

Background: Ionizing radiation can induce DNA damage in nonirradiated (N-IR) cells via nontargeted effects (NTE).
Results: TNF-α and IL-1α mediate NTE in N-IR bone marrow-derived EPCs, and neutralizing TNF-α diminishes NTE in WT and p55 knock-out BM-EPCs.
Conclusion: TNF-TNFR2/p75 signaling alters accumulation of inflammatory cytokines that attenuate NTE in N-IR EPCs.
Significance: TNFR2/p75 may represent a gene target for mitigation of delayed RBR in BM-EPCs.
Keywords: Cytokine, DNA Damage Response, Radiation Biology, Receptors, Tumor Necrosis Factor (TNF), Nontargeted Effects
Abstract
TNF-α, a pro-inflammatory cytokine, is highly expressed after being irradiated (IR) and is implicated in mediating radiobiological bystander responses (RBRs). Little is known about specific TNF receptors in regulating TNF-induced RBR in bone marrow-derived endothelial progenitor cells (BM-EPCs). Full body γ-IR WT BM-EPCs showed a biphasic response: slow decay of p-H2AX foci during the initial 24 h and increase between 24 h and 7 days post-IR, indicating a significant RBR in BM-EPCs in vivo. Individual TNF receptor (TNFR) signaling in RBR was evaluated in BM-EPCs from WT, TNFR1/p55KO, and TNFR2/p75KO mice, in vitro. Compared with WT, early RBR (1–5 h) were inhibited in p55KO and p75KO EPCs, whereas delayed RBR (3–5 days) were amplified in p55KO EPCs, suggesting a possible role for TNFR2/p75 signaling in delayed RBR. Neutralizing TNF in γ-IR conditioned media (CM) of WT and p55KO BM-EPCs largely abolished RBR in both cell types. ELISA protein profiling of WT and p55KO EPC γ-IR-CM over 5 days showed significant increases in several pro-inflammatory cytokines, including TNF-α, IL-1α (Interleukin-1 alpha), RANTES (regulated on activation, normal T cell expressed and secreted), and MCP-1. In vitro treatments with murine recombinant (rm) TNF-α and rmIL-1α, but not rmMCP-1 or rmRANTES, increased the formation of p-H2AX foci in nonirradiated p55KO EPCs. We conclude that TNF-TNFR2 signaling may induce RBR in naïve BM-EPCs and that blocking TNF-TNFR2 signaling may prevent delayed RBR in BM-EPCs, conceivably, in bone marrow milieu in general.
Introduction
Radiation-induced bystander responses (RBR)2 are defined as the induction of biological effects in cells that were not directly traversed by an ionized particle but were affected because of their proximity to cells that were directly exposed to radiation (1–7). RBR effects are well documented in vitro in variety of cell cultures (8–11). These responses have been shown by various methodologies, such as media transfer experiments (12, 13), co-cultures of irradiated (IR) and nonirradiated (N-IR) cells (14, 15), microbeam studies (16), and animal models in vivo (11). It has been proposed that RBR is mediated by an initiating event near the cell surface that activates and integrates numerous intracellular signaling pathways followed by activation of transcription factors and expression of genes that mediate RBR (7). Based on the previous investigations, it is evident that there appears to be a significant cell specificity in both the ability to induce the RBR (11) and the ability to receive the secreted signals (8). This suggests that in addition to the ability of IR cells to release cytokines, chemokines, and growth factors, the ligand-receptor interaction on N-IR cells may also play an important role in propagation of the bystander response (3, 8–10).
Low linear energy transfer radiation, such as γ-irradiation (γ-IR), has been reported to induce a bystander effect in glioblastoma cells (3). A more recent report found no evidence for low linear energy transfer induction of bystander responses in normal human fibroblast and colon carcinoma cells (17). Therefore, it is apparent that in addition to many factors that may influence bystander responses, including but not limited to production and release of inflammatory cytokines and chemokines, such as TNF-α, IL-1α, and others (9), there is a large intrinsic variability for bystander responses in different primary and tumor cells. Full body low dose radiation such as x-ray and γ-IR has been found to induce apoptotic and immunological responses in various organ and tissues, including bone marrow (18). The acute phase is usually characterized by neutrophil infiltration of the affected area, whereas macrophages are responsible for the phagocytic clearance of the apoptotic cells (19, 20). It was shown that phagocytosis of IR-induced apoptotic cells can activate macrophages, leading to their induction of an inflammatory response in the surrounding tissue (21). This is mediated by a release of various cytokines, superoxide, and nitric oxide (8). All of which are capable of causing tissue damage (22) by signaling through pro-apoptosis mediator TNF-α, Fas ligand, nitric oxide, and superoxide (23, 24).
TNF-α is a pro-inflammatory cytokine whose expression is known to be highly up-regulated in many tissues and cells after IR (23, 25). TNF-α is a 17-kDa polypeptide that specifically binds and exerts its function via two cell surface receptors, TNFR1 (p55) and TNFR2 (p75). Each TNF receptor has been shown to activate distinct signaling pathways with a small degree of overlap (26, 27). Functions of TNFR1/p55 have been well studied and described (28, 29). TNFR1/p55 is responsible for signaling a variety of responses predominantly cytotoxic, such as apoptosis and cell death, but also regulates inflammatory responses including cytokine secretion (30–33). In contrast, TNFR2/p75 is generally pro-survival and pro-angiogenic and responsible for cell protective effects of TNF but regulates inflammatory signaling as well (30, 31, 33–35). Both TNF receptors are ubiquitously expressed on nearly all cell types, but the p75 receptor is predominantly expressed by lymphoid cells as well as other hematopoietic and endothelial lineage cells, including endothelial progenitor cells (EPCs) (27, 36, 37). TNF induces inflammation via activation of transcription factor NF-κB and its downstream targets: COX-2, MMP1, IL-1α, IL-1β, IL-6, IL-8, IL-33, insulin growth factor 1 (IGF-1), and TNF itself, along with many other cytokines (9). Many of these cytokines, chemokines, and inflammatory enzymes (e.g. COX-2) are implicated in mediating RBR in variety of cells (38). However, the role of TNF receptors, p55 or p75, in regulating RBR in endothelial lineage cells, specifically in EPCs, is largely unknown.
A growing body of evidence indicates that neovascularization involves both the proliferation of local endothelial cells (ECs) as well as mobilization, recruitment and proliferation of the EPCs (39–43). EPCs have been shown to be proliferating clonally and capable of migrating and differentiating into ECs (44–48). In various animal models (48–52) and human clinical trials (53–56), it has been shown that transplantation of EPCs leads to migration and homing of these cells to the areas of ischemic injury such as acute myocardial infarction. Improved neovascularization and increased blood flow were observed in surgically induced hind limb ischemia model with infusion of ex vivo expanded EPCs (39, 41, 48, 54, 57). In GFP bone marrow-transplanted wild type mice using the hind limb ischemia model, our laboratory has shown that 60–70% of GFP-positive cells mobilized from the BM to ischemic limbs were BM-derived endothelial lineage cells (35). Thus, without being bound to a particular theory, if EPCs are critical to endothelial maintenance and repair, radiation-induced EPC dysfunction could impair the recovery after ischemic injury or trauma.
We hypothesized that inhibition of TNF-ligand-receptor interactions may alter TNF-mediated downstream signaling, thereby affecting regulation of inflammatory cytokines and chemokines. Increased levels of IR-induced cytokines, chemokines, and growth factors may then augment nontargeted effects in nearby cells not traversed directly by ionizing radiation, as well as in cells and tissues distant from the initial irradiation site, hence propagating bystander responses.
EXPERIMENTAL PROCEDURES
Animal Model
Mice were used in this study following a protocol approved by the Steward St. Elizabeth's Medical Center Institutional Animal Care and Use Committee. Mice used in this study were all 8–12 weeks old. They included WT (C57BL/6J-control of the mixed C57BL/6 and 129 background strains defined by the vendor as N10F34, meaning that these two strains were backcrossed 10 times (N10 indicates the number of backcross generations) and inbred 34 times (F34 is number of filial or inbreeding generations), TNFR2/p75 KO (B6.129S2-Tnfrsf1btm1Mwm/J) and TNFR1/p55KO (B6.129-Tnfrsf1atm1Mak/J) were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were fed standard laboratory chow diet (Harlan Teklad), given water access ad libitum, and kept in the temperature-controlled and light-controlled (12-h light/dark cycles) environment.
Isolation, Culture, and Characterization of Bone Marrow-derived Endothelial Progenitor Cells
Young WT mice were euthanized, and EPCs were isolated from mononuclear faction of bone marrow cells using density gradient centrifugation and cultured in 6-well dishes (Corning Inc., Corning, NY) on 22-mm × 22 mm square glass coverslips (Fisher HealthCare, Houston, TX) coated with 0.2% gelatin (Sigma). EPCs were expanded ex vivo in selective EBM2 growth medium (Lonza, Walkersville, PA) supplemented with growth factor as described previously (35, 41, 58). Upon attaining 70–80% confluence, cells were trypsinized using 0.25% Trypsin (Genesee Scientific, San Diego, CA) and processed for FACS analysis using EPC markers.
Immunofluorescent Characterization of Bone Marrow-derived Endothelial Progenitor Cells
BM-EPCs were expanded ex vivo to ∼70–80% confluence on glass coverslips as described in the paragraph above. To characterize our bone marrow-derived cell cultures, glass coverslips on which EPCs were cultured were fixed in 4% paraformaldehyde (FD NeuroTechnologies Inc., Columbia, MD) for 15 min at room temperature and washed with ice-cold 1× PBS (MediaTech, Herndon, VA) for 5 min. Fixed cells were permeabilized with 0.1% Triton X-100 (Sigma) for 15 min at room temperature and washed three times in 1× PBS for 5 min. To determine the stem or progenitor nature and to confirm endothelial cell lineage of cells in our ex vivo expanded BM marrow-derived cultures, cells on coverslips were triple-stained with rat anti-mouse Sca-1 (catalog no. sc-52601; Santa Cruz Biotechnology Inc., Dallas, TX) or rat anti-mouse c-kit (catalog no. 553868; BD Pharmingen, San Jose, CA) and biotinylated isolectin-B4 (catalog no. I21414; Invitrogen) along with TopRo3 (catalog no. T3605; Invitrogen) to visualize nuclei. Alexa 488 goat anti-rat (catalog no. A11006; Invitrogen) secondary antibody was used for both Sca-1 and c-kit, whereas Alexa 594-labeled streptavidin (catalog no. S11227; Invitrogen) was used as secondary antibody for Isolectin-B4. In addition, to determine the purity and a possible hematopoietic “contamination” of our BM-EPCs cultures, we have performed immunofluorescent staining of our BM-derived cultures with a panel of mouse hematopoietic anti-mouse antibodies for: Alexa 488-conjugated rat anti-mouse Gr1/Ly-6G (neutrophils) (catalog no. 108419; BioLegend, San Diego, CA), rat anti-mouse F4/80 (macrophages and blood monocytes) (catalog no. 123101; BioLegend), B220 (B lymphocytes), CD3ϵ (T lymphocytes), TER-119 (erythrocytes and erythroid precursors) (catalog no. 88–7774-75; eBiosciences, San Diego, CA), and endothelial cell marker Isolectin-B4 (Invitrogen) (staining), as well as uptake while still in culture of the acetylated low density lipoprotein, labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate (DiI-ac-LDL; catalog no. BT-902; Biomedical Technologies Inc., Stoughton, MA). Please note that DiI-ac-LDL is reported to label both vascular endothelial cells and bone marrow-derived macrophages. Alexa 488 goat anti-rat (Invitrogen) secondary antibody was used for F4/80, whereas FITC-labeled streptavidin (catalog no. 11-4317-87; eBiosciences) was used as secondary antibody for staining mouse hematopoietic lineage panel, and Alexa 594-labeled streptavidin (Invitrogen) was used as secondary antibody for Isolectin-B4. Corresponding IgG antibodies were also used as negative control to confirm the specificity of the primary antibodies. Images were obtained using laser scanning confocal microscope (LSM 510 Meta; Zeiss, Thornwood, NY) at ×20 magnification.
Irradiation and Dosimetry
Prior to full body IR, each animal was placed individually into a rectangular polypropylene box with multiple air holes (3 mm in diameter) for a stress-free environment. Typically eight unanesthetized mice were irradiated simultaneously. All γ-IR experiments were performed at Steward St Elizabeth's Medical Center using a Cesium (137Cs) source irradiator to yield a total single full body dose of 1 Gy at an average dose rate of 46.6 cGy/min.
BM-EPC Culture from Full Body Irradiated WT Mice
Young WT mice were exposed to 1-Gy full body γ-IR as described above. Both N-IR control and γ-IR WT mice were euthanized 30 min, 24 h, or 7 days post-IR. EPCs were isolated from mononuclear fraction of bone marrow cells and expanded ex vivo until the cells attained ∼70% confluence as described previously (35, 41, 58). Upon attaining confluence, cells were fixed on glass coverslips and processed for p-H2AX immunostaining.
Media Transfer Experiments in WT BM-EPCs
Two sets of ∼70% confluent EPCs from WT mice were derived from each animal. Within each preparation, one set of EPCs were γ-IR with 1 Gy. The conditioned media (CM) from irradiated EPCs (IR-CM) were collected at 30 min, 5 h, and 24 h post-IR. The second set of cells was not irradiated and was used as naïve (e.g. nonirradiated) EPCs for media transfer study. The medium was changed in all wells, including control N-IR wells, with fresh 3 ml of EBM2 media on the day of study, and EPCs were incubated in fresh media for 1 h prior to IR. IR-CM was filtered through a sterile 0.22-μm membrane syringe filter (Corning Inc.), and 2 ml of IR-CM collected from EPCs 30 min, 5 h, and 24 h post-IR was added to N-IR EPCs. Control media from N-IR cells (N-IR-CM) were also collected and filtered similarly. After 24 h of incubation with IR-CM and N-IR-CM, naïve EPCs at each time point were fixed and stained for the formation and decay of p-H2AX foci.
Media Transfer Experiments in WT, p55KO, and p75KO BM-EPCs
Two sets of subconfluent ex vivo expanded EPCs from WT, p55KO, and p75KO mice were prepared from the same animal for each genotype. At ∼70% confluence, one set of 6-well dishes of WT, p55KO, and p75KO EPCs were γ-IR with 1 Gy and CM from irradiated EPCs (IR-CM) of all genotypes were collected before and 1 h, 5 h, 24 h, 3 days, and 5 days post-IR. The second set of N-IR WT, p55KO, and p75KO cells was used as corresponding genotype naïve EPCs for media transfer study. The medium was changed in all wells, including control N-IR wells, with fresh 3 ml of EBM2 media on the day of study, and EPCs were incubated in fresh media for 1 h prior to IR. IR-CM was filtered through a sterile 0.22-μm membrane syringe filter (Corning Inc.), and 2 ml of IR-CM collected from WT, p55KO, and p75KO EPCs at 1 h, 5 h, 24 h, 3 days, and 5 days post-IR was added to corresponding genotype naïve N-IR EPCs. Control media from N-IR WT, p55KO, and p75KO EPCs were also collected and filtered similarly. After 24 h of incubation with IR-CM and N-IR-CM collected from WT, p55KO, and p75KO EPCs at 1 h, 5 h, 24 h, 3 days, and 5 days post-IR, naïve EPCs on the coverslips were fixed and processed for p-H2AX immunostaining. In addition, both IR-CM and N-IR-CM from all three genotypes were collected and saved for ELISA profiling. WT, p55KO, and p75KO IR EPC pellets were harvested at each time point and snap-frozen for future processing. All studies involved three biological replicates of WT, p75KO, and p55KO mice for each time point.
Media Transfer Experiments in WT and p55KO BM-EPCs Post-TNF-α Neutralization
As described in the previous section, two sets of subconfluent ex vivo expanded EPCs from WT and p55KO mice were prepared from same animal, and one set was γ-IR with 1 Gy. At various time points before and 1 h, 5 h, 24 h, 3 days, and 5 days post-IR CM from irradiated WT and p55KO EPCs were collected and filtered as described earlier. CM was incubated with TNF-α neutralizing antibody (catalog no. 506309; BioLegend) at a final concentration of 10 ng/ml for 1 h at room temperature and then transferred on to corresponding genotype naïve N-IR EPCs at respective time points. Control media from N-IR WT and p55KO EPCs were also treated the same way. After 24 h of incubation of N-IR EPCs with IR-CM and N-IR-CM collected from respective genotypes at 1 h, 5 h, 24 h, 3 days, and 5 days post-IR, naïve EPCs on the coverslips were fixed and processed for p-H2AX immunostaining.
Immunostaining, Imaging, and Analysis
To assess the formation and decay of p-H2AX foci in CM-treated and in untreated WT, p55KO, and p75KO EPCs treated with CM from corresponding genotype γ-IR EPCs, glass coverslips on which EPCs from each genotype were cultured were fixed in 4% paraformaldehyde (FD NeuroTechnologies Inc.) for 15 min at room temperature and washed with ice-cold 1× PBS (MediaTech) for 5 min. Fixed cells were permeabilized with 0.1% Triton X-100 (Sigma) for 15 min at room temperature and washed three times in 1× PBS for 5 min. Primary anti-p-H2AX rabbit monoclonal antibody (catalog no. 9718S; Cell Signaling Technology, Danvers, MA) and Alexa 488 goat anti-rabbit secondary antibody (catalog no. A11008; Invitrogen) were used to assay p-H2AX foci formation and decay over time. Topro-3 was used to visualize nuclei (catalog no. T3605; Invitrogen). Images were obtained using laser scanning confocal microscope (LSM 510 Meta; Zeiss) at ×100 magnification. Cells with apoptotic features or micronuclei were not considered for p-H2AX analysis. Data were obtained from three replicate samples for both IR and N-IR treatment conditions at each time point totaling 300–400 cells each. Using a computer-assisted image analysis algorithm based on pixel and color distribution, the p-H2AX foci were evaluated by quantifying all cells with ≥1 p-H2AX foci. Graphs were plotted for mean foci/cell and for a percentage of cells with a given number (N) of p-H2AX foci.
ELISA
Aliquots of the IR-CM and N-IR-CM collected from WT and p55KO EPCs at 1 h, 5 h, 24 h, 3 days, and 5 days post-IR were processed for mouse multiplex cytokine ELISA according to the manufacturer's protocols (Signosis, Sunnyvale, CA). The following 14 cytokines, chemokines, and growth factors were analyzed: IL-1α, IL-1β, IL-6, monocyte chemoattractant protein 1 (MCP-1), RANTES, microphage inflammatory protein 1α (MIP-1α), granulocyte colony-stimulating factor (G-CSF), granulocyte macrophage colony-stimulating factor (GM-CSF), EGF, IGF-1, VEGF, stem cell factor, basic fibroblast growth factor, and TNF-α. The plates were read using Tecan Spectra model 96-well microplate reader (MTX Lab Systems, Vienna, VA). The ELISA assay was also performed after TNF-α neutralization of γ-IR conditioned for three cytokines: TNF-α, IL-1α, and IL-1β.
Mouse Recombinant Cytokine and Chemokine Treatments
To determine the role of TNF-α, IL-1α, MCP-1, and RANTES in the formation of p-H2AX foci p55KO EPCs were treated with various concentrations of these mouse recombinant (rm) proteins. Recombinant, lyophilized IL-1α, MCP-1, and RANTES (catalog nos. D-61112, D-64030, and D-6413; PromoKine, Heidelberg, Germany) were reconstituted with sterile, ultrapure water to a stock concentration of 0.1 mg/ml and stored as aliquots according to the manufacturer's recommendations. Recombinant protein for TNF-α used for the study was available as a 0.5 mg/ml stock from the vendor (catalog no. 34–8321-82; eBiosciences). Based on the results of multiplex ELISA of CM from 1 Gy γ-IR p55KO EPCs, naïve p55KO EPCs were treated with the following concentrations of single recombinant protein: IL-1α, 290 and 580 pg/ml; MCP-1, 580, 1160, and 2900 pg/ml; and RANTES, 600 and 1500 pg/ml, for 24 h under normal growth conditions. Based on our previously published work, we used three significantly different concentrations for TNF-α: two angiogenic (0.1 and 1 ng/ml) and one cytotoxic (40 ng/ml) (35). Formation and decay of p-H2AX foci were quantified as described under “Immunostaining, Imaging, and Analysis.”
Statistical Analysis
All results are expressed as means ± S.E. Statistical analysis was performed using one-way analysis of variance (Stat View software; SAS Institute Inc., Gary, NC). Differences were considered significant at p < 0.05.
RESULTS
Characterization of ex Vivo Expanded BM-EPC Culture
We have previously characterized and published our BM-EPCs cultures (35). Briefly, in our previous work, BM-EPCs were stained with β-gal (biological EC marker cells were grown from Tie2/LacZ mice) and c-kit (stem/progenitor cell marker), and we demonstrated that 95–100% of cells by days 4 and 6 were double positive for both markers (35). Two additional markers for endothelial cell lineage, Isolectin-B4 and Flk-1, also showed similar results by day 6 in culture (35). Despite extensive EPC culture characterization over the years, we have tested but have not published a possibility that at early stages of ex vivo BM-EPC selection, there may be hematopoietic “contamination” of the BM-EPC culture. Accordingly, we performed immunofluorescent staining of our BM-derived cultures with antibodies for c-kit and Sca-1 (stem/progenitor cell markers) combined with the endothelial cell marker Isolectin-B4. Nearly 100% of cells were double positive for c-kit/Isolectin-B4, and ∼95% were positive for Sca-1/Isolectin-B4, confirming stem/progenitor nature and endothelial lineage of cells in our BM-derived cultures (Fig. 1A).
FIGURE 1.

Characterization of bone marrow derived EPCs. A, top row, representative fluorescent confocal images of BM-EPCs triple-stained with c-kit (green, stem/progenitor cell marker), Isolectin-B4 (red, endothelial cell marker), and TopRo-3 (blue, to visualize nuclei) on day 5 after initial plating. Bottom row, representative fluorescent confocal images of BM-EPCs double stained with Sca-1 (green, stem/progenitor cell marker), Isolectin-B4 (red), and TopRo-3 (blue, to visualize nuclei) on day 5 after initial plating. The far right panels in both rows are the triple overlay. These double c-kit/Isolectin-B4 (+) and Sca-1/Isolectin-B4 (+) cells, presumably EPCs, constituted nearly 100% of cells on day 5, respectively, indicating that by this time most if not all of BM-derived cells were identified as EPCs by double (+) staining with EC marker and two progenitor markers (c-kit and Sca-1). B, representative confocal images of BM-EPCs expanded ex vivo for 5 days and stained for hematopoietic lineage markers: Gr1 (to identify neutrophils), F4/80 (to identify macrophages), Isolectin-B4, and the uptake of DiI-ac-LDL while in culture (to confirm EC lineage of the cells). Graphic representation of the percentage (+) cells in BM-EPC cultures for Gr1, F4/80, Isolectin-B4, and DiI-ac-LDL. C, representative phase contrast (×20) images of ex vivo expanded BM-EPC cultures on days 3 and 5 to demonstrate colony-specific expansion of EPCs on day 3 and relative morphologic homogeneity of EPC cultures on days 3 and 5 after initial plating. D, in vitro tube-like structure formation assay on VEGF-reduced Matrigel to confirm EC function of EPCs in BM-derived cultures.
To determine whether our BM-EPCs cultures may contain other lineage specific hematopoietic cells, we have performed immunofluorescent staining of our BM-derived cultures with antibodies for Gr1/Ly-6G (neutrophils), F4/80 (macrophages and blood monocytes), CD45R/B220 (B lymphocytes), CD3ϵ (T lymphocytes), TER-119 (erythrocytes and erytroid precursors), and endothelial cell marker Isolectin-B4 (staining), as well as uptake while still in culture of the acetylated low density lipoprotein labeled with DiI-ac-LDL. Please note that DiI-ac-LDL is reported to label both vascular endothelial cells and bone marrow-derived macrophages (59–61). On day 5 no staining was detected for B220, Cd3ϵ, and TER-119, suggesting that our culture was free of B and T lymphocyte and erythroid precursor “contamination.” On day 5, there was a negligible 1.17 ± 0.7% positivity for Gr1 (neutrophils) staining (Fig. 1B, upper left panel). Further, ∼19% of cells were positive for F4/80 staining (Fig. 1B, upper right panel), a macrophage-specific marker, suggesting that by day 5 approximately one-fifth of the cells in our culture could be macrophages/monocytes. However, several lines of evidence from different groups have suggested that various macrophage markers, such as F4/80, Mac-3, CD68, or CD11b, may be expressed in bone marrow-derived cultures during selection and especially during maturation/differentiation (62, 63). More importantly, Modarai et al. (62), using a “chimera” animal model where the BM of WT mice were transplanted with GFP/Tie2 BM cells (Tie2 is a tyrosine kinase receptor that is expressed on endothelial cells), have shown that many of BM-derived Tie2/GFP-expressing cells that were recruited into the thrombus during its resolution also expressed Mac-3, CD68, and/or F4/80, suggesting that these Tie2-positive cells have retained macrophage phenotype. These authors suggested that these cells with macrophage phenotype could be a population of plastic stem cells, but this was not validated experimentally.
Phase contrast images (×20) of ex vivo expanded BM-EPCs on days 3 and 5 showed that already by day 3 after initial plating, EPC clusters formed well defined colonies (Fig. 1C). By both 3 and 5 days, there was no significant morphological differences observed in EPC culture, and by day 5 cells attained ∼70% confluence (Fig. 1C). To further confirm endothelial lineage of cells in our culture, we performed an additional functional characterization using tube-like structure formation assay. EPCs collected on day 5 and reseeded to 4-well chambers coated with phenol-free and VEGF-reduced Matrigel formed tube-like structures starting at 16 h after reseeding (Fig. 1D), confirming an important EC functional characteristic of BM-EPCs. Phase contrast images (×20) of EPCs in culture at 3 and 5 days after initial plating were taken using Nikon Eclipse TE 200 microscope (Nikon Instruments Inc., Melville, NY).
Within 24 h the Decay of p-H2AX Foci in BM-EPCs Is Slow, and the Number of p-H2AX Foci Is Doubled by 7 Days
DNA damage-induced p-H2AX foci occur specifically at sites of double-strand breaks, and the time-dependent decline in p-H2AX foci correlates well with double-strand break repair (64). We sought to determine the effect of low dose full body γ-IR on the formation and decay of p-H2AX foci in BM-EPCs from WT mice. BM-EPCs were isolated 30 min, 24 h, and 7 days after full body γ-IR and selected in the culture ex vivo for 60 h. In control, N-IR EPCs there was a negligible number of p-H2AX (+) EPCs over a 7-day period (Fig. 2A). Compared with N-IR EPCs, analysis of γ-IR samples revealed a significant >20-fold increase in the p-H2AX foci at 30 min (0.27 ± 0.15 versus 5.8 ± 1.2 foci/cell, p < 0.0003) that was followed by an ∼43% reduction that was statistically not significant (5.8 ± 1.2 versus 3.3 ± 1.1 foci/cell, p = NS) at 24 h post-IR (Fig. 2A). However, compared with the 24-h time point, the number of p-H2AX foci/cell at 7 days was increased more than 200% (3.3 ± 1.1 versus 6.8 ± 1.4, p < 0.006), to the level of the pH2AX foci at 30 min (Fig. 2A).
FIGURE 2.

Slow decay and increased p-H2AX foci in ex vivo EPCs by day 7 after full body 1 Gy γ-irradiation and increased bystander responses demonstrated by BM-EPCs in vitro with increase in mean p-H2AX foci/cell over time. A, graphic representation of mean p-H2AX foci/cell after 60 h in the selective EPC culture media from WT mice at 30 min (black bars), 24 h (gray bars), and 7 days (white bars) after full body irradiation with 1 Gy γ-IR compared with respective N-IR controls. In control N-IR ex vivo expanded EPCs, there was no change over 7 days in p-HA2X foci: 0.27 ± 0.15 versus 0.27 ± 0.15 and 0.29 ± 0.08, p = NS, all comparisons. Graphs represent data pooled from three independent biological samples treated under similar conditions. B, foci distribution plot of percentage of WT EPCs with an N of p-H2AX foci for 30 min, 24 h, and 7 days post-IR treatment. C, representative images for p-H2AX immunostaining (yellow) and Topro-3-stained nuclei (red) in naïve WT EPCs in vitro, treated with IR-CM medium transferred at 30 min, 5 h, and 24 h post-IR of WT EPCs with 1-Gy γ-IR compared with respective controls after 24 h treatment with CM. D, graphic representation of mean p-H2AX foci/cell after 24-h treatment of naïve WT EPCs with IR-CM medium from WT EPCs at 30 min (black bars), 5 h (gray bars), and 24 h (white bars) after 1-Gy γ-IR. In control N-IR CM-treated EPCs, there was no change over 24 h in p-HA2X foci: 0.7 ± 0.1 versus 0.8 ± 0.2 and 0.8 ± 0.2, p = NS, all comparisons. In IR-CM-treated EPCs, there was a significant increase in p-H2AX foci when comparing 30 min versus 24 h: 1 ± 0.2 versus 3.2 ± 0.5, p < 0.0001, and 5 h versus 24 h 1.4 ± 0.3 versus 3.2 ± 0.46, p < 0.001. The graphs represent data pooled from three independent biological samples treated under similar conditions. E, foci distribution plot of percentage of naïve WT EPCs after IR-CM transfer with a given number (N) of foci count from 0–15 foci for 30-min, 5-h, and 24-h treatment conditions. F, for better visualization of distribution of EPCs with N foci, the graph provided shows the distribution after excluding the cells with 0 foci (1–15 foci).
Analysis of p-H2AX foci quantity distribution at each time point showed that ∼5% of BM-EPCs showed an increase in the percentage of cells with ≥18–22 p-H2AX foci/cell on day 7 (Fig. 2B, dotted line). These data indicate that decay of p-H2AX foci in WT BM-EPCs ex vivo is slow within the first 24 h, which may be indicative of delayed or inefficient radiation-induced DNA damage repair. In addition, significant increase in mean foci/cell on day 7 post-IR along with an increase in the percentage of p-H2AX foci/cell may be indicative of significant radiobiological bystander responses in BM-EPCs (3, 4, 6, 7, 65).
BM-EPCs Exhibit Significant Bystander Responses in Vitro
To determine whether BM-EPCs show evidence of a bystander response in media transfer experiments in vitro, N-IR WT EPC cells were treated with CM collected from γ-IR WT EPCs. Formation of p-H2AX foci over 24 h post-treatment was used to evaluate the bystander effect. In control N-IR media-treated EPCs, there was negligible number of detectable p-H2AX (Fig. 2, C and D). There was no significant change in the mean foci/cell when comparing 30-min and 5-h IR-CM-treated cells (1 ± 0.2 versus 1.4 ± 0.3, p = NS) (Fig. 2, C and D). BM-EPCs treated with 24 h IR-CM had significant 320% (p < 0.0001) and 228% (p < 0.001) increases in the mean number of foci/cell when compared with N-IR EPCs treated with 30-min and 5-h IR-CM, respectively (Fig. 2, C and D). Quantification of p-H2AX foci distribution with a given number (N) of foci/cell showed an increase in the percentage of cells with more than 6–15 p-H2AX foci/cell 24 h after addition IR-CM (Fig. 2, E and F), suggesting that BM-EPCs exhibit significant bystander responses in vitro.
TNF Ligand-Receptor Interactions Modify Formation of p-H2AX Foci in BM-EPCs in Vitro
It has been reported that early IR-induced vascular damage can be diminished by anti-TNF antibody (66), and elevated tissue TNF levels after IR are associated with significant increase in p-H2AX foci and genomic instability (67). Moreover, TNFR2/p75 signaling was suggested to have protective effects against IR-induced demyelination in the brain (68). Therefore, we sought to examine whether TNF ligand-receptor interactions mediate RBR in BM-EPCs. To determine the involvement of TNFRs, media transfer experiments were carried out with in N-IR BM-EPCs isolated from WT, TNFR1/p55, and TNFR2/p75KO animals. Both p55KO and p75KO EPCs treated with 1 h IR-CM showed a significant increase in the mean pH2AX foci/cell with respect to the WT cells (p < 0.002 and p < 0.0001, respectively) (Fig. 3). For clarity of data presentation, only cells with ≥1 p-H2AX positive foci were considered for the graphs presented in Fig. 3.
FIGURE 3.

TNF ligand-receptor interactions modify formation of p-H2AX foci in BM-EPCs in vitro. The analysis of p-H2AX formation and decay was performed for every time point where we included all cells with ≥1 p-H2AX foci and mean foci/cell was plotted. Graphic representation of mean p-H2AX foci/cell 24 h after treatment of naïve WT, p55KO, and p75KO EPCs with IR-CM medium collected from respective WT, p55KO, and p75KO EPCs at 1 h, 5 h, 24 h, 3 days, and 5 days after 1-Gy γ-IR. pH2AX foci/cell treated with 1 h IR-CM in WT versus p75KO and p55KO: 4.3 ± 0.4 versus 6.9 ± 1 and 8.5 ± 0.9, p < 0.002 and p < 0.0001, respectively; pH2AX foci/cell treated with 5 h IR-CM in WT versus p75KO and p55KO: 6.9 ± 0.9 versus 7.7 ± 0.6 and 8.5 ± 0.9, p = NS, both comparisons; pH2AX foci/cell treated with 24 h IR-CM in WT versus p75KO and p55KO: 9 ± 0.8 versus 3.7 ± 0.5 and 4.8 ± 0.6, p < 0.0001, both comparisons; pH2AX foci/cell treated with 3-day IR-CM in WT versus p75KO and p55KO: 7.6 ± 0.8 versus 7.3 ± 1.5 and 7.8 ± 0.7, p = NS, both comparisons; pH2AX foci/cell treated with 5-day IR-CM in WT versus p75KO and p55KO: 3.8 ± 0.4 versus 5.9 ± 0.8 and 8.5 ± 1, p < 0.04 and p < 0.0001, as well as p < 0.03 when comparing p75KO versus p55KO. There was a steady increase over time in the formation of p-H2AX foci in N-IR p55KO treated with 1-, 3-, and 5-day IR-CM collected from corresponding genotype EPCs: 4.8 ± 0.6 versus 7.8 ± 0.7 versus 8.5 ± 1; p < 0.002 for day 1 versus day 3, p < 0.0001 for day 1 versus day 5, p = NS for day 3 versus day 5. There was a similar increase, however, followed by a decrease in p75KO EPCs: 3.7 ± 0.5 versus 7.3 ± 1.5 versus 5.9 ± 0.8: p < 0.003 for day 1 versus day 3, p < 0.04 for day 1 versus day 5, p = NS for day 3 versus day 5. The graphs represent data pooled from three independent biological samples treated under similar conditions.
There was no significant difference in the formation of p-HA2X foci in N-IR WT, p55KO, and p75KO EPC treated with 5-h IR-CM (Fig. 3). Compared with WT, in the absence TNF receptors (p55 or p75), there was at least 2-fold decrease (p < 0.0001) in the formation of p-H2AX foci/cell in N-IR p55KO and p75KO EPCs treated for 24 h with 1-day IR-CM from the respective genotype EPCs (Fig. 3). This was not the case for the WT cells that exhibited the peak increase (9.0 ± 0.8 p-H2AX foci/cell) post-treatment with 1-day IR-CM media. There was no difference in the formation of p-HA2X foci between N-IR p55KO and p75KO EPC treated with 1-day IR-CM (Fig. 3). These findings indicate that in γ-IR EPCs, the presence of both TNF receptors (p55 and p75) is necessary for 1-day CM to increase the formation of p-H2AX foci in corresponding genotype N-IR EPCs, suggesting that by blocking either p55 or p75 TNF receptors, one could inhibit formation of p-H2AX foci in N-IR BM-EPCs within a day after radiation exposure.
Although there was no significant difference in the formation of p-H2AX foci in N-IR WT, p55KO, and p75KO EPC treated with 3-day IR-CM (Fig. 3), over 5-day period IR-CM-treated WT N-IR EPCs showed a significant decrease (p < 0.0001) in the mean p-H2AX foci/cell (Fig. 3). In contrast, there was a significant, steady increase over time in the formation of p-H2AX foci in N-IR p55KO treated with 1-, 3-, and 5-day IR-CM from corresponding genotype EPCs and, to a lesser degree, in p75KO EPCs (Fig. 3). Both WT and p75KO EPCs treated with 5-day IR-CM exhibited a decreasing trend in mean p-H2AX foci/cell from 3 to 5 days with a small but significant (p < 0.04) difference between them at 5 days (Fig. 3). There was a much larger >2-fold increase in the mean p-H2AX foci/cell for N-IR p55KO EPCs treated with 5-day IR-CM compared with both p75KO and WT EPCs (p < 0.02 and p < 0.0001, respectively) with p55KO EPCs having the highest and WT having the lowest mean p-H2AX foci/cell (Fig. 3). Foci distribution of the percentage of EPCs with N foci count (data not shown) showed an increase of 0.5–2.2% of p55KO EPCs with 14–27 foci at 5 h and 5 days compared with respective control, whereas peaks for WT and p75KO EPCs at 1 and 3 days demonstrated a decreasing trend normalizing to respective controls by day 5. These findings indicate that TNF signaling via TNFR1/p55 and TNFR2/p75 is necessary for development of early RBR in N-IR naïve EPCs. More importantly, our results demonstrate a significant increase of delayed bystander response seen in naïve p55KO EPCs at the 5-day time point when compared with WT and p75KO EPCs, suggesting that signaling through the remaining TNFR2/p75 in p55KO cells plays an important role in mediating radiobiological bystander responses in BM-EPCs. Because of a significant and continued (from days 1 to 5) increase in delayed bystander response in p55KO BM-EPCs, we decide to focus our next set of studies on p55KO BM-EPCs.
Neutralization of TNF-α in γ-IR-CM Resulted in Significant Decrease in the Formation p-H2AX Foci in TNFR1/p55KO EPCs
Because of the role of elevated TNF levels post-IR in tissue resulting in increased p-H2AX foci formation (67), we determined the effects of TNF-α neutralization in IR-CM on p-H2AX foci formation in vitro for both p55KO and WT EPCs over 5 days. Media transfer experiments were performed in N-IR BM-EPCs from both WT and TNFR1/p55KO mice, wherein the IR-CM at respective time points were filtered and then incubated in TNF-α neutralizing antibody before transferring on to respective naïve N-IR BM-EPCs.
Nonirradiated p55KO EPCs treated with 1-h TNF-α neutralized IR-CM showed a significant increase in the mean pH2AX foci/cell with respect to N-IR WT EPCs (p < 0.05) (Fig. 4, red dotted line versus solid blue line). There was no significant difference in the formation of p-HA2X foci between N-IR WT and p55KO EPCs treated with respective 5 h, 24 h, 3 days, and 5 days of TNF-α neutralized IR-CM (Fig. 4). Even though N-IR WT EPCs treated with TNF-α neutralized IR-CM still showed a gradual increase in p-H2AX foci formation over time with the peak increase at 24 h, it was not significant compared with the number of p-H2AX foci in p55KO EPCs at this time point (Fig. 4). Foci distribution of naïve p55KO EPCs with N number of foci with and without TNF-α neutralization of 1 Gy γ-IR conditioned media at various time points: before (control) and 1 h, 5 h, 24 h, 3 days, and 5 days after, is presented (Fig. 5, A–F). Taken together with the data of mean p-H2AX foci/cell, the distribution of the N number of foci/cell confirms a significant decrease in foci formation after TNF-α neutralization at 1 h, 5 h, and 5 days.
FIGURE 4.

Neutralization of TNF-α in IR-CM resulted in significantly decreased p-H2AX foci formation in TNFR1/p55KO and WT EPCs in vitro. Please note that for clarity of the comparison between the formation p-H2AX foci in EPCs treated with IR-CM media with or without TNF neutralization data points for WT and p55KO, EPCs from Fig. 3 are overplayed in Fig. 4 again. Shown is a graphic representation of mean p-H2AX foci/cell post 24-h treatment of naïve WT and p55KO with IR-CM medium collected and treated with TNF-α neutralizing antibody from respective WT and p55KO EPCs at 1 h, 5 h, 24 h, 3 days, and 5 days post-1-Gy γ-IR. pH2AX foci/cell treated with 1 h IR-CM in WT versus p55KO: 2.4 ± 0.2 versus 3.7 ± 0.6, p < 0.05; pH2AX foci/cell treated with 5 h IR-CM in WT versus p55KO: 4.3 ± 0.5 versus 3.6 ± 0.5, p = NS; pH2AX foci/cell treated with 24 h IR-CM in WT versus p55KO: 5.0 ± 0.5 versus 4.2 ± 0.6, p = NS; pH2AX foci/cell treated with 3-day IR-CM in WT versus p55KO: 3.2 ± 0.4 versus 4.9 ± 0.8, p < 0.03; pH2AX foci/cell treated with 5-day IR-CM in WT versus p55KO: 3.5 ± 0.3 versus 3.5 ± 0.3, p = NS. Treatment of IR-CM with TNF neutralizing antibody decreased the formation of p-H2AX foci at all time points examined in both WT and p55KO EPCs. However, the most significant decreases for WT EPCs were observed at 5 h, 24 h, and 3 days, and those for p55KO EPCs were at 1 h, 5 h, 3 days, and 5 days. Graphs represent data pooled from three independent biological samples treated under similar conditions.
FIGURE 5.

Foci distribution of naïve p55KO EPCs with the number of foci (N) with and without TNF-α neutralization after 1 Gy γ-IR and media transfer at various time points: before (control) and 1 h, 5 h, 24 h, 3 days, and 5 days after. A–F, foci distribution of naïve p55KO EPCs with the number of foci (N) upon treatment with IR-CM from p55KO (red bars and dashed lines) and with IR-CM from p55KO EPCs after TNF neutralization (green bars and dashed lines).
The continuous increase of p-H2AX foci formation over time in N-IR p55KO treated with 1-, 3-, and 5-day IR-CM from corresponding genotype EPCs (Fig. 4, gray dotted line) was significantly inhibited in p55KO EPCs treated with IR-CM after incubation with TNF-α neutralizing antibody before the media transfer (Fig. 4). These significant decreases in the formation of p-H2AX foci in N-IR p55KO EPCs treated with TNF-α neutralized IR-CM at all time points examined substantiate the significant role of TNF-TNFR2/p75 signaling axis in delayed radiobiological responses in p55KO EPCs. Moreover, the incubation of γ-IR-CM from WT and p55KO EPCs with TNF-α neutralizing antibody showed that the formation of p-H2AX foci was significantly inhibited not only in p55KO but also in WT BM-EPCs up to 3 days, indicating that inhibition of TNF-α may represent a therapeutic modality for the prevention of early and intermediate radiobiological bystander responses in WT tissue.
Increased Radiation Induced Accumulation of Cytokines and Growth Factors in TNFR1/p55KO EPCs in Vitro
Radiation-mediated effects converge with increased levels of various cytokines and chemokines in that both generate reactive oxygen and nitrogen species that may lead to inflammation (69). In the cell growth media of p55KO EPCs, we sought to determine the effect of γ-IR on secretion and accumulation of cytokines, chemokines, and growth factors, such as TNF-α, IL-1α, IL-1β, IL-6, MCP-1, MIP-1α, G-CSF, GM-CSF, EGF, VEGF, etc., all of which are known to be elevated within minutes to hours after ionizing radiation and other exogenous signals without the need of de novo protein synthesis (8).
Conditioned media from γ-IR WT and p55KO EPCs were collected before (control) and 1 h, 5 h, 24 h, 3 days, and 5 days post-IR and processed for multiplex (14-gene) ELISA. There was no significant difference in TNF-α level in IR-CM from p55KO and WT EPCs at 1 h and up to 3 days. However, compared with WT EPCs, there was a significant 175% increase (p < 0.04) in TNF-α level in p55KO EPCs on day 5 (Fig. 6A). Compared with the media from IR WT EPCs, conditioned media from IR p55KO EPCs showed significant (200–1600%) increases in the secretion of IL-1α, IL-1β, RANTES, MIP-1α, MCP-1, G-CSF, GM-CSF, and stem cell factor as compared with IR-CM WT media (*, p < 0.05; **, p < 0.001; and ***, p < 0.0009) (Fig. 6, B, C, E–G, and L–N). There was no significant difference in the concentrations of IGF1, VEGF, and basic fibroblast growth factor between γ-IR-CM-treated WT and p55KO (Fig. 6, I–K), except a small but statistically significant (p < 0.05) increase in VEGF concentration at day 3 in p55KO versus WT IR-CM. In addition to comparing increases in the concentration of cytokines and growth factors between IR-CM of WT and p55KO EPCs at each time point, we also analyzed the kinetics of changes for each protein over time when compared with corresponding genotype N-IR control levels (Table 1, A and B).
FIGURE 6.

Radiation induced increase in the accumulation of cytokines, chemokines, and growth factors in WT and p55KO EPCs in vitro. A, TNF-α levels (pg/ml) measured in IR-CM growth media from WT and p55KO EPCs after 1 Gy γ-IR at 1 h, 5 h, 24 h, 3 days, and 5 days. B–G, cytokine and chemokine concentrations (pg/ml) measured in IR-CM from WT and p55KO EPCs after 1-Gy γ-IR at 1 h, 5 h, 24 h, 3 days, and 5 days. H–N, growth factor concentrations (pg/ml) measured in IR-CM from WT and p55KO EPCs after 1-Gy γ-IR at 1 h, 5 h, 24 h, 3 days, and 5 days. Graphs represent data pooled from three independent biological samples treated under similar conditions. Statistical significance between WT and p55KO EPCs at each time point is denoted as follows: *, p < 0.05 to p < 0.01; **, p < 0.009 to p < 0.001; and ***, p < 0.0009.
TABLE 1.
Statistical analysis of accumulation of cytokines, chemokines, and growth factors in WT and p55KO γ-IR EPC media in control versus all other time points
A, statistical analysis of protein concentrations using ELISA for different cytokines and chemokine and growth factors of γ-IR-CM from WT EPCs compared with respective control N-IR medium. Categorized into cytokines, chemokines, and growth factors showed that in WT EPC medium most statistical significance at any time point compared with control was mostly growth factors with the exception of just two cytokines, IL-1α and MCP-1. B, statistical analysis of protein concentrations using ELISA for different cytokines, chemokine, and growth factors of γ-IR-CM from p55KO EPCs compared to respective control N-IR medium. Compared with WT, IR-CM from p55KO EPCs demonstrated significantly higher levels of predominantly cytokine at any time point compared with control with the exception of just one growth factor, G-CSF. Red font indicates cytokines and chemokines that were significantly different in IR-CM from WT versus p55KO EPCs, and blue font indicates growth factors that were significantly different in IR-CM from WT versus p55KO EPCs.
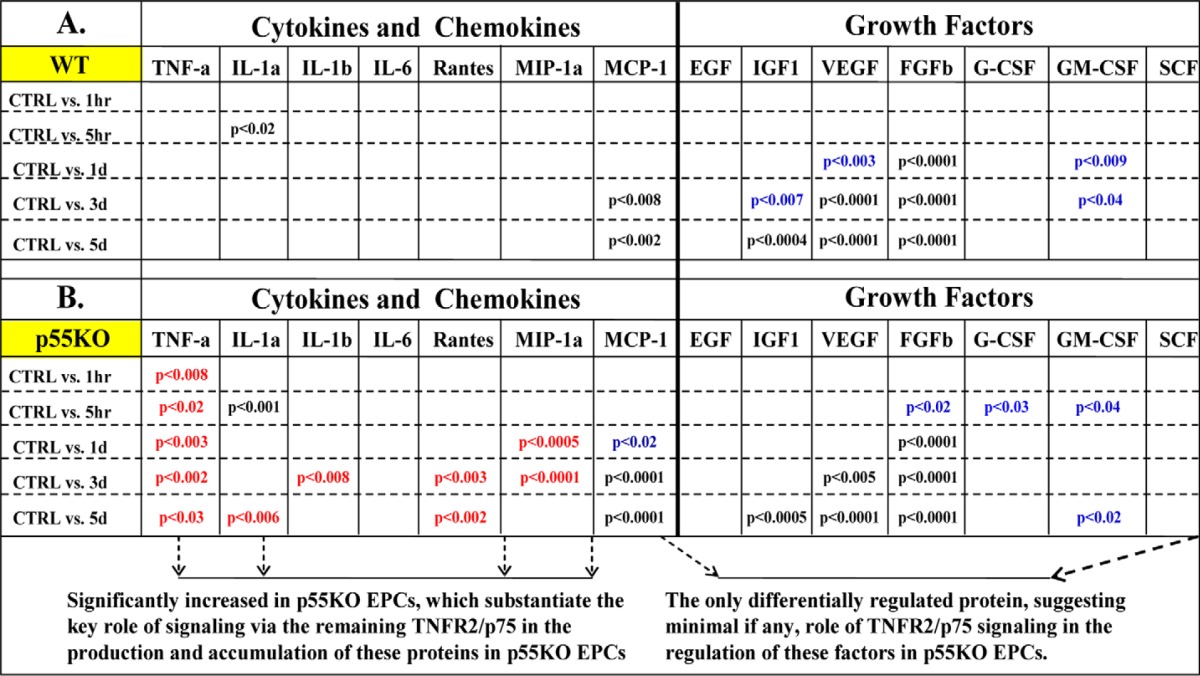
This analysis revealed that in WT IR-CM, statistically significant increases that were observed over 5 days were primarily in growth factors (e.g. IGF-1, VEGF, basic fibroblast growth factor, GM-CSF and G-CSF) (Table 1A). In contrast, in p55KO IR-CM, statistically significant increases (when comparing control levels with any one time point post-IR) were predominantly in cytokine levels such as TNF-α, IL-1α, IL-1β, RANTES, MIP-1α, and MCP-1 (Table 1B). Taken together, our findings in multiplex ELISA studies suggest that the signaling through TNFR2/p75 in irradiated TNFR1/p55KO EPCs may lead to significant increases in the accumulation of the several cytokines and chemokines such as IL-1α, IL-1β, RANTES, and MIP-1α (with the exception of MCP-1 at 3 and 5 days and IL-1α at 5 h, which were also increased in WT irradiated EPCs). Please note that cytokine levels for TNF-α, IL-1α, and IL-1β after neutralization of TNF-α in IR-CM were not detectable in our ELISA assay.
Treatment with Mouse Recombinant TNF-α and IL-1α, but Not MCP-1 or RANTES Increases the Formation of p-H2AX Foci in N-IR TNFR1/p55KO EPCs in Vitro
Based on our findings that p55KO EPCs exhibited continuous increases in the levels of TNF-α, IL-1α, RANTES, and MCP-1 between days 1 and 5 (Fig. 6, A, B, E, and G, and Table 1) and the continuous increase in the mean p-H2AX foci/cell in N-IR p55KO EPCs treated with 1–5-day p55KO EPC IR-CM (Figs. 3, dotted line, and 4, gray dotted line), we sought to determine whether treatment of N-IR p55KO EPCs with mouse recombinant IL-1α, MCP-1, RANTES, and TNF-α could lead to the formation of p-H2AX foci. N-IR p55KO EPCs were treated with mouse recombinant TNF-α (100, 1,000, and 40,000 pg/ml), IL-1α (290 and 580 pg/ml), MCP-1 (580, 1,160, and 2,900 pg/ml), or RANTES (600 and 1,500 pg/ml). For clarity of data presentation, only cells with ≥1 p-H2AX positive foci were considered for the graphs presented in Fig. 7A. The data with inclusion of cells with zero p-H2AX foci were also quantified and plotted, which showed a similar trend in mean p-H2AX foci formation between treatment conditions (not shown).
FIGURE 7.

p-H2AX foci formation increased in nonirradiated p55KO EPCs in vitro after treatment with mouse recombinant TNF-α and IL-1α. A, graphic representation of mean p-H2AX foci/cell after 24-h treatment of naïve p55KO EPCs with various concentrations of rm IL-1α (red bars), rmRANTES (green bars), rmMCP-1 (blue bars), and rmTNF-α (yellow bars) compared with control (CTRL) p55KO EPCs (black bar). The graphs represent data pooled from three identically treated independent biological samples. B, foci distribution of naïve p55KO EPCs with the number of foci (N) after treatment with various concentrations of rmTNF-α protein in vitro for 24 h. Almost 0.5–2.55% and 0.5–1% of cells treated with 100 pg/ml (red bars and dashed line), 1,000 pg/ml (green bars and dashed line), and 40,000 pg/ml (blue bars and dashed line) of TNF-α had 9–18 and 19–31 foci/cell, respectively, when compared with control p55KO EPCs (black bars and dashed line), which had no more than 0.5% cells with a maximum of 13 and 15 p-H2AX foci/cell. Treatment with 40,000 pg/ml of TNF-α resulted in a maximum of 31 foci/cell in 0.5% of EPCs. C, foci distribution of naïve p55KO EPCs with number of foci (N) upon treatment with 290 pg/ml (green bars) and 580 pg/ml (red bars) concentrations of rmIL-1α protein in vitro for 24 h. Mouse recombinant IL-1α treatment resulted in >4% of cells with 9–18 foci/cell, whereas control p55KO EPCs (black bars) had no more than 0.5% cells with a maximum of 13 and 15 p-H2AX foci/cell. At the same time, 0.5–3% of p55KO EPCs had a maximum of 19–37 foci/cell in 580 pg/ml and 19–51 foci/cell in 290 pg/ml treatment conditions.
After 24-h treatment, N-IR p55KO EPCs were stained with anti-p-H2AX antibodies. Quantification showed that the mean p-H2AX foci count of MCP-1 and RANTES were not significantly different from control, which had a mean of 2.2 ± 0.2 p-H2AX foci/cell, with the exception of MCP-1 at 1160 pg/ml (p < 0.03, mean foci count of 3.7 ± 0.5) (Fig. 7A). TNF-treated N-IR p55KO EPCs showed significant increases (p < 0.0001) in mean p-H2AX foci/cell at all concentrations (with mean p-H2AX foci in TNF-treated ranging from 4 ± 0.3 to 6 ± 0.6) compared with the control, MCP-1, and RANTES (Fig. 7A). IL-1α-treated p55KO EPCs showed the greatest increase in p-H2AX foci/cell with mean foci counts of 10.1 ± 0.9 at 290 pg/ml and 11.1 ± 0.7 at 580 pg/ml and were significantly (p < 0.0001) different from all tested mouse recombinant proteins (Fig. 7A). Analysis of p-H2AX foci distribution of EPCs with more than 3 foci showed that in control p55KO EPCs, less than 1–1.3% of cells had a maximum of 408 p-H2AX foci/cell, and less than 0.5% of cells had 13 and 15 p-H2AX foci/cell (Fig. 7, B and C). Whereas in TNF-α- and IL-1α-treated p55KO EPCs >2% and >4% of cells had 9–18 foci/cell, respectively (Fig. 7, B and C). Remarkably, 0.5–1% of cells had as many as 19–31 foci/cell for TNF-treated cells (Fig. 7B), and as many as 0.5–3% of cells had 19–51 foci/cell for IL-1α-treated p55KO EPCs (Fig. 7C). These findings suggest that TNF-α and IL-1α but not MCP-1 and RANTES significantly increase the formation of p-H2AX foci in naïve BM-derived TNFR1/p55KO EPCs.
DISCUSSION
Hematopoietic EPCs originate from the bone marrow and are part of the subpopulation of hematopoietic stem cells, which have been shown to be sensitive to radiobiological bystander response (20, 24, 70). EPCs are recruited to areas of neovascularization in the process of vasculogenesis to repair tissues with ischemia and cardiovascular diseases (44, 48, 57). EPCs are critical also to endothelial maintenance, and thus it is possible that radiation-induced EPC dysfunction could contribute to the pathogenesis of coronary and peripheral vascular diseases. Studies have demonstrated that in patients with CV risk factors, the number and migratory ability of EPCs isolated from peripheral blood is reduced (71), and EPC function is impaired (72). In 2006, Fadini et al. (73) demonstrated that early subclinical atherosclerosis in healthy subjects arises because of depletion of CD34+/KDR+ EPCs. In addition, a strong inverse correlation was reported between the number of circulating EPCs, vascular function, and the subject's combined Framingham cardiovascular factor score (74). Furthermore, measurements of flow-mediated brachial artery reactivity also revealed a significant relation between endothelial function and the number of EPCs, supporting a role for BM-EPCs in the maintenance of endothelial integrity (75).
Dose response curves for γ-radiation have been found through medium transfer experiments using ranges of 0.01–5.0 Gy on a human epithelial cell line. The degree of bystander effect seemed to saturate at ranges 0.03–0.05 Gy as measured by clonogenic death (76, 77). In experiments using γ-radiation, a low linear energy transfer radiation, it has been found that not all cell types have the same bystander response to radiation. In experiments with keratinocytes (epithelial cells), fibroblasts, and radio-sensitive carcinoma cells, fibroblasts did not show induction of bystander effect through irradiated conditioned media transfer (17, 76, 77). In particular, the role of the bystander responses in bone marrow-derived EPC remains largely unknown.
Radiation-induced chromosomal instability was demonstrated in the bone marrow for up to 24 months after full body irradiation with either x-rays or neutrons, indicating that chromosomal instability can be initiated and maintained in vivo (78, 79). In addition, it was shown for myeloid and lymphoid bone marrow stem and progenitor cells that after space flight, the numbers of these cells are reduced to just one-half of their normal levels (80), suggesting that EPCs may be similarly reduced in the normal EPC population after space flights. Unfortunately, neither data on EPC survival or mobilization, nor DNA damage responses of EPCs during and after space flights nor after lower doses of terrestrial radiation are currently available.
Current understanding of low dose space and terrestrial radiation and its biological effects is that direct damage of DNA in the nucleus causes cell death and mutations (3). However, there have been numerous studies, especially in the past two decades, suggesting that radiation can cause damage in nonirradiated cells through radiobiological effects currently not fully understood (81, 82). The term “bystander effect” was used to describe the ability of a cell affected by radiation to cause damage in other cells not directly traversed by the initial radiation (83). This radiobiological bystander effect was observed in radiation dosage as low as 0.31 mGy in an experiment by Nagasawa and Little (84), where CHO cells were irradiated with α-particles in G1 phase and measured for the induction of sister chromatid exchanges. Results showed that 30% of cells had increased sister chromatid exchanges, even though only 1% of cell nuclei were traversed by α-particles.
It is well studied in the literature that ionizing radiation at high doses can suppress the immune system; however, the response at low doses is less well known. Present risk assessment of high and low dose radiation is extrapolated from epidemiological studies of atomic bomb survivors in Hiroshima and Nagasaki; however, there is some uncertainty in the assessment of risks at lower doses (11, 85). Full body low dose radiation is known to induce apoptotic and immunological responses in the organ tissues, including the bone marrow (18, 86, 87). Full body low dose radiation in mice has been found to enhance the function of macrophages (and CD8+ T cells) in several studies (18–20). Current evidence suggests that the immune response is enhanced at lower doses and influences the production and the release of inflammatory cytokines and chemokines such as TNF-α, IL-1α, and others (18). The main goal of this study is to elucidate the mechanisms of the signal transmission for IR-induced radiobiological bystander responses in bone marrow-derived EPCs and determine the role of TNF ligand-receptor interaction in mediating RBR in these cells.
TNF-α is thought to be an important factor in the immune response of nonirradiated cells. ELISA of lung fibroblasts irradiated with low doses of α-particles showed a dosage-dependent increase in IL-8 mRNA levels from 30 min to 24 h post-irradiation (88). IL-8 expression is under the control of cis-elements of nuclear factor NF-κB, which is associated with the major pro-inflammatory pathway for TNF-α (89). TNF-α was also directly implicated as one of the damaging signals in in vivo bystander experiments, as well as Fas ligand, nitric oxide, and super oxide (24, 90). Hematopoietic cells from bone marrow were treated with radiation and measured for cells in sub-G0 region as a screening method for dead/damaged cells. The experiment showed an increasing trend in the number of sub-G0 cells at the 1-, 2-, and 3-h time points post-irradiation. When TNF-α neutralizing antibody was added to the irradiated bone marrow medium, the number of sub-G0 cells was significantly decreased in all three time points (24, 90). Reduction was also noted for Fas ligand neutralizing antibody, which might be another signal of interest.
Our findings here complement a recent gene expression study in directly irradiated and bystander cells that revealed NF-κB as the dominant transcription factor in mediating bystander responses (91, 92). TNF is one of the key cytokines that activates the NF-κB (93). In turn, NF-κB activation may lead to increased expression of NF-κB-dependent genes (94) in irradiated and N-IR bystander cells. These include NF-κB-dependent cytokines IL-6 and IL-8 (16), and cytokines that induce the NF-κB signaling pathway via paracrine or autocrine mechanisms: IL-1β, TNF-α, and IL-33 (11, 17).
Our findings, taken together with previously published work, strongly suggest that by blocking TNFR2/p75 signaling one can reduce production of growth factors and cytokines after ionizing radiation via reduced activation of NF-κB and other stress response transcription factors. Moreover, our findings indicate that unopposed signaling via TNFR2/p75 in TNFR1/p55KO EPCs plays a significant role in inhibiting early and increasing delayed (5 days) formation of p-H2AX foci, which correlate well with the induction of double-strand breaks (95, 96).
In summary, in BM-EPCs in vivo and ex vivo we report (a) slow decay of p-H2AX foci after full body 1 Gy γ-IR over 24 h and increase over 7 days and (b) significant bystander responses in BM-EPCs in media transfer experiments. In BM-derived WT, TNFR2/p75KO, and TNFR1/p55KO EPCs ex vivo, we report that (a) compared with WT, in the absence of either of TNF receptors (p55 or p75), there is a significant decrease in the formation of p-H2AX foci at 5 and 24 h after adding IR-conditioned medium to naïve BM-EPCs, indicating that TNF and signaling via either of TNF receptors is necessary for development of radiobiological bystander responses in N-IR radiated BM-EPC; (b) compared with WT, in the absence of the either of TNF receptors (p55 or p75), there is a significant increase in the formation of p-H2AX foci on days 1, 3, and 5 after adding IR-conditioned medium to naïve BM-EPCs, indicating that TNF and signaling via either of TNF receptors increases delayed bystander responses in nonirradiated BM-EPCs; and (c) continuous increase in the number (N) p-H2AX foci/cell between 1 and 5 days in naïve TNFR1/p55KO BM-EPCs may indicate that unopposed (by p55) signaling via TNFR2/p75 in TNFR1/p55KO EPCs plays a significant role in delayed bystander responses. The specificity of the role of TNF in mediating bystander responses in BM-EPCs was confirmed in two experiments: (a) the formation of p-H2AX foci was decreased more than twice in EPCs treated with γ-IR conditioned media after neutralizing TNF, and (b) treatment of nonirradiated naïve EPCs with recombinant murine TNF led to significant increase in the formation of p-H2AX.
We conclude that TNF plays a significant role in mediating radiobiological bystander responses in BM-EPCs, and these effects may be regulated (decreased or increased) through modification of TNF signaling via TNFR1/p55 or TNFR2/p75. Possible implications are that blocking TNF or the signaling of TNF through one of its two receptors may be used to prevent radiation-induced delayed bystander effects in naïve “nonhit” BM-EPC. We further suggest that blocking/neutralizing TNFR2/p75 (versus TNFR1/p55) signaling may be a more effective strategy to inhibit the formation of p-H2AX foci in non-IR radiated EPCs and conceivably other cells in bone marrow milieu.
This material is based upon work supported by the National Aeronautic and Space Administration under Grant No. NNJ10ZSA001N and AHA Grant No. 14GRNT18860032 to D. A. G. and in part by grants from AHA 10GRNT4710003 and NHLBI HL106098 to X. Y., NIH HL091983 to R. K. and NASA NNX11AK26G to Dr. L. Hlatky.
- RBR
- radiobiological bystander response
- IR
- irradiated/irradiation
- N-IR
- nonirradiated
- EPC
- endothelial progenitor cell
- BM-EPC
- bone marrow-derived EPC
- CM
- conditioned media
- RANTES
- regulated on activation, normal T cell expressed and secreted
- TNFR
- TNF receptor
- γ-IR
- γ-irradiation
- EC
- endothelial cell
- DiI-ac-LDL
- 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate
- MCP-1
- monocyte chemoattractant protein 1
- MIP-1α
- microphage inflammatory protein 1α
- G-CSF
- granulocyte colony-stimulating factor
- GM-CSF
- granulocyte macrophage colony-stimulating factor
- IGF-1
- insulin growth factor 1
- rm
- murine recombinant
- NS
- not significant.
REFERENCES
- 1. Azzam E. I., de Toledo S. M., Little J. B. (2001) Direct evidence for the participation of gap junction-mediated intercellular communication in the transmission of damage signals from α-particle irradiated to nonirradiated cells. Proc. Natl. Acad. Sci. U.S.A. 98, 473–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Azzam E. I., de Toledo S. M., Little J. B. (2003) Expression of CONNEXIN43 is highly sensitive to ionizing radiation and other environmental stresses. Cancer Res. 63, 7128–7135 [PubMed] [Google Scholar]
- 3. Lorimore S. A., Coates P. J., Wright E. G. (2003) Radiation-induced genomic instability and bystander effects: inter-related nontargeted effects of exposure to ionizing radiation. Oncogene 22, 7058–7069 [DOI] [PubMed] [Google Scholar]
- 4. Little J. B. (2006) Cellular radiation effects and the bystander response. Mutat. Res. 597, 113–118 [DOI] [PubMed] [Google Scholar]
- 5. Little J. B. (2006) Lauriston S. Taylor Lecture: Nontargeted effects of radiation: implications for low-dose exposures. Health Phys. 91, 416–426 [DOI] [PubMed] [Google Scholar]
- 6. Morgan W. F., Sowa M. B. (2007) Non-targeted bystander effects induced by ionizing radiation. Mutat. Res. 616, 159–164 [DOI] [PubMed] [Google Scholar]
- 7. Hei T. K., Zhou H., Ivanov V. N., Hong M., Lieberman H. B., Brenner D. J., Amundson S. A., Geard C. R. (2008) Mechanism of radiation-induced bystander effects: a unifying model. J. Pharm. Pharmacol. 60, 943–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schaue D., Kachikwu E. L., McBride W. H. (2012) Cytokines in radiobiological responses: a review. Radiat. Res. 178, 505–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ivanov V. N., Zhou H., Ghandhi S. A., Karasic T. B., Yaghoubian B., Amundson S. A., Hei T. K. (2010) Radiation-induced bystander signaling pathways in human fibroblasts: a role for interleukin-33 in the signal transmission. Cell. Signal. 22, 1076–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hubackova S., Krejcikova K., Bartek J., Hodny Z. (2012) IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine “bystander senescence.” Aging 4, 932–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morgan W. F. (2003) Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiation-induced genomic instability and bystander effects in vitro. Radiat. Res. 159, 567–580 [DOI] [PubMed] [Google Scholar]
- 12. Yang H., Asaad N., Held K. D. (2005) Medium-mediated intercellular communication is involved in bystander responses of x-ray-irradiated normal human fibroblasts. Oncogene 24, 2096–2103 [DOI] [PubMed] [Google Scholar]
- 13. Yang H., Anzenberg V., Held K. D. (2007) The time dependence of bystander responses induced by iron-ion radiation in normal human skin fibroblasts. Radiat. Res. 168, 292–298 [DOI] [PubMed] [Google Scholar]
- 14. Mitchell S. A., Marino S. A., Brenner D. J., Hall E. J. (2004) Bystander effect and adaptive response in C3H 10T(1/2) cells. Int. J. Radiat. Biol. 80, 465–472 [DOI] [PubMed] [Google Scholar]
- 15. Yang H., Magpayo N., Held K. D. (2011) Targeted and non-targeted effects from combinations of low doses of energetic protons and iron ions in human fibroblasts. Int. J. Radiat. Biol. 87, 311–319 [DOI] [PubMed] [Google Scholar]
- 16. Burdak-Rothkamm S., Short S. C., Folkard M., Rothkamm K., Prise K. M. (2007) ATR-dependent radiation-induced γH2AX foci in bystander primary human astrocytes and glioma cells. Oncogene 26, 993–1002 [DOI] [PubMed] [Google Scholar]
- 17. Sowa M. B., Goetz W., Baulch J. E., Pyles D. N., Dziegielewski J., Yovino S., Snyder A. R., de Toledo S. M., Azzam E. I., Morgan W. F. (2010) Lack of evidence for low-LET radiation induced bystander response in normal human fibroblasts and colon carcinoma cells. Int. J. Radiat. Biol. 86, 102–113 [DOI] [PubMed] [Google Scholar]
- 18. Pandey R., Shankar B. S., Sharma D., Sainis K. B. (2005) Low dose radiation induced immunomodulation: effect on macrophages and CD8+ T cells. Int. J. Radiat. Biol. 81, 801–812 [DOI] [PubMed] [Google Scholar]
- 19. Wright E. G., Coates P. J. (2006) Untargeted effects of ionizing radiation: implications for radiation pathology. Mutat. Res. 597, 119–132 [DOI] [PubMed] [Google Scholar]
- 20. Lorimore S. A., Coates P. J., Scobie G. E., Milne G., Wright E. G. (2001) Inflammatory-type responses after exposure to ionizing radiation in vivo: a mechanism for radiation-induced bystander effects? Oncogene 20, 7085–7095 [DOI] [PubMed] [Google Scholar]
- 21. Mungrue I. N., Gros R., You X., Pirani A., Azad A., Csont T., Schulz R., Butany J., Stewart D. J., Husain M. (2002) Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J. Clin. Invest. 109, 735–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kojda G., Laursen J. B., Ramasamy S., Kent J. D., Kurz S., Burchfield J., Shesely E. G., Harrison D. G. (1999) Protein expression, vascular reactivity and soluble guanylate cyclase activity in mice lacking the endothelial cell nitric oxide synthase: contributions of NOS isoforms to blood pressure and heart rate control. Cardiovasc. Res. 42, 206–213 [DOI] [PubMed] [Google Scholar]
- 23. Meng Y., Beckett M. A., Liang H., Mauceri H. J., van Rooijen N., Cohen K. S., Weichselbaum R. R. (2010) Blockade of tumor necrosis factor α signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 70, 1534–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burr K. L., Robinson J. I., Rastogi S., Boylan M. T., Coates P. J., Lorimore S. A., Wright E. G. (2010) Radiation-induced delayed bystander-type effects mediated by hemopoietic cells. Radiat. Res. 173, 760–768 [DOI] [PubMed] [Google Scholar]
- 25. Sherman M. L., Datta R., Hallahan D. E., Weichselbaum R. R., Kufe D. W. (1991) Regulation of tumor necrosis factor gene expression by ionizing radiation in human myeloid leukemia cells and peripheral blood monocytes. J. Clin. Invest. 87, 1794–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tartaglia L. A., Weber R. F., Figari I. S., Reynolds C., Palladino M. A., Jr., Goeddel D. V. (1991) The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc. Natl. Acad. Sci. U.S.A. 88, 9292–9296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Higuchi Y., McTiernan C. F., Frye C. B., McGowan B. S., Chan T. O., Feldman A. M. (2004) Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor-α-induced cardiomyopathy. Circulation 109, 1892–1897 [DOI] [PubMed] [Google Scholar]
- 28. Chen G., Goeddel D. V. (2002) TNF-R1 signaling: a beautiful pathway. Science 296, 1634–1635 [DOI] [PubMed] [Google Scholar]
- 29. Ihnatko R., Kubes M. (2007) TNF signaling: early events and phosphorylation. Gen. Physiol. Biophys. 26, 159–167 [PubMed] [Google Scholar]
- 30. Jacobsen S. E., Jacobsen F. W., Fahlman C., Rusten L. S. (1994) TNF-α, the great imitator: role of p55 and p75 TNF receptors in hematopoiesis. Stem Cells 12, 111–128 [PubMed] [Google Scholar]
- 31. Barbara J. A., Van ostade X., Lopez A. (1996) Tumour necrosis factor-α (TNF-α): the good, the bad and potentially very effective. Immunol. Cell Biol. 74, 434–443 [DOI] [PubMed] [Google Scholar]
- 32. Pastorino F., Mumbengegwi D. R., Ribatti D., Ponzoni M., Allen T. M. (2008) Increase of therapeutic effects by treating melanoma with targeted combinations of c-myc antisense and doxorubicin. J. Control. Release 126, 85–94 [DOI] [PubMed] [Google Scholar]
- 33. Sasi S. P., Yan X., Enderling H., Park D., Gilbert H. Y., Curry C., Coleman C., Hlatky L., Qin G., Kishore R., Goukassian D. A. (2012) Breaking the “harmony” of TNF-α signaling for cancer treatment. Oncogene 31, 4117–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vandenabeele P., Declercq W., Beyaert R., Fiers W. (1995) Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 5, 392–399 [DOI] [PubMed] [Google Scholar]
- 35. Goukassian D. A., Qin G., Dolan C., Murayama T., Silver M., Curry C., Eaton E., Luedemann C., Ma H., Asahara T., Zak V., Mehta S., Burg A., Thorne T., Kishore R., Losordo D. W. (2007) Tumor necrosis factor-α receptor p75 is required in ischemia-induced neovascularization. Circulation 115, 752–762 [DOI] [PubMed] [Google Scholar]
- 36. Ksontini R., MacKay S. L., Moldawer L. L. (1998) Revisiting the role of tumor necrosis factor α and the response to surgical injury and inflammation. Arch. Surg. 133, 558–567 [DOI] [PubMed] [Google Scholar]
- 37. Brockhaus M., Schoenfeld H. J., Schlaeger E. J., Hunziker W., Lesslauer W., Loetscher H. (1990) Identification of two types of tumor necrosis factor receptors on human cell lines by monoclonal antibodies. Proc. Natl. Acad. Sci. U.S.A. 87, 3127–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou H., Ivanov V. N., Gillespie J., Geard C. R., Amundson S. A., Brenner D. J., Yu Z., Lieberman H. B., Hei T. K. (2005) Mechanism of radiation-induced bystander effect: role of the cyclooxygenase-2 signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 102, 14641–14646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Asahara T., Murohara T., Sullivan A., Silver M., van der Zee R., Li T., Witzenbichler B., Schatteman G., Isner J. M. (1997) Isolation of putative progenitor endothelial cells for angiogenesis. Science 275, 964–967 [DOI] [PubMed] [Google Scholar]
- 40. Lin Y., Weisdorf D. J., Solovey A., Hebbel R. P. (2000) Origins of circulating endothelial cells and endothelial outgrowth from blood. J. Clin. Invest. 105, 71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kalka C., Masuda H., Takahashi T., Kalka-Moll W. M., Silver M., Kearney M., Li T., Isner J. M., Asahara T. (2000) Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc. Natl. Acad. Sci. U.S.A. 97, 3422–3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shi Q., Rafii S., Wu M. H., Wijelath E. S., Yu C., Ishida A., Fujita Y., Kothari S., Mohle R., Sauvage L. R., Moore M. A., Storb R. F., Hammond W. P. (1998) Evidence for circulating bone marrow-derived endothelial cells. Blood 92, 362–367 [PubMed] [Google Scholar]
- 43. Asahara T., Masuda H., Takahashi T., Kalka C., Pastore C., Silver M., Kearne M., Magner M., Isner J. M. (1999) Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ. Res. 85, 221–228 [DOI] [PubMed] [Google Scholar]
- 44. Ribatti D. (2007) The discovery of endothelial progenitor cells. An historical review. Leuk. Res. 31, 439–444 [DOI] [PubMed] [Google Scholar]
- 45. Urbich C., Dimmeler S. (2004) Endothelial progenitor cells: characterization and role in vascular biology. Circ. Res. 95, 343–353 [DOI] [PubMed] [Google Scholar]
- 46. Folkman J., Shing Y. (1992) Angiogenesis. J. Biol. Chem. 267, 10931–10934 [PubMed] [Google Scholar]
- 47. Risau W. (1995) Differentiation of endothelium. FASEB J. 9, 926–933 [PubMed] [Google Scholar]
- 48. Yang J., Ii M., Kamei N., Alev C., Kwon S. M., Kawamoto A., Akimaru H., Masuda H., Sawa Y., Asahara T. (2011) CD34+ cells represent highly functional endothelial progenitor cells in murine bone marrow. PLoS One 6, e20219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gill M., Dias S., Hattori K., Rivera M. L., Hicklin D., Witte L., Girardi L., Yurt R., Himel H., Rafii S. (2001) Vascular trauma induces rapid but transient mobilization of VEGFR+AC133+ endothelial precursor cells. Circ. Res. 88, 167–174 [DOI] [PubMed] [Google Scholar]
- 50. Shintani S., Murohara T., Ikeda H., Ueno T., Honma T., Katoh A., Sasaki K., Shimada T., Oike Y., Imaizumi T. (2001) Mobilization of endothelial progenitor cells in patients with acute myocardial infarction. Circulation 103, 2776–2779 [DOI] [PubMed] [Google Scholar]
- 51. Kawamoto A., Gwon H. C., Iwaguro H., Yamaguchi J. I., Uchida S., Masuda H., Silver M., Ma H., Kearney M., Isner J. M., Asahara T. (2001) Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation 103, 634–637 [DOI] [PubMed] [Google Scholar]
- 52. Kocher A. A., Schuster M. D., Szabolcs M. J., Takuma S., Burkhoff D., Wang J., Homma S., Edwards N. M., Itescu S. (2001) Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat. Med. 7, 430–436 [DOI] [PubMed] [Google Scholar]
- 53. Fischer-Rasokat U., Assmus B., Seeger F. H., Honold J., Leistner D., Fichtlscherer S., Schächinger V., Tonn T., Martin H., Dimmeler S., Zeiher A. M. (2009) A pilot trial to assess potential effects of selective intracoronary bone marrow-derived progenitor cell infusion in patients with nonischemic dilated cardiomyopathy: final 1-year results of the transplantation of progenitor cells and functional regeneration enhancement pilot trial in patients with nonischemic dilated cardiomyopathy. Circ. Heart Fail. 2, 417–423 [DOI] [PubMed] [Google Scholar]
- 54. Tateishi-Yuyama E., Matsubara H., Murohara T., Ikeda U., Shintani S., Masaki H., Amano K., Kishimoto Y., Yoshimoto K., Akashi H., Shimada K., Iwasaka T., Imaizumi T. (2002) Therapeutic angiogenesis for patients with limb ischaemia by autologous transplantation of bone-marrow cells: a pilot study and a randomised controlled trial. Lancet 360, 427–435 [DOI] [PubMed] [Google Scholar]
- 55. Schächinger V., Assmus B., Britten M. B., Honold J., Lehmann R., Teupe C., Abolmaali N. D., Vogl T. J., Hofmann W. K., Martin H., Dimmeler S., Zeiher A. M. (2004) Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction: final one-year results of the TOPCARE-AMI Trial. J. Am. Coll. Cardiol. 44, 1690–1699 [DOI] [PubMed] [Google Scholar]
- 56. van Ramshorst J., Bax J. J., Beeres S. L., Dibbets-Schneider P., Roes S. D., Stokkel M. P., de Roos A., Fibbe W. E., Zwaginga J. J., Boersma E., Schalij M. J., Atsma D. E. (2009) Intramyocardial bone marrow cell injection for chronic myocardial ischemia: a randomized controlled trial. JAMA 301, 1997–2004 [DOI] [PubMed] [Google Scholar]
- 57. Iwaguro H., Yamaguchi J., Kalka C., Murasawa S., Masuda H., Hayashi S., Silver M., Li T., Isner J. M., Asahara T. (2002) Endothelial progenitor cell vascular endothelial growth factor gene transfer for vascular regeneration. Circulation 105, 732–738 [DOI] [PubMed] [Google Scholar]
- 58. Asahara T., Takahashi T., Masuda H., Kalka C., Chen D., Iwaguro H., Inai Y., Silver M., Isner J. M. (1999) VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 18, 3964–3972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goldstein J. L., Ho Y. K., Basu S. K., Brown M. S. (1979) Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. U.S.A. 76, 333–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stein O., Stein Y. (1980) Bovine aortic endothelial cells display macrophage-like properties towards lipoacetylated 125I-labelled low density lipoprotein. Biochim. Biophys. Acta 620, 631–635 [DOI] [PubMed] [Google Scholar]
- 61. Voyta J. C., Via D. P., Butterfield C. E., Zetter B. R. (1984) Identification and isolation of endothelial cells based on their increased uptake of acetylated-low density lipoprotein. J. Cell Biol. 99, 2034–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Modarai B., Burnand K. G., Sawyer B., Smith A. (2005) Endothelial progenitor cells are recruited into resolving venous thrombi. Circulation 111, 2645–2653 [DOI] [PubMed] [Google Scholar]
- 63. Lee C. M., Hu J. (2013) Cell density during differentiation can alter the phenotype of bone marrow-derived macrophages. Cell Biosci. 3, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rothkamm K., Löbrich M. (2003) Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl. Acad. Sci. U.S.A. 100, 5057–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rzeszowska-Wolny J., Przybyszewski W. M., Widel M. (2009) Ionizing radiation-induced bystander effects, potential targets for modulation of radiotherapy. Eur. J. Pharmacol. 625, 156–164 [DOI] [PubMed] [Google Scholar]
- 66. Ansari R., Gaber M. W., Wang B., Pattillo C. B., Miyamoto C., Kiani M. F. (2007) Anti-TNFA (TNF-α) treatment abrogates radiation-induced changes in vacular density and tissue oxygenation. Radiat. Res. 167, 80–86 [DOI] [PubMed] [Google Scholar]
- 67. Westbrook A. M., Wei B., Hacke K., Xia M., Braun J., Schiestl R. H. (2012) The role of tumour necrosis factor-α and tumour necrosis factor receptor signalling in inflammation-associated systemic genotoxicity. Mutagenesis 27, 77–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Daigle J. L., Hong J. H., Chiang C. S., McBride W. H. (2001) The role of tumor necrosis factor signaling pathways in the response of murine brain to irradiation. Cancer Res. 61, 8859–8865 [PubMed] [Google Scholar]
- 69. Bubici C., Papa S., Dean K., Franzoso G. (2006) Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Oncogene 25, 6731–6748 [DOI] [PubMed] [Google Scholar]
- 70. Wright E. G. (1998) Radiation-induced genomic instability in haemopoietic cells. Int. J. Radiat. Biol. 74, 681–687 [DOI] [PubMed] [Google Scholar]
- 71. Vasa M., Fichtlscherer S., Adler K., Aicher A., Martin H., Zeiher A. M., Dimmeler S. (2001) Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation 103, 2885–2890 [DOI] [PubMed] [Google Scholar]
- 72. Tepper O. M., Galiano R. D., Capla J. M., Kalka C., Gagne P. J., Jacobowitz G. R., Levine J. P., Gurtner G. C. (2002) Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 106, 2781–2786 [DOI] [PubMed] [Google Scholar]
- 73. Fadini G. P., Coracina A., Baesso I., Agostini C., Tiengo A., Avogaro A., de Kreutzenberg S. V. (2006) Peripheral blood CD34+KDR+ endothelial progenitor cells are determinants of subclinical atherosclerosis in a middle-aged general population. Stroke 37, 2277–2282 [DOI] [PubMed] [Google Scholar]
- 74. Hill J. M., Zalos G., Halcox J. P., Schenke W. H., Waclawiw M. A., Quyyumi A. A., Finkel T. (2003) Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. New Engl. J. Med. 348, 593–600 [DOI] [PubMed] [Google Scholar]
- 75. Loomans C. J., de Koning E. J., Staal F. J., Rookmaaker M. B., Verseyden C., de Boer H. C., Verhaar M. C., Braam B., Rabelink T. J., van Zonneveld A. J. (2004) Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes 53, 195–199 [DOI] [PubMed] [Google Scholar]
- 76. Mothersill C., Seymour C. (1997) Medium from irradiated human epithelial cells but not human fibroblasts reduces the clonogenic survival of unirradiated cells. Int. J. Radiat. Biol. 71, 421–427 [DOI] [PubMed] [Google Scholar]
- 77. Mothersill C., Seymour C. B. (1998) Cell-cell contact during γ irradiation is not required to induce a bystander effect in normal human keratinocytes: evidence for release during irradiation of a signal controlling survival into the medium. Radiat. Res. 149, 256–262 [PubMed] [Google Scholar]
- 78. Watson G. E., Lorimore S. A., Macdonald D. A., Wright E. G. (2000) Chromosomal instability in unirradiated cells induced in vivo by a bystander effect of ionizing radiation. Cancer Res. 60, 5608–5611 [PubMed] [Google Scholar]
- 79. Watson G. E., Pocock D. A., Papworth D., Lorimore S. A., Wright E. G. (2001) In vivo chromosomal instability and transmissible aberrations in the progeny of haemopoietic stem cells induced by high- and low-LET radiations. Int. J. Radiat. Biol. 77, 409–417 [DOI] [PubMed] [Google Scholar]
- 80. Todd P., Pecaut M. J., Fleshner M. (1999) Combined effects of space flight factors and radiation on humans. Mutat. Res. 430, 211–219 [DOI] [PubMed] [Google Scholar]
- 81. Mothersill C., Seymour R. J., Seymour C. B. (2006) Increased radiosensitivity in cells of two human cell lines treated with bystander medium from irradiated repair-deficient cells. Radiat. Res. 165, 26–34 [DOI] [PubMed] [Google Scholar]
- 82. Averbeck D. (2010) Non-targeted effects as a paradigm breaking evidence. Mutat. Res. 687, 7–12 [DOI] [PubMed] [Google Scholar]
- 83. Djordjevic B. (2000) Bystander effects: a concept in need of clarification. BioEssays 22, 286–290 [DOI] [PubMed] [Google Scholar]
- 84. Nagasawa H., Little J. B. (1992) Induction of sister chromatid exchanges by extremely low doses of α-particles. Cancer Res. 52, 6394–6396 [PubMed] [Google Scholar]
- 85. Pierce D. A., Preston D. L. (2000) Radiation-related cancer risks at low doses among atomic bomb survivors. Radiat. Res. 154, 178–186 [DOI] [PubMed] [Google Scholar]
- 86. van Os R., Konings A. W., Down J. D. (1993) Compromising effect of low dose-rate total body irradiation on allogeneic bone marrow engraftment. Int. J. Radiat. Biol. 64, 761–770 [DOI] [PubMed] [Google Scholar]
- 87. Seed T. M., Fritz T. E., Tolle D. V., Jackson W. E., 3rd (2002) Hematopoietic responses under protracted exposures to low daily dose γ irradiation. Adv. Space Res. 30, 945–955 [DOI] [PubMed] [Google Scholar]
- 88. Narayanan P. K., LaRue K. E., Goodwin E. H., Lehnert B. E. (1999) α particles induce the production of interleukin-8 by human cells. Radiat. Res. 152, 57–63 [PubMed] [Google Scholar]
- 89. Sethi G., Sung B., Aggarwal B. B. (2008) TNF: a master switch for inflammation to cancer. Front. Biosci. 13, 5094–5107 [DOI] [PubMed] [Google Scholar]
- 90. Rastogi S., Coates P. J., Lorimore S. A., Wright E. G. (2012) Bystander-type effects mediated by long-lived inflammatory signaling in irradiated bone marrow. Radiat. Res. 177, 244–250 [DOI] [PubMed] [Google Scholar]
- 91. Amundson S. A., Do K. T., Vinikoor L. C., Lee R. A., Koch-Paiz C. A., Ahn J., Reimers M., Chen Y., Scudiero D. A., Weinstein J. N., Trent J. M., Bittner M. L., Meltzer P. S., Fornace A. J., Jr. (2008) Integrating global gene expression and radiation survival parameters across the 60 cell lines of the National Cancer Institute Anticancer Drug Screen. Cancer Res. 68, 415–424 [DOI] [PubMed] [Google Scholar]
- 92. Ghandhi S. A., Yaghoubian B., Amundson S. A. (2008) Global gene expression analyses of bystander and α particle irradiated normal human lung fibroblasts: synchronous and differential responses. BMC Med. Genomics 1, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Coward W. R., Okayama Y., Sagara H., Wilson S. J., Holgate S. T., Church M. K. (2002) NF-κB and TNF-α: a positive autocrine loop in human lung mast cells? J. Immunol. 169, 5287–5293 [DOI] [PubMed] [Google Scholar]
- 94. Pahl H. L. (1999) Activators and target genes of Rel/NF-κB transcription factors. Oncogene 18, 6853–6866 [DOI] [PubMed] [Google Scholar]
- 95. Sharma A., Singh K., Almasan A. (2012) Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol 920, 613–626 [DOI] [PubMed] [Google Scholar]
- 96. Valdiglesias V., Giunta S., Fenech M., Neri M., Bonassi S. (2013) γH2AX as a marker of DNA double-strand breaks and genomic instability in human population studies. Mutat. Res. 753, 24–40 [DOI] [PubMed] [Google Scholar]