

Background: Genetic defects affecting subunits of protein complexes are presumed to generate identical diseases in mammals.
Results: Two mouse mutants in genes belonging to the BLOC-1 complex have divergent brain and pigmentation phenotypes.
Conclusion: Genetic defects affecting subunits of a complex manifest by partially overlapping clinical features.
Significance: Disease resulting from mutations in protein complexes may generate a wide range of clinically presentations.
Keywords: Adaptor Proteins, Mouse Genetics, Protein Complexes, Schizophrenia, Synapses
Abstract
Post-mortem analysis has revealed reduced levels of the protein dysbindin in the brains of those suffering from the neurodevelopmental disorder schizophrenia. Consequently, mechanisms controlling the cellular levels of dysbindin and its interacting partners may participate in neurodevelopmental processes impaired in that disorder. To address this question, we studied loss of function mutations in the genes encoding dysbindin and its interacting BLOC-1 subunits. We focused on BLOC-1 mutants affecting synapse composition and function in addition to their established systemic pigmentation, hematological, and lung phenotypes. We tested phenotypic homogeneity and gene dosage effects in the mouse null alleles muted (Bloc1s5mu/mu) and dysbindin (Bloc1s8sdy/sdy). Transcripts of NMDA receptor subunits and GABAergic interneuron markers, as well as expression of BLOC-1 subunit gene products, were affected differently in the brains of Bloc1s5mu/mu and Bloc1s8sdy/sdy mice. Unlike Bloc1s8sdy/sdy, elimination of one or two copies of Bloc1s5 generated indistinguishable pallidin transcript phenotypes. We conclude that monogenic mutations abrogating the expression of a protein complex subunit differentially affect the expression of other complex transcripts and polypeptides as well as their downstream effectors. We propose that the genetic disruption of different subunits of protein complexes and combinations thereof diversifies phenotypic presentation of pathway deficiencies, contributing to the wide phenotypic spectrum and complexity of neurodevelopmental disorders.
Introduction
Genetic polymorphisms in the gene encoding dysbindin are risk factors for schizophrenia onset and associate with cognitive and neuroanatomical differences in normal individuals (1–13). Dysbindin (Bloc1s8) is a subunit of the cytosolic hetero-octamer referred to as BLOC-1 (the biogenesis of lysosome-related organelles complex 1). This complex consists of Bloc1s1–8 subunits. Analysis of dysbindin and its closely interacting BLOC-1 subunit mutants reveals a common set of autosomal recessive phenotypes in vertebrates and invertebrates. These phenotypes result from defective trafficking to lysosome-related organelles affecting systemic processes as well as pre- and post-synaptic neuronal compartments (13–16). For example, the sandy mouse null allele affecting dysbindin polypeptide expression, Bloc1s8sdy/sdy, impairs synaptic vesicle composition and function including glutamatergic and GABA-dependent neurotransmission (12, 13, 17–20). Similarly, dysbindin and Bloc1s1 mutants in Drosophila exhibit impaired neurotransmission, behavior and presynaptically abrogated glutamatergic synaptic homeostasis (21–24). Neuronal phenotypes have not been systematically explored in null mutations affecting other BLOC-1 complex subunits. In contrast, pigment dilution, pulmonary fibrosis, and bleeding diathesis, which constitute the core recessive systemic phenotypes of BLOC-1 mutations in mammals, are common to Bloc1s8sdy/sdy and four alleles affecting the expression of Bloc1s4 (reduced pigmentation), Bloc1s5 (muted), Bloc1s6 (pallid), and Bloc1s7 (cappuccino), respectively (14, 16, 25–30). The commonality of systemic phenotypes among these five mouse mutants is attributed to the idea long held by us and others in the field that each of these mutations exerts equal effects on BLOC-1 architecture, function, and downstream effectors (12, 14, 31).
Here we demonstrate that Bloc1s5 and Bloc1s8 mutations exert differential effects on BLOC-1 complex function and downstream effectors. We explored the type and magnitude of neuronal phenotypes associated with single and double copy Bloc1s5 muted or Bloc1s8 sandy null alleles present in mice of identical genetic background. We identified neuronal transcriptional phenotypes whose quality and/or magnitude differ between these alleles. Transcriptional phenotypes were sensitive to the genetic dosage of mutant alleles. Our results support the concept that genetic mutations in dysbindin and its interacting BLOC-1 subunits generate only partially overlapping neuronal phenotypes in neurotransmitter systems implicated in the pathogenesis of schizophrenia.
MATERIALS AND METHODS
Reagents
Mouse anti-pallidin was a gift from Dr. Esteban Dell'Angelica (UCLA, Los Angeles, CA) (29). Rabbit anti-VAMP-2 and VGAT were purchased from Synaptic Systems (Göttingen, Germany). VAMP7 monoclonal antibody was a generous gift of Dr. Andrew Peden (University of Sheffield, UK). Dysbindin 1A and 1C were detected with the antibody PA3111 (32) Mouse mutants have been previously described (17, 33, 34). Bloc1s5mu/mu mice in CHMU background (CHMU/LeJ, stock number 000293) were backcrossed by at least six generations with C57B6 mice obtained from The Jackson Laboratory (Bar Harbor, ME). Bloc1s5mu/mu were a gift of Dr. R. Swank (Roswell Park Cancer Institute, Buffalo, NY). Bloc1s8sdy/sdy (sandy) and Bloc1s6pa/pa were also in C57B6 genetic background. Sandy mice were previously described (35). Mouse genotyping was performed by PCR of genomic DNA with the primers forward muted (ctatgaagagtgacgagctgt) and reverse muted (agcagtaggattctctcagg).
Mouse and Human Subjects
All mice were bred in-house following institutional animal care and use committee-approved protocols. Human post-mortem tissue derived from samples of U.S. citizens autopsied at the Hospital of the University of Pennsylvania as approved by the institutional review board at that university. Autopsy consent from next of kin or legal guardian was obtained in all cases. For most cases, consent was granted in writing before death and always confirmed after death. Ethics committee at the University of Pennsylvania approved the consent procedures. To keep post-mortem delays to a minimum when written consent had not been obtained before death, verbal consent was obtained as witnessed by a third party and documented by the physician making the request. Written records of the consent for autopsy were archived. These procedures for written and verbal consent are standard medical practice in the United States.
Brain Sections, Immunohistochemistry, and Microscopy
Detailed procedures for mouse tissue preparation, indirect immunofluorescence microscopy, and quantification procedures were described in our previous work (17, 33, 36). Briefly, brains were obtained from mice 6–8 weeks postnatal. Animals were anesthetized with ketamine and then transcardially perfused with Ringer's solution followed by fixative (4% paraformaldehyde with 0.1% glutaraldehyde). Brains were post-fixed in 4% paraformaldehyde for 12–18 h followed by sectioning on a vibratome into 60-μm-thick sections and stored in antifreeze (0.1 m sodium phosphate monobasic, 0.1 m sodium phosphate dibasic heptahydrate, 30% ethylene glycol, 30% glycerol) at −20 °C. Vibrotome sections containing the hippocampus were incubated in 1% sodium borohydride. Tissue was blocked for 60 min (5% normal horse serum, 1% BSA, and 0.3% Triton X-100). Brain sections were incubated in primary antibody overnight (anti-Pallidin 1:200 with anti-Vamp2 1:000 V2, 1% normal horse serum and 1% BSA). The following day, tissue was incubated in a secondary antibody for 60 min (1% normal horse serum and 1% BSA, 1:500 anti-mouse 488 and anti-rabbit 568) (Invitrogen). Finally, brain sections were incubated for 30 min in cupric sulfate (3.854 w/v ammonium acetate, 1.596 w/v cupric sulfate, pH 5). Tissue sections were mounted on slides with Vectashield (Vector Laboratories). Confocal microscopy of immunofluorescent samples was performed with an Anxiovert 100 m (Carl Zeiss) coupled to an argon laser, a HeNe1 laser, and a titanium sapphire laser. Z-stacks were acquired using Plan Apochromat 20×/0.5 dry objective. The emission filters used for fluorescence imaging were BP 505–530 and LP 560. The images were acquired with ZEN software (Carl Zeiss).
Melanin Measurements Procedure
Procedures were performed according to Hoyle et al. (37). Briefly, a solution containing a (9:1 ratio of Solune 350 (PerkinElmer Life Sciences) and water was added to hair samples at a ratio of 250 μl/mg of hair. Samples were vortexed for 1 min, heated to 95 °C for 30 min, cooled, vortexed for 2 min, heated to 95 °C for 15 min and then brought to room temperature. Next, 400 μl of the sample were transferred to a tube containing 600 μl of Solune 350/water mixture. Samples were again vortexed and heated to 95 °C for 15 min. Then they were centrifuged for 10 min at 13,000 × g to remove debris. Absorbance at 500 nm was measured for each sample. The standard curve was obtained by dissolving purified melanin from Sepia officinalis (Sigma-Aldrich). Sepia melanin was processed as hair samples. Dilutions were made from the stock solution using Solune 350/water solution.
Quantitative Real Time PCR
Control and mutant cortical and hippocampal regions were dissected from young adult animals between postnatal days 42 and 52 sacrificed by CO2 narcosis. Tissue was flash frozen and TRIzol-extracted (Invitrogen), and isolated RNA was reverse transcribed into cDNA using SuperScript III first strand synthesis (Invitrogen). PCR amplifications were performed on a LightCycler480 real time plate reader using LightCycler 480 SYBR Green reagents (Roche).
Statistical and Bioinformatic Analyses
Statistical analyses were performed with a KaleidaGraph v4.03 (Synergy, Reading, PA).
RESULTS
Bloc1s5 Muted Mouse Models Reveal Unique Dysbindin and Pigment Dilution Phenotypes
Our studies were prompted by the identification of unexpected differences in dysbindin polypeptide composition in brains of mice carrying null alleles of Bloc1s5 (muted, mu) and Bloc1s8 (sandy, sdy). Dysbindin immunoblot detected two dysbindin immunoreactive polypeptides of ∼35 and ∼50 kDa in human and wild type mouse brain (Fig. 1). We have termed these bands dysbindin 1A and 1C (Fig. 1) (32). The identity of these immunoreactive bands as Bloc1s8 dysbindin polypeptides was confirmed by their absence in dysbindin null Bloc1s8sdy/sdy brain tissue (Fig. 1A, lanes 5 and 6 versus lanes 3 and 4 from samples of wild type littermates). Similarly, we observed a drastic reduction of dysbindin 1A in mouse brain lacking the BLOC-1 subunit muted (Bloc1s5mu/mu). In contrast, dysbindin 1C expression remained unaffected in Bloc1s5 null mice (Fig. 1A, compare lanes 7 and 8 with lanes 9 and 10). We performed quantitative real time PCR to examine the expression of dysbindin 1C by an alternative approach. The only AceView-predicted transcript encoding a Bloc1s8 dysbindin polypeptide with a putative molecular mass of dysbindin 1C is bSep07 (Fig. 1B). The bSep07 mRNA was proportionally reduced in hippocampus from single or double copy loss of Bloc1s8 (Fig. 1C). In contrast, the bSep07 mRNA remained unaltered in Bloc1s5 muted hippocampus. Thus, the dysbindin 1C polypeptide and the bSep07 mRNA expression are spared in the Bloc1s5 muted hippocampal formation.
FIGURE 1.

Dysbindin polypeptides are susceptible to BLOC-1 deficiencies differentially. A, hippocampal homogenates of human brain (lanes 1 and 2), control C57Bl/6J mice (lanes 3 and 4), Bloc1s8sdy/sdy C57BL/6J littermates (lanes 5 and 6), Bloc1s5mu/mu on a CHMU mouse background (lanes 7 and 8), and its wild type littermates were analyzed by immunoblot (IB) using antibody PA3111 against dysbindin. Dysbindin 1A and 1C refer to previously described polypeptides likely generated by differential splicing. B, diagram of two AceView predicted transcripts encoding polypeptides matching the molecular mass (MW) of either dysbindin-1A or -1C. C, Bloc1s8 bSep07 transcript from adult wild type, Bloc1s5+/mu, Bloc1s5mu/mu, Bloc1s8+/sdy, and Bloc1s8sdy/sdy mice hippocampi were analyzed by quantitative RT-PCR. The box plot depicts relative mRNA content. Asterisks indicate p < 0.0001 as compared with wild type. Bloc1s8+/sdy and Bloc1s8sdy/sdy are different with a p < 0.005. No significant differences were found among Bloc1s5 mutants (n = 3 animals with determinations in duplicate). Significance was determined with one-way analysis of variance followed by Bonferroni's multiple comparisons.
We hypothesized that this difference in dysbindin polypeptide expression between Bloc1s5mu/mu and Bloc1s8sdy/sdy could reflect wider differences in molecular and systemic phenotypes among BLOC-1 null mutations. To test this hypothesis, we generated mice carrying the Bloc1s5mu/mu and Bloc1s8sdy/sdy alleles on identical genetic backgrounds. These mutant alleles were originally isolated in CHMU and DBA/2J backgrounds, respectively (16, 28). We confirmed the presence of mutant alleles by sequencing PCR fragments encompassing the ETn transposon insertion in the Bloc1s5 (Fig. 2, A and B) and the deletion in the Bloc1s8 loci in C57B mouse strains (data not shown). Additionally, BLOC-1 subunit mutations are visually characterized by pigment dilution. This phenotype was readily evident in C57Bl/6J mice carrying double copy BLOC-1 null mutations: Bloc1s5mu/mu, Bloc1s6pa/pa, and Bloc1s8sdy/sdy (Fig. 2C). Pigment dilution was similar in all these mutant mice as determined by quantitation of hair melanin (Fig. 2D). Notably, we observed striking differences in the pigmentation phenotype among single copy BLOC-1 mutations. Bloc1s6+/pa and Bloc1s8+/sdy pigmentation were indistinguishable from wild type animals (Fig. 2, C and D). In contrast, Bloc1s5+/mu mouse pigmentation decreased to 50% of wild type hair melanin content (Fig. 2, C and D), demonstrating an allele-specific genetic dosage effect in one of their characteristic systemic phenotypes.
FIGURE 2.
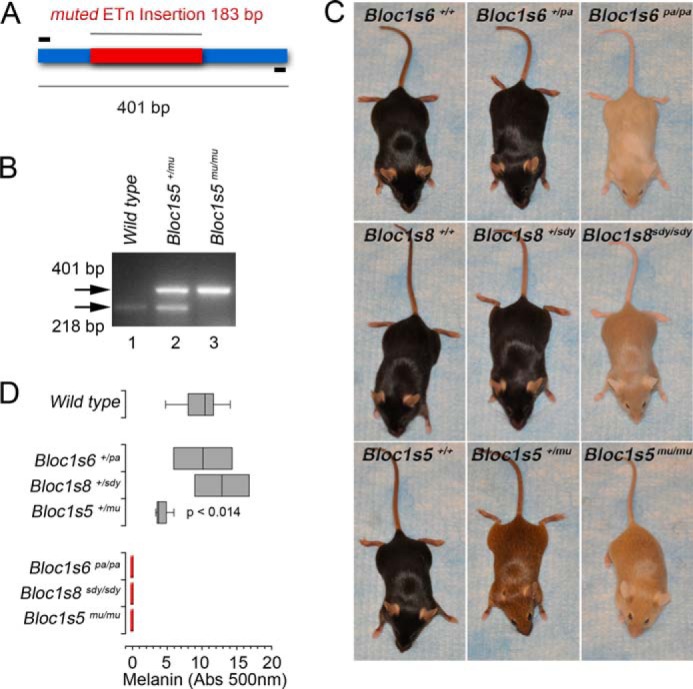
Gene dosage differentiates BLOC-1 null alleles pigment dilution phenotypes. All studies were performed in the C57B background. A, diagram of the ETn transposon insertion (red) in the Bloc1s5 locus. Primers used to generate predicted amplicons are in black. B, PCR of genomic DNA from wild type (lane 1), Bloc1s5+/mu (lane 2), and Bloc1s5mu/mu mice (lane 3). C, images depict pigment dilution phenotypes affecting single and double copy loss null alleles in Bloc1s5 (muted, mu), Bloc1s6 (pallid, pa), and Bloc1s8 (sandy, sdy). D, hair melanin determinations in mice depicted in C (n = 10 wild type, 3 double copy loss, and 2 single copy loss mice; one-way analysis of variance followed by Dunnett's Multiple Comparisons).
Bloc1s5 Muted Affects the Expression of BLOC-1 Complex in Hippocampus and Cortex
We predicted that Bloc1s5+/mu pigmentation phenotype could emerge from a compound effect of compromised expression of alternative BLOC-1 subunits in addition to Bloc1s5 muted. This hypothesis stems from the prior notion that single copy loss of any single BLOC-1 subunit is not sufficient to yield a phenotype. With no precedent to indicate otherwise, we tested whether Bloc1s5+/mu could be mimicking a BLOC-1 null to produce the observed coat color discoloration. To test this hypothesis, we focused on the hippocampal formation of Bloc1s5 mutant mice. We chose the hippocampal formation because this brain region seems to be particularly sensitive to BLOC-1 levels. For example, the schizophrenia-associated dysbindin reduction is highly penetrant in the hippocampus of schizophrenia patients (32, 38). In addition, the hippocampal formation of Bloc1s5mu/mu and other BLOC-1 mutants prominently display BLOC-1 null phenotypes or phenotypes associated with regulators of BLOC-1 expression such as Mecp2 (17, 33, 34, 36, 39).
We determined the expression of Bloc1s6 pallidin by confocal immunofluorescence microscopy of pallidin in the dentate gyrus of adult Bloc1s5+/mu and Bloc1s5mu/mu mice. We used a Bloc1s6 pallidin antibody whose immunoreactivity is absent in Bloc1s6pa/pa brains (29, 39). VAMP2 immunoreactivity was used as a marker of presynaptic tissue and a control counterstain. In the dentate gyrus, Bloc1s6 pallidin immunoreactivity was significantly reduced in mice with a loss of both copies of the gene encoding Bloc1s5, yet Bloc1s5mu/+ dentate just showed a trend to reduced Bloc1s6 pallidin (Fig. 3, A and B). We further examined Bloc1s6 pallidin levels with a more quantitative approach. Immunoblot demonstrated that Bloc1s6 pallidin content is similarly decreased to 50% of wild type levels in the hippocampi of both Bloc1s5+/mu and Bloc1s5mu/mu mice (Fig. 3, C and D).
FIGURE 3.
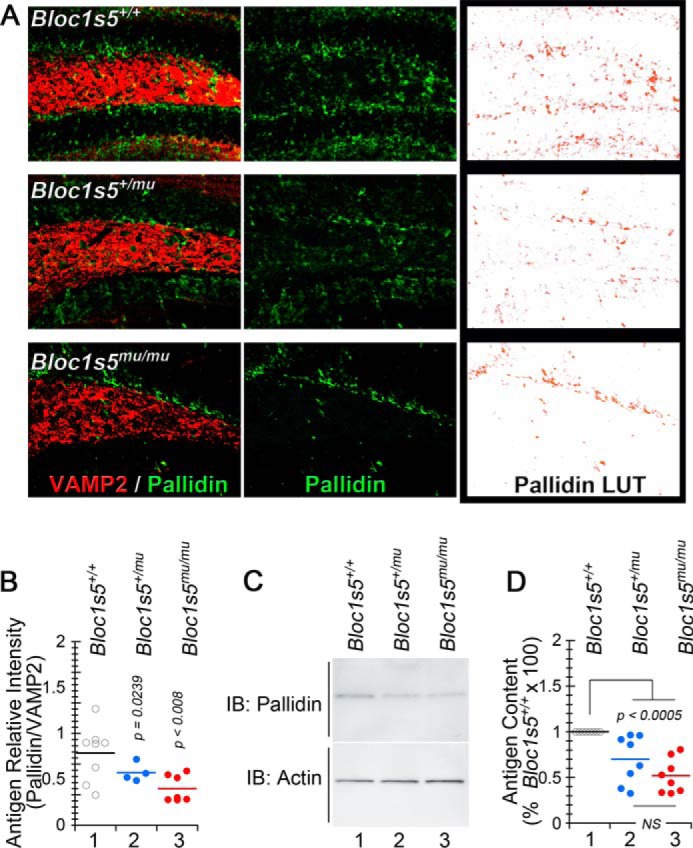
Bloc1s5 muted affects the expression of the BLOC-1 complex polypeptide pallidin. A, hippocampal sections from wild type, Bloc1s5+/mu, and Bloc1s5mu/mu mice were stained with antibodies against the BLOC-1 subunit pallidin and the synaptic vesicle marker VAMP2. The right panels present look-up tables (LUT) to highlight differences in pallidin expression among genotypes. B, a quantitation of pallidin immunoreactivity expressed as ratios between pallidin and VAMP2. Each dot represents an independent staining performed in four wild type and Bloc1s5mu/mu hippocampi or two Bloc1s5+/mu hippocampi. C and D, immunoblot determinations of pallidin and their quantitation in mouse hippocampi (n = 3 animals and 3 independent determinations; one-way analyses of variance followed by Dunnett's multiple comparison was used in B and D). IB, immunoblot.
It is well established that BLOC-1 subunit genetic defects decrease polypeptide expression of the other BLOC-1 complex constituents (16, 25–29). This effect is thought to result from degradation of unassembled BLOC-1 polypeptides. Thus, we tested whether pallidin protein reductions observed in Bloc1s5+/mu and Bloc1s5mu/mu hippocampi were due to only post-translational mechanisms or whether altered transcript levels could account for the similar reduction of pallidin protein levels. We measured Bloc1s6 pallidin transcripts in the adult hippocampal formation as well as the cortex of Bloc1s5+/mu and Bloc1s5mu/mu mice by quantitative real time PCR (Fig. 4, A and B, rows 1 and 2). Pallidin Bloc1s6 transcript levels were reduced in the hippocampal formation of both Bloc1s5+/mu (Fig. 4A, rows 1 and 2) and Bloc1s5mu/mu mice (Fig. 4B, rows 1 and 2), whereas we saw no changes in the expression of dysbindin Bloc1s8 mRNA. Similarly, pallidin Bloc1s6 transcript content was half of control adult cerebral cortex in Bloc1s5+/mu (Fig. 4C, rows 9 and 10) and Bloc1s5mu/mu mice (Fig. 4D, rows 9 and 10). Bloc1s5 muted mRNA was 50% of the wild type levels in Bloc1s5+/mu cortex (Fig. 4A, rows 7 and 8) and was undetectable in Bloc1s5mu/mu cortex (Fig. 4C, rows 7 and 8). Thus, mRNA levels precisely matched the genotype of Bloc1s5 mutant mice, demonstrating the fidelity of mRNA determinations.
FIGURE 4.
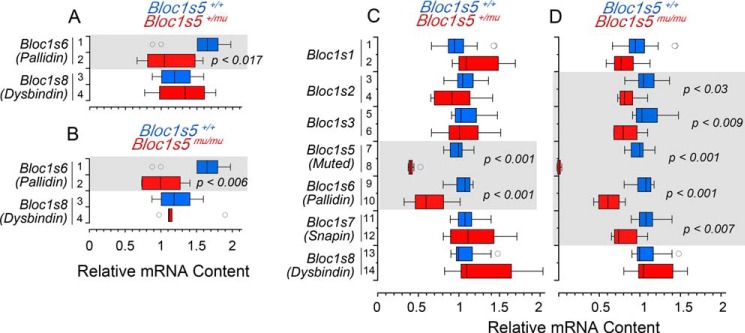
Bloc1s5 muted affects the expression of the BLOC-1 complex transcripts. BLOC-1 subunit transcripts from adult wild type, Bloc1s5+/mu, and Bloc1s5mu/mu mice hippocampi were analyzed by quantitative RT-PCR. A box plot depicts relative mRNA content in hippocampus (A and B) and cortex (C and D). Red boxes in A and C depict results for Bloc1s5+/mu, and red boxes in B and D for Bloc1s5mu/mu, respectively. Wild type is depicted in blue in all panels. Transcripts are listed to the left of graphs in italics (n = 3 animals with determinations in duplicate; one-way analysis of variance followed by Dunnett's multiple comparisons).
The unexpected effect of the Bloc1s5 muted allele on the expression Bloc1s6 pallidin messages prompted us to test whether the Bloc1s5 muted allele affected the expression of other BLOC-1 subunit mRNAs. Bloc1s5 and 6 were the only transcripts whose expression was reduced in Bloc1s5+/mu brain tissue (Fig. 4C). In contrast, we observed a significant decrease in the content of six of the eight BLOC-1 complex subunits, Bloc1s2, 3, 5, 6, and 7 (Fig. 4D, rows 3–12), in Bloc1s5mu/mu tissue. Decreased mRNA levels were more pronounced for Bloc1s5 muted and Bloc1s6 pallidin transcripts in Bloc1s5mu/mu tissue (Fig. 4D, rows 7–10). Unlike the double copy Bloc1s5 muted mutation, double copy null Bloc1s6 pallid (pa) and Bloc1s8 sandy (sdy) had no effect on neuronal Bloc1s5 muted and Bloc1s6 pallidin mRNA expression (Fig. 5). These results indicate that mutations in loci encoding BLOC-1 subunits differ in molecular phenotypes related to the expression of transcripts encoding BLOC-1 subunits. Moreover, these findings uncover a hitherto unknown effect of mutations in protein traffic complexes in the levels of their own subunit encoding transcripts.
FIGURE 5.
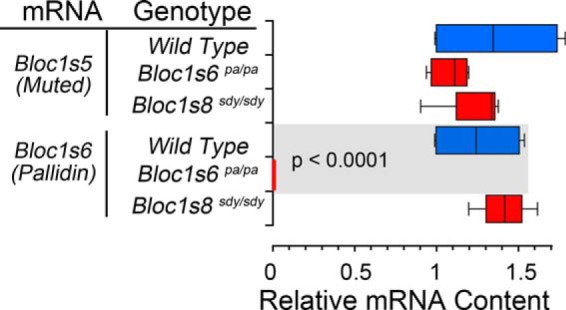
Pallidin and muted transcripts are not affected by a null Bloc1s8 allele. BLOC-1 subunit transcripts from adult wild type, Bloc1s6pa/pa, and Bloc1s8sdy/sdy mice hippocampi were analyzed by quantitative RT-PCR. The box plot depicts relative mRNA content. Red boxes depict relative mRNA content for mutant alleles. Wild type is depicted in blue in all panels. Transcripts are listed to the far left of graph in italics. (n = 3 animals with determinations in at least duplicate; one-way analysis of variance followed by Dunnett's multiple comparisons).
Bloc1s5mu/mu and Bloc1s8sdy/sdy Mouse Hippocampi Diverge in Glutamatergic Transcriptional Profiles
Differences in BLOC-1 subunits transcriptional profiles in Bloc1s5mu/mu and Bloc1s8sdy/sdy mutants predict that downstream effectors of the BLOC-1 complex should be differentially affected by these alleles. To test whether phenotypic divergence between Bloc1s5mu/mu and Bloc1s8sdy/sdy extends to effectors downstream of BLOC-1, we measured the expression of mRNAs encoding glutamatergic and GABAergic markers in Bloc1s5mu/mu and Bloc1s8sdy/sdy mouse hippocampal formations. We chose these neurotransmitter systems because they are implicated in the pathogenesis of schizophrenia (40–45). NMDA receptor subunit transcripts were selected as markers of glutamatergic neurotransmission as mutations in Bloc1s8 modify the expression of NMDA receptor mRNA and alter NMDA subcellular distribution and function of receptors in neurons (18, 46, 47). We compared expression of mRNAs encoding NMDA receptor NR1 subunit and NR2 isoforms in Bloc1s5mu/mu, Bloc1s+/mu, Bloc1s8sdy/sdy, and Bloc1s8+/sdy hippocampal formations.
Similar to previous reports of NR1 transcripts in sandy prefrontal cortex, we found that NR1 mRNA is decreased in Bloc1s8sdy/sdy hippocampus (Fig. 6B, rows 1 and 2) (18). In addition, mRNA levels of all NR2 isoforms were significantly reduced in Bloc1s8sdy/sdy as compared with wild type hippocampal formations (Fig. 6B, rows 3–10). In contrast, Bloc1s8+/sdy hippocampal formations had increased in NR2A and NR2B mRNAs without effects on other receptor subunits (Fig. 6A, rows 3–6). Vamp2 transcript levels encoding the synaptic vesicle protein synaptobrevin 2 were used as mRNA loading controls in all Bloc1s8+/sdy and Bloc1s8sdy/sdy studies (Fig. 6, A and B, rows 11 and 12). We contrasted the NMDA receptor transcriptional signature observed in Bloc1s8sdy/sdy and Bloc1s8+/sdy hippocampi with that of Bloc1s5mu/mu and Bloc1s5+/mu (Fig. 6, C and D). Bloc1s5mu/mu tissue altered the expression of all NMDA receptor mRNAs tested (Fig. 6D, rows 1–10). However, phenotypic overlap between Bloc1s5mu/mu and Bloc1s8sdy/sdy was restricted to only two of the five NMDA receptor subunits analyzed, NR2C and D (Fig. 6D, rows 7–10). Transcripts encoding these two subunits were reduced both in Bloc1s5mu/mu and Bloc1s8sdy/sdy (Fig. 6D, rows 7–10). Furthermore, and in contrast to Bloc1s8+/sdy hippocampi, Bloc1s5+/mu did not affect expression of any receptor subunit mRNAs (Fig. 6C). Syp transcript levels encoding the synaptic vesicle protein synaptophysin were used as controls in all Bloc1s5+/mu and Bloc1s5mu/mu determinations (Fig. 6, C and D, rows 11 and 12). These results demonstrate that mutations to different BLOC-1 subunit genes differentially affect NMDA receptor subunit expression in the hippocampus, with marked changes in Bloc1s8 mice but moderate effects in Bloc1s5 animals.
FIGURE 6.
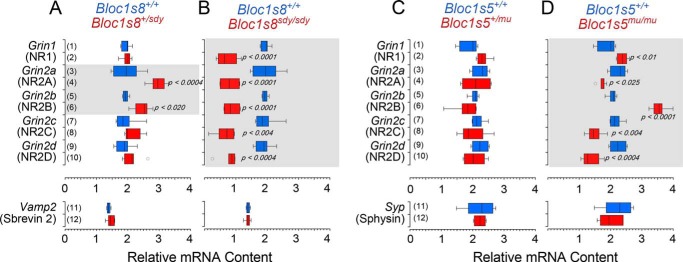
Bloc1s8sdy/sdy and Bloc1s5mu/mu differentially affects the expression of NMDA receptor subunit transcripts. NMDA receptor subunit transcripts from adult wild type, Bloc1s8+/sdy, and Bloc1s8sdy/sdy mice hippocampi (A and B); Bloc1s5+/mu, and Bloc1s5mu/mu mice hippocampi (C and D) were analyzed by quantitative RT-PCR. A box plot depicts relative mRNA content with red boxes in A and C depicting single copy loss and red boxes in B and D depicting double copy loss alleles, respectively. Wild type is depicted in blue in all panels. Transcripts encoding the synaptic vesicle proteins VAMP2 and synaptophysin (Sphysin) were used as loading controls. Transcripts are listed to the left of graphs in italics (n = 3 animals with determinations in at least duplicate; one-way analysis of variance followed by Dunnett's multiple comparisons).
Bloc1s5mu/mu and Bloc1s8sdy/sdy Mouse Hippocampi Diverge in GABAergic Transcriptional Profiles
Homeostatic plasticity maintains circuit excitability in face of perturbations. Set point is restored by changes in gene expression in functionally opposing neurotransmitter systems (48, 49). Thus, we reasoned that the NMDA receptor phenotypic divergence between Bloc1s5mu/mu and Bloc1s8sdy/sdy should be accompanied by divergent and proportional changes in GABAergic neurotransmission markers. Parvalbumin levels are reduced in GABAergic interneurons of the Bloc1s8sdy/sdy hippocampal formation (19). We measured parvalbumin and a panel of GABAergic internerneuron subpopulation markers by quantitative real time PCR using (Fig. 7). We observed a reduction in the mRNA level for parvalbumin in Bloc1s8sdy/sdy hippocampal formations, an observation consistent with the reported reduced density of parvalbumin positive cells in Bloc1s8sdy/sdy tissue (Fig. 7A, rows 3 and 4). Similar results were obtained with markers of GABA-positive interneurons such as the glutamate decarboxylase of 65 kDa (Gad2), GABA transporter 2 (Slc6a13), vesicular GABA transporter (VGAT, Slc32a1), the neuropeptide somatostatin (Sst; Fig. 7A, rows 1–10), or interneuron enriched transcription factors such as Arx, Npas1, and Lhx6 (Fig. 7A, rows 11–16) (50–52). In contrast with Bloc1s8sdy/sdy hippocampi, transcript levels of GABA interneurons remained unaltered in Bloc1s5mu/mu tissue except for Arx and Npas1 transcripts, which were less pronouncedly reduced (Fig. 7B, rows 11–14). There were no significant changes in transcript levels in Bloc1s8+/sdy and Bloc1s5+/mu hippocampi (data not shown). mRNA loading controls were similar across genotypes as measured by transcripts encoding the synaptic vesicle protein synaptophysin (Sphysin, Syp; Fig. 7, A and B, rows 17 and 18). We confirmed these differences in GABAergic phenotypes by measuring the protein levels of the synaptic vesicle GABA transporter (VGAT; Fig. 7, C–E). Similar to what we observed with VGAT mRNA levels, the VGAT polypeptide was drastically reduced in Bloc1s8sdy/sdy hippocampus (Fig. 7, C and E). In contrast, VGAT polypeptide expression in Bloc1s5mu/mu tissue was not different from wild type controls (Fig. 7, D and E). Importantly, the BLOC-1-sensistive cargo and synaptic vesicle SNARE VAMP7 was reduced in Bloc1s5mu/mu (Fig. 7, D and E), demonstrating that the Bloc1s5mu/mu mutation generated phenotypes in the organelle where VGAT resides. This VAMP7 polypeptide decrease occurred, even though VAMP7 transcripts remained unaffected in Bloc1s5mu/mu hippocampi (VAMP7; Fig. 7F). These findings demonstrate that mutations in Bloc1s8 profoundly affect the expression of GABAergic markers, a phenotype that diverges from mutations in Bloc1s5. Our results provide evidence that Bloc1s5mu/mu and Bloc1s8sdy/sdy modify the hippocampal transcriptome downstream of the BLOC-1 complex in different ways (see summary in Fig. 8). We suggest that different mutations in BLOC-1 subunits influence neuronal circuit physiology in distinctive yet partially overlapping ways.
FIGURE 7.

Bloc1s8sdy/sdy and Bloc1s5mu/mu differentially affect the expression of GABAergic interneuron markers. A and B, transcripts encoding markers of GABAergic interneurons from adult wild type, Bloc1s8sdy/sdy (A) and Bloc1s5mu/mu (B) hippocampi were analyzed by quantitative RT-PCR. The box plot depicts relative mRNA content with red boxes indicating double copy loss alleles. Wild type is depicted in blue in all panels. Transcripts encoding the synaptic vesicle protein synaptophysin (Sphysin) were used as loading controls. Transcripts are listed to the left of graphs in italics (n = 3 animals with determinations in at least duplicate; one-way analysis of variance followed by Dunnett's multiple comparisons). C–E, immunoblot (IB) determinations of VGAT in wild type and mutant mouse hippocampi and their quantitation normalized to actin. F, VAMP7 and VAMP2 transcript determinations in wild type and Bloc1s5mu/mu hippocampi. Bloc1s5mu/+ transcript levels are not shown (n = 5 animals; one-way analysis of variance followed by Dunnett's multiple comparisons).
FIGURE 8.

Summary of transcriptional phenotypes in Bloc1s8sdy/sdy and Bloc1s5mu/mu brains. Diagram depicts the transcriptional phenotypes in BLOC-1 null brains. Bloc1s8* denotes the putative dysbindin 1C transcript bSept07. Colored boxes depict no changes (gray), down-regulation (blue), or up-regulation (orange) as compared with wild type C57BL/6J tissue.
DISCUSSION
Much of what we know about the function of dysbindin and its interacting BLOC-1 complex subunits comes from the study of genetic mutations in mice. Phenotypic homogeneity has been a hallmark of BLOC-1 subunit mutant alleles (14, 16, 25–29). This assertion is supported by the shared systemic phenotypes associated to these mutants, as well as the severely decreased protein expression of multiple BLOC-1 subunits common to dysbindin and other BLOC-1 subunits mutations (14, 16, 25–30). Here we genetically tested the extent of phenotypic homogeneity in neuronal cells. We demonstrate divergences and allele-specific gene dosage effects in neuronal phenotypes associated with Bloc1s5 muted and Bloc1s8 sandy mutations in neurons. Phenotypic divergence encompasses expression of BLOC-1 subunit gene products, NMDA receptor subunits transcripts, and GABAergic interneurons markers (Fig. 8). These transcripts and polypeptides highlight neurotransmitter mechanisms and cell types implicated by schizophrenia pathogenesis hypotheses (40–43). Our mouse genetic results support the dosage balance hypothesis, which predicts the emergence of distinct phenotypes among mutations affecting different subunits of a protein complex (53, 54).
The mechanism by which mutations in dysbindin and interacting BLOC-1 subunits generate common as well as divergent phenotypes is presently unknown. However, we speculate that phenotypes common to all BLOC-1 complex subunits mutations require its normal quaternary structure. In contrast, divergent phenotypes may be caused by remnants of the BLOC-1 complex left after uneven protein down-regulation of the octamer. As such, BLOC-1 down-regulation remnants should have different composition/stoichiometry among mutations affecting the dysbindin-BLOC-1 complex subunits to account for phenotypic divergence. The extent of degradation of BLOC-1 complex subunits seems divergent among mutations affecting different BLOC-1 complex subunits, although this has not been quantitatively assessed (16, 25–30). Our data support the remnant-derived phenotype hypothesis as exemplified by the effects that the Bloc1s5mu/mu and Bloc1s8sdy/sdy mutants exert upon the expression of dysbindin 1C in mouse brain. The remnant-derived phenotype hypothesis makes two predictions. First, it suggests that some mutation-associated phenotypes could be semidominant or partially expressed in single copy loss mutations. This is the case for the hippocampal pallidin transcript decrease and pigmentation reduction in Bloc1s5+/mu, as well as the effect of Bloc1s8+/sdy on NR2A and B transcripts. Second, it predicts the counterintuitive notion that phenotypes in a null mutation affecting one BLOC-1 complex subunit could be ameliorated by genetic deficiencies in a second subunit of the complex. Additionally, divergent phenotypes may reflect loss of function of roles played by individual dysbindin-BLOC-1 subunits occurring outside the octameric dysbindin-BLOC-1 complex. This is the case of Bloc1s7 snapin that engages in molecular interactions independent of dysbindin-BLOC-1 complex (55–59). The complexity of the divergences in transcriptional phenotypes extends beyond differences in just the mRNAs affected. NR1 and NR2B are decreased in Bloc1s8sdy/sdy, yet these same mRNAs are up-regulated in Bloc1s5mu/mu. This suggests different pathways affecting these messages in these two BLOC-1 null phenotypes. The molecular determinants defining the type of transcript affected and whether they are up- or down-regulated in BLOC-1 null alleles remains to be explored.
Our findings also have implications for neurodevelopmental disorders such as schizophrenia. Dysbindin protein expression is decreased in the brain of 80% of schizophrenia patients tested thus far (32, 38). The cause(s) of this dysbindin down-regulation remains unknown. However, it is unlikely that DTNBP1 polymorphisms associated with schizophrenia risk alone may explain dysbindin protein reduction. The frequency of these DTNBP1 disease-associated small nucleotide polymorphisms is too low to account for the highly penetrant dysbindin protein down-regulation phenotype observed in schizophrenia populations (60). Dysbindin levels depend on protein expression of other BLOC-1 complex subunits, a mechanism considered post-translational. Likely, ubiquitin ligases like TRIM32 may down-regulate dysbindin in the absence of other BLOC-1 subunits (61). This untested model is analogous to the mechanism that accounts for the degradation of the adaptor complex AP-3 after one of its subunit encoding genes is mutated (62). Protein complexes frequently respond with multiprotein down-regulation of many of their constituents after genetic mutation of only one of their components (62–65). En bloc degradation of the BLOC-1 complex does not consider transcriptional contributions. Our results add an unsuspected and novel layer of complexity as BLOC-1 subunit transcripts encoded by nonmutated genes are modified by specific mutations in BLOC-1 subunits. We demonstrate down-regulation of four BLOC-1 transcripts, other than Bloc1s5 mRNA, in Bloc1s5mu/mu brain tissue. Most notably, there is a concurrent and similar drop in pallidin transcript and polypeptide levels in both single and double copy Bloc1s5 muted mutants. Pallidin transcript levels are normal in Bloc1s8 mutants irrespective of gene dosage. However, phenotypes associated with loss of one gene copy are not restricted to Bloc1s5+/mu brain. They at least include pigment dilution in Bloc1s5+/mu skin and NR2A-B transcripts in Bloc1s8+/sdy brain. Thus, allele and gene dosage-specific effects in a protein complex such as BLOC-1 could be a general mechanism to generate phenotypic diversity among individuals with mutations in different loci belonging to a pathway. Single copy losses of either one gene or chromosomal segment are important genetic risk factors for diverse neurodevelopmental disorders ranging from schizophrenia to autism spectrum disorder (66–70). These genetic defects are also genetic modifiers of human cognition (71). The phenotypic spectrum in these single copy loss variations is quite wide. On one hand, the same genetic defect generates more than one disorder. On the other hand, the same disorder is produced by single copy loss of different loci (72–74). Mechanisms by which these single copy losses generate neurodevelopmental and cognition defects are not understood. However, it is likely that multiple molecular mechanisms may account for their pathogenic effect. We propose that common single copy loss of genetic loci associated to schizophrenia (44, 45), such as chromosome 22q11 deletion syndrome (75), could expand and diversify their clinical presentation spectrum by altering the stability of messages and/or polypeptides belonging to macromolecular complexes.
Acknowledgments
We are indebted to the Faundez laboratory members for comments.
Footnotes
This work was supported, in whole or in part, by National Institutes of Health Grants GM077569 and NS42599 (to V. F.), R01 MH072880 and PP MH064045 (to K. T.), and Emory University Integrated Cellular Imaging Microscopy Core and Viral Cores of the Emory Neuroscience NINDS Core Facilities Grant P30NS055077. This work was also supported by funds from the Children's Hospital of Atlanta (CHOA) Children's Center for Neuroscience (to V. F.).
REFERENCES
- 1. Straub R. E., Jiang Y., MacLean C. J., Ma Y., Webb B. T., Myakishev M. V., Harris-Kerr C., Wormley B., Sadek H., Kadambi B., Cesare A. J., Gibberman A., Wang X., O'Neill F. A., Walsh D., Kendler K. S. (2002) Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am. J. Hum. Genet. 71, 337–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Van Den Bogaert A., Schumacher J., Schulze T. G., Otte A. C., Ohlraun S., Kovalenko S., Becker T., Freudenberg J., Jönsson E. G., Mattila-Evenden M., Sedvall G. C., Czerski P. M., Kapelski P., Hauser J., Maier W., Rietschel M., Propping P., Nöthen M. M., Cichon S. (2003) The DTNBP1 (dysbindin) gene contributes to schizophrenia, depending on family history of the disease. Am. J. Hum. Genet. 73, 1438–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bray N. J., Preece A., Williams N. M., Moskvina V., Buckland P. R., Owen M. J., O'Donovan M. C. (2005) Haplotypes at the dystrobrevin binding protein 1 (DTNBP1) gene locus mediate risk for schizophrenia through reduced DTNBP1 expression. Hum. Mol. Genet. 14, 1947–1954 [DOI] [PubMed] [Google Scholar]
- 4. Ayalew M., Le-Niculescu H., Levey D. F., Jain N., Changala B., Patel S. D., Winiger E., Breier A., Shekhar A., Amdur R., Koller D., Nurnberger J. I., Corvin A., Geyer M., Tsuang M. T., Salomon D., Schork N. J., Fanous A. H., O'Donovan M. C., Niculescu A. B. (2012) Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol. Psychiatry 17, 887–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cerasa A., Quattrone A., Gioia M. C., Tarantino P., Annesi G., Assogna F., Caltagirone C., De Luca V., Spalletta G. (2011) Dysbindin C-A-T haplotype is associated with thicker medial orbitofrontal cortex in healthy population. NeuroImage 55, 508–513 [DOI] [PubMed] [Google Scholar]
- 6. Luciano M., Miyajima F., Lind P. A., Bates T. C., Horan M., Harris S. E., Wright M. J., Ollier W. E., Hayward C., Pendleton N., Gow A. J., Visscher P. M., Starr J. M., Deary I. J., Martin N. G., Payton A. (2009) Variation in the dysbindin gene and normal cognitive function in three independent population samples. Genes Brain Behav. 8, 218–227 [DOI] [PubMed] [Google Scholar]
- 7. Markov V., Krug A., Krach S., Jansen A., Eggermann T., Zerres K., Stöcker T., Shah N. J., Nöthen M. M., Treutlein J., Rietschel M., Kircher T. (2010) Impact of schizophrenia-risk gene dysbindin 1 on brain activation in bilateral middle frontal gyrus during a working memory task in healthy individuals. Human Brain Mapping 31, 266–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Markov V., Krug A., Krach S., Whitney C., Eggermann T., Zerres K., Stöcker T., Shah N. J., Nöthen M. M., Treutlein J., Rietschel M., Kircher T. (2009) Genetic variation in schizophrenia-risk-gene dysbindin 1 modulates brain activation in anterior cingulate cortex and right temporal gyrus during language production in healthy individuals. NeuroImage 47, 2016–2022 [DOI] [PubMed] [Google Scholar]
- 9. Mechelli A., Viding E., Kumar A., Pettersson-Yeo W., Fusar-Poli P., Tognin S., O'Donovan M. C., McGuire P. (2010) Dysbindin modulates brain function during visual processing in children. NeuroImage 49, 817–822 [DOI] [PubMed] [Google Scholar]
- 10. Tognin S., Viding E., McCrory E. J., Taylor L., O'Donovan M. C., McGuire P., Mechelli A. (2011) Effects of DTNBP1 genotype on brain development in children. J. Child Psychol. Psychiatry 52, 1287–1294 [DOI] [PubMed] [Google Scholar]
- 11. Wolf C., Jackson M. C., Kissling C., Thome J., Linden D. E. (2011) Dysbindin-1 genotype effects on emotional working memory. Mol. Psychiatry 16, 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mullin A. P., Gokhale A., Larimore J., Faundez V. (2011) Cell biology of the BLOC-1 complex subunit dysbindin, a schizophrenia susceptibility gene. Mol. Neurobiol. 44, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghiani C. A., Dell'Angelica E. C. (2011) Dysbindin-containing complexes and their proposed functions in brain: from zero to (too) many in a decade. ASN Neuro. 3, e00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei M. L. (2006) Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 19, 19–42 [DOI] [PubMed] [Google Scholar]
- 15. Ghiani C. A., Starcevic M., Rodriguez-Fernandez I. A., Nazarian R., Cheli V. T., Chan L. N., Malvar J. S., de Vellis J., Sabatti C., Dell'Angelica E. C. (2010) The dysbindin-containing complex (BLOC-1) in brain: developmental regulation, interaction with SNARE proteins and role in neurite outgrowth. Mol. Psychiatry 15, 204–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li W., Zhang Q., Oiso N., Novak E. K., Gautam R., O'Brien E. P., Tinsley C. L., Blake D. J., Spritz R. A., Copeland N. G., Jenkins N. A., Amato D., Roe B. A., Starcevic M., Dell'Angelica E. C., Elliott R. W., Mishra V., Kingsmore S. F., Paylor R. E., Swank R. T. (2003) Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat. Genet. 35, 84–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larimore J., Tornieri K., Ryder P. V., Gokhale A., Zlatic S. A., Craige B., Lee J. D., Talbot K., Pare J. F., Smith Y., Faundez V. (2011) The schizophrenia susceptibility factor dysbindin and its associated complex sort cargoes from cell bodies to the synapse. Mol. Biol. Cell 22, 4854–4867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karlsgodt K. H., Robleto K., Trantham-Davidson H., Jairl C., Cannon T. D., Lavin A., Jentsch J. D. (2011) Reduced dysbindin expression mediates N-methyl-d-aspartate receptor hypofunction and impaired working memory performance. Biol. Psychiatry 69, 28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carlson G. C., Talbot K., Halene T. B., Gandal M. J., Kazi H. A., Schlosser L., Phung Q. H., Gur R. E., Arnold S. E., Siegel S. J. (2011) Dysbindin-1 mutant mice implicate reduced fast-phasic inhibition as a final common disease mechanism in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 108, E962–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jentsch J. D., Trantham-Davidson H., Jairl C., Tinsley M., Cannon T. D., Lavin A. (2009) Dysbindin modulates prefrontal cortical glutamatergic circuits and working memory function in mice. Neuropsychopharmacology 34, 2601–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheli V. T., Daniels R. W., Godoy R., Hoyle D. J., Kandachar V., Starcevic M., Martinez-Agosto J. A., Poole S., DiAntonio A., Lloyd V. K., Chang H. C., Krantz D. E., Dell'Angelica E. C. (2010) Genetic modifiers of abnormal organelle biogenesis in a Drosophila model of BLOC-1 deficiency. Hum. Mol. Genet. 19, 861–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dickman D. K., Tong A., Davis G. W. (2012) Snapin is critical for presynaptic homeostatic plasticity. J. Neurosci. 32, 8716–8724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dickman D. K., Davis G. W. (2009) The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science 326, 1127–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shao L., Shuai Y., Wang J., Feng S., Lu B., Li Z., Zhao Y., Wang L., Zhong Y. (2011) Schizophrenia susceptibility gene dysbindin regulates glutamatergic and dopaminergic functions via distinctive mechanisms in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 108, 18831–18836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gwynn B., Martina J. A., Bonifacino J. S., Sviderskaya E. V., Lamoreux M. L., Bennett D. C., Moriyama K., Huizing M., Helip-Wooley A., Gahl W. A., Webb L. S., Lambert A. J., Peters L. L. (2004) Reduced pigmentation (rp), a mouse model of Hermansky-Pudlak syndrome, encodes a novel component of the BLOC-1 complex. Blood 104, 3181–3189 [DOI] [PubMed] [Google Scholar]
- 26. Ciciotte S. L., Gwynn B., Moriyama K., Huizing M., Gahl W. A., Bonifacino J. S., Peters L. L. (2003) Cappuccino, a mouse model of Hermansky-Pudlak syndrome, encodes a novel protein that is part of the pallidin-muted complex (BLOC-1). Blood 101, 4402–4407 [DOI] [PubMed] [Google Scholar]
- 27. Huang L., Kuo Y. M., Gitschier J. (1999) The pallid gene encodes a novel, syntaxin 13-interacting protein involved in platelet storage pool deficiency. Nat. Genet. 23, 329–332 [DOI] [PubMed] [Google Scholar]
- 28. Zhang Q., Li W., Novak E. K., Karim A., Mishra V. S., Kingsmore S. F., Roe B. A., Suzuki T., Swank R. T. (2002) The gene for the muted (mu) mouse, a model for Hermansky-Pudlak syndrome, defines a novel protein which regulates vesicle trafficking. Hum. Mol. Genet. 11, 697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Starcevic M., Dell'Angelica E. C. (2004) Identification of snapin and three novel proteins (BLOS1, BLOS2, and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J. Biol. Chem. 279, 28393–28401 [DOI] [PubMed] [Google Scholar]
- 30. Yang Q., He X., Yang L., Zhou Z., Cullinane A. R., Wei A., Zhang Z., Hao Z., Zhang A., He M., Feng Y., Gao X., Gahl W. A., Huizing M., Li W. (2012) The BLOS1-interacting protein KXD1 is involved in the biogenesis of lysosome-related organelles. Traffic 13, 1160–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dell'Angelica E. C. (2009) AP-3-dependent trafficking and disease: the first decade. Curr. Opin. Cell Biol. 21, 552–559 [DOI] [PubMed] [Google Scholar]
- 32. Talbot K., Louneva N., Cohen J. W., Kazi H., Blake D. J., Arnold S. E. (2011) Synaptic dysbindin-1 reductions in schizophrenia occur in an isoform-specific manner indicating their subsynaptic location. PLoS One 6, e16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Newell-Litwa K., Chintala S., Jenkins S., Pare J. F., McGaha L., Smith Y., Faundez V. (2010) Hermansky-Pudlak protein complexes, AP-3 and BLOC-1, differentially regulate presynaptic composition in the striatum and hippocampus. J. Neurosci. 30, 820–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Newell-Litwa K., Salazar G., Smith Y., Faundez V. (2009) Roles of BLOC-1 and adaptor protein-3 complexes in cargo sorting to synaptic vesicles. Mol. Biol. Cell 20, 1441–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cox M. M., Tucker A. M., Tang J., Talbot K., Richer D. C., Yeh L., Arnold S. E. (2009) Neurobehavioral abnormalities in the dysbindin-1 mutant, sandy, on a C57BL/6J genetic background. Genes Brain Behav. 8, 390–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gokhale A., Larimore J., Werner E., So L., Moreno-De-Luca A., Lese-Martin C., Lupashin V. V., Smith Y., Faundez V. (2012) Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex 1. J. Neurosci. 32, 3697–3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoyle D. J., Rodriguez-Fernandez I. A., Dell'angelica E. C. (2011) Functional interactions between OCA2 and the protein complexes BLOC-1, BLOC-2, and AP-3 inferred from epistatic analyses of mouse coat pigmentation. Pigment Cell Melanoma Res. 24, 275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Talbot K., Eidem W. L., Tinsley C. L., Benson M. A., Thompson E. W., Smith R. J., Hahn C. G., Siegel S. J., Trojanowski J. Q., Gur R. E., Blake D. J., Arnold S. E. (2004) Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J. Clin. Invest. 113, 1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Larimore J., Ryder P. V., Kim K. Y., Ambrose L. A., Chapleau C., Calfa G., Gross C., Bassell G. J., Pozzo-Miller L., Smith Y., Talbot K., Park I. H., Faundez V. (2013) MeCP2 regulates the synaptic expression of a Dysbindin-BLOC-1 network component in mouse brain and human induced pluripotent stem cell-derived neurons. PLoS One 8, e65069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carlsson A., Waters N., Holm-Waters S., Tedroff J., Nilsson M., Carlsson M. L. (2001) Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu. Rev. Pharmacol. Toxicol. 41, 237–260 [DOI] [PubMed] [Google Scholar]
- 41. Gonzalez-Burgos G., Lewis D. A. (2012) NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr. Bull. 38, 950–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tsai G., Coyle J. T. (2002) Glutamatergic mechanisms in schizophrenia. Annu. Rev. Pharmacol. Toxicol. 42, 165–179 [DOI] [PubMed] [Google Scholar]
- 43. van Os J., Kapur S. (2009) Schizophrenia. Lancet 374, 635–645 [DOI] [PubMed] [Google Scholar]
- 44. Purcell S. M., Moran J. L., Fromer M., Ruderfer D., Solovieff N., Roussos P., O'Dushlaine C., Chambert K., Bergen S. E., Kähler A., Duncan L., Stahl E., Genovese G., Fernández E., Collins M. O., Komiyama N. H., Choudhary J. S., Magnusson P. K., Banks E., Shakir K., Garimella K., Fennell T., DePristo M., Grant S. G., Haggarty S. J., Gabriel S., Scolnick E. M., Lander E. S., Hultman C. M., Sullivan P. F., McCarroll S. A., Sklar P. (2014) A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fromer M., Pocklington A. J., Kavanagh D. H., Williams H. J., Dwyer S., Gormley P., Georgieva L., Rees E., Palta P., Ruderfer D. M., Carrera N., Humphreys I., Johnson J. S., Roussos P., Barker D. D., Banks E., Milanova V., Grant S. G., Hannon E., Rose S. A., Chambert K., Mahajan M., Scolnick E. M., Moran J. L., Kirov G., Palotie A., McCarroll S. A., Holmans P., Sklar P., Owen M. J., Purcell S. M., O'Donovan M. C. (2014) De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tang T. T., Yang F., Chen B. S., Lu Y., Ji Y., Roche K. W., Lu B. (2009) Dysbindin regulates hippocampal LTP by controlling NMDA receptor surface expression. Proc. Natl. Acad. Sci. U.S.A. 106, 21395–21400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saggu S., Cannon T. D., Jentsch J. D., Lavin A. (2013) Potential molecular mechanisms for decreased synaptic glutamate release in dysbindin-1 mutant mice. Schizophr. Res. 146, 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Turrigiano G. G. (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Davis G. W. (2013) Homeostatic signaling and the stabilization of neural function. Neuron 80, 718–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tricoire L., Pelkey K. A., Erkkila B. E., Jeffries B. W., Yuan X., McBain C. J. (2011) A blueprint for the spatiotemporal origins of mouse hippocampal interneuron diversity. J. Neurosci. 31, 10948–10970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Winden K. D., Oldham M. C., Mirnics K., Ebert P. J., Swan C. H., Levitt P., Rubenstein J. L., Horvath S., Geschwind D. H. (2009) The organization of the transcriptional network in specific neuronal classes. Mol. Syst. Biol. 5, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liodis P., Denaxa M., Grigoriou M., Akufo-Addo C., Yanagawa Y., Pachnis V. (2007) Lhx6 activity is required for the normal migration and specification of cortical interneuron subtypes. J. Neurosci. 27, 3078–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Birchler J. A., Veitia R. A. (2012) Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc. Natl. Acad. Sci. U.S.A. 109, 14746–14753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Veitia R. A., Bottani S., Birchler J. A. (2008) Cellular reactions to gene dosage imbalance: genomic, transcriptomic and proteomic effects. Trends Genet. 24, 390–397 [DOI] [PubMed] [Google Scholar]
- 55. Ye X., Cai Q. (2014) Snapin-mediated BACE1 retrograde transport is essential for its degradation in lysosomes and regulation of APP processing in neurons. Cell Rep. 6, 24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou B., Cai Q., Xie Y., Sheng Z. H. (2012) Snapin recruits dynein to BDNF-TrkB signaling endosomes for retrograde axonal transport and is essential for dendrite growth of cortical neurons. Cell Rep. 2, 42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pan P. Y., Tian J. H., Sheng Z. H. (2009) Snapin facilitates the synchronization of synaptic vesicle fusion. Neuron 61, 412–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen M., Lucas K. G., Akum B. F., Balasingam G., Stawicki T. M., Provost J. M., Riefler G. M., Jörnsten R. J., Firestein B. L. (2005) A novel role for snapin in dendrite patterning: interaction with cypin. Mol. Biol. Cell 16, 5103–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rüder C., Reimer T., Delgado-Martinez I., Hermosilla R., Engelsberg A., Nehring R., Dörken B., Rehm A. (2005) EBAG9 adds a new layer of control on large dense-core vesicle exocytosis via interaction with Snapin. Mol. Biol. Cell 16, 1245–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Talbot K., Ong W. Y., Blake D. J., Tang D., Louneva N., Carlson G. C., Arnold S. E. (2009) Dysbindin-1 and its protein family, with special attention to the potential role of dysbindin-1 in neuronal functions and the pathophysiology of schizophrenia. In Handbook of Neurochemistry and Molecular Neurobiology (Kantrowitz J., ed) pp. 107–241, Springer Science, New York [Google Scholar]
- 61. Locke M., Tinsley C. L., Benson M. A., Blake D. J. (2009) TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum. Mol. Genet. 18, 2344–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dell'Angelica E. C., Shotelersuk V., Aguilar R. C., Gahl W. A., Bonifacino J. S. (1999) Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the β3A subunit of the AP-3 adaptor. Mol. Cell 3, 11–21 [DOI] [PubMed] [Google Scholar]
- 63. Peden A. A., Rudge R. E., Lui W. W., Robinson M. S. (2002) Assembly and function of AP-3 complexes in cells expressing mutant subunits. J. Cell Biol. 156, 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jia D., Gomez T. S., Metlagel Z., Umetani J., Otwinowski Z., Rosen M. K., Billadeau D. D. (2010) WASH and WAVE actin regulators of the Wiskott-Aldrich syndrome protein (WASP) family are controlled by analogous structurally related complexes. Proc. Natl. Acad. Sci. U.S.A. 107, 10442–10447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wu L., Candille S. I., Choi Y., Xie D., Jiang L., Li-Pook-Than J., Tang H., Snyder M. (2013) Variation and genetic control of protein abundance in humans. Nature 499, 79–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bassett A. S., Scherer S. W., Brzustowicz L. M. (2010) Copy number variations in schizophrenia: critical review and new perspectives on concepts of genetics and disease. Am. J. Psychiatry 167, 899–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Doherty J. L., O'Donovan M. C., Owen M. J. (2012) Recent genomic advances in schizophrenia. Clin. Genet. 81, 103–109 [DOI] [PubMed] [Google Scholar]
- 68. Malhotra D., Sebat J. (2012) CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148, 1223–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rapoport J. L., Giedd J. N., Gogtay N. (2012) Neurodevelopmental model of schizophrenia: update 2012. Mol. Psychiatry 17, 1228–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stefansson H., Ophoff R. A., Steinberg S., Andreassen O. A., Cichon S., Rujescu D., Werge T., Pietiläinen O. P., Mors O., Mortensen P. B., Sigurdsson E., Gustafsson O., Nyegaard M., Tuulio-Henriksson A., Ingason A., Hansen T., Suvisaari J., Lonnqvist J., Paunio T., Børglum A. D., Hartmann A., Fink-Jensen A., Nordentoft M., Hougaard D., Norgaard-Pedersen B., Böttcher Y., Olesen J., Breuer R., Möller H. J., Giegling I., Rasmussen H. B., Timm S., Mattheisen M., Bitter I., Réthelyi J. M., Magnusdottir B. B., Sigmundsson T., Olason P., Masson G., Gulcher J. R., Haraldsson M., Fossdal R., Thorgeirsson T. E., Thorsteinsdottir U., Ruggeri M., Tosato S., Franke B., Strengman E., Kiemeney L. A., Melle I., Djurovic S., Abramova L., Kaleda V., Sanjuan J., de Frutos R., Bramon E., Vassos E., Fraser G., Ettinger U., Picchioni M., Walker N., Toulopoulou T., Need A. C., Ge D., Yoon J. L., Shianna K. V., Freimer N. B., Cantor R. M., Murray R., Kong A., Golimbet V., Carracedo A., Arango C., Costas J., Jonsson E. G., Terenius L., Agartz I., Petursson H., Nothen M. M., Rietschel M., Matthews P. M., Muglia P., Peltonen L., St Clair D., Goldstein D. B., Stefansson K., Collier D. A. (2009) Common variants conferring risk of schizophrenia. Nature 460, 744–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stefansson H., Meyer-Lindenberg A., Steinberg S., Magnusdottir B., Morgen K., Arnarsdottir S., Bjornsdottir G., Walters G. B., Jonsdottir G. A., Doyle O. M., Tost H., Grimm O., Kristjansdottir S., Snorrason H., Davidsdottir S. R., Gudmundsson L. J., Jonsson G. F., Stefansdottir B., Helgadottir I., Haraldsson M., Jonsdottir B., Thygesen J. H., Schwarz A. J., Didriksen M., Stensbøl T. B., Brammer M., Kapur S., Halldorsson J. G., Hreidarsson S., Saemundsen E., Sigurdsson E., Stefansson K. (2014) CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505, 361–366 [DOI] [PubMed] [Google Scholar]
- 72. Craddock N., Owen M. J. (2010) The Kraepelinian dichotomy: going, going. but still not gone. Br. J. Psychiatry 196, 92–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Moreno-De-Luca A., Myers S. M., Challman T. D., Moreno-De-Luca D., Evans D. W., Ledbetter D. H. (2013) Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. 12, 406–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mullin A. P., Gokhale A., Moreno-De-Luca A., Sanyal S., Waddington J. L., Faundez V. (2013) Neurodevelopmental disorders: mechanisms and boundary definitions from genomes, interactomes and proteomes. Transl. Psychiatry 3, e329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Karayiorgou M., Simon T. J., Gogos J. A. (2010) 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat. Rev. Neurosci. 11, 402–416 [DOI] [PMC free article] [PubMed] [Google Scholar]