI] [PubMed] [Google Scholar]
- 37. Shen Y., Wang Y., Chen Z., Wang D., Wang X., Jin M., Bai C. (2011) Role of aquaporin 5 in antigen-induced airway inflammation and mucous hyperproduction in mice. J. Cell. Mol. Med. 15, 1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hardin J. A., Wallace L. E., Wong J. F., O'Loughlin E. V., Urbanski S. J., Gall D. G., MacNaughton W. K., Beck P. L. (2004) Aquaporin expression is downregulated in a murine model of colitis and in patients with ulcerative colitis, Crohn's disease and infectious colitis. Cell Tissue Res. 318, 313–323 [DOI] [PubMed] [Google Scholar]
- 39. Auguste K. I., Jin S., Uchida K., Yan D., Manley G. T., Papadopoulos M. C., Verkman A. S. (2007) Greatly impaired migration of implanted aquaporin-4-deficient astroglial cells in mouse brain toward a site of injury. FASEB J. 21, 108–116 [DOI] [PubMed] [Google Scholar]
- 40. Nishino T., Devuyst O. (2008) Clinical application of aquaporin research: aquaporin-1 in the peritoneal membrane. Pflugers Arch. 456, 721–727 [DOI] [PubMed] [Google Scholar]
- 41. Li L., Zhang H., Verkman A. S. (2009) Greatly attenuated experimental autoimmune encephalomyelitis in aquaporin-4 knockout mice. BMC Neurosci. 10, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ko S. B., Mizuno N., Yatabe Y., Yoshikawa T., Ishiguro H., Yamamoto A., Azuma S., Naruse S., Yamao K., Muallem S., Goto H. (2009) Aquaporin 1 water channel is overexpressed in the plasma membranes of pancreatic ducts in patients with autoimmune pancreatitis. J. Med. Invest. 56, 318–321 [DOI] [PubMed] [Google Scholar]
- 43. Huysseune S., Kienlen-Campard P., Hébert S., Tasiaux B., Leroy K., Devuyst O., Brion J. P., De Strooper B., Octave J. N. (2009) Epigenetic control of aquaporin 1 expression by the amyloid precursor protein. FASEB J. 23, 4158–4167 [DOI] [PubMed] [Google Scholar]
- 44. Mobasheri A., Moskaluk C. A., Marples D., Shakibaei M. (2010) Expression of aquaporin 1 (AQP1) in human synovitis. Anat. Anz. 192, 116–121 [DOI] [PubMed] [Google Scholar]
- 45. Moftakhar P., Lynch M. D., Pomakian J. L., Vinters H. V. (2010) Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 69, 1201–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hansel N. N., Sidhaye V., Rafaels N. M., Gao L., Gao P., Williams R., Connett J. E., Beaty T. H., Mathias R. A., Wise R. A., King L. S., Barnes K. C. (2010) Aquaporin 5 polymorphisms and rate of lung function decline in chronic obstructive pulmonary disease. PloS ONE 5, e14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adamzik M., Frey U. H., Möhlenkamp S., Scherag A., Waydhas C., Marggraf G., Dammann M., Steinmann J., Siffert W., Peters J. (2011) Aquaporin 5 gene promoter: 1364A/C polymorphism associated with 30-day survival in severe sepsis. Anesthesiology 114, 912–917 [DOI] [PubMed] [Google Scholar]
- 48. Mao K., Chen S., Chen M., Ma Y., Wang Y., Huang B., He Z., Zeng Y., Hu Y., Sun S., Li J., Wu X., Wang X., Strober W., Chen C., Meng G., Sun B. (2013) Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 23, 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu P., Wen Z., Shi X., Li Y., Fan L., Xiang M., Li A., Scott M. J., Xiao G., Li S., Billiar T. R., Wilson M. A., Fan J. (2013) Hemorrhagic shock augments Nlrp3 inflammasome activation in the lung through impaired pyrin induction. J. Immunol. 190, 5247–5255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oliveira R. P., Velasco I., Soriano F. G., Friedman G. (2002) Clinical review: Hypertonic saline resuscitation in sepsis. Crit. Care 6, 418–423 [DOI] [PMC free article] [PubMed] [Google Scholar]