

Background: Periaxin and AHNAK nucleoprotein 2 contain a poorly conserved PDZ domain.
Results: The crystal structures for the PDZ domains were determined.
Conclusion: Both PDZ domains form intertwined domain-swapped homodimers.
Significance: The structures have implications for the organization of complexes involving the periaxin/AHNAK family.
Keywords: Crystal Structure, Myelin, Protein Domains, Protein Structure, Protein-Protein Interactions, X-ray Scattering, PDZ Domain, Dimerization, Domain Swapping
Abstract
Periaxin (PRX) is an abundant protein in the peripheral nervous system, with an important role in myelination. PRX participates in large molecular complexes, most likely through the interactions of its N-terminal PSD-95/Discs-large/ZO-1 (PDZ)-like domain. We present the crystal structures of the PDZ-like domains from PRX and its homologue AHNAK nucleoprotein 2 (AHNAK2). The unique intertwined, domain-swapped dimers provide a structural basis for the homodimerization of both proteins. The core of the homodimer is formed by a 6-stranded antiparallel β sheet, with every other strand from a different chain. The AHNAK2 PDZ domain structure contains a bound class III ligand peptide. The binding pocket is preformed, and the peptide-PDZ interactions have unique aspects, including two salt bridges and weak recognition of the peptide C terminus. Tight homodimerization may be central to the scaffolding functions of PRX and AHNAK2 in molecular complexes linking the extracellular matrix to the cytoskeletal network.
Introduction
The PDZ (PSD-95/Discs-large/ZO-1) domain is one of the most common modular interaction domains, mediating protein-protein interactions by binding to the C terminus, or to an internal sequence, of a target protein. PDZ domains consist of 80–90 amino acid residues and normally fold into six β strands and two α helices. Peptide ligands bind into a pocket lined by helix α2, strand β2, and the β1–β2 loop, leading to the formation of an antiparallel β sheet between the ligand and the PDZ domain (1–3). Usually, the C-terminal carboxylate group of the ligand interacts directly with the backbone of the β1–β2 loop, containing a conserved XΦGΦ sequence (also called the GLGF motif); X denotes any amino acid and Φ is a hydrophobic residue. Based on binding specificity, PDZ domains can be divided into 3 classes; class I PDZ domains bind to the C-terminal sequence (S/T)-X-Φ-COOH, class II PDZ domains to Φ-X-Φ-COOH, and class III PDZ domains to (D/E)-X-Φ-COOH (4).
Periaxin (PRX)2 plays a significant role in myelination of the peripheral nervous system (5, 6), comprising 16% of peripheral nervous system myelin protein by weight (7). Genetic defects in PRX result in demyelinating peripheral neuropathies, such as Charcot-Marie-Tooth and Dejerine-Sottas diseases, indicating a crucial role for PRX in the normal development of the peripheral nervous system (8–10). PRX is also a member of cytoskeletal complexes in lens fibers, and it is considered to function in maturation, packing, and membrane organization of lens fiber cells (11).
Two major isoforms of PRX, L-PRX and S-PRX, of 1461 and 147 amino acids, respectively, are expressed by myelinating Schwann cells. During myelination, L-PRX is first localized in the adaxonal plasma membrane and later, in the abaxonal plasma membrane (5, 6). S-PRX is uniformly distributed in the cytoplasm and the nucleus of the Schwann cell, and a function for S-PRX in regulating mRNA splicing has been suggested (12).
Both PRX isoforms share their 127 N-terminal residues, including a predicted PDZ domain (13). The function of the PRX PDZ domain is unknown, but it is essential for PRX dimerization (14). Through its interaction with dystrophin-related protein 2 (Drp2), L-PRX is a member of the periaxin-Drp2-dystroglycan complex. The PRX PDZ domain is apparently not required in this interaction (14). Except for Drp2, no other binding partners for PRX have been reported, and the ligand of the PRX PDZ domain is, hence, also unknown.
The homology of PRX to other proteins is very low; essentially, the only conserved domain is the PDZ domain (15). Even this domain is poorly conserved, and the only homologues with sequence identity >30% in this region are the giant AHNAK proteins. The PDZ domain of PRX has the highest sequence identity, 57%, with AHNAK2. PRX and the AHNAK proteins form a unique subfamily of PDZ proteins that are likely to have similar functions in complexes linking the extracellular matrix to the cytoskeleton (12).
AHNAKs are giant proteins (molecular mass >600 kDa) expressed in all muscular cells (16). AHNAK1 (also called desmoyokin) is involved in cytoarchitecture and calcium signaling by directly interacting with several proteins, such as dysferlin, S100B, and calpain3 (17–19). It is assumed that AHNAK2 localizes at similar sites as AHNAK1 and has similar functions (20). Both AHNAK1 and AHNAK2 can be divided into 3 regions, i.e. an N-terminal PDZ-like region and a C-terminal region with a nuclear localization signal, separated by a large central repeat region (21). Dimerization of AHNAK1 has been reported (12). For both AHNAK1 and PRX, splicing produces two isoforms, i.e. L-AHNAK1/S-AHNAK1 and L-PRX/S-PRX (12).
Here, we report the first crystal structures for PRX and AHNAK2. Their PDZ domains exhibit uniquely intertwined dimers, with extensive three-dimensional domain swapping. Longer constructs of PRX were also characterized in solution and observed to be mainly disordered outside the PDZ domain. The structures of the PRX and AHNAK2 PDZ domains implicate an intriguing mechanism for the formation of stable homodimers. Based on strong interactions in the crystal state, similar dimerization of both proteins is likely to occur also in vivo.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The production of recombinant PRX PDZ domain variants has been described (22). Briefly, the constructs were expressed as His-tagged variants in Escherichia coli and purified using nickel affinity chromatography. The affinity tag was removed, and the final purification step consisted of size exclusion chromatography. The constructs that were produced include residue ranges 1–147 (full-length S-PRX), 1–127 (the region common to S- and L-PRX), 14–127, 14–117, and 14–107.
A codon-optimized synthetic gene of human AHNAK2 (residues 108–203) (accession number NM_138420; locus tag NM_138420), encoding a hexahistidine tag and a tobacco etch virus protease cleavage site at the N terminus of the predicted AHNAK2 PDZ domain, subcloned into pETM11, was purchased from MWG Eurofins. The protein was overexpressed in E. coli Rosetta (DE3) cells in ZYM-5052 autoinduction medium (23), containing 50 μg/ml of kanamycin and 34 μg/ml of chloramphenicol, at 37 °C for 7 h. Protein purification was done as for the PRX PDZ domain (22).
Small-angle X-ray Scattering
Synchrotron small-angle x-ray scattering (SAXS) data for the PRX constructs were collected on beamline P12 of EMBL/DESY at PETRA III (Hamburg, Germany). Samples were prepared at 2–20 mg/ml in 50 mm Tris-HCl (pH 7.5), 100 mm NaCl. Data processing and analysis were performed with the ATSAS package (24). PRIMUS (25) was used for processing and GNOM (26) for estimating the distance distribution. DAMMIN (27), DAMMIF (28), and GASBOR (29) were used for ab initio model building with either dummy residues or chain-like assemblies. BUNCH (30) was used to build hybrid models based on the crystal structure and ab initio built N- and C-terminal extensions. Different models were superimposed using SUPCOMB (31). CRYSOL (32) was used to compare the crystal structure with SAXS data. The molecular weight of the samples was calculated by comparing the forward scattering intensity, I(0), to that of freshly prepared BSA.
Crystallography and Structure Determination
Crystallization of the PRX PDZ domain (residues 14–107) has been described (22). Briefly, crystals were obtained in 30% PEG 2000MME, 0.15 m KBr after 1 day at 4 °C. New higher resolution native data were collected at the P13 EMBL/DESY beamline at PETRA III (Hamburg, Germany). A tungsten-derivatized PRX PDZ crystal was prepared by soaking in 5 mm (NH4)2WS4 for 2 days. The preparation of a xenon-derivatized crystal was executed at BESSY (Berlin, Germany). A crystal was picked and incubated in the xenon chamber at 200 p.s.i. for 8 min. Diffraction data for the derivatized crystals were collected on beamline BL14.1 at BESSY.
Crystals of the AHNAK2 PDZ domain were similarly obtained with sitting-drop vapor diffusion using a 35 mg/ml protein stock in 50 mm Tris (pH 7.5), 100 mm NaCl, with a well solution consisting of 10% PEG8000 and 50 mm KH2PO4. X-ray diffraction data were collected on the EMBL/DESY beamline P13 at PETRA III (Hamburg).
All data were processed using XDS (33). The phasing of PRX, which was solved first, did not work either by molecular replacement or by the bromide single-wavelength anomalous dispersion method. Thus, various derivatization experiments were performed, and data from a crystal soaked with tungsten showed a strong anomalous signal. In addition, crystals incubated in the xenon chamber also exhibited an anomalous signal from xenon. Combining these two datasets with native diffraction data, phasing was successful. Phasing by MIRAS, followed by automatic model building, was done with AutoSol in Phenix (34). The structure solution of the AHNAK2 PDZ domain was done by molecular replacement using the partially refined PRX PDZ structure as a search model in Phaser (35). Model building was done using COOT (36) and refinement using phenix.refine (37). Molprobity (38) was used for structure validation.
Static Light Scattering
The oligomeric state of the PRX and AHNAK2 constructs in solution (50 mm Tris, pH 7.5, 100 mm NaCl) was studied using static light scattering. Samples of the protein variants used for crystal structure determination were run through a size exclusion column with an Äkta Purifier (GE Healthcare), and refractive index and light scattering were subsequently measured online using the Optilab rEX and miniDAWN TREOS instruments (Wyatt). Data were analyzed with ASTRA software (Wyatt). The run was similarly performed in the presence of 6 m urea.
Circular Dichroism Spectroscopy
Synchrotron radiation circular dichroism spectra were measured on beamline CD1 at ISA, University of Århus (Denmark). The buffer was 20 mm sodium phosphate (pH 7.5). Each sample was scanned from 280 to 170 nm in 100-μm quartz cuvettes at a concentration of 1 mg/ml. Each spectrum was measured 3 times, and the corresponding buffer spectrum was subtracted. Deconvolution was carried out on the Dichroweb server (39).
Urea titration of both proteins was done using a Chirascan Plus (Applied Photophysics) instrument, in a 0.5-mm cuvette. PRX (0.6 mg/ml) was in 10 mm Tris (pH 7.5), 20 mm NaCl, and AHNAK2 (0.2 mg/ml) in H2O. Spectra were also measured in the presence of up to 6 m urea.
Native PAGE
The native PAGE samples of PRX and AHNAK2 PDZ domains were prepared at different concentrations of SDS and urea in 50 mm Tris (pH 7.5), 100 mm NaCl. The gel was run in 25 mm Tris, 195 mm glycine (pH 8.5) at 4 °C for 2 h and Coomassie stained.
RESULTS
Dimerization of PRX and AHNAK2 N-terminal Domains in Solution
The recombinant PRX PDZ domain was a folded dimer, as evidenced by CD spectroscopy, size exclusion chromatography, and static light scattering (Fig. 1). The PDZ domain of AHNAK2 was also homodimeric in solution (Fig. 1D). CD spectroscopy indicated both proteins were stable in 1 m urea, but started unfolding in 3 m urea (Fig. 1B). Static light scattering proved that the dimers observed in solution were dissociated into unfolded monomers at 6 m urea (Fig. 1, C and D). Native gel electrophoresis further showed that the mobility of neither protein was affected by urea concentrations up to 1 m (Fig. 1E), whereas changes in mobility were seen with SDS. Hence, the dimers are stable in solution at relatively high concentrations of urea.
FIGURE 1.
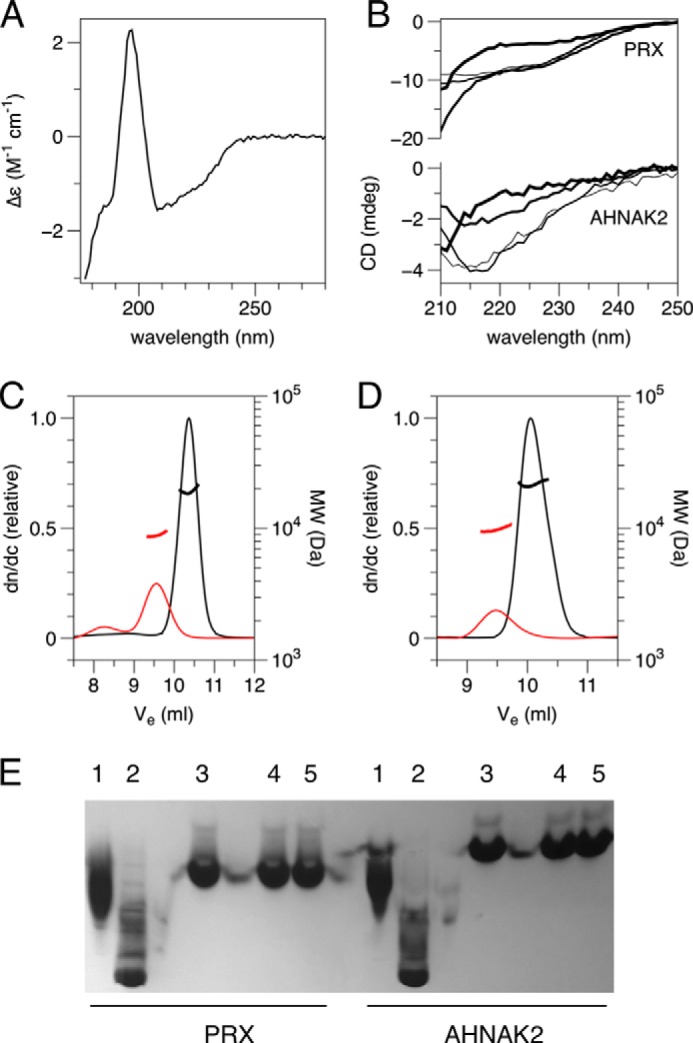
Characterization of the folding and dimerization of the PDZ domains. A, synchrotron radiation circular dichroism spectrum of PRX construct 14–107. Deconvolution of the spectrum suggests 19% α helix and 28% β sheet (calculated from structure: 16% α helix, 37% β sheet). B, urea titration of PRX and AHNAK2 folding. From thinnest to thickest lines: 0, 1, 3, and 6 m urea. C and D, size exclusion chromatography/static light scattering for PRX (C) and AHNAK2 (D) PDZ domains. Red, 6 m urea; black, 0 m urea. The calculated molecular masses in buffer, 19.1 kDa for PRX(14–107) and 21.8 kDa for AHNAK2, indicate homodimer formation, whereas the molecular mass is reduced to that of a monomer in urea. E, analysis of mild denaturants by native PAGE. 1, 0.05% SDS; 2, 0.5% SDS; 3, no additives; 4, 0.1 m urea; 5, 1 m urea.
Three-dimensional Domain-swapped Dimeric Structure of the PRX PDZ Domain
The crystal structure of the PRX PDZ domain was solved at 2.7-Å resolution (Table 1). Three monomers exist in one asymmetric unit, and the structure presents unique dimeric folding, involving a high degree of domain swapping and intertwining between the two monomers (Fig. 2).
TABLE 1.
Statistics of x-ray data collection and structure refinement
Numbers in parentheses refer to the highest-resolution shell.
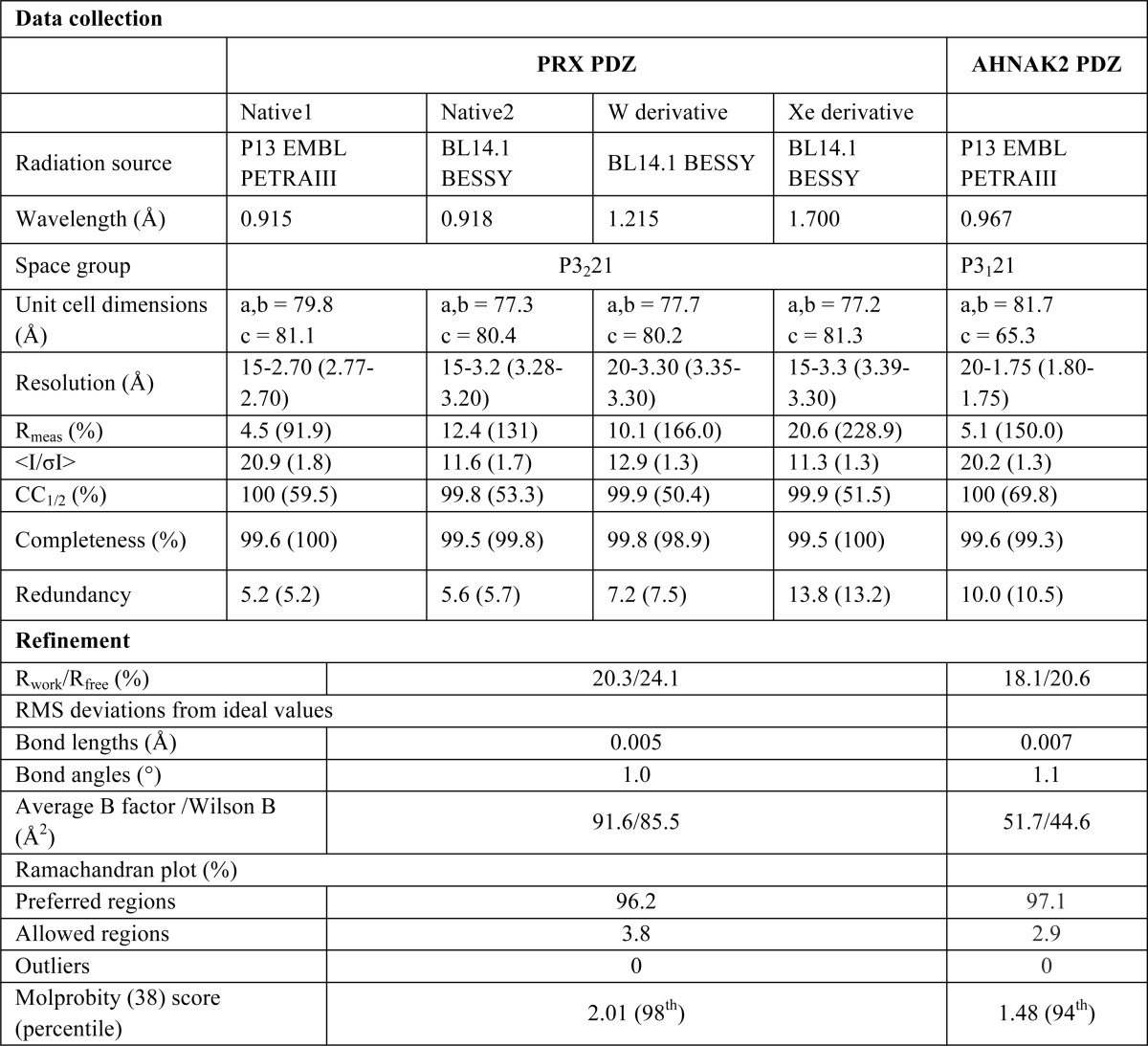
FIGURE 2.
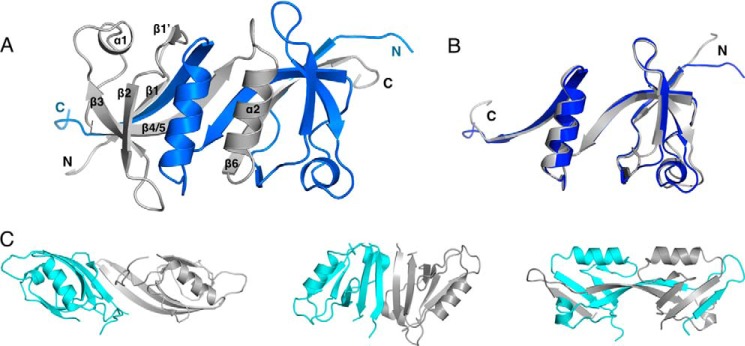
Crystal structure of the PRX PDZ domain and comparison with other dimeric PDZ domains. A, the overall structure shows intertwined homodimerization. Blue and gray colors denote different monomer chains, and the termini and secondary structure elements are labeled. B, the two chains from the PRX homodimer superimposed. C, other homodimeric PDZ structures. The structures of the PDZ domain of Shank1 (left) (42) and GRIP1 (middle) (41) display two intact monomers forming homodimer, whereas domain PDZ2 of ZO-1 (right) (40) swaps strands β1 and β2.
The PRX PDZ domain displays 5 major β strands and 2 α helices in one monomer (Fig. 2B). Compared with other PDZ domains, which generally have 6 β strands, the PRX PDZ domain does not have separate strands β4 and β5, but a long β4/5 strand, and the β4-β5 loop is missing. The loss of this loop results in the change of direction of α2 and β6 in the overall monomer structure. These units further intertwine with the neighboring monomer and vice versa, and two monomers form a domain-swapped homodimer (Fig. 2).
Homo- and heterodimerization of PDZ domains have been observed. However, only the ZO-1 PDZ2 domain was reported to be a domain-swapped homodimer (40), whereas the PDZ domains from Shank1 and GRIP1 (glutamate receptor interacting protein 1) form homodimers without domain swapping (41, 42). The PRX PDZ domain exhibits a dimerization mode distinct from any other known structure (Fig. 2C). Although in ZO-1 PDZ2, a domain-swapped homodimer is formed through swapping the N-terminal β1 and β2 strands with the neighboring molecule, much more extensive domain swapping is present in PRX, via the interchange of α2 and β6 between two monomers (Fig. 2).
In the PRX dimer, a 6-stranded antiparallel β sheet is formed by the two chains, with the two β4/5 strands in the center (Fig. 3). Every second β strand comes from a different chain, resulting in a highly intertwined structure for the homodimer. This sheet is formed of strands β1, β4/5, and β6 of the A and B chains, in the order β1(A)-β6(B)-β4/5(A)-β4/5(B)-β6(A)-β1(B). In addition, the α2 helices are side by side in the dimer structure.
FIGURE 3.
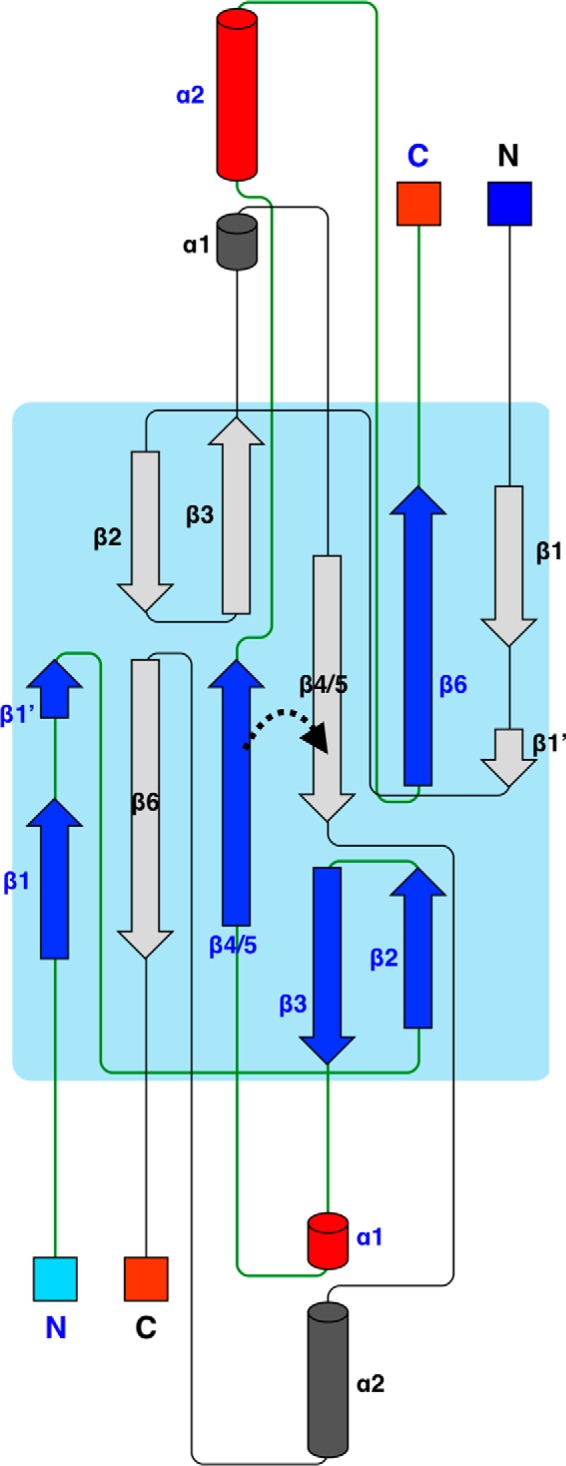
Secondary structure topology. Topology diagram of the PRX PDZ domain dimer is shown, and the two chains are labeled in black and blue. The turn between strands β4 and β5 in canonical, monomeric PDZ domains is indicated with a dashed arrow. The topology of AHNAK2 is identical.
Between the α2 helices from the two chains, π-π stacking is present between the side chains of Phe75 and Tyr90. This stacking also includes Pro89 and Phe72; in addition, there is a buried hydrophobic core (Fig. 4A). Another unique conformation is present on the β1 strand. In PRX, β1 ends at Glu22, and a bulge is observed from Thr23 to Ala25. Gln26–Gly28 acquire an additional small β strand conformation, β1′, by backbone hydrogen bonding with Tyr90–Val92 on strand β6 of the neighboring monomer. This bulge is formed due to the insertion of one residue into the β1 strand in PRX (Fig. 4B). The side chain of Thr23 makes the missing hydrogen bond to the neighboring strand.
FIGURE 4.
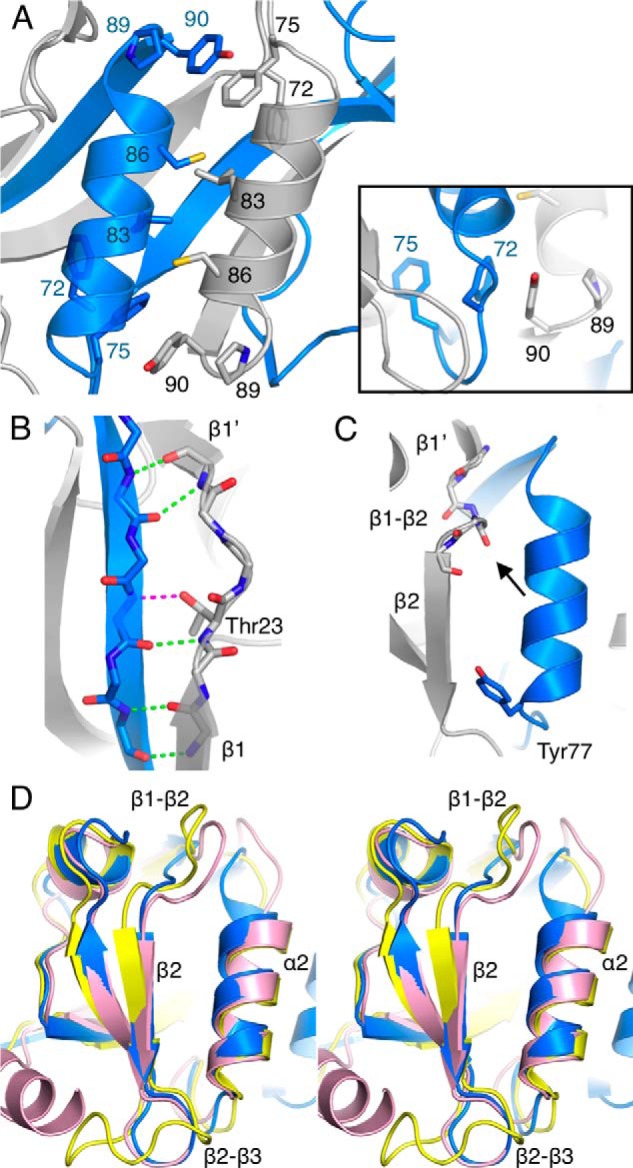
Structural details of PRX. A, arrangement of the two α2 helices involves hydrophobic interactions and aromatic stacking. Inset, an aromatic stacking network is found at both ends of the two α2 helices. B, hydrogen bonding at the bulge between strands β1 and β1′. The bond between the Thr23 side chain and the neighboring strand is in magenta. C, the peptide binding site of PRX. The backbone carbonyl of Val29 points toward the binding site (arrow), and Tyr77 defines the other end of the binding pocket. D, comparison (in stereo) of loop conformations near the peptide binding site. Blue, PRX; yellow, synaptojanin-2-binding protein (SYNJ2BP) (Protein Data Bank entry 2JIN); pink, ZO-1 PDZ3 (58).
The two peptide binding sites of the PRX homodimer are composed between helix α2 and strand β2 from different chains. Because the first residue of the α2 helix is Tyr, the PRX PDZ domain can be categorized into class III and may specifically bind to a C-terminal (D/E)-X-Φ-COOH motif on the ligand protein (43). A backbone peptide group between Val29 and Ser30, expected to recognize the target ligand C terminus, is flipped compared with earlier PDZ domain structures (Fig. 4C). As a result, the carbonyl moiety points toward the expected position of the ligand peptide carboxyl terminus.
As expected from sequence alignments, the PRX sequence 28GVSGI32, homologous to the GLGF motif, is located in the C-terminal carboxylate binding β1–β2 loop (Fig. 4, C and D). In PRX, this loop is 2–6 amino acids shorter than in other PDZ structures. Also loop β2–β3 is shorter than in most other PDZ structures (Fig. 4D). In addition, the C-terminal end of helix α2 in PRX leans slightly toward β2. Due to these factors, the apparent volume of the peptide binding pocket of the PRX PDZ domain is small compared with other PDZ domains. However, it is possible that the peptide binding pocket opens up when a binding partner enters.
Solution Structure
The solution structures of several constructs of the PRX PDZ domain were modeled based on SAXS data (Fig. 5). The forward scattering intensity indicated quantitative homodimer formation by each construct in solution (Table 2). The data for PRX14–107, which was also successfully used for crystallization, suggested compact folding, whereas the longer constructs exhibited more extended structures. The latter was also exemplified by normalized Kratky plots (Fig. 5C) (44).
FIGURE 5.

Solution structures of PRX PDZ constructs. A, SAXS scattering curves. The coloring in panels A–C and E is as follows: black, 1–147; blue, 1–127; magenta, 14–127; green, 14–117; orange, 14–107. B, distance distribution functions. C, normalized Kratky plots; the theoretical position of the maximum (x = 1.73, y = 1.1) for a globular folded particle is indicated by the cross. The movement of this maximum toward higher values is a sign of increasing relative disorder. D, fit (black) of the PRX PDZ domain crystal structure on the SAXS data (red) from the same construct. E, chain-like (GASBOR) models of all PRX constructs. F, a model for full-length S-PRX(1–147) built using the crystal structure (green) and the BUNCH software. Scattering data for constructs 1–147 and 14–127 were simultaneously fitted. The built N- and C-terminal extensions are light blue.
TABLE 2.
SAXS analysis of PRX PDZ domain constructs
| Construct |
||||||
|---|---|---|---|---|---|---|
| 14-107 (crystal) | 14-107 | 14-117 | 14-127 | 1-127 | 1-147 | |
| Monomer molecular mass (kDa) | 10.3 | 10.3 | 11.3 | 12.4 | 14.0 | 15.9 |
| Rg (nm) (Guinier) | 1.80 | 2.01 | 2.29 | 2.55 | 3.31 | 3.39 |
| Dmax (nm) | 6.3 | 6.5 | 9.0 | 10.0 | 13.0 | 14.0 |
| Mass from I(0) (kDa)a | 18 | 21 | 25 | 34 | 41 | |
| Porod volume (nm3) | 38 | 54 | 56 | 82 | 89 | |
| Chi, model vs data | ||||||
| Dammin | 1.05 | 0.93 | 0.93 | 0.78 | 0.75 | |
| Dammif | 1.05 | 0.93 | 0.96 | 0.80 | 0.77 | |
| Gasbor | 1.12 | 1.06 | 1.05 | 1.19 | 1.12 | |
| Bunchb | 1.24 | 1.53 | ||||
| Crysol | 1.30 | |||||
a Mass in solution was calculated compared to BSA.
b Bunch was run with the simultaneous fitting of two datasets.
The crystal structure fit the SAXS data very well (Fig. 5D), indicating highly similar conformations in the crystal state and in solution. The N- and C-terminal extensions gradually increased the dimensions of PRX (Table 2), suggesting they are elongated. Three-dimensional models of the various constructs confirmed the above findings, and N- and C-terminal extensions can be distinguished in the models (Fig. 5, E and F).
The AHNAK2 PDZ Domain Structure and Peptide Binding
To shed light on the uniqueness of the PRX PDZ domain homodimer structure, we also determined the structure of its closest homologue. The crystal structure of the human AHNAK2 PDZ domain was refined at 1.75-Å resolution, with a dimer in the asymmetric unit (Table 1). The overall structure of the AHNAK2 PDZ domain displays exactly the same domain-swapped homodimer as PRX (Fig. 6, A and B), indicating that PRX and AHNAK2 form a distinct subgroup of the PDZ domain family, with a unique, previously unseen way of intertwined folding. However, strand β2 of AHNAK2 is leaning more toward strand β3 (Fig. 6A); thus, the peptide binding pocket is larger than in PRX.
FIGURE 6.

AHNAK2 PDZ domain structure. A, superposition of AHNAK2 (pink) and PRX (gray) PDZ domains. The C terminus of a symmetry related molecule is found in one binding site of AHNAK2 (blue). The binding site is more open in AHNAK2 than in PRX, also in the unliganded site (arrow). B, superposition of the two chains in the AHNAK2 dimer. C, details of peptide binding by the AHNAK2 PDZ domain. Hydrogen bonds are indicated by green dashed lines. D, B factor plot of the two chains indicates increased flexibility in the binding site without the peptide. Largest differences are seen in helix α2 from one chain and strands β2 and β3 from the other chain (arrows). E, the peptide binding site between α2 and β2 in AHNAK2 (pink) and PAR-6 (green) (59). Note the distinct conformations of the β1–β2 loop. The bound peptide ligands are shown in orange and gray, respectively, and the hydrogen bonds between the terminal carboxyl group and backbone amide groups as dashed lines (AHNAK-2, magenta; PAR-6, green). The arrow points to the carbonyl group of Ala123.
A fortuitous feature in the AHNAK2 structure is the binding of the C terminus of a crystallographic symmetry mate in one, but not the other, peptide binding pocket of the homodimer (Fig. 6C); hence, both the unliganded and liganded binding sites were trapped in the same crystal. The sequence of the bound C terminus is Glu-Glu-Trp-Ala-COOH (residues 200–203). Thus, the AHNAK2 PDZ domain can be categorized into class III, as also expected from its sequence. Three backbone hydrogen bonds are present between strand β2 and the ligand (Fig. 6C). In addition, several side chain hydrogen bonds are present (Fig. 6C). For example, Glu200 forms a salt bridge with Lys176 from helix α2. The side chains of Glu201 and Lys139 (from β3) also interact through a salt bridge. Lys176 and Lys139 correspond to Arg82 and Arg45 in the PRX PDZ domain, implying that similar salt bridge interactions are available for positions P−2 and P−3 in a PRX ligand peptide. The side chain of the C-terminal Ala residue sits in a hydrophobic pocket lined by the side chains of AHNAK2 residues Tyr126 and Val128 (from strand β2), as well as Leu175, Leu178, and Gln179 (from helix α2).
An interesting conformation in the peptide binding site of AHNAK2 is found in the peptide bond between Ala123 and Ser124. In most other peptide-bound PDZ domain structures, the corresponding peptide bond is oriented so that the NH group interacts with the C-terminal carboxylate of the ligand. This interaction is missing in AHNAK2 (Fig. 6, C and E). The same backbone conformation is also found in the empty peptide binding pocket, as well as in the unliganded PRX PDZ domain (Fig. 4C). Thus, this unexpected conformation is a common property of these homologous PDZ domains, and it is not affected by ligand binding. This segment is in the β1–β2 loop, at the conserved GLGF motif (GVSGI in PRX, GASGY in AHNAK2) for carboxylate binding. The Ser side chain in this motif in AHNAK2 makes a hydrogen bond to the ligand C terminus (Fig. 6C); this residue is also conserved in PRX and may be involved in ligand recognition by both proteins.
To investigate conformational changes induced by ligand peptide binding, the structures of the two AHNAK2 monomers were superimposed (Fig. 6B). The two binding sites are nearly identical; some differences are found in temperature factors. The peptide-bound pocket, formed of strand β2 from chain A and helix α2 from chain B, has lower B factors than the empty pocket (Fig. 6D). The ligand-PDZ domain interaction decreases the dynamics of α2, β2, and β3, although does not affect the binding site conformation.
DISCUSSION
Both PRX and the AHNAK proteins participate in large protein complexes at the plasma membrane, linking the extracellular matrix to the cytoskeleton. Although evidence exists for dimerization in these proteins (12, 14), the molecular details have been missing. We provide here a structural basis for the dimeric assembly of PRX/AHNAK, and the surprising intertwined folding of these two PDZ domains is likely to be relevant to the overall architecture and stability of the respective multiprotein complexes, including the EPPD (ezrin, periplakin, periaxin, and desmoyokin/AHNAK)) complex in lens fibers and the dystrophin glycoprotein complex in Schwann cells.
It has been assumed that the PRX PDZ domain is canonical (45, 46). A new category of homodimeric PDZ domain structures is now provided by PRX and AHNAK2, which have a distinct domain-swapped fold compared with other known PDZ domain structures. By default, the dimerization also dictates that there are two peptide binding sites, with opposite orientations, next to each other in this PDZ assembly, which may be relevant for building large ordered protein complexes involving PRX/AHNAK. Experimental evidence has been presented for PDZ domain-mediated dimerization of both proteins before; PRX dimerization was shown by yeast two-hydrid experiments, pulldowns from nerve lysates, affinity chromatography, and co-immunoprecipitations from mammalian cells overexpressing PRX constructs (14). On the other hand, AHNAK was previously shown to form detergent-resistant homo- and heterodimers (between S- and L-AHNAK) (12). Hence, the tight homodimers observed here are likely to be of relevance in a cellular environment.
Despite the low homology to even the closest relatives (Fig. 7A), a comparison of sequences pinpoints unique features of PRX and AHNAK2, which correlate with their structures. The insertion of one residue into strand β1 in both proteins is related to the formation of a bulge and division of β1 into two short strands. On the other hand, most PDZ domains have a glycine residue in the β4–β5 turn; in PRX and AHNAK2, this residue is deleted, and a long continuous β4/5 strand is formed. In general, shortening hinge loops in domain-swapped proteins tends to lead to oligomerization (47, 48), and making such loops longer may generate monomers instead of domain-swapped dimers (48, 49). The folding pathways of PDZ domains have been studied before, and strands β1, β4, and β6 were shown to comprise a folding nucleus for the PTP-BL second PDZ domain (50). The corresponding strands in PRX and AHNAK2 form the 6-stranded antiparallel β sheet between the two intertwined chains. It is possible that interactions within this β sheet act as determinants for homodimeric folding.
FIGURE 7.

Comparison of PRX and AHNAK2 sequence and binding cavity. A, structural sequence alignment of PRX and AHNAK2 PDZ domains with two of their closest sequence homologues in PDB, the human SYNJ2BP (Protein Data Bank entry 2JIN) and human ZO-1 PDZ3 (Protein Data Bank entry 3TSV) (58). The secondary structures of the PDZ domains from AHNAK2 and SYNJ2BP are shown above and below the alignment, respectively. The image was prepared using Espript (60). The positions of the insertion in β1 (green) and the deletion in β4/5 (red) are indicated with triangles above the alignment. B, structures of internal peptide-binding PDZ domains. The PDZ domains are colored cyan, the binding ligands yellow, and the hydrogen bonds green. Note the varying conformations of the β1–β2 loop. Left, the PAR-6 PDZ domain with the PALS1 peptide (53). Instead of the terminal carboxylate of P0, the side chain and backbone carbonyl of Asp(P+1) form hydrogen bonds with the GLGF loop. Middle, α-1 syntropin PDZ domain with neuronal nitric-oxide synthase (52). The carbonyl group of P0 interacts with two backbone nitrogen atoms. Right, the Dishevelled-2 PDZ domain with N1 inhibitory peptide (54). The backbone conformation of P0 of the ligand is unique. The carboxylate in the Asp(P+1) side chain is located at a position corresponding to a canonical peptide C terminus, interacting with the GLGF loop. C, estimation of peptide ligand binding pocket volumes of AHNAK2 (red), PRX (blue), and SYNJ2BP (magenta) using POVME (61). The pocket volumes were 912, 576, and 1200 Å3, respectively. The liganded and unliganded pockets in the AHNAK2 homodimer are essentially identical, excluding large scale induced fitting of the peptide.
Although limited data exist on domain swapping in PDZ domains, it is relatively common in the protein universe. Intertwined structures, on the other hand, are much more rare; domain swapping normally would involve the swap of a single secondary structure element. One example of an intertwined domain-swapped protein is the dimeric Ig domain of RelB (51). Usually, domain swapping involves secondary structure elements at the N or C terminus of the protein (48), and the case of PRX/AHNAK2 is unique, due to the fact that the two chains for a large part wrap around each other.
The protein-ligand backbone interactions in the liganded AHNAK2 structure are weaker than in other liganded PDZ structures, i.e. only 3 hydrogen bonds are present, whereas in general, more than 4 hydrogen bonds exist in other complexes. The conformation of the carboxylate-binding loop in PRX/AHNAK2 suggests it may not be optimal for recognizing the C terminus of a ligand, because a backbone carbonyl group points toward the ligand carboxyl group. It is possible that this arrangement is more suitable for internal peptide motif binding, in which case the flipped carbonyl group could interact with a backbone amide from a ligand. The available structures of non-C-terminal peptide complexes of PDZ domains (Fig. 7B), on the other hand, each imply different mechanisms of recognition (52–54). It should be mentioned, however, that structural information on PDZ domains binding non-C-terminal sequences is very scarce.
PRX and AHNAK2 are likely to regulate the organization of multimolecular complexes. The liganded AHNAK2 PDZ structure shows that the PDZ domains with this intertwined homodimeric folding can interact with target sequences similarly to canonical PDZ domains. Intriguingly, the volume of the peptide binding pocket in PRX and AHNAK2 differs 2-fold, whereas the corresponding volume for a canonical PDZ domain lies between these two (Fig. 7C). The fact that both binding sites in the AHNAK2 dimer are very similar suggests that no large-scale changes occur upon peptide binding; hence, it is possible that the binding determinants for PRX and AHNAK2 are different. Ligands for these PDZ domains have not been discovered yet. The PRX and AHNAK2 PDZ domain structures provide starting points in the search for their binding partners. Likely possibilities include recognition of class III target sequences or internal peptide motifs, or combinations of both.
The significance of the homodimerization of PRX and AHNAK2 may lie in assembling the matrix lining the intracellular side of membrane; i.e. the PRX basic domain binds to the Drp2 spectrin-like domain. Then, the spectrin-like, WW, and ZZ domains of Drp2 interact with other proteins, such as dystroglycan, utrophin, or Dp116, to make a large complex. The stable homodimerization of two domain-swapped PDZ monomers certainly would be one of the secure points in such a large complex, and the symmetry of the dimer would assist to arrange the complex. A corresponding scenario is likely to occur for AHNAKs, which are involved in similar complexes (20, 55). In fact, both PRX and AHNAK are members of the EPPD complex in lens fiber cells (56), where they may even function together. Interestingly, PRX is also required for the hexagonal packing of lens fibers (11). The domain organization of PRX and AHNAKs, as well as the structures of their dimerization domains, may be signs of these proteins being involved in macromolecular complexes with similar assembly pathways (57).
Taken together, the structures of PRX and AHNAK2 PDZ domains reveal unique folding properties, with extensive three-dimensional domain swapping in an intertwined manner. Our data provide insights into identifying binding partners for these domains, which are unknown to date, and thus, will lead to a better understanding of the cellular functions of PRX and the AHNAK proteins.
Acknowledgments
Beamtime and beamline support at BESSY, ISA, and DESY are gratefully acknowledged.
This work was supported by grants from the Hamburg Research and Science Foundation (Germany), the Academy of Finland, and the Sigrid Jusélius Foundation (Finland) (to P. K.).
The atomic coordinates and structure factors (codes 4CMZ and 4CN0) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PRX
- periaxin
- SYNJ2BP
- synaptojanin-2 binding protein
- EPPD
- ezrin/periplakin/periaxin/desmoyokin
- SAXS
- small-angle X-ray scattering
- GRIP1
- glutamate receptor interacting protein 1.
REFERENCES
- 1. Ivarsson Y. (2012) Plasticity of PDZ domains in ligand recognition and signaling. FEBS Lett. 586, 2638–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee H. J., Zheng J. J. (2010) PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun. Signal. 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nourry C., Grant S. G., Borg J. P. (2003) PDZ domain proteins: plug and play! Sci. STKE 2003, RE7. [DOI] [PubMed] [Google Scholar]
- 4. Kalyoncu S., Keskin O., Gursoy A. (2010) Interaction prediction and classification of PDZ domains. BMC Bioinformatics 11, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gillespie C. S., Sherman D. L., Blair G. E., Brophy P. J. (1994) Periaxin, a novel protein of myelinating Schwann cells with a possible role in axonal ensheathment. Neuron 12, 497–508 [DOI] [PubMed] [Google Scholar]
- 6. Scherer S. S., Xu Y. T., Bannerman P. G., Sherman D. L., Brophy P. J. (1995) Periaxin expression in myelinating Schwann cells: modulation by axon-glial interactions and polarized localization during development. Development 121, 4265–4273 [DOI] [PubMed] [Google Scholar]
- 7. Patzig J., Jahn O., Tenzer S., Wichert S. P., de Monasterio-Schrader P., Rosfa S., Kuharev J., Yan K., Bormuth I., Bremer J., Aguzzi A., Orfaniotou F., Hesse D., Schwab M. H., Möbius W., Nave K. A., Werner H. B. (2011) Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. J. Neurosci. 31, 16369–16386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gillespie C. S., Sherman D. L., Fleetwood-Walker S. M., Cottrell D. F., Tait S., Garry E. M., Wallace V. C., Ure J., Griffiths I. R., Smith A., Brophy P. J. (2000) Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron 26, 523–531 [DOI] [PubMed] [Google Scholar]
- 9. Guilbot A., Williams A., Ravisé N., Verny C., Brice A., Sherman D. L., Brophy P. J., LeGuern E., Delague V., Bareil C., Mégarbané A., Claustres M. (2001) A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot-Marie-Tooth disease. Hum. Mol. Genet. 10, 415–421 [DOI] [PubMed] [Google Scholar]
- 10. Takashima H., Boerkoel C. F., De Jonghe P., Ceuterick C., Martin J. J., Voit T., Schröder J. M., Williams A., Brophy P. J., Timmerman V., Lupski J. R. (2002) Periaxin mutations cause a broad spectrum of demyelinating neuropathies. Ann. Neurol. 51, 709–715 [DOI] [PubMed] [Google Scholar]
- 11. Maddala R., Skiba N. P., Lalane R., 3rd., Sherman D. L., Brophy P. J., Rao P. V. (2011) Periaxin is required for hexagonal geometry and membrane organization of mature lens fibers. Dev. Biol. 357, 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Morrée A., Droog M., Grand Moursel L., Bisschop I. J., Impagliazzo A., Frants R. R., Klooster R., van der Maarel S. M. (2012) Self-regulated alternative splicing at the AHNAK locus. FASEB J. 26, 93–103 [DOI] [PubMed] [Google Scholar]
- 13. Dytrych L., Sherman D. L., Gillespie C. S., Brophy P. J. (1998) Two PDZ domain proteins encoded by the murine periaxin gene are the result of alternative intron retention and are differentially targeted in Schwann cells. J. Biol. Chem. 273, 5794–5800 [DOI] [PubMed] [Google Scholar]
- 14. Sherman D. L., Fabrizi C., Gillespie C. S., Brophy P. J. (2001) Specific disruption of a schwann cell dystrophin-related protein complex in a demyelinating neuropathy. Neuron 30, 677–687 [DOI] [PubMed] [Google Scholar]
- 15. Han H., Myllykoski M., Ruskamo S., Wang C., Kursula P. (2013) Myelin-specific proteins: a structurally diverse group of membrane-interacting molecules. Biofactors 39, 233–241 [DOI] [PubMed] [Google Scholar]
- 16. Shtivelman E., Cohen F. E., Bishop J. M. (1992) A human gene (AHNAK) encoding an unusually large protein with a 1.2-microns polyionic rod structure. Proc. Natl. Acad. Sci. U.S.A. 89, 5472–5476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gentil B. J., Delphin C., Mbele G. O., Deloulme J. C., Ferro M., Garin J., Baudier J. (2001) The giant protein AHNAK is a specific target for the calcium- and zinc-binding S100B protein: potential implications for Ca2+ homeostasis regulation by S100B. J. Biol. Chem. 276, 23253–23261 [DOI] [PubMed] [Google Scholar]
- 18. Huang Y., Laval S. H., van Remoortere A., Baudier J., Benaud C., Anderson L. V., Straub V., Deelder A., Frants R. R., den Dunnen J. T., Bushby K., van der Maarel S. M. (2007) AHNAK, a novel component of the dysferlin protein complex, redistributes to the cytoplasm with dysferlin during skeletal muscle regeneration. FASEB J. 21, 732–742 [DOI] [PubMed] [Google Scholar]
- 19. Zacharias U., Purfürst B., Schöwel V., Morano I., Spuler S., Haase H. (2011) Ahnak1 abnormally localizes in muscular dystrophies and contributes to muscle vesicle release. J. Muscle Res. Cell Motil. 32, 271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Komuro A., Masuda Y., Kobayashi K., Babbitt R., Gunel M., Flavell R. A., Marchesi V. T. (2004) The AHNAKs are a class of giant propeller-like proteins that associate with calcium channel proteins of cardiomyocytes and other cells. Proc. Natl. Acad. Sci. U.S.A. 101, 4053–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marg A., Haase H., Neumann T., Kouno M., Morano I. (2010) AHNAK1 and AHNAK2 are costameric proteins: AHNAK1 affects transverse skeletal muscle fiber stiffness. Biochem. Biophys. Res. Commun. 401, 143–148 [DOI] [PubMed] [Google Scholar]
- 22. Han H., Kursula P. (2013) Preliminary crystallographic analysis of the N-terminal PDZ-like domain of periaxin, an abundant peripheral nerve protein linked to human neuropathies. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 804–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 24. Konarev P. V., Petoukhov M. V., Volkov V. V., Svergun D. I. (2006) ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 26. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 27. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petoukhov M. V., Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kozin M. B., Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 32. Svergun D., Barberato C., Koch M. H. J. (1995) CRYSOL-a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 33. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen V. B., Arendall W. B., 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Whitmore L., Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fanning A. S., Lye M. F., Anderson J. M., Lavie A. (2007) Domain swapping within PDZ2 is responsible for dimerization of ZO proteins. J. Biol. Chem. 282, 37710–37716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Im Y. J., Park S. H., Rho S. H., Lee J. H., Kang G. B., Sheng M., Kim E., Eom S. H. (2003) Crystal structure of GRIP1 PDZ6-peptide complex reveals the structural basis for class II PDZ target recognition and PDZ domain-mediated multimerization. J. Biol. Chem. 278, 8501–8507 [DOI] [PubMed] [Google Scholar]
- 42. Im Y. J., Lee J. H., Park S. H., Park S. J., Rho S. H., Kang G. B., Kim E., Eom S. H. (2003) Crystal structure of the Shank PDZ-ligand complex reveals a class I PDZ interaction and a novel PDZ-PDZ dimerization. J. Biol. Chem. 278, 48099–48104 [DOI] [PubMed] [Google Scholar]
- 43. Sheng M., Sala C. (2001) PDZ domains and the organization of supramolecular complexes. Annu. Rev. Neurosci. 24, 1–29 [DOI] [PubMed] [Google Scholar]
- 44. Durand D., Vivès C., Cannella D., Pérez J., Pebay-Peyroula E., Vachette P., Fieschi F. (2010) NADPH oxidase activator p67phox behaves in solution as a multidomain protein with semi-flexible linkers. J. Struct. Biol. 169, 45–53 [DOI] [PubMed] [Google Scholar]
- 45. Kursula P. (2001) The current status of structural studies on proteins of the myelin sheath (review). Int. J. Mol. Med. 8, 475–479 [PubMed] [Google Scholar]
- 46. Kursula P. (2008) Structural properties of proteins specific to the myelin sheath. Amino Acids 34, 175–185 [DOI] [PubMed] [Google Scholar]
- 47. Green S. M., Gittis A. G., Meeker A. K., Lattman E. E. (1995) One-step evolution of a dimer from a monomeric protein. Nat. Struct. Biol. 2, 746–751 [DOI] [PubMed] [Google Scholar]
- 48. Liu Y., Eisenberg D. (2002) 3D domain swapping: as domains continue to swap. Protein Sci. 11, 1285–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Albright R. A., Mossing M. C., Matthews B. W. (1996) High-resolution structure of an engineered Cro monomer shows changes in conformation relative to the native dimer. Biochemistry 35, 735–742 [DOI] [PubMed] [Google Scholar]
- 50. Gianni S., Geierhaas C. D., Calosci N., Jemth P., Vuister G. W., Travaglini-Allocatelli C., Vendruscolo M., Brunori M. (2007) A PDZ domain recapitulates a unifying mechanism for protein folding. Proc. Natl. Acad. Sci. U.S.A. 104, 128–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang D. B., Vu D., Ghosh G. (2005) NF-κB RelB forms an intertwined homodimer. Structure 13, 1365–1373 [DOI] [PubMed] [Google Scholar]
- 52. Hillier B. J., Christopherson K. S., Prehoda K. E., Bredt D. S., Lim W. A. (1999) Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science 284, 812–815 [PubMed] [Google Scholar]
- 53. Penkert R. R., DiVittorio H. M., Prehoda K. E. (2004) Internal recognition through PDZ domain plasticity in the Par-6-Pals1 complex. Nat. Struct. Mol. Biol. 11, 1122–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang Y., Appleton B. A., Wiesmann C., Lau T., Costa M., Hannoush R. N., Sidhu S. S. (2009) Inhibition of Wnt signaling by Dishevelled PDZ peptides. Nat. Chem. Biol. 5, 217–219 [DOI] [PubMed] [Google Scholar]
- 55. De Seranno S., Benaud C., Assard N., Khediri S., Gerke V., Baudier J., Delphin C. (2006) Identification of an AHNAK binding motif specific for the Annexin2/S100A10 tetramer. J. Biol. Chem. 281, 35030–35038 [DOI] [PubMed] [Google Scholar]
- 56. Straub B. K., Boda J., Kuhn C., Schnoelzer M., Korf U., Kempf T., Spring H., Hatzfeld M., Franke W. W. (2003) A novel cell-cell junction system: the cortex adhaerens mosaic of lens fiber cells. J. Cell Sci. 116, 4985–4995 [DOI] [PubMed] [Google Scholar]
- 57. Marsh J. A., Hernández H., Hall Z., Ahnert S. E., Perica T., Robinson C. V., Teichmann S. A. (2013) Protein complexes are under evolutionary selection to assemble via ordered pathways. Cell 153, 461–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nomme J., Fanning A. S., Caffrey M., Lye M. F., Anderson J. M., Lavie A. (2011) The Src homology 3 domain is required for junctional adhesion molecule binding to the third PDZ domain of the scaffolding protein ZO-1. J. Biol. Chem. 286, 43352–43360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Peterson F. C., Penkert R. R., Volkman B. F., Prehoda K. E. (2004) Cdc42 regulates the Par-6 PDZ domain through an allosteric CRIB-PDZ transition. Mol. Cell 13, 665–676 [DOI] [PubMed] [Google Scholar]
- 60. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]
- 61. Durrant J. D., de Oliveira C. A., McCammon J. A. (2011) POVME: an algorithm for measuring binding-pocket volumes. J. Mol. Graph Model 29, 773–776 [DOI] [PMC free article] [PubMed] [Google Scholar]