

Background: Monomeric C-reactive protein (mCRP) may contribute to atherogenesis by inducing endothelial activation.
Results: mCRP induces much weaker endothelial cell (EC) responses from the basolateral than from the apical surfaces of ECs.
Conclusion: Tissue-associated mCRP likely contributes little to EC activation.
Significance: Topological localization is an important factor that defines the contribution of inflammatory mediators to chronic vascular inflammation.
Keywords: Atherosclerosis, Endothelial Dysfunction, Inflammation, Innate Immunity, Pentraxins, C-Reactive Protein, Polarized Activation
Abstract
The activation of endothelial cells (ECs) by monomeric C-reactive protein (mCRP) has been implicated in contributing to atherogenesis. However, the potent proinflammatory actions of mCRP on ECs in vitro appear to be incompatible with the atheroprotective effects of mCRP in a mouse model. Because mCRP is primarily generated within inflamed tissues and is rapidly cleared from the circulation, we tested whether these discrepancies can be explained by topological differences in response to mCRP within blood vessels. In a Transwell culture model, the addition of mCRP to apical (luminal), but not basolateral (abluminal), surfaces of intact human coronary artery EC monolayers evoked a significant up-regulation of MCP-1, IL-8, and IL-6. Such polarized stimulation of mCRP was observed consistently regardless of EC type or experimental conditions (e.g. culture of ECs on filters or extracellular matrix-coated surfaces). Accordingly, we detected enriched lipid raft microdomains, the major surface sensors for mCRP on ECs, in apical membranes, leading to the preferential apical binding of mCRP and activation of ECs through the polarized induction of the phospholipase C, p38 MAPK, and NF-κB signaling pathways. Furthermore, LPS and IL-1β induction of EC activation also exhibited topological dependence, whereas TNF-α did not. Together, these results indicate that tissue-associated mCRP likely contributes little to EC activation. Hence, topological localization is an important, but often overlooked, factor that determines the contribution of mCRP and other proinflammatory mediators to chronic vascular inflammation.
Introduction
C-reactive protein (CRP)3 is a major human acute phase reactant that is highly conserved during evolution (1). The serum level of CRP is closely associated with the risk and prognosis of many chronic inflammatory diseases, including cardiovascular disease (2) and cancer (3). However, it is still hotly debated whether this protein plays any significant role in the underlying pathological processes (1, 3, 4). This may be due to the following reasons. First, the 2-3 orders of nonspecific fluctuations in the serum level of CRP appear to be incompatible with a role of CRP in the fine regulation of chronic inflammation (1). Second, large-scale genetic epidemiologic studies do not support a causal association of CRP with these diseases (5, 6). Third, contradictory findings have been reported in both in vitro and animal models regarding the proinflammatory and atherogenic activities of CRP (7).
Recently, we identified a recurrent promoter mutation and markedly enhanced expression of CRP in human cancers, indicating that CRP may be a potential driver in tumorigenesis and, hence, plays an important role in the regulatory network of inflammation (8). Moreover, continuous efforts from several groups, including ours, have led to the notion that the discordant actions attributed to CRP can be explained by distinct activities and localizations of CRP isoforms as well as their interplay (7, 9, 10). Thus, circulating CRP is secreted by hepatocytes as a homopentamer and primarily exhibits anti-inflammatory activities (9). In inflammatory loci, however, pentameric CRP undergoes sequential conformational changes, including dissociation into subunits (i.e. mCRP) (11–14), followed by reduction of the intrasubunit disulfide bond (15), to express its full proinflammatory potential. Therefore, mCRP is proposed to be the major CRP isoform that functions as a regulator of local inflammation, whereas native pentameric CRP may serve as the precursor of mCRP and a systemic marker of inflammation (7, 9, 10).
Because both reduced and Cys-mutated mCRP are potent activators of endothelial cells (ECs) (15, 16), it appears plausible that mCRP would contribute to the initiation of atherosclerosis, in which EC dysfunction is one of the earliest events (17). However, subcutaneous administration of Cys-mutated mCRP into ApoE−/− mice was found to reduce plaque formation (18). ECs are highly polarized cells with compositionally and functionally distinct plasma membrane domains, i.e. apical and basolateral membranes (19, 20). It is worth noting that, in in vitro experiments, mCRP is usually added to the apical surface of cultured EC. This would mimic the luminal localization of mCRP, whereas the generation of mCRP primarily occurs within inflamed tissues (7, 10), wherein only the basolateral surface of ECs is accessible. Thus, we investigated the impact of distinct localizations of mCRP with respect to EC on EC function and detected profound EC responses only with apically applied, Cys-mutated mCRP.
EXPERIMENTAL PROCEDURES
Reagents
Human native pentameric CRP (purity >99%, purified from ascites) was purchased from The BindingSite (Birmingham, UK, catalog no. BP300.X). Acylated Cys-mutated mCRP was prepared as described previously (15, 16). The two cysteines of this mutant were replaced with alanines. Proteins were dialyzed to remove NaN3 and passed through Detoxi-Gel columns (Thermo Fisher Scientific, Rockford, IL, catalog no. 20344) to remove endotoxin when necessary. 2.5 μg/ml polymyxin B (Sigma, catalog no. P4932) was included in all EC response experiments to exclude possible confounding effects from residual endotoxin.
Clearance of CRP Isoforms from the Circulation
Cys-mutated mCRP or CRP (2.5 mg protein/kg) was injected into male C57BL/6 mice (3–4 mice/group) via the tail veins. Mice were anesthetized to an immobile state by exposure to a cotton applicator with diethyl ether. Blood samples were collected from the retro-orbital sinus into anticoagulation tubes at the indicated time points. After centrifugation for 15 min, the concentrations of CRP/mCRP in the supernatants were determined with a specific ELISA (12). At the end of the experiments, the mice were injected intraperitoneally with pentobarbital sodium (100 mg/kg), followed by cervical dislocation. The experiments conformed to the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health and were conducted according to the protocols approved by the Ethics Committee of Animal Experiments of Lanzhou University.
Transwell Culture of Endothelial Cells
Primary human aortic endothelial cells (HAEC) (Cascade Biologics, Portland, OR; catalog no. C-006-5C; lot nos. 5C0619, 5C1143, and 765093), human umbilical vein endothelial cells (Cascade Biologics, catalog no. C-003-5C, lot no. 4C0218), and human coronary artery endothelial cells (HCAEC) (Cell Applications, San Diego, CA; catalog no. 300-05a; lot nos. 2463, 2827, and 2139; Lifeline Cell Technology, Frederick, MD; catalog no. FC-0032; lot no. 01181) were cultured as described previously (12, 15). For the coating of the Transwell membrane filters, rat tail collagen I (Sigma, 36 μg/filter, catalog no. C7661) in 70% ethanol were air-dried on each filter. Alternatively, filters were incubated with 0.1 mg/ml fibronectin (BD Biosciences, catalog no. 354008) overnight. After washing with PBS, ECs (at passages 4–6) were seeded in the Transwell at a density of 105/filter for 8 h before washing to remove non-adherent cells. The culture media were changed every 2 days and used upon confluence. In addition to staining with crystal violet, the intactness of the EC monolayers was also evaluated by measuring the transendothelial electrical resistance by Millicell-ERS (Millipore, Billerica, MA).
Endothelial Cell Responses
Cys-mutated mCRP or other molecules were added to either the apical or basolateral chambers of the Transwell with an intact EC monolayer for 4 h at 37 °C. IL-8, IL-6, and monocyte chemoattractant protein (MCP) 1 concentrations in conditioned media were determined by commercial ELISA kits (BD Biosciences; catalog nos. 555244, 555220, and 555179) (12, 15). Cytokine mRNA levels were measured by quantitative PCR using SYBR Premix DimerEraser (Takara, Shiga, Japan, catalog no. DRR091C) in a CFX96 real-time thermal cycler (Bio-Rad). The mRNA levels of cytokines were normalized to that of β-actin. The primer sequences used were as follows: MCP-1, 5′-TGGACCACCTGGACAAGCAAACC-3′ (forward) and 5′-TCCGGGAAAGCTAGGGGAAAATAAGT-3′ (reverse); IL-8, 5′-CTCCAAACCTTTCCACCCC-3′ (forward) and 5′-CCACAACCCTCTGCACCCA-3′ (reverse); IL-6, 5′-GAGGAGACTTGCCTGGTGAA-3′ (forward) and 5′-TTGGGTCAGGGGTGGTTATT-3′ (reverse); and β-actin, 5′-CGTGGACATCCGCAAAGAC-3′ (forward) and 5′-CTCAGGAGGAGCAATGATCTTGA-3′ (reverse).
Fluorescence Imaging
For colocalization analysis of Cys-mutated mCRP and lipid rafts, HCAECs were cultured on collagen-coated coverslips to ∼80% confluence. After washing with cold PBS, lipid rafts on cell surfaces were marked with Alexa Fluor 594-cholera toxin subunit B (Invitrogen, catalog no. C22842) at 4 °C for 20 min. Cells were then incubated with 20 μg/ml Cys-mutated mCRP for an additional 30 min, followed by treatment with a Cytofix/Cytoperm fixation/permeabilization kit (BD Biosciences, catalog no. 554714) according to the instructions of the manufacturer. After extensive washing and blocking with 0.5% BSA, cells were incubated sequentially with mCRP mAb 3H12 and a DyLight 488-labeled secondary antibody (Abcam, Cambridge, UK, catalog no. ab96871). Nuclei were counterstained with DAPI (Sigma, 300 nm, 5 min, catalog no. D9542). Samples were examined by FluoView FV1000 confocal microscopy (Olympus, Tokyo, Japan). For analysis of the polarized binding of Cys-mutated mCRP and the distribution of lipid rafts, HCAECs cultured on collagen-coated CM filters were used with a similar protocol for sample preparation.
Flow Cytometry
Confluent HCAEC monolayer on collagen-coated CM filters were incubated with Cys-mutated mCRP (20 μg/ml) in apical or basolateral chambers at 4 °C for 30 min. Cells were incubated with anti-mCRP mAb 3H12 for 30 min and then with a DyLight 650-labeled secondary antibody (Abcam, catalog no. ab96874) for 30 min at room temperature. After counterstaining with 50 μg/ml propidium iodide (PI) for 15 min, suspended cells were examined with a BD LSRFortessa flow cytometer.
Statistical Analysis
Data are presented as mean ± S.E. Statistical analysis was performed by the two-tailed Student's t test or one-way analysis of variance with Tukey's post hoc test. Values of p < 0.05 were considered significant.
RESULTS
Blood-borne, Cys-mutated mCRP Is Cleared Rapidly from the Circulation
Previous studies have demonstrated the presence of mCRP in blood vessels (21) and human atherosclerotic plaques (13, 15). However, mCRP may also be generated in the circulation, for example upon binding of CRP to blood-borne microparticles (22) or activated platelets (11, 13, 23). Although non-reduced mCRP is cleared rapidly from the circulation of mice within minutes (24), it is uncertain whether this also applies to mCRP whose intrasubunit disulfide bond is broken. Thus, we administered Cys-mutated mCRP into mice intravenously and found that only ∼10% of the injected protein remained in blood after 10 min (Fig. 1A). By contrast, over 70% of injected CRP could be recovered from the blood 30 min post-administration. Thus, these results suggest that the majority of mCRP, regardless of the redox state, is tissue-associated in vivo and can only access the basolateral surface of ECs in intact vessels.
FIGURE 1.

Cys-mutated mCRP activates HCAECs in a polarized manner. A, time-dependent clearance of Cys-mutated mCRP from the circulation. CRP or Cys-mutated mCRP was injected intravenously into male C57BL/6 mice (3–4 mice/group) at a dose of 2.5 mg protein/kg. Blood samples were collected at the indicated time points, and the plasma concentrations of proteins were determined with a specific ELISA. The half-life of Cys-mutated mCRP in blood was calculated to be less than 2 min. B, schematic of the Transwell EC culture system for polarized EC activation. Confluent monolayers of HCAECs and HAECs on CM filters were visualized by crystal violet staining. C, electron microscopy imaging of Cys-mutated mCRP. D, cytokine expression induced by apical and basolateral stimulation of Cys-mutated mCRP. HCAEC monolayers on collagen-coated CM filters were incubated with 20 μg/ml Cys-mutated mCRP added to either the apical or basolateral chambers for 4 h at 37 °C. HCAEC monolayers without mCRP treatment served as controls. mRNA levels of MCP-1, IL-8, and IL-6 were quantified by quantitative PCR (n = 4–8). *, p < 0.05. Apical addition of Cys-mutated mCRP resulted in much stronger cytokine up-regulation in HCAECs over basolateral addition.
Polarized Induction of EC Responses by Cys-mutated mCRP
We next examined whether the effects of Cys-mutated mCRP on ECs depend on topological localizations by using the Transwell model, in which EC monolayers were cultured on porous membrane filters with their apical and basolateral surfaces exposed to different chambers (Fig. 1B). The pore size of the filters (0.4 μm) does not hinder the interaction of mCRP with the basolateral surface of ECs (Fig. 1C). As expected, the addition of Cys-mutated mCRP to the apical chamber resulted in a significant up-regulation of MCP-1, IL-8, and IL-6 in HCAECs after 4 h of incubation. By contrast, much weaker responses were evoked by the addition of Cys-mutated mCRP to the basolateral surface (Fig. 1D). These polarized effects of Cys-mutated mCRP were also observed with HAEC and human umbilical vein endothelial cell monolayers cultured on different types of filters with or without coating with either collagen or fibronectin (Fig. 2). Control experiments showed that prior treatment of Cys-mutated mCRP with proteinase K, which would degrade mCRP but leave LPS intact, abrogated HCAEC responses to Cys-mutated mCRP (data not shown), thus demonstrating that the observed effects were due to mCRP instead of LPS contamination. We also prepared reduced mCRP by treating native CRP with urea/DTT and confirmed that this mCRP preparation activated HCAECs and HAECs only when applied apically (data not shown).
FIGURE 2.

The polarized EC activation by Cys-mutated mCRP does not depend on EC type or culture conditions. A, the filter types used in the experiments. B–F, Cys-mutated mCRP induced polarized responses in HCAEC monolayers on collagen-coated CM filters (n = 6–8) (B); human umbilical vein endothelial cell (HUVEC) monolayers on collagen-coated PET filters (n = 3) (C); and HAEC monolayers on fibronectin-coated HA filters (n = 3) (D), collagen-coated CM filter (n = 4) (E), and collagen-coated PET filters (n = 3–4) (F). Following incubation for 4 h with 20 μg/ml Cys-mutated mCRP added apically or basolaterally, conditioned culture media from both chambers were collected, and MCP-1 and IL-8 levels were determined with specific ELISAs. EC monolayers without mCRP treatment served as controls. *, p < 0.05; **, p < 0.01.
Reduced responses to basolaterally added Cys-mutated mCRP to activate ECs cannot be attributed to the small difference in the extent of surface accessibility because up to 80% of the filter area is decorated by pores, according to the specifications of the manufacturer. Moreover, anisomycin induced comparable HCAEC responses regardless of whether it was added to the apical or basolateral chambers (Fig. 3A). Because anisomycin is a small molecule that can cross cell membranes freely and potently activates MAPK, the non-polarized stimulation of ECs by anisomycin verifies the integrity of our experimental system and indicates that the observed polarized effects of Cys-mutated mCRP were not due to artifacts. Further experiments revealed a similar topological dependence of EC activation by LPS (Fig. 3B) and IL-1β (Fig. 3C) but not by TNF-α (Fig. 3D). Taken together, we conclude that polarized EC stimulation is not a universal phenomenon but, rather, a specific one for selective mediators.
FIGURE 3.

Polarized and non-polarized activation of HCAECs by other inflammatory mediators. HCAEC monolayers on CM filters were activated for 4 h by apically or basolaterally added anisomycin (100 ng/ml, n = 3) (A), LPS (100 ng/ml, n = 5) (B), IL-1β (10 ng/ml, n = 5) (C), or TNF-α (1 ng/ml, n = 3) (D). HCAEC monolayers without stimulation served as controls. The expression of MCP-1, IL-8, and IL-6 mRNA were determined by quantitative PCR. *, p < 0.05.
Apically Enriched Lipid Rafts on ECs Mediate the Polarized Effects of Cys-mutated mCRP
We have identified cholesterol-enriched lipid raft microdomains as the major cell surface sensors that mediate the actions of apically added reduced or Cys-mutated mCRP on ECs (15, 25). Accordingly, Cys-mutated mCRP colocalized with lipid rafts marked with cholera toxin subunit B on the surface of HCAECs (Fig. 4A), and depletion of cholesterol resulted in diminished HCAEC responses to both apically and basolaterally added Cys-mutated mCRP (Fig. 4B). These findings indicate that the actions of Cys-mutated mCRP on both apical and basolateral surfaces depend on intact lipid raft microdomains. By contrast, disruption of lipid rafts did not affect the polarized EC activation by LPS (Fig. 4C), likely because of the unusual intracellular localization of TLR4 in HCAECs (26). Of note, monocytes express TLR4 on their surfaces (26, 27), and, consistent with a previous report (27), disruption of lipid rafts fully inhibited the effects of LPS on monocytic cell lines (data not shown). Further experiments showed that lipid rafts were highly enriched in the apical membranes of HCAECs (Fig. 4D) and that the apical binding of Cys-mutated mCRP was over 4-fold stronger than the basolateral binding, as assessed by both FACS analysis and fluorescence imaging (Fig. 5). Therefore, the preferential apical binding and activation of Cys-mutated mCRP are most likely due to the polarized distribution of lipid rafts on ECs.
FIGURE 4.
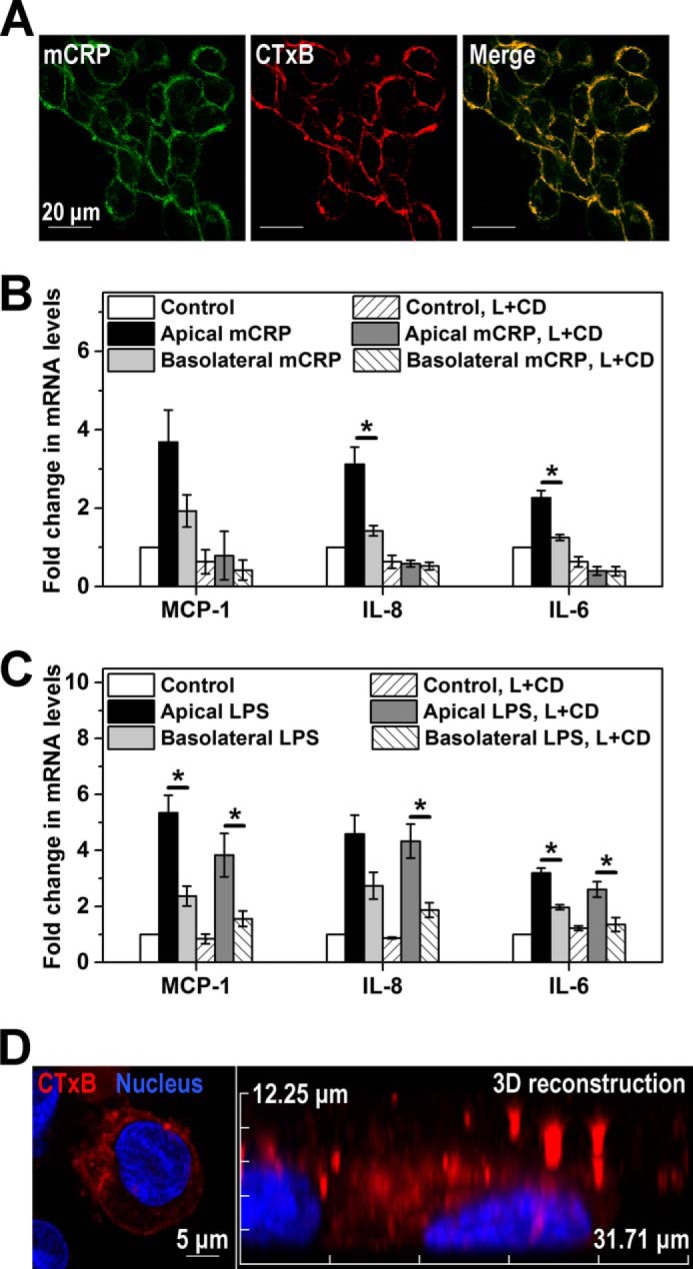
Lipid rafts are mCRP sensors and are enriched in the apical membranes of HCAECs. A, confocal imaging of the colocalization of Cys-mutated mCRP and membrane lipid rafts on HCAECs. Cells were cultured on coverslips and incubated sequentially with the raft marker, Alexa Fluor 594-cholera toxin subunit B, and Cys-mutated mCRP. The latter was visualized by mAb 3H12 and a DyLight 488-labeled secondary antibody. Nuclei were counterstained with DAPI. B and C, cholesterol depletion abrogated HCAEC responses to Cys-mutated mCRP (B) but did not affect the polarized actions of LPS (C). HCAEC cells were pretreated with a combination of lovastatin (L) and methyl-cyclodextrin (CD), followed by stimulation with Cys-mutated mCRP. HCAEC monolayers without stimulation served as controls. MCP-1, IL-8, and IL-6 mRNA expression were quantified with quantitative PCR (n = 3). D, confocal imaging of the asymmetric distribution of lipid rafts on HCAECs cultured on collagen-coated CM filters. Three-dimensional models of cholera toxin subunit B-labeled cells were constructed by x-y sections with a 0.5-μm z axis step. One representative x-z section of a three-dimensional cell model is shown.
FIGURE 5.

Preferential interaction of Cys-mutated mCRP with the apical membranes of HCAECs. A, confluent HCAEC monolayers on CM filters were incubated with Cys-mutated mCRP (20 μg/ml) added to the apical or basolateral chamber at 4 °C for 30 min. mCRP binding was detected with 3H12 mAb and a DyLight 650-labeled secondary antibody. Cells were detached, counterstained with propidium iodide, and analyzed with flow cytometry. HCAEC monolayers without binding served as controls. B, confluent HCAEC monolayers were incubated with FITC-Cys-mutated mCRP (20 μg/ml) added to the apical or basolateral chamber for 30 min at 37 °C in the presence of 10 mm NaN3. Following extensive washing, cells were lysed, and fluorescence intensity was measured (n = 5). *, p < 0.05. C, a representative x-z section of a three-dimensional HCAEC model bound with Cys-mutated mCRP. The model was constructed with a series of 1-μm-thick sections along the z axis.
Polarized Activation of the PLC and p38 MAPK Signaling Pathways by Cys-mutated mCRP
The EC responses to Cys-mutated mCRP have been reported to be partly dependent on p38 MAPK signaling (16). To further dissect the intracellular pathways evoked by Cys-mutated mCRP, we first activated HCAECs cultured in 96-well plates with Cys-mutated mCRP in the presence of specific pharmacological inhibitors (Fig. 6A). The results showed that the effects of Cys-mutated mCRP were essentially abrogated by the NF-κB inhibitor Bay11-7082 and partially suppressed by the p38 MAPK inhibitor SB20385 and the phospholipase C (PLC) inhibitor U73122. By contrast, blockade of MEK1/2 with U0126, of c-Raf-1 with GW5074, and of Syk with piceatannol was without effect. Concurrent inhibition of PLC and p38 MAPK, however, fully blocked HCAEC responses to Cys-mutated mCRP. Together, these results suggest that Cys-mutated mCRP induces parallel activation of PLC and p38 MAPK signaling, converging on the activation of NF-κB.
FIGURE 6.

The PLC/p38 MAPK → NF-κB signaling axis mediates the polarized activation of HCAECs by Cys-mutated mCRP. A, HCAECs were cultured in 96-well plates and pretreated with pharmacological inhibitors, followed by stimulation for 4 h with Cys-mutated mCRP. MCP-1 and IL-8 release in culture supernatants was determined with specific ELISAs and normalized to their respective controls with only inhibitor treatment. The inhibitors used included the NF-κB inhibitor Bay11-7082 (10 μm, n = 5), the PLC inhibitor U73122 (10 μm, n = 8), the p38 MAPK inhibitor SB20358 (20 μm, n = 6), the Syk inhibitor piceatannol (10 μm, n = 6), the MEK inhibitor U0126 (10 μm, n = 6), and the Raf1 inhibitor GW5074 (20 μm, n = 6). Suppression of NF-κB, PLC, and p38 MAPK abrogated or significantly reduced HCAEC responses to Cys-mutated mCRP. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus control. B–D, HCAEC monolayers cultured on collagen-coated CM filters were challenged by Cys-mutated mCRP in the presence of absence of one of the indicated inhibitors (n = 3–4). The effects of apically added Cys-mutated mCRP were inhibited completely by the NF-κB inhibitor Bay11-7082 (B), affected mildly by the p38 MAPK inhibitor SB20358 (C), and inhibited significantly by the PLC inhibitor U73122 (D). The basolateral activation, however, was abrogated by either Bay11-7082 or U73122 but not affected by SB20358. *, p < 0.05; **, p < 0.01.
In the Transwell system, the responses of HCAECs to Cys-mutated mCRP were still exclusively dependent on downstream NF-κB signaling because Bay11-7082 completely inhibited the effects of Cys-mutated mCRP, regardless of whether it was added to the apical or basolateral chambers (Fig. 6B). By contrast, the contribution of p38 MAPK appeared to be much less evident in this model, with moderate or little suppression by SB20385 of the HCAEC responses to the apical and basolateral addition of Cys-mutated mCRP, respectively (Fig. 6C). It is worth noting that the PLC inhibitor U73122 partially reduced the effects of apically applied Cys-mutated mCRP but completely inhibited those of basolaterally applied mCRP (Fig. 6D). These would indicate that the Cys-mutated mCRP signals through the phospholipase C → NF-κB axis from the basolateral surface instead of induction of the phospholipase C/p38 MAPK and NF-κB pathway from the apical surface (Fig. 7).
FIGURE 7.

Schematic of the polarized EC activation by reduced mCRP. Reduced mCRP interacts with ECs through lipid raft microdomains on both apical and basolateral surfaces. The enrichment of lipid rafts in apical membranes would, thus, dictate the preferential apical activation of ECs by reduced mCRP through polarized induction of the PLC, p38 MAPK, and NF-κB signaling pathways. PLC may activate NF-κB through PKC. Of note, mCRP is generated primarily in inflamed tissues and would, therefore, access only the basolateral surfaces of ECs. Significant accumulation of circulating mCRP may, however, occur in acute inflammation, where it could amplify the inflammatory response via apical activation of ECs.
DISCUSSION
The apical and basolateral surfaces of ECs face blood and tissues, respectively (19, 20). ECs respond strongly to apical but weakly to basolateral stimulation of Cys-mutated or reduced mCRP, suggesting that tissue-resident mCRP may be inefficient in activating ECs. Because mCRP would be generated primarily within inflamed tissues (13–15, 28–31) and the half-life of blood-borne mCRP is rather short (Ref. 24 and this study), it appears conceivable that circulating mCRP does not play a significant role in direct induction of EC dysfunction, at least in the setting of chronic local inflammation (e.g. atherogenesis). Moreover, tissue-associated mCRP, irrespective of its redox state (15), may exert antiatherosclerotic actions, in particular at initial stages of plaque development, by promoting opsonic activation of the early complement pathway and non-inflammatory clearance of damaged cells (14, 28, 32, 33) and by inhibiting foam cell formation (34, 35). Overall, these would underlie the seemingly counterintuitive findings of the in vivo atheroprotective effects of Cys-mutated mCRP (18).
The polarized response of ECs to Cys-mutated mCRP may be due to the distinct cellular sensors engaged. However, our results are incompatible with such a scenario because Cys-mutated mCRP activates ECs through cholesterol-enriched lipid raft microdomains on both apical and basolateral membranes. By contrast, the asymmetric distribution of lipid rafts on EC surfaces seems to be the key determinant. Lipid rafts are enriched in the apical membranes but depleted in the basolateral membranes, tightly correlating with the preferential apical binding of Cys-mutated mCRP. Consequently, this may lead to differential induction of intracellular pathways as the result of distinct interaction strength and polarized localization of receptors (e.g. CD16 (16)) or signaling components. Indeed, Cys-mutated mCRP signals through p38 MAPK and PLC to NF-κB from the apical surfaces but only activates the PLC → NF-κB pathway from the basolateral surfaces (Fig. 7). Therefore, the absence of the concurrent p38 MAPK signaling may account for the weak EC responses to basolaterally applied Cys-mutated mCRP.
Besides Cys-mutated mCRP, LPS and IL-1β, but not TNF-α, also exhibited polarized EC stimulation. These data highlight the fact that topological localization of certain mediators within the vessels is an important factor that determines their contributions to vascular inflammation and, hence, may to some extent account for the inconsistencies between in vitro and in vivo findings. Our results identify spatial localization as a novel determinant for the highly context-dependent actions of CRP isoforms within the vessels. Tissue-resident mCRP may primarily act as a pattern recognition molecule to facilitate non-inflammatory complement activation (14, 15, 32) and macrophage clearance (34, 35) within vessels undergoing chronic inflammation. However, in acute vascular inflammation, mCRP might be present in the circulation for an extended period of time as a consequence of endothelial damage, enhanced conversion, or transportation by microparticles (22) or activated platelets (11, 13, 23). Ultimately, these would lead to amplification of the inflammatory responses by further activating ECs (15, 16), platelets (36), and neutrophils (37). In summary, our data reveals an additional layer of regulation of the actions of mCRP on ECs by topological localization, consistent with the apical enrichment of lipid rafts, the primary endothelial sensors of mCRP.
Acknowledgments
We thank Jing Zhao and Li-Ping Guan for technical assistance.
This work was supported by Ministry of Science and Technology of China grant 2011CB910500; by National Natural Science Foundation of China grants 30930024, 31222015, 31270813, 31170696; and by Ministry of Education of China grants PCSIRT: IRT1137, 121108, 20100211110006, and lzujbky-2010-k03.
- CRP
- C-reactive protein
- mCRP
- monomeric C-reactive protein
- EC
- endothelial cell
- HAEC
- human aortic endothelial cell
- HCAEC
- human coronary artery endothelial cell
- MCP
- monocyte chemoattractant protein
- PLC
- phospholipase C.
REFERENCES
- 1. Pepys M. B., Hirschfield G. M. (2003) C-reactive protein: a critical update. J. Clin. Invest. 111, 1805–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Singh S. K., Suresh M. V., Voleti B., Agrawal A. (2008) The connection between C-reactive protein and atherosclerosis. Ann. Med. 40, 110–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Allin K. H., Nordestgaard B. G. (2011) Elevated C-reactive protein in the diagnosis, prognosis, and cause of cancer. Crit. Rev. Clin. Lab. Sci. 48, 155–170 [DOI] [PubMed] [Google Scholar]
- 4. Yousuf O., Mohanty B. D., Martin S. S., Joshi P. H., Blaha M. J., Nasir K., Blumenthal R. S., Budoff M. J. (2013) High-sensitivity C-reactive protein and cardiovascular disease: a resolute belief or an elusive link? J. Am. Coll. Cardiol. 62, 397–408 [DOI] [PubMed] [Google Scholar]
- 5. Allin K. H., Nordestgaard B. G., Zacho J., Tybjaerg-Hansen A., Bojesen S. E. (2010) C-reactive protein and the risk of cancer: a Mendelian randomization study. J. Natl. Cancer Inst. 102, 202–206 [DOI] [PubMed] [Google Scholar]
- 6. Zacho J., Tybjaerg-Hansen A., Jensen J. S., Grande P., Sillesen H., Nordestgaard B. G. (2008) Genetically elevated C-reactive protein and ischemic vascular disease. N. Engl. J. Med. 359, 1897–1908 [DOI] [PubMed] [Google Scholar]
- 7. Ma X., Ji S. R., Wu Y. (2013) Regulated conformation changes in C-reactive protein orchestrate its role in atherogenesis. Chinese Sci. Bull. 58, 1642–1649 [Google Scholar]
- 8. Wang M.-Y., Zhou H.-H., Zhang S.-C., Hui F., Zhu W., Su H.-X., Guo H.-Y., Li X.-W., Ji S.-R., Wu Y. (2014) Recurrent mutations at C-reactive protein promoter SNP position −286 in human cancers. Cell Res. 24, 505–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Filep J. G. (2009) Platelets affect the structure and function of C-reactive protein. Circ. Res. 105, 109–111 [DOI] [PubMed] [Google Scholar]
- 10. Eisenhardt S. U., Thiele J. R., Bannasch H., Stark G. B., Peter K. (2009) C-reactive protein: how conformational changes influence inflammatory properties. Cell Cycle 8, 3885–3892 [DOI] [PubMed] [Google Scholar]
- 11. Molins B., Peña E., de la Torre R., Badimon L. (2011) Monomeric C-reactive protein is prothrombotic and dissociates from circulating pentameric C-reactive protein on adhered activated platelets under flow. Cardiovasc. Res. 92, 328–337 [DOI] [PubMed] [Google Scholar]
- 12. Ji S. R., Wu Y., Zhu L., Potempa L. A., Sheng F. L., Lu W., Zhao J. (2007) Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRP(m). FASEB J. 21, 284–294 [DOI] [PubMed] [Google Scholar]
- 13. Eisenhardt S. U., Habersberger J., Murphy A., Chen Y. C., Woollard K. J., Bassler N., Qian H., von Zur Muhlen C., Hagemeyer C. E., Ahrens I., Chin-Dusting J., Bobik A., Peter K. (2009) Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ. Res. 105, 128–137 [DOI] [PubMed] [Google Scholar]
- 14. Mihlan M., Blom A. M., Kupreishvili K., Lauer N., Stelzner K., Bergström F., Niessen H. W., Zipfel P. F. (2011) Monomeric C-reactive protein modulates classic complement activation on necrotic cells. FASEB J. 25, 4198–4210 [DOI] [PubMed] [Google Scholar]
- 15. Wang M. Y., Ji S. R., Bai C. J., El Kebir D., Li H. Y., Shi J. M., Zhu W., Costantino S., Zhou H. H., Potempa L. A., Zhao J., Filep J. G., Wu Y. (2011) A redox switch in C-reactive protein modulates activation of endothelial cells. FASEB J. 25, 3186–3196 [DOI] [PubMed] [Google Scholar]
- 16. Khreiss T., József L., Potempa L. A., Filep J. G. (2004) Conformational rearrangement in C-reactive protein is required for proinflammatory actions on human endothelial cells. Circulation 109, 2016–2022 [DOI] [PubMed] [Google Scholar]
- 17. Ross R. (1999) Atherosclerosis: an inflammatory disease. N. Engl. J. Med. 340, 115–126 [DOI] [PubMed] [Google Scholar]
- 18. Schwedler S. B., Amann K., Wernicke K., Krebs A., Nauck M., Wanner C., Potempa L. A., Galle J. (2005) Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation 112, 1016–1023 [DOI] [PubMed] [Google Scholar]
- 19. Muller W. A., Gimbrone M. A., Jr. (1986) Plasmalemmal proteins of cultured vascular endothelial cells exhibit apical-basal polarity: analysis by surface-selective iodination. J. Cell Biol. 103, 2389–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mehta D., Malik A. B. (2006) Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279–367 [DOI] [PubMed] [Google Scholar]
- 21. Diehl E. E., Haines G. K., 3rd, Radosevich J. A., Potempa L. A. (2000) Immunohistochemical localization of modified C-reactive protein antigen in normal vascular tissue. Am. J. Med. Sci. 319, 79–83 [DOI] [PubMed] [Google Scholar]
- 22. Habersberger J., Strang F., Scheichl A., Htun N., Bassler N., Merivirta R. M., Diehl P., Krippner G., Meikle P., Eisenhardt S. U., Meredith I., Peter K. (2012) Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc. Res. 96, 64–72 [DOI] [PubMed] [Google Scholar]
- 23. de la Torre R., Peña E., Vilahur G., Slevin M., Badimon L. (2013) Monomerization of C-reactive protein requires glycoprotein IIb-IIIa activation: pentraxins and platelet deposition. J. Thromb. Haemost. 11, 2048–2058 [DOI] [PubMed] [Google Scholar]
- 24. Motie M., Schaul K. W., Potempa L. A. (1998) Biodistribution and clearance of 125I-labeled C-reactive protein and 125I-labeled modified C-reactive protein in CD-1 mice. Drug Metab. Dispos. 26, 977–981 [PubMed] [Google Scholar]
- 25. Ji S. R., Ma L., Bai C. J., Shi J. M., Li H. Y., Potempa L. A., Filep J. G., Zhao J., Wu Y. (2009) Monomeric C-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB J. 23, 1806–1816 [DOI] [PubMed] [Google Scholar]
- 26. Dunzendorfer S., Lee H. K., Soldau K., Tobias P. S. (2004) Toll-like receptor 4 functions intracellularly in human coronary artery endothelial cells: roles of LBP and sCD14 in mediating LPS responses. FASEB J. 18, 1117–1119 [DOI] [PubMed] [Google Scholar]
- 27. Triantafilou M., Miyake K., Golenbock D. T., Triantafilou K. (2002) Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J. Cell Sci. 115, 2603–2611 [DOI] [PubMed] [Google Scholar]
- 28. Lauer N., Mihlan M., Hartmann A., Schlötzer-Schrehardt U., Keilhauer C., Scholl H. P., Charbel Issa P., Holz F., Weber B. H., Skerka C., Zipfel P. F. (2011) Complement regulation at necrotic cell lesions is impaired by the age-related macular degeneration-associated factor-H His402 risk variant. J. Immunol. 187, 4374–4383 [DOI] [PubMed] [Google Scholar]
- 29. Schwedler S. B., Guderian F., Dämmrich J., Potempa L. A., Wanner C. (2003) Tubular staining of modified C-reactive protein in diabetic chronic kidney disease. Nephrol. Dial. Transplant. 18, 2300–2307 [DOI] [PubMed] [Google Scholar]
- 30. Slevin M., Matou-Nasri S., Turu M., Luque A., Rovira N., Badimon L., Boluda S., Potempa L., Sanfeliu C., de Vera N., Krupinski J. (2010) Modified C-reactive protein is expressed by stroke neovessels and is a potent activator of angiogenesis in vitro. Brain Pathol. 20, 151–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Strang F., Scheichl A., Chen Y. C., Wang X., Htun N. M., Bassler N., Eisenhardt S. U., Habersberger J., Peter K. (2012) Amyloid plaques dissociate pentameric to monomeric C-reactive protein: a novel pathomechanism driving cortical inflammation in Alzheimer's disease? Brain Pathol. 22, 337–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ji S. R., Wu Y., Potempa L. A., Liang Y. H., Zhao J. (2006) Effect of modified C-reactive protein on complement activation: a possible complement regulatory role of modified or monomeric C-reactive protein in atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 26, 935–941 [DOI] [PubMed] [Google Scholar]
- 33. Mihlan M., Stippa S., Józsi M., Zipfel P. F. (2009) Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting factor H. Cell Death Differ. 16, 1630–1640 [DOI] [PubMed] [Google Scholar]
- 34. Schwedler S. B., Hansen-Hagge T., Reichert M., Schmiedeke D., Schneider R., Galle J., Potempa L. A., Wanner C., Filep J. G. (2009) Monomeric C-reactive protein decreases acetylated LDL uptake in human endothelial cells. Clin. Chem. 55, 1728–1731 [DOI] [PubMed] [Google Scholar]
- 35. Ji S. R., Wu Y., Potempa L. A., Qiu Q., Zhao J. (2006) Interactions of C-reactive protein with low-density lipoproteins: implications for an active role of modified C-reactive protein in atherosclerosis. Int. J. Biochem. Cell Biol. 38, 648–661 [DOI] [PubMed] [Google Scholar]
- 36. Molins B., Peña E., Vilahur G., Mendieta C., Slevin M., Badimon L. (2008) C-Reactive protein isoforms differ in their effects on thrombus growth. Arterioscler. Thromb. Vasc. Biol. 28, 2239–2246 [DOI] [PubMed] [Google Scholar]
- 37. Khreiss T., József L., Potempa L. A., Filep J. G. (2005) Loss of pentameric symmetry in C-reactive protein induces interleukin-8 secretion through peroxynitrite signaling in human neutrophils. Circ. Res. 97, 690–697 [DOI] [PubMed] [Google Scholar]