

Background: hPso4 has been implicated in the resistance to interstrand cross-linking DNA-damaging agents.
Results: hPso4 complex is involved in normal replication, and it has a role in DNA repair by homologous recombination through the regulation of DNA strand resection.
Conclusion: hPso4 has a function in DNA double strand break repair by homologous recombination.
Significance: hPso4 complex levels could predict sensitivity of cancer cells to PARP inhibitors.
Keywords: BRCA1, DNA Damage, DNA Repair, DNA Replication, Homologous Recombination
Abstract
Psoralen 4 (Pso4) is an evolutionarily conserved protein that has been implicated in a variety of cellular processes including RNA splicing and resistance to agents that cause DNA interstrand cross-links. Here we show that the hPso4 complex is required for timely progression through S phase and transition through the G2/M checkpoint, and it functions in the repair of DNA lesions that arise during replication. Notably, hPso4 depletion results in delayed resumption of DNA replication after hydroxyurea-induced stalling of replication forks, reduced repair of spontaneous and hydroxyurea-induced DNA double strand breaks (DSBs), and increased sensitivity to a poly(ADP-ribose) polymerase inhibitor. Furthermore, we show that hPso4 is involved in the repair of DSBs by homologous recombination, probably by regulating the BRCA1 protein levels and the generation of single strand DNA at DSBs. Together, our results demonstrate that hPso4 participates in cell proliferation and the maintenance of genome stability by regulating homologous recombination. The involvement of hPso4 in the recombinational repair of DSBs provides an explanation for the sensitivity of Pso4-deficient cells to DNA interstrand cross-links.
Introduction
PSO4 was initially identified as one of several yeast genes required for resistance to DNA interstrand cross-links (ICLs)3 induced by psoralen in combination with ultraviolet light (1). Subsequent studies revealed that Pso4 is identical to Prp19, a protein required for spliceosome integrity and completion of the splicing reaction (2). The Pso4 protein harbors an N-terminal U-box ubiquitin E3 ligase domain and C-terminal WD40 repeats, and there is evidence that it is a functional ubiquitin ligase (3, 4). Similar to other E3 ligases, the human homolog of Prp19/Pso4 (hPrp19/hPso4) exists in a multiprotein complex consisting of seven subunits, hPrp19/hPso4, CDC5L, PRL1, AD002, SPF27, CTNNBL1, and HSP73) (5). hPso4 maintains the overall domain structure of the Saccharomyces cerevisiae protein with most of the other complex subunits being conserved from yeast to humans. The architecture of the Pso4 complex has been described in relation to its function in pre-mRNA splicing (5, 6).
The human homolog of Pso4 has also been implicated in DNA repair, directly participating in the repair of ICL substrates in vitro in a cell-free system that requires DNA synthesis, which suggests a splicing-independent role (7). hPso4 has also been linked to S phase checkpoint activation in response to UV light, and the hPso4 complex subunit CDC5L has been shown to interact with the ATM and Rad3-related (ATR) kinase, which is a key mediator of the checkpoint response (8). Although genetic studies in both yeast and mammalian cells have identified several pathways for the repair of ICLs (9), hPso4 has not been directly implicated in any of these pathways.
In this report, we describe a role for hPso4 in cell cycle regulation and DNA double strand break (DSB) repair. Specifically, we show that the hPso4 complex plays important roles in S phase progression in unperturbed cells, recovery from hydroxyurea (HU)-induced replication stress, and the repair of DSBs that result from collapsed replication forks. In accord with HR being the major pathway that repairs replication-associated DSBs, we demonstrate a role for the hPso4 complex in this repair pathway, an association with the breast cancer type 1 susceptibility protein (BRCA1) A complex, and a requirement for generating single stranded DNA that is a prerequisite for HR to proceed.
EXPERIMENTAL PROCEDURES
Cell Lines and siRNA Treatment
U2OS, HEK293, and A549 cells were obtained from the American Type Culture Collection (ATCC) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and a mixture of antibacterial and antifungal antibiotics. Cells were incubated at 37 °C in 5% CO2. Pools of four siRNAs specific for hPso4 and CDC5L were obtained from Dharmacon (Smart Pool) and transfected into the cells using Lipofectamine RNAi/Max reagent according to the manufacturer's instructions (Invitrogen). siRNA was delivered at 100 nm concentration in two transfections 24 h apart. An siRNA targeting the 3′-UTR of hPso4/PRPF19 mRNA, to decrease the endogenous but not the overexpressed protein, with the sense strand sequence of CAGTAGAGGGGTAGAATTA was obtained from Dharmacon. Scrambled siRNA controls were used in all experiments.
Antibodies and Plasmids
Rabbit hPso4 and p-RPA H4/H8 (S4/S8) antibodies were obtained from Bethyl. Mouse CDC5L, proliferative cell nuclear antigen (PCNA), and Rad51 antibodies were from Santa Cruz Biotechnology. Mouse BRCA2 and BRCA-1(Ab-1) antibodies were from Millipore. BrdU antibody was from BD Biosciences. hPso4 DNA was amplified from a placenta cDNA library and cloned into the pcDNA3.1 vector. The wild type hPso4 gene was mutated by deleting three conserved amino acid residues in the U-box using a site-directed mutagenesis kit from Agilent. All clones were verified by Sanger sequencing in the Keck-University of New Mexico Genomics Shared Resource.
Immunofluorescence Staining
U2OS cells growing on glass coverslips were pre-extracted with 0.3% Triton X-100 solution and fixed in 3% paraformaldehyde. After blocking with 4% bovine serum albumin (BSA) in PBS, cells were incubated with antibodies against PCNA (PC-10; Santa Cruz Biotechnology) and Pso4 (Bethyl) for 1 h, washed, and then incubated with Alexa Fluor 633-tagged donkey anti-rabbit and 488-tagged goat anti-mouse secondary antibodies (Invitrogen). After 1 h, coverslips were washed and mounted on slides with 4′,6-diamidino-2-phenylindole (DAPI) in Vectashield mounting medium. For DSB assessment, γH2AX foci were visualized using mouse anti-γH2AX (Millipore) and Alexa Fluor goat anti-mouse antibodies. Two hundred cells were counted for each experiment. Slides were visualized on a Zeiss Meta Confocal microscope equipped with an oil-immersed 60× objective at the University of New Mexico Cancer Center Fluorescence Microscopy and Cell Imaging Shared Resource.
Cell Cycle Analysis
U2OS cells were treated with control siRNAs or siRNAs against hPso4 or CDC5L and subsequently synchronized with a double thymidine block (first incubation with 2 mm thymidine for 17 h, then released into a complete medium for 12 h, followed by another thymidine incubation for 17 h). After the second thymidine treatment, the cells were washed with PBS, and a fresh complete medium was added. Cells were collected over time, fixed with 70% EtOH, and treated with RNase H, propidium iodide at 50 μg/ml concentration, and analyzed by Caliber FACS Scan for DNA content.
BrdU Incorporation
U2OS cells were incubated with siRNA against hPso4 or scrambled siRNA for 72 h, followed by the addition of 2 mm HU for 4 h. After washing, cells were pulsed with 10 μm BrdU for 20 min and fixed with 70% EtOH. DNA was denatured with 3 n HCl and subsequently neutralized with 0.1 m sodium borate (Na2B4O7). After blocking with BSA, BrdU incorporated into genomic DNA was detected with mouse anti-BrdU antibody (BD Biosciences) and goat anti-mouse Alexa Fluor 488 antibody. Washed cells were analyzed by FACS for BrdU content against side scatter.
Cell Viability Assays
Cells were treated with two rounds of control or hPso4 siRNA and distributed into the wells of a 96-well plate. After 48 h, HU or the poly(ADP-ribose) polymerase (PARP) inhibitor ABT-888 (Abbott) was added at the specified concentrations. Cells were analyzed 72 h later for viability utilizing the Cell Titer 96® NonRadioactive Cell Proliferation Assay (MTT) from Promega according to the manufacturer's instructions. To determine radiation sensitivity, cells were treated with the appropriate siRNA for 72 h, distributed into a 96-well plate, and exposed to ionizing radiation from a cesium 137 source at a range of doses. The cells were evaluated for viability 48 h later using the MTT assay.
Immunoprecipitation (IP) and Western Blotting
Cells were collected with IP lysis buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol (DTT), 0.5% Nonidet P-40, with a mixture of protease and phosphatase inhibitors (Sigma)), sonicated, and centrifuged at high speed for 30 min. Samples were precleared and incubated with protein G-conjugated magnetic beads (Dynabeads; Invitrogen) that had been bound to the corresponding antibodies. The beads were washed extensively with the IP lysis buffer and PBS and boiled in Laemmli sample buffer. For Rap80 IP, nuclear extracts were used. These were obtained by swelling cells with hypotonic buffer (10 mm HEPES-KOH, pH 7.9, 10 mm KCl, 0.1 mm EDTA, and 1 mm DTT) for 10 min, followed by the addition of Nonidet P-40 to a final concentration of 0.5%. After vortexing for 2 min, nuclear pellets were collected, lysed with IP lysis buffer, and sonicated. For Western blot analysis, proteins were separated on SDS-polyacrylamide gels, transferred to PVDF membranes, and blocked with 4% milk or 2% BSA before primary antibodies were added. Antibody-antigen complexes were detected using horseradish peroxidase (HRP)-conjugated secondary antibodies and the enhanced chemiluminescence (ECL) substrate from Thermo Scientific.
The HR Reporter Assay
HeLa-DR-GFP and U2OS-DR-GFP cells harboring an integrated reporter substrate to measure HR substrate were kindly provided by D. Weinstock. These cells were subjected to the indicated siRNA in a 12-well plate format and 48 h later transfected with a plasmid encoding I-SceI with another round of siRNA using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). Cells were analyzed 72 h later for GFP expression by FACS at the University of New Mexico Cancer Center Flow Cytometry and High Throughput Shared Resource.
RT-PCR
After two rounds of siRNA for 72 h, total RNA was extracted using the RNeasy mini kit (Qiagen). cDNA was synthesized from equal amounts of mRNA using the reverse transcriptase SuperScript III (Invitrogen). Triplicates of cDNA samples were assembled in a 96-well plate. Real-time PCR was conducted utilizing Maxima Cybergreen master mix (Thermo Scientific), and the amplification reaction was followed on the Step One Plus real-time PCR machine (Applied Biosystems). The results were normalized to mRNA levels of 18S and β-actin.
RESULTS
Human Pso4 Is Required for Cell Proliferation and Co-localizes with the Replication Clamp PCNA
To investigate the cellular roles of hPso4, we decreased its levels by siRNA. In initial studies, we observed that siRNA against hPso4 but not a scrambled siRNA markedly reduced the proliferation of two human cell lines (Fig. 1, A and B). Moreover, we were unable to obtain viable clones of hPso4 knockdown cells in clonogenic survival assays. To determine whether this growth defect was due to the direct involvement of hPso4 in cell proliferation, we examined the subcellular localization of hPso4 and found that it co-localized with the DNA replication clamp PCNA in both unperturbed cells and in cells treated with HU (Fig. 1, C–E), suggesting that hPso4 may directly participate in DNA replication. We observed hPso4 foci even when PCNA protein levels were depleted by siRNA (Fig. 1, F and G). This could be due to incomplete depletion of PCNA protein or to a PCNA-independent recruitment of hPso4 to replication origins. No interaction between hPso4 and PCNA could be demonstrated by IP (data not shown). Subsequently, we investigated the effect of hPso4 depletion on cell cycle progression in HeLa cells arrested at the G1-S boundary by a double thymidine block. We noted that the knockdown of hPso4 resulted in an equivalent reduction in the level of CDC5L and vice versa (see Figs. 5F and 7), supporting previous reports that the two proteins function together in a complex (5, 6). Depletion of either hPso4 or CDC5L resulted in a slower progression through S phase after release from the block (Fig. 2). In addition, cells depleted of these factors entered mitosis later than cells treated with scrambled siRNA and remained in the G2/M phase 24 h after release from cell cycle arrest. In contrast, most cells treated with scrambled siRNA had passed through mitosis and progressed into a new cycle by this time (Fig. 2).
FIGURE 1.

Effects of hPso4 knock-down on cell proliferation and co-localization of hPso4 with PCNA. A and B, cell proliferation of U20S cells (A) and HEK293 cells (B), treated with either hPso4 siRNA (open circles) or scrambled siRNA (filled circles). The level of protein knockdown is shown in the panels below each experiment. C, co-localization of hPso4 with PCNA in untreated U20S cells. D, the same in U2OS cells treated with 2 mm HU for 4 h. Upper left, PCNA antibody (green); lower left, hPso4 antibody; upper right, DAPI staining of DNA; lower right, overlay of images. E, percentage of cells with co-localized hPso4 and PCNA foci before and after HU addition. F, PCNA depletion not eliminating the hPso4 foci. G, PCNA and hPso4 foci co-localization when scrambled siRNA was used. Error bars, S.E.
FIGURE 5.

HR defects in cells depleted of hPso4 units. Levels of HR were determined by utilizing the DR-GFP substrate and counting the number of GFP-positive cells after the addition of I-SceI plasmid. A, the structure of the integrated substrate. For details on the methods please refer to “Experimental Procedures.” A–E, flow cytometric analysis of the GFP-positive cells after incubation with I-SceI (A) and scrambled siRNA (B), hPso4 siRNA (C), BRCA2 siRNA (D), and Rad51 siRNA (E). F, effects of the used siRNAs on the protein levels. G, representation of the percentage of GFP-positive cells after each siRNA treatment. H, depletion of each of the hPso4 complex units resulting in defective HR in HeLa-DR-GFP cells. I, conserved U-box sequence that was mutated by deleting the amino acid residues shown in red. J, rescue of the HR phenotype with siRNA-resistant hPso4 vector, either wild type or mutant. BRCA1 siRNA was used as a positive control. EV, empty vector; error bars, S.E.
FIGURE 7.

Interaction of the hPso4 and BRCA1 complexes and the effect of hPso4 complex knockdown on BRCA1 protein levels and p-RPA generation. A and B, immunoprecipitation of CDC5L by NBA1 antibody in HEK293 cells (A) or U2OS cells (B). C, immunoprecipitation of CDC5L by RAP80. D, hPso4 immunoprecipitated by NBA1 antibody. E, depleting hPso4 by siRNA leading to a reduced BRCA1 protein level and a decrease in RPA phosphorylation after HU in HEK293. F, depletion of CDC5L resulting in similar findings in HEK293 cells. G and H, same phenotype also observed in U2OS cells after the depletion of hPso4 (G) or CDC5L (H). I, decrease in BRCA1, CDC5L protein levels detected after hPso4 siRNA administration rescued by siRNA-resistant clones of hPso4 (WT untagged, WT FLAG-tagged, and three amino acids-deleted U-box mutant).
FIGURE 2.

Kinetics of cell cycle progression in hPso4 complex-depleted cells. HeLa cells were treated with scrambled siRNA (A), CDC5L siRNA (B), or hPso4 siRNA (C), and analyzed for cell cycle progression. U2OS cells were synchronized at G1/S by a double thymidine block and then released into serum-rich media at time 0 (72 h after addition of siRNA). The histograms show cell cycle progression for 24 h after the release by measuring DNA content with propidium iodide (x axis) versus cell count (y axis). The protein levels after knockdown are shown in the top panel.
hPso4 Contributes to HU Resistance and Fork Progression after Release from HU
Many of the proteins associated with replication foci are involved in the cellular response to replication stress that may occur due to internal or external factors. To address the potential role of hPso4 in the cellular response to replication stress, we examined the effects of knocking down hPso4 on the sensitivity to HU, which causes replication stress by depleting deoxynucleotide pools. As shown in Fig. 3A, the osteosarcoma U2OS cells with reduced levels of hPso4 were significantly more sensitive to chronic HU exposure compared with control cells. We measured this HU sensitivity by utilizing the MTT method because hPso4-depleted cells failed to generate colonies as we alluded to above. Under chronic exposure to HU the replication forks of U2OS cells collapse, whereas transient exposure results in fork stalling (10). To investigate the role of hPso4 in fork restart after stalling, we treated cells with HU for 4 h and then measured BrdU incorporation after the removal of HU. Under these conditions, depletion of hPso4 resulted in a reduced rate of BrdU incorporation (Fig. 3, B, C, and E), indicating that hPso4 contributes to replication fork stability and/or replication restart.
FIGURE 3.
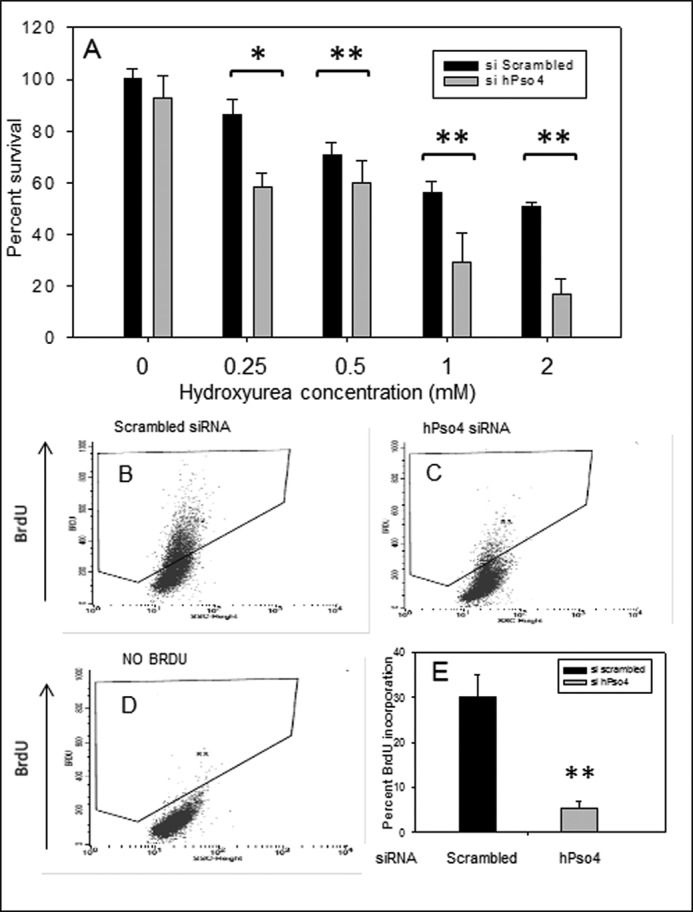
HU sensitivity and BrdU incorporation in cells depleted of hPso4. A, viability of U2OS cells was measured by the MTT assay in response to increasing concentrations of HU in the presence of hPso4 or scrambled siRNA. B and C, BrdU incorporation into DNA of replicating U2OS was measured after release from fork stalling induced by 4 h of 2 mm HU, in the presence of scrambled siRNA (B) or hPso4 siRNA (C). D, no BrdU (negative control) sample that was stained with the same antibody (anti-BrdU antibody). y axis is BrdU, x axis is side scatter. E, representation of the percentage of BrdU-positive cells with data collected from three different experiments. Statistical differences were determined using Student's t test with α values of 0.05 and 0.01 (*, p < 0.05; **, p < 0.01). Error bars, S.E.
hPso4 Depletion Results in Increased Levels of Spontaneous and HU-induced DSBs
Replication fork collapse resulting from chronic HU exposure generates DSBs (10, 11), which are marked by the phosphorylated histone variant H2AX (γH2AX) (12). To examine the possible role of hPso4 in the repair of HU-induced DSBs, U20S cells treated with scrambled or hPso4 siRNAs were incubated with HU for 16 h and then allowed to recover for 18 h in the absence of the drug. Although residual γH2AX-positive cells were present in the U20S cell population treated with scrambled siRNA after recovery from HU exposure (Fig. 4A), there were significantly more γH2AX-positive cells in the hPso4-depleted cell population (Fig. 4, B and C). Interestingly, we also observed a significant increase in the number of γH2AX-positive cells in the untreated hPso4-depleted cell population (Fig. 4C). Together, our results demonstrate that hPso4 not only contributes to the maintenance of genome integrity in response to replication stress but also during unperturbed DNA replication.
FIGURE 4.

Genome instability associated with hPso4 knockdown. A and B, DSBs were measured by counting the number of γH2AX-positive cells after recovery from HU. siRNA was added to U2OS cells for 36 h followed by incubation with 2 mm HU for 16 h. After washing HU, cells were allowed to recover for 18 h and were fixed and stained with γH2AX antibody. Cells with residual γH2AX foci (>6) were counted. Residual γH2AX foci after recovery from HU in cells treated with scrambled siRNA (A) or hPso4 siRNA (B). C, representation shows the percentage of γH2AX-positive cells before and after the addition of HU. Means of three experiments are shown with the differences determined using Student's t test. (**, p < 0.01). Error bars, S.D. The level of the protein knockdown is shown in the lower right panel.
Depletion of hPso4 Results in a Defect in the Repair of DSBs by HR
The elevated levels of DSBs in hPso4-depleted cells may arise because of reduced replication fork stability and/or reduced repair of DSBs. Because HR plays a major role in the repair of DSBs resulting from collapsed replication forks (10, 13), we asked whether hPso4 was involved in this repair process using derivatives of U2OS and HeLa cells harboring a substrate that interrogates HR (DR-GFP) (14). This substrate contains two copies of the gene encoding GFP, one of which is interrupted by the I-SceI restriction enzyme recognition sequence whereas the other is truncated at both the 5′- and 3′-ends (Fig. 5A). If a DSB introduced at the I-SceI site is repaired by the predominant gene conversion type of HR, a functional GFP gene is produced. As expected, only a few GFP-positive cells were observed in the absence of I-SceI, but the number of GFP-positive cells increased dramatically after expression of I-SceI, reflecting the repair of DSBs by HR. Depletion of hPso4 or CDC5L in either U20S (Fig. 5, B, C, and G) or HeLa cells (Fig. 5H) resulted in a significant decrease in the number of GFP-positive cells after expression of I-SceI, albeit not to the same extent as when the levels of the key HR proteins, BRCA2 and Rad51, were reduced by siRNA (Fig. 5, D, E, and G). Knockdown of PLRG1 or BCAS2, two subunits of the hPso4 complex, also resulted in a significant reduction in HR (Fig. 5H). The HR defect in hPso4-depleted cells was rescued by transfection with a plasmid encoding an siRNA-resistant version of hPso4 (Fig. 5J), indicating that the phenotype was specific to the decrease in Pso4 levels. To examine the role of the E3 ligase activity of hPso4 in HR, we deleted three residues in the hPso4 catalytic U-box: valine 17, serine 18, and proline 19, which are conserved from yeast to human (Fig. 5I). Notably, leucine 15 in yeast Pso4 (which is equivalent to valine 17 in human Pso4) is required for U-box dimerization and yeast viability (15). The HR defect in hPso4-depleted cells was rescued by transfection with a plasmid encoding an siRNA-resistant version of hPso4 lacking the conserved U-box residues (Fig. 5J), suggesting that hPso4 function in HR is independent of its E3 ligase activity.
hPso4 Depletion Sensitizes Cells to Ionizing Radiation (IR) and to a PARP Inhibitor
To further confirm the contribution of hPso4 to the repair of DSBs, we examined the effect of hPso4 depletion on cellular sensitivity to IR. Whereas non-homologous end-joining (NHEJ)-deficient cells display the highest sensitivity to IR, HR-deficient cells also show significant susceptibility to DNA damage induced by IR (16). Consistent with the above-demonstrated role in HR, hPso4-depleted cells exhibited increased sensitivity to IR compared with control cells at high doses of radiation (Fig. 6A). In addition to IR sensitivity, HR-defective cells and tumors display high sensitivity to PARP inhibitors, and impaired HR has been utilized as a predictor of response to this class of antineoplastic agents (17). One proposed mechanism of this sensitivity is an increase in the rate of DNA single strand lesions by PARP inhibitors, resulting in DSBs when replication forks encounter such lesions. Because of the significant defect in HR observed with the depletion of the hPso4 complex, we investigated whether this depletion sensitized cells to killing by PARP inhibitors. We used a validated PARP inhibitor (ABT-888) that has entered cancer clinical trials. Depletion of either hPso4 or its partner protein CDC5L resulted in hypersensitivity of the U2OS osteosarcoma and the A549 lung cancer cell lines to the PARP inhibitor, ABT-888 (Fig. 6, B–D). Together, these results provide strong evidence for hPso4 complex involvement in the recombinational repair of replication-associated DSBs.
FIGURE 6.

Sensitivity to hPso4 complex-depleted cells to IR and a PARP inhibitor. A, radiation sensitivity of hPso4-depleted U2OS cells versus control cells as measured by the MTT method. 72 h after the addition of siRNA, cells were exposed to the specified doses of IR, and viability was checked 48 h later by the MTT assay as described under “Experimental Procedures.” B, PARP inhibitor (ABT-888) sensitivity in the setting of decreased hPso4 protein level utilizing the same MTT assay in the A549 lung cancer line. The cells were exposed to siRNA for 72 h followed by the incubation with ABT-888 for another 72 h. C and D, similar trends of PARP inhibitor sensitivity when depleting hPso4 (C) or CDC5L (D) in U2OS cells. Statistical differences were determined using Student's t test with α values of 0.05 and 0.01 (*, p < 0.05; **, p < 0.01). Error bars, S.E.
hPso4 Complex Interacts with BRCA1-A Complex and Regulates BRCA1 Protein Levels
To gain mechanistic insight into the role of the hPso4 complex in HR, we focused on its potential interaction with the BRCA1-A complex because of the importance of BRCA1 in HR and large scale proteomic data linking the hPso4 and BRCA1-A complexes. Specifically, peptides from NBA1, a subunit of the BRCA1-A complex, were recovered from a CDC5L pulldown experiment (18), and subunits of the hPso4 complex were identified as associated proteins of NBA1, and RAP80, which is another subunit of the BRCA1-A complex (19, 20). Using antibodies that immunoprecipitate endogenous NBA1 and RAP80 proteins, we detected an association between these proteins and CDC5L and hPso4 in extracts from two different cell lines in addition to the expected association with BRCA1 (Fig. 7, A–D). The amounts of CDC5L associated with subunits of the BRCA1-A complex were similar in both the presence and absence of HU, suggesting that there is a constitutive interaction between the BRCA1-A and hPso4 complexes.
Next, we asked whether the hPso4 complex regulates the function of the BRCA1 protein in HR. To this end, we observed that depletion of either hPso4 or CDC5L resulted in significantly lower steady-state levels of BRCA1 protein (Fig. 7, E–I). This effect was a direct consequence of hPso4 protein depletion, as the transfection of a siRNA-resistant hPso4 clone, or one that has the same three-amino acid deletion in the U-box as reported above, rescued to a large extent the decrease in BRCA1 protein level (Fig. 7I). It is known that BRCA1 protein levels are regulated in a cell cycle-dependent manner, increasing during S phase and peaking in G2 phase (21). The observed reduction in BRCA1 levels does not appear to be due to effects on the cell cycle, as depletion of hPso4 did not result in a decrease in cells in S and G2/M phases when asynchronized cells were analyzed (Fig. 8). hPso4 protein levels also increased during S and G2 phases, attesting to a role in replication and HR (Fig. 8D). One important function of BRCA1 in HR is the recruitment of the CTIP exonuclease, which is required for the production of single stranded DNA (ssDNA) at DSBs (22). RPA binds to this ssDNA and then becomes phosphorylated (23), and p-RPA levels reflect the extent of ssDNA generation. Similar to BRCA1 knockdown (24, 25), depletion of either hPso4 or CDC5L resulted in decreased replication protein A phosphorylation following HU treatment (Fig. 7, E–H). The same phenotype was observed after knocking down hPso4 complex subunits by different siRNAs that target dissimilar regions of the mRNAs which argues against off-target effects of the used siRNAs (Fig. 7, E–H). To gain mechanistic insight into how the hPso4 complex regulates BRCA1 protein levels, we measured BRCA1 mRNA levels in cells depleted of either hPso4 or CDC5L. As shown in Fig. 9D, we observed a modest decrease in the level of BRCA1 mRNA in cells depleted of the hPso4 complex units compared with the control. The association between the hPso4 and BRCA1 complexes suggests a functional interaction at the protein level that goes beyond the regulation of BRCA1 mRNA. To investigate the impact of hPso4 on BRCA1 protein stability, we blocked new protein synthesis by cycloheximide and followed BRCA1 protein levels at multiple time points. As shown in Fig. 9, BRCA1 is a relatively stable protein, and its level does not decrease until after several hours of cycloheximide addition. We could not follow longer time points because cellular death was observed at 24 h of incubation with cycloheximide in hPso4-depleted cells. We observed that the levels of BRCA1 protein declined faster in cells depleted of hPso4, which suggests that hPso4 contributes to the stability of BRCA1. Overall, our data show that the hPso4 complex participates in the initial end-resection step of HR, possibly by regulating the protein levels of BRCA1.
FIGURE 8.

Cell cycle phase distribution of asynchronized hPso4-depleted cells. A, U2OS cells treated with scrambled siRNA. x axis, propidium iodide; y axis, cell count. B, U2OS cells treated with hPso4 siRNA. C, representation of percentage of cells in each phase of the cell cycle. D, change in hPso4 protein levels as cells enter the cell cycle after serum deprivation. Cells were synchronized at G1 by serum starvation and then released by serum addition and collected at the indicated time points. Error bars, S.E.
FIGURE 9.

hPso4 regulates the mRNA and protein stability of BRCA1. A and B, BRCA1 protein levels detected after the incubation with cycloheximide (50 μg/ml) following the addition of two rounds of scrambled siRNA (A) or hPso4 (B). C, representation of the decline of BRCA1 protein from A and B normalized to actin. D, BRCA1 mRNA levels as measured by RT-PCR after the depletion of hPso4, CDC5L, and BRCA1 and compared with the control. Error bars, S.D. (*, p < 0.05, **, p < 0.01).
DISCUSSION
The essential yeast PSO4 gene is required for mRNA splicing and resistance to agents that cause DNA damage, in particular those that generate ICL lesions. Similarly, the human homolog of Pso4 also appears to be involved in DNA repair and damage response, but its role has not been characterized at the molecular level. There is, however, emerging evidence that the function of hPso4 in ICL repair is independent of its role in mRNA splicing (7, 8). Furthermore, the involvement of PLRG1, a subunit of the hPso4 complex, in cellular proliferation also appears to be independent of the role of this complex in mRNA splicing (26). Deletion of either the PLRG1 or hPSO4 gene causes lethality in mice at an early embryonic stage (26, 27). Mouse embryonic fibroblasts generated by conditional inactivation of the PLRG1 gene failed to progress through S phase in response to serum stimulation but were not defective in the splicing of mRNAs encoding multiple cell cycle regulators, including p53, cyclin D1, and cyclin E1 (26). In accord with the murine studies, human cells depleted of either hPso4 or CDC5L failed to form colonies in our hands and as reported by others (8). Interestingly, in short term assays, these cells exhibited prolonged G2/M arrest, a similar phenotype to that of an Schizosaccharomyces pombe cdc5 strain (28), in addition to a defect in S phase progression. hPso4-depleted cells also displayed increased sensitivity to HU. Together, these findings suggest that the hPso4 complex has a direct role in cell proliferation that may be independent of its function in mRNA splicing. Two genes encoding subunits of the hPso4 complex, CDC5L and breast cancer amplified sequence 2 (BCAS2, which encodes the subunit SPF27), are amplified in osteosarcoma, and breast cancer, respectively (29, 30). This amplification of hPso4 complex genes may confer a proliferative advantage by promoting cell cycle progression in the face of activated checkpoints, a phenomenon that occurs in early stages of carcinogenesis (31).
In support of the idea that the hPso4 complex directly participates in cell proliferation, we observed that hPso4 localized to replication foci in unperturbed and HU-treated cells. The reduced recovery of DNA synthesis after transient exposure to hydroxyurea and the higher steady-state levels of DSBs before and after HU treatment in hPso4-depleted cells suggest that this complex contributes to replication fork stability and/or the repair of replication-associated DSBs. This is consistent with the results of a whole genome siRNA screen that identified the hPso4 and CDC5L transcripts among the hits that contribute to the repair of spontaneous DSBs (32). There is now compelling evidence that HR plays an essential role in the repair of DSBs that arise during DNA replication, even in the absence of exogenous DNA damage (13, 33). The increased sensitivity of hPos4-depleted cells to a PARP inhibitor that causes DSBs in a replication-dependent manner and the defect in the repair of site-specific DSBs by HR in these cells provide strong evidence that the hPso4 complex participates in the repair of replication-associated DSBs via a homology-dependent pathway. A previous study suggested a role of hPso4 in NHEJ based on a two-step assay that measures plasmid rejoining in human cells coupled to chromatin integration of this plasmid (34). The defect observed in this assay could be due to genuine defect in NHEJ, versus a problem with plasmid integration into chromatin. hPso4 may be required for both pathways of DSB repair similar to other DNA repair proteins such as the MRN complex (35) and BRCA1 (36). However, the primary function of HR in the repair of ICL lesions (9, 37) and the role of hPso4 in HR as we demonstrated provide an explanation for the main sensitivity of Pso4 mutant or depleted cells to ICL agents.
In the recombinational repair of a DSB, a key step is end resection to generate a 3′-DNA single strand that invades a homologous duplex. The BRCA1 protein plays a role in end resection by recruiting the CTIP nuclease, which initiates early steps in this process. In accord with proteomic studies (18–20), we detected an association between the BRCA1-A and the hPso4 complexes. The steady-state levels of BRCA1 protein were reduced in cells depleted of hPso4, a result of both lower BRCA1 mRNA levels and reduced BRCA1 protein stability. We observed decreased RPA phosphorylation after HU treatment in hPso4 complex-depleted cells, an indicator of decreased ssDNA generation. Because RPA-bound ssDNA represents an early signal for checkpoint activation, our findings provide an explanation for the defective ATR activation and reduced Chk1 phosphorylation in cells depleted of either hPso4 or CDC5L (8). Interestingly, CDC5L depletion also results in decreased levels of another protein, Fanconi D2 (8), which is also involved in the recombinational repair of DSBs.
Unlike most E3 ubiquitin ligases that assemble lysine 48 ubiquitin chains and target substrates for proteasome-mediated degradation, hPso4 appears to assemble atypical lysine 63 ubiquitin chains4 (38), which enhance protein-protein interactions and may protect substrates from Lys-48 ubiquitin chain-mediated degradation (39). In an attempt to determine whether the E3 ligase activity of hPso4 is required for HR we deleted key conserved amino acid residues within the Pso4 U-box, but this did not disrupt the function of hPso4 in HR and in preserving BRCA1 protein levels. Although this result argues that the E3 ligase activity is not required for its HR function, it is possible that the amino acid changes did not inactivate hPso4 catalytic activity. In the absence of a validated substrate for hPso4 E3 ligase, it is difficult to assess the impact of this deletion on U-box activity. Other studies have reported similar difficulties in completely ablating the ligase function of the structurally similar RING finger by changing just one or a few amino acids. A pertinent example is the case of the RING finger of BRCA1 where one study has found that an amino acid change in this RING finger, which disrupts its interaction with all ubiquitin E2 ligases, does not affect its role in HR or tumor suppression (40), thereby suggesting that the BRCA1 E3 ligase is not involved in HR. In contrast, others argue that the BRCA1 E3 ligase activity is required for HR and that this same amino acid change within the BRCA1 RING finger does not fully inactivate the E3 ligase activity (41). Nonetheless, our results provide compelling evidence that the hPso4 complex participates in the recombinational repair of DSBs, which provides a mechanistic explanation for the sensitivity of yeast pso4 mutants and hPso4-depleted cells to agents that cause ICLs (1, 7, 8).
The development of PARP inhibitors to target a specific DNA repair defect in hereditary breast cancer (42, 43) has stimulated an interest in identifying or generating a status of defective HR in sporadic cancers to widen the therapeutic application of these drugs. Our studies have uncovered a new function of a multiprotein complex involved in the repair of DSBs by HR that could represent such a therapeutic target to impair HR and render cancer cells susceptible to PARP inhibitors.
Acknowledgments
We thank D. Weinstock for providing the reporter cell lines and M. A. Osley and A. Tomkinson for a critical review of the paper.
This work was supported, in whole or in part, by National Institutes of Health Grant 5K01HL103182-03 (to M. S.).
M. Abbas, I. Shanmugam, M. Bsaili, R. Hromas, and M. Shaheen, unpublished data.
- ICL
- interstrand cross-link
- ATR
- ATM and Rad3-related
- BRCA1
- breast cancer type 1 susceptibility protein
- DSB
- double strand break
- HR
- homologous recombination
- HU
- hydroxyurea
- IP
- immunoprecipitation
- IR
- ionizing radiation
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PARP
- poly(ADP-ribose) polymerase
- PCNA
- proliferative cell nuclear antigen
- Pso4
- psoralen 4
- RPA
- replication protein A
- NHEJ
- nonhomologous end-joining.
REFERENCES
- 1. Brendel M., Henriques J. A. (2001) The Pso mutants of Saccharomyces cerevisiae comprise two groups: one deficient in DNA repair and another with altered mutagen metabolism. Mutat. Res. 489, 79–96 [DOI] [PubMed] [Google Scholar]
- 2. Grey M., Düsterhöft A., Henriques J. A., Brendel M. (1996) Allelism of PSO4 and PRP19 links pre-mRNA processing with recombination and error-prone DNA repair in Saccharomyces cerevisiae. Nucleic Acids Res. 24, 4009–4014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ohi M. D., Vander Kooi C. W., Rosenberg J. A., Chazin W. J., Gould K. L. (2003) Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat. Struct. Biol. 10, 250–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hatakeyama S., Yada M., Matsumoto M., Ishida N., Nakayama K. I. (2001) U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem. 276, 33111–33120 [DOI] [PubMed] [Google Scholar]
- 5. Grote M., Wolf E., Will C. L., Lemm I., Agafonov D. E., Schomburg A., Fischle W., Urlaub H., Lührmann R. (2010) Molecular architecture of the human Prp19/CDC5L complex. Mol. Cell. Biol. 30, 2105–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chan S. P., Kao D. I., Tsai W. Y., Cheng S. C. (2003) The Prp19p-associated complex in spliceosome activation. Science 302, 279–282 [DOI] [PubMed] [Google Scholar]
- 7. Zhang N., Kaur R., Lu X., Shen X., Li L., Legerski R. J. (2005) The Pso4 mRNA splicing and DNA repair complex interacts with WRN for processing of DNA interstrand cross-links. J. Biol. Chem. 280, 40559–40567 [DOI] [PubMed] [Google Scholar]
- 8. Zhang N., Kaur R., Akhter S., Legerski R. J. (2009) Cdc5L interacts with ATR and is required for the S-phase cell-cycle checkpoint. EMBO Rep. 10, 1029–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deans A. J., West S. C. (2011) DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 11, 467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Petermann E., Orta M. L., Issaeva N., Schultz N., Helleday T. (2010) Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 37, 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hanada K., Budzowska M., Davies S. L., van Drunen E., Onizawa H., Beverloo H. B., Maas A., Essers J., Hickson I. D., Kanaar R. (2007) The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 14, 1096–1104 [DOI] [PubMed] [Google Scholar]
- 12. Sedelnikova O. A., Pilch D. R., Redon C., Bonner W. M. (2003) Histone H2AX in DNA damage and repair. Cancer Biol. Ther. 2, 233–235 [DOI] [PubMed] [Google Scholar]
- 13. Petermann E., Helleday T. (2010) Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell. Biol. 11, 683–687 [DOI] [PubMed] [Google Scholar]
- 14. Pierce A. J., Johnson R. D., Thompson L. H., Jasin M. (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 13, 2633–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vander Kooi C. W., Ohi M. D., Rosenberg J. A., Oldham M. L., Newcomer M. E., Gould K. L., Chazin W. J. (2006) The Prp19 U-box crystal structure suggests a common dimeric architecture for a class of oligomeric E3 ubiquitin ligases. Biochemistry 45, 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Groth P., Orta M. L., Elvers I., Majumder M. M., Lagerqvist A., Helleday T. (2012) Homologous recombination repairs secondary replication induced DNA double-strand breaks after ionizing radiation. Nucleic Acids Res. 40, 6585–6594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shaheen M., Allen C., Nickoloff J. A., Hromas R. (2011) Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood 117, 6074–6082 [DOI] [PubMed] [Google Scholar]
- 18. Llères D., Denegri M., Biggiogera M., Ajuh P., Lamond A. I. (2010) Direct interaction between hnRNP-M and CDC5L/PLRG1 proteins affects alternative splice site choice. EMBO Rep. 11, 445–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang B., Hurov K., Hofmann K., Elledge S. J. (2009) NBA1, a new player in the BRCA1-A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 23, 729–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sowa M. E., Bennett E. J., Gygi S. P., Harper J. W. (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Choudhury A. D., Xu H., Baer R. (2004) Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. J. Biol. Chem. 279, 33909–33918 [DOI] [PubMed] [Google Scholar]
- 22. Yun M. H., Hiom K. (2009) CtIP-BRCA1 modulates the choice of DNA double-strand break repair pathway throughout the cell cycle. Nature 459, 460–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fotedar R., Roberts J. M. (1992) Cell cycle regulated phosphorylation of RPA-32 occurs within the replication initiation complex. EMBO J. 11, 2177–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feng Z., Zhang J. (2012) A dual role of BRCA1 in two distinct homologous recombination mediated repair in response to replication arrest. Nucleic Acids Res. 40, 726–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schlegel B. P., Jodelka F. M., Nunez R. (2006) BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res. 66, 5181–5189 [DOI] [PubMed] [Google Scholar]
- 26. Kleinridders A., Pogoda H. M., Irlenbusch S., Smyth N., Koncz C., Hammerschmidt M., Brüning J. C. (2009) PLRG1 is an essential regulator of cell proliferation and apoptosis during vertebrate development and tissue homeostasis. Mol. Cell. Biol. 29, 3173–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fortschegger K., Wagner B., Voglauer R., Katinger H., Sibilia M., Grillari J. (2007) Early embryonic lethality of mice lacking the essential protein SNEV. Mol. Cell. Biol. 27, 3123–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nurse P., Thuriaux P., Nasmyth K. (1976) Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 146, 167–178 [DOI] [PubMed] [Google Scholar]
- 29. Lu X. Y., Lu Y., Zhao Y. J., Jaeweon K., Kang J., Xiao-Nan L., Ge G., Meyer R., Perlaky L., Hicks J., Chintagumpala M., Cai W. W., Ladanyi M., Gorlick R., Lau C. C., Pati D., Sheldon M., Rao P. H. (2008) Cell cycle regulator gene CDC5L, a potential target for 6p12-p21 amplicon in osteosarcoma. Mol. Cancer Res. 6, 937–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maass N., Rösel F., Schem C., Hitomi J., Jonat W., Nagasaki K. (2002) Amplification of the BCAS2 gene at chromosome 1p13.3–21 in human primary breast cancer. Cancer Lett. 185, 219–223 [DOI] [PubMed] [Google Scholar]
- 31. Bartkova J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J. M., Lukas C., Ørntoft T., Lukas J., Bartek J. (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 [DOI] [PubMed] [Google Scholar]
- 32. Paulsen R. D., Soni D. V., Wollman R., Hahn A. T., Yee M. C., Guan A., Hesley J. A., Miller S. C., Cromwell E. F., Solow-Cordero D. E., Meyer T., Cimprich K. A. (2009) A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 35, 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Budzowska M., Kanaar R. (2009) Mechanisms of dealing with DNA damage-induced replication problems. Cell Biochem. Biophys. 53, 17–31 [DOI] [PubMed] [Google Scholar]
- 34. Beck B. D., Park S. J., Lee Y. J., Roman Y., Hromas R. A., Lee S. H. (2008) Human Pso4 is a metnase (SETMAR)-binding partner that regulates metnase function in DNA repair. J. Biol. Chem. 283, 9023–9030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Williams G. J., Lees-Miller S. P., Tainer J. A. (2010) Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair 9, 1299–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang E. S., Xia F. (2010) BRCA1 16 years later: DNA damage-induced BRCA1 shuttling. FEBS J. 277, 3079–3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Long D. T., Räschle M., Joukov V., Walter J. C. (2011) Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 333, 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Song E. J., Werner S. L., Neubauer J., Stegmeier F., Aspden J., Rio D., Harper J. W., Elledge S. J., Kirschner M. W., Rape M. (2010) The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 24, 1434–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Popov N., Schülein C., Jaenicke L. A., Eilers M. (2010) Ubiquitylation of the amino terminus of Myc by SCF(β-TrCP) antagonizes SCF(Fbw7)-mediated turnover. Nat. Cell Biol. 12, 973–981 [DOI] [PubMed] [Google Scholar]
- 40. Reid L. J., Shakya R., Modi A. P., Lokshin M., Cheng J. T., Jasin M., Baer R., Ludwig T. (2008) E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc. Natl. Acad. Sci. U.S.A. 105, 20876–20881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Silver D. P., Livingston D. M. (2012) Mechanisms of BRCA1 tumor suppression. Cancer Discovery 2, 679–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Farmer H., McCabe N., Lord C. J., Tutt A. N., Johnson D. A., Richardson T. B., Santarosa M., Dillon K. J., Hickson I., Knights C., Martin N. M., Jackson S. P., Smith G. C., Ashworth A. (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 [DOI] [PubMed] [Google Scholar]
- 43. Bryant H. E., Schultz N., Thomas H. D., Parker K. M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N. J., Helleday T. (2005) Specific killing of BRCA2-deficient tumors with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 [DOI] [PubMed] [Google Scholar]