

Background: Necroptosis is a regulated signaling pathway leading to necrotic cell death.
Results: Genetic mapping identified that down-regulation of the deubiquitinase CYLD confers resistance to necroptosis in a wild-derived mouse strain.
Conclusion: Different strains of inbred mice regulate cell death pathways using distinct mechanisms.
Significance: Genetic diversity of wild-derived mice underlies phenotypic diversity, which can identify novel mechanisms of regulation in cell death signaling.
Keywords: Deubiquitination, Gene Regulation, Mouse Genetics, Necrosis (Necrotic Death), Toll-like Receptor (TLR), Forward Genetic Mapping, Wild-derived Mice
Abstract
Pathogen recognition by the innate immune system initiates the production of proinflammatory cytokines but can also lead to programmed host cell death. Necroptosis, a caspase-independent cell death pathway, can contribute to the host defense against pathogens or cause damage to host tissues. Receptor-interacting protein (RIP1) is a serine/threonine kinase that integrates inflammatory and necroptotic responses. To investigate the mechanisms of RIP1-mediated activation of immune cells, we established a genetic screen on the basis of RIP1-mediated necroptosis in wild-derived MOLF/EiJ mice, which diverged from classical laboratory mice over a million years ago. When compared with C57BL/6, MOLF/EiJ macrophages were resistant to RIP1-mediated necroptosis induced by Toll-like receptors. Using a forward genetic approach in a backcross panel of mice, we identified cylindromatosis (CYLD), a deubiquitinase known to act directly on RIP1 and promote necroptosis in TNF receptor signaling, as the gene conferring the trait. We demonstrate that CYLD is required for Toll-like receptor-induced necroptosis and describe a novel mechanism by which CYLD is down-regulated at the transcriptional level in MOLF/EiJ macrophages to confer protection from necroptosis.
Introduction
Macrophages and other phagocytes contribute to the first tier of defense against an invading pathogen by producing proinflammatory cytokines such as TNF and IL-6 to potentiate activation of the innate immune system and prime B and T cell responses (1). As a result of excessive inflammation or as an alternative to it, programmed cell death pathways can be activated to cause apoptosis or necrosis through specific cell signaling pathways (2). Necroptosis (also called programmed necrosis) is a caspase-independent cell death mechanism underlying certain host-pathogen interactions. Necroptotic cell death has been shown to drive systemic inflammatory response syndrome and sepsis (3), whereas pathogens such as vaccinia virus and CMV block caspases via expression of caspase inhibitors, requiring necroptosis for efficient clearance by the host (4, 5). In each case, the ability for a host to control infection is determined, in part, by how effectively macrophages either activate an immune response or restrict the spread of infection by undergoing cell death.
To understand how macrophages regulate the balance between necroptosis and cytokine production, we examined signaling molecules shared by these pathways downstream of TLR2 activation. RIP1, a serine/threonine death domain-containing kinase, is directly involved in both inflammatory and necroptotic responses, and posttranslational modification of RIP1 through multiple types of ubiquitination (6) and caspase 8-mediated cleavage (7–9) regulates its activation. It is well established in the context of TNF receptor 1 (TNFR1) that signaling RIP1 modified by lysine 63-linked polyubiquitination and linear ubiquitination promotes signaling to NF-κB, activating prosurvival and proinflammatory signals as a scaffold protein without the use of its kinase activity (10, 11). By contrast, deubiquitinated RIP1 is a prodeath molecule that triggers RIP1 kinase-dependent necroptosis through RIP3 and the pseudokinase mixed-lineage kinase domain-like (10, 12–15). A deubiquitinase that drives necroptosis is CYLD, which specifically removes lysine 63 and linear ubiquitin chains from its targets (16). CYLD promotes necroptosis through its deubiquitinase activity on RIP1 (17–19), and cleavage to inactivate CYLD is a critical regulatory function of caspase 8 to prevent necroptosis (18). Chemical blockade of caspase activity or down-regulation of caspase 8 predisposes cells to die by necroptosis because of the loss of the inhibitory activity of caspase 8 on CYLD and RIP1 (20–22).
In a forward genetic approach, we used the wild-derived inbred MOLF/EiJ (MOLF) mouse strain to interrogate RIP1 function and regulation of necroptosis in this mouse strain when compared with a classical inbred mouse strain such as C57BL/6 (B6). Our work is part of an ongoing effort to genetically map regulators of the inflammatory response in wild-derived mice. This mouse model, which is an evolutionarily divergent representative of the Mus musculus molossinus subspecies, allows us to exploit a reservoir of diverse phenotypes in the context of innate immune signaling (23–25). Furthermore, in some instances, MOLF inflammatory responses more closely resemble human responses downstream of TLRs, rendering this model highly relevant to human health (26). Our approach allows for the discovery of naturally occurring phenotypic differences between wild-derived strains and classical inbred strains.
To identify mechanisms by which necroptosis is regulated in wild-derived mice, we established an unbiased genetic screen that is dependent on RIP1 kinase activity. Macrophages from the wild-derived MOLF/Ei mouse strain were resistant to TLR-mediated necroptosis compared with C57BL/6 (B6). Linkage analysis identified Cyld as the gene conferring resistance of MOLF macrophages to necroptosis. In MOLF macrophages, we describe a novel mechanism of acute down-regulation of CYLD through differential pausing or stalling of RNA polymerase II (Pol II) at the Cyld promoter, thereby resulting in low levels of CYLD at the mRNA and protein levels in activated MOLF macrophages. Such down-regulation of this pronecroptotic protein as a result of decreased transcription of CYLD prevents necroptosis in MOLF peritoneal macrophages.
EXPERIMENTAL PROCEDURES
Mice and Isolation of Primary Cells
The C57BL/6, MOLF/Ei, TNFR1−/−(C57BL/6-Tnfrsf1atm1Imx), IL1R1−/−(B6.129S7-Il1r1tm1Imx), and NOD2−/−(B6.129S1-Nod2tm1Flv) strains were obtained from The Jackson Laboratory. Mice were housed in a pathogen-free facility at Tufts University School of Medicine. All experiments with mice were performed in accordance within regulations and with the approval of the Tufts University Institutional Animal Care and Use Committee. Peritoneal macrophages were isolated from 6- to 10-week-old mice. Mice were injected with 1 ml of 3% thioglycollate, and cells were collected by peritoneal lavage with cold PBS. Cells were resuspended, plated, and stimulated 4 h after plating in DMEM with 10% FBS (Atlas Biologicals and Atlanta Biologicals), 1% Pen-Strep (Invitrogen) at 37 °C in 5% CO2. Bone marrow cells were isolated by washing femurs with cold RPMI medium. Cells were centrifuged for 10 min at 1000 rpm. Cell pellets were resuspended in BMDM differentiating medium (RPMI medium with l-glutamine, 20% FBS, 28% L-cell conditioned medium, and 1% Pen-Strep) and cultured for 7–8 days at 37 °C in 5% CO2 to mature to macrophages.
Cell Lines
HEK293 and L929 cells were obtained from Dr. R. Isberg (Tufts University) and grown in DMEM, 10% FBS, and 1% Pen-Strep or RPMI medium, 10% FBS, and 1% Pen-Strep, respectively. Immortalized RIP1−/− fetal liver-derived macrophages and immortalized RIP3−/− BMDM were obtained from Dr. K. Fitzgerald (University of Massachusetts) and grown in DMEM, 10% FBS, and 1% Pen-Strep. RAW264.7 macrophages were obtained from the ATCC and cultured in DMEM, 10% FBS, and 1% Pen-Strep.
Reagents
Salmonella minnesota Re595 LPS was purchased from Enzo Biochem Inc. Poly I:C-LMW, Imiquimod (R837), CL-097, and lipoteichoic acid (purified from Staphylococcus aureus) were purchased from Invivogen. CpG oligodeoxynucleotides were synthesized by Integrated DNA Technologies. Z-VAD(OMe)-FMK was purchased from Bachem (catalog no. N-1560). Necrostatin 1 was purchased from Sigma. SM164 was obtained from Dr. A. Degterev (Tufts University). GAPDH antibody (catalog no. 2118) was purchased from Cell Signaling Technology. CYLD antibody (clone E10) was purchased from Santa Cruz Biotechnology.
Genetic Mapping and Analysis
Peritoneal macrophages were elicited as described above from N2 or F2 mice. Genomic DNA was isolated from all mouse tails using a DirectPCR (Viagen) or DNeasy blood and tissue kit (Qiagen). Genome-wide genotyping was performed by PCR of known microsatellite markers per chromosome using primers obtained from Jackson Laboratories Mouse Genome Informatics. Genotyping PCR reactions were performed using JumpStart Red Taq (Sigma), and PCR products were electrophoresed on 3.5% agarose gels. Quantitative trait locus analysis was performed using QTXb software.
DNA Sequencing
Genomic DNA or cDNA was amplified using Phusion high-fidelity DNA polymerase (New England BioLabs). PCR products were electrophoresed on a 1.2% agarose gel and extracted using a QIAquick gel extraction kit (Qiagen). DNA was sequenced from gene-specific primers using an ABI 3130XL DNA sequencer at the Tufts University Core Facility.
Phenotypic Assays
Peritoneal macrophages were plated in 96-well plates at a density of 5 × 104 cells/well. Cell viability was measured 18 h post-stimulation using a CellTiter-Glo luminescence ATP viability assay (Promega), and data were presented as a percentage of the signal from treated cells to cells in medium with vehicle (DMSO). For ELISA, BMDM were plated at a density of 1 × 105 cells/well in a 96-well plate or 2 × 105 cells/well in a 48-well plate. After stimulation, TNF protein concentrations measured from cell-free supernatants using ELISA kits purchased from R&D Systems (catalog no. DY410).
RNA Preparation and Analysis
Cells were lysed with TRIzol (Invitrogen) or TriPure (Roche) isolation reagent, and RNA was extracted according to the instructions of the manufacturer. For 5′ rapid amplification of cDNA ends, mRNA was extracted and amplified using the GeneRacer kit (Invitrogen) according to the protocol of the manufacturer. A gene-specific primer within CYLD was used for initial RNA priming (CACAGTGTCCAGGGCAGAACTAAAAGCA), and the GGCCCTGGATGCCTTTCTTCTTTCCA and TTAGGTGGAGTATTTTCTTCTACTAC primers were used to amplify CYLD mRNA species in combination with forward primers amplifying from the RNA oligo added by the GeneRacer kit. A ribonuclease protection assay was performed using the RPA II ribonuclease protection assay kit (Ambion) according to the protocol of the manufacturer. Primers used to generate the probe for RPA were as follows: CYLD, GTCTAGCGAGAACAGATTCCAC (forward) and ATCTCTAAACCTTCCTTTTCCA (reverse). For Northern blot hybridization, total RNA was electrophoresed on a 1.2% agarose gel, transferred to a Hybond-N membrane (Amersham Biosciences-GE) by the capillary method. After prehybridization with QuikHyb (Stratagene), the membrane was hybridized with a Cyld-specific radioactive probe amplified using the primers GATTCCTTTTAGCAGAGCAG (forward) and AATTGTACTTTCAACACTTGC (reverse) and exposed to film overnight. To generate cDNA, reverse transcription was performed using M-MuLV reverse transcriptase, RNase inhibitor, random primers 9 (for qPCR) or oligo(dT) (for cloning), and dNTPs (New England BioLabs). cDNA was analyzed for mRNA expression levels using SYBR Green and TaqMan probe-based gene expression analysis with the following custom, full-length, specific CYLD primer/probe: GCTCAGTGACGTGCTAGCT (forward) and GCGTGCCCCTGAAAGTTC (reverse), Probe(FAM)-CTGGAACTGGAAGATGAA (Applied Biosystems). A Gapdh-specific TaqMan probe (catalog no. 4352339) was used to normalize mRNA expression.
Protein Lysis and Western Blot Analysis
To prepare whole-cell lysates, cells were lysed on ice with a cytoplasmic lysis buffer (50 mm Tris (pH 8), 150 mm NaCl, 2 mm EDTA, 1% Triton X-100, 1 mm sodium vanadate, and 10 mm NaF) supplemented with Halt protease inhibitor mixture (Thermo Fisher Scientific) for 10 min. Lysates were centrifuged for 10 min at 13,000 rpm at 4 °C. Cleared lysates were mixed with Laemmli sample buffer and boiled for 10 min. Protein lysates were resolved on a 4–12% gradient BisTris SDS gel (NuPAGE, Invitrogen) and transferred to a nitrocellulose membrane. After incubation with protein-specific antibodies, chemiluminescence was detected using ECL substrate (Thermo Fisher Scientific).
Lentiviral Knockdown/Overexpression
shRNA plasmids in the pLKO.1 vector were purchased from Open Biosystems (Thermo Scientific). A GFP-targeting shRNA hairpin was used as a control (Addgene, catalog no.12273). The Cyld-targeting shRNA vector was clone ID TRCN0000030991. The CYLD overexpression construct was cloned from cDNA into a pLEX lentiviral expression vector. Lentiviral particles were generated by transfecting the shRNA expression construct (pLKO.1 construct) with the packaging constructs psPAX2 (lentivirus packaging) and pMD2.G (VSV G) into HEK293 cells using X-tremeGENE9 DNA transfection reagent (Roche). Supernatants were collected 48 and 72 h after transfection, pooled, and filtered using a 45-μm filter. BMDM and RAW264.7 cells were transduced by incubation with lentiviral supernatant for 24 h. Infected clones were selected by culturing with 3 μg/ml (BMDM) or 5 μg/ml (RAW) puromycin for 48–72 h.
siRNA Transfection
Following peritoneal lavage, macrophages were plated and allowed to adhere for 4 h prior to transfection. Peritoneal macrophages were transfected with 100 nm siRNA using the N-TER nanoparticle siRNA transfection system (Sigma). Cells were transfected for 24 h before cell death assays were initiated. Gene-specific and control siRNA duplexes were obtained from Integrated DNA Technologies with the following sequences: siCYLD, GGUUUAGAGAUAAUGAUUGGAAAGA; negative control, CGUUAAUCGCGUAUAAUACGCGUAT.
Chromatin Immunoprecipitation
To prepare chromatin, 1 × 107 peritoneal macrophages from MOLF and B6 were stimulated with LPS for 2 h. Cells were cross-linked with 1% formaldehyde for 10 min at room temperature. The reaction was stopped by adding glycine to a final concentration of 0.125 m and incubating for 5 min. After washing twice with cold PBS, cells were resuspended and centrifuged at 2000 rpm for 5 min at 4 °C. Cells were rinsed once with 1 ml of PBS with protease inhibitors and then resuspended in cold lysis buffer (5 mm PIPES (pH 8), 85 mm KCl, and 0.5% Nonidet P-40) and incubated for 10 min on ice. After centrifugation at 5000 rpm for 5 min at 4 °C to pellet the nuclei and discard the supernatant, the nuclei were resuspended in nucleus lysis buffer (50 mm Tris-Cl (pH 8.1), 10 mm EDTA, and 1% SDS) with protease inhibitors and incubated on ice for 10 min. Samples were then diluted with ChIP dilution buffer (1% Triton X-100, 2 mm EDTA, 150 mm NaCl, and 20 mm Tris-HCl (pH 8.1)) and protease inhibitors. Chromatin was sonicated with a Branson 350 sonifier. After sonication, samples were microfuged at 14,000 rpm for 10 min at 4 °C, and supernatants were kept. Lysates were precleared with agarose beads for 1–2 h. After microfugation to remove agarose, a small fraction of supernatant was kept for input DNA control. Agarose·anti-RNA Pol II antibody N-20 (catalog no. sc-899X) complex was added to the sample, diluting the SDS to 0.1% using ChIP dilution buffer, and incubated overnight at 4 °C. The following day, samples were washed for 30 min in wash buffer 1 (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-Cl (pH 8), and 150 mm NaCl), for 30 min in wash buffer 2 (identical to wash buffer 1 but with 500 mm NaCl), and for 30 min in radioimmune precipitation assay buffer (50 mm HEPES (pH 7.6), 1 mm EDTA, 0.7% sodium deoxycholate, 1% Nonidet P-40, and 0.5 m LiCl). The three wash steps were repeated. Samples were washed twice with 1 ml of cold Tris-EDTA (pH 7.6). Next, 100 μl of 1% SDS and 0.1 m NaHCO3 were added to the ChIP sample and input control and incubated for 30 min at room temperature. Solutions were then incubated for 9 h at 65 °C, purified with a QIAquick spin kit (Qiagen), treated with RNase mixture, and the DNA amounts in the input and immunoprecipitation were quantified using Nanodrop 2000 (Thermo Scientific). Quantitative PCR was performed using SYBR Green (as described above) using primers in the Cyld promoter with the following sequences: (−700), AGGTAGAGGGTGGAAAGC (forward) and CTCTTTCTGTCCTCAGATGG (reverse; (−300), CCCACCCTAGTTTTGACAGC (forward) and GCCAGTGTCATGTCACCATT (reverse); (TSS), CGGATCTCTGAATACGATTGG (forward) and CAGCCGAACCTGCTACCTG (reverse); (+300), ACGAGCTTCTGACGTCAACC (forward) and CCCTGTGGTGCAGGAGTC (reverse); and (+700), TTATGGTCAGCCTTGGAGGA (forward) and GCAGCTGACCCAGATGAACT (reverse).
Statistical Analysis
Values are presented as mean ± S.E. or S.D. Statistical significance was determined using Student's t test.
RESULTS
Establishing a Genetic Screen for TLR-induced Necroptosis
Necroptosis is a caspase-independent cell death pathway activated during host-pathogen interactions and can be activated by RIP homotypic interaction motif domain-containing TLR3 and TLR4 in the presence of caspase inhibitors (21, 27). To investigate whether MyD88-dependent TLRs can also mediate necroptosis, B6 thioglycollate-elicited peritoneal macrophages were stimulated ex vivo with synthetic TLR agonists (TLR3, polyI:C, TLR7, imiquimod (IMQ), or CL-097) or purified bacterial components (TLR2, lipoteichoic acid, TLR4, or LPS) in the presence of the pan-caspase inhibitor zVAD. In keeping with previous findings (21, 27, 28), TLR3 and TLR4 induced RIP1 kinase-dependent necroptosis (Fig. 1A). TRIF-independent, MyD88-dependent TLR2, TLR7, and TLR9 also activated caspase-independent cell death, which was blocked by the specific inhibitor of RIP1 kinase activity Necrostatin 1 (Nec-1) (17).
FIGURE 1.
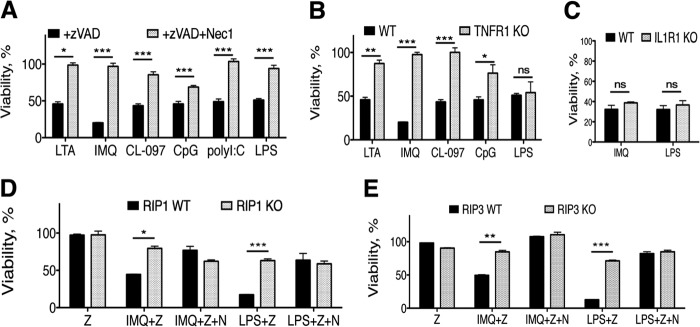
TLR-induced necroptosis in macrophages can be cell-intrinsic or require TNFR1. Shown are B6 (A), TNFR1−/− (B), or IL1R1−/− (C) peritoneal macrophages stimulated for 18 h with lipoteichoic acid (LTA) (2 μg/ml), IMQ (5 μg/ml), CL-097 (1 μg/ml), CpG (200 nm), polyI:C (10 μg/ml), or LPS (100 ng/ml) in the presence of zVAD (12.5 μm) with or without the RIP1 kinase inhibitor Nec-1 (30 μm). Immortalized macrophage cell lines lacking RIP1 (D) or RIP3 (E) were stimulated with TLR agonists in the presence of zVAD (Z) (100 or 50 μm, respectively) with or without Nec-1 (N). Cell viability was measured by ATP levels using the CellTiter-Glo assay (Promega). Values are a percentage of viable treated cells relative to cells in medium with vehicle (DMSO), shown as mean ± S.E. Shown is one representative experiment of at least three independent experiments. Statistical significance was determined by two-tailed Student's t test. *, p < 0.05; **, p < 0.001; ***, p < 0.0001; ns, not significant.
To determine whether an intrinsic signaling cascade mediates MyD88-dependent necroptosis, like TRIF-dependent necroptosis, or whether it requires signaling through the TNF receptor (TNFR1), we stimulated TNFR1−/− peritoneal macrophages with TLR agonists in the presence of zVAD. TLRs that are entirely MyD88-dependent (TLR2, TLR7, and TLR9) required TNFR1 to induce necroptosis, whereas TLRs that can engage TRIF did not require TNFR1 (Fig. 1B). IL-1R1 deficiency had no impact on cell death (Fig. 1C). Because TRIF-dependent necroptosis is activated via an intrinsic mechanism, whereas activation of MyD88-dependent necroptosis requires TNFR1, these data suggest that autocrine production of TNF is necessary for MyD88-dependent, but not for TRIF-dependent, TLR-mediated necroptosis, as described recently using TNF-deficient macrophages (27).
Necroptosis can be activated in the presence (12) or absence (4) of RIP1. To determine whether TLR-mediated necroptosis required RIP1 and RIP3, macrophage cell lines deficient in these genes were stimulated with TLR agonists in the presence of zVAD. Both RIP1−/− and RIP3−/− macrophages were protected from necroptosis induced by MyD88- and TRIF-dependent TLRs (Fig. 1, D and E). These data demonstrate that TLR stimulation in the presence of caspase inhibitors requires both RIP1 kinase activity and RIP3 to induce necroptotic cell death, in agreement with previous reports (21, 27). Although TLRs that signal through the adapter MyD88 require TNFR1, TRIF-mediated necroptosis does not (27). Although the TNRF1- and TRIF-dependent signaling cascades differ, they likely share many regulatory and effector mechanisms. Furthermore, these data establish a molecular basis for a novel genetic screen we employed in primary mouse macrophages.
RIP1 Kinase Activity Promotes Necroptosis in B6 and Cytokine Production MOLF Macrophages
Next, we tested whether macrophages from parental strains (B6 and MOLF) displayed a different susceptibility to necroptosis induced by TLRs. As we reported previously, MOLF macrophages are more robust producers of some proinflammatory cytokines, such as IL-6, compared with B6 macrophages because of preferential expression of the proinflammatory isoform of IRAK2 (29). However, the MOLF signaling network incorporates negative regulators of TLR signaling that are not expressed in B6 macrophages, such IRAK1BP1, to dampen some of these proinflammatory effects (24, 30). Because of their genetic diversity, proinflammatory disposition, and distinct TLR signaling network, we examined the susceptibility of MOLF macrophages to TLR-induced necroptosis under conditions in which macrophages from the classical inbred strain B6 undergo cell death. Specifically, peritoneal macrophages were activated with the TLR4 agonist LPS or the TLR7 agonist IMQ in the presence or absence of zVAD. Although B6 macrophages were susceptible to cell death induced by TLR stimulation in the presence of zVAD, MOLF macrophages were resistant (Fig. 2A). Importantly, this difference was dependent on the kinase activity of RIP1 because kinase inhibition with Nec-1 significantly increased survival of B6. Macrophages from either strain activated without zVAD showed no decrease in viability. Furthermore, peritoneal macrophages from F1 (B6xMOLF) hybrids showed intermediate susceptibility, suggesting a codominant inheritance pattern amenable to classical genetic analysis (Fig. 2B).
FIGURE 2.
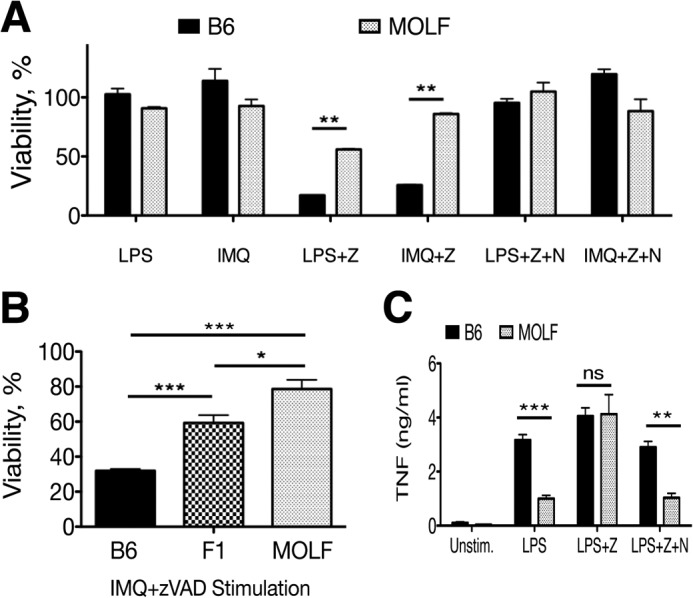
MOLF peritoneal macrophages are resistant to TLR-induced necroptosis. A, B6 and MOLF peritoneal macrophages stimulated for 18 h with LPS (100 ng/ml) or IMQ (5 μg/ml) with and without zVAD (Z) (12.5 μm) and Nec-1 (N) (30 μm). B, B6, F1 (B6xMOLF), and MOLF peritoneal macrophages stimulated with IMQ and zVAD. Cell viability was measured relative to cells kept in medium with vehicle (DMSO). C, B6 and MOLF bone marrow-derived macrophages were stimulated with LPS (10 ng/ml) with and without zVAD (25 μm) and with or without Nec-1. Unstim, unstimulated. TNF production was measured by ELISA after 18 h. Shown are means for biological triplicates in a representative experiment. Error bars are mean ± S.E. (A and B) or S.D. (C). Data are representative of at least three independent experiments. Statistical significance was determined by two-tailed Student's t test. *, p < 0.05; **, p < 0.001; ***, p < 0.0001; ns, not significant.
Next, we assessed the contribution of RIP1 kinase to cytokine responses of LPS-stimulated B6 and MOLF macrophages. In response to stimulation with LPS, B6 BMDM produced twice as much TNF as MOLF BMDM. This difference was abrogated upon addition of zVAD, which increased TNF production in MOLF to the same level as B6 (Fig. 2C). However, addition of the RIP1 kinase inhibitor Nec-1 reversed the effect of zVAD in MOLF while only slightly reducing B6 TNF production, suggesting a larger contribution of RIP1 kinase activity to cytokine production in MOLF than in B6, which could be relevant to improved survival of MOLF macrophages.
Notably, the differential effect of RIP1 on necroptosis or TNF production was observed only in the presence of zVAD. Because Nec-1 had a minor effect on MOLF macrophage survival but dramatically increased B6 viability, we conclude that the kinase activity of RIP1 has a more prominent role in promoting necroptosis in B6 than MOLF macrophages. In contrast, the kinase activity of RIP1 affects TNF production in MOLF but not B6 macrophages, suggesting different roles for RIP1 between the strains during inflammatory responses. In B6, the engagement of RIP1 as a kinase leads to necroptosis, whereas in MOLF, it functions to promote TNF production. Taken together, these data lead us to propose a model in which B6 and MOLF employ RIP1 kinase activity differently, which can be used to map genetic traits that require RIP1 kinase activity.
Resistance to Necroptosis in MOLF Macrophages Is Linked to Cyld
Forward genetic analysis provides the opportunity to uncover genes that contribute to a given trait using an unbiased approach. We mapped the resistance to necroptosis trait on a panel of 67 N2 mice (B6x(B6xMOLF)) by using peritoneal macrophages stimulated with the TLR7 agonist imiquimod and zVAD to induce necroptosis. All phenodeviants were genotyped across the entire genome, and the association between the genotype and the phenotype, the logarithm of odds, was calculated. Resistance to necroptosis was mapped to two distinct loci, confirming that this phenotype is a complex trait conferred by multiple genes and is subject to quantitative trait linkage analysis. A locus on chromosome 8 demonstrated the strongest linkage to the trait (logarithm of odds = 6.7), and the MOLF allele at a marker within the region of the highest linkage (D8Mit51) conferred resistance to cell death (Fig. 3A). The second locus (logarithm of odds = 2.89) is located on chromosome 1 in a 38-Mb region, and the MOLF allele also promotes resistance to necroptosis.
FIGURE 3.

Resistance to necroptosis in MOLF peritoneal macrophages is linked to Cyld. A, viability of peritoneal macrophages from 67 N2 mice after stimulation with IMQ+zVAD grouped by genotype at D8Mit51 (marker within the peak of highest linkage). B, NOD2−/− and B6 (WT) peritoneal macrophages were stimulated for 18 h with IMQ (5 μg/ml) or LPS (100 ng/ml) and zVAD (Z) (12.5 μm) ± Nec-1 (N) (30 μm). Viability was measured by CellTiter-Glo ATP assay. C, B6 and MOLF peritoneal macrophages stimulated for 8 h with IMQ (5 μg/ml), IMQ+zVAD (12.5 μm), or IMQ+zVAD+Nec-1 (30 μm). TNF production in supernatants was measured by ELISA. Unstim, unstimulated. D, B6 and MOLF peritoneal macrophages stimulated for 18 h with LPS (100 ng/ml) or IMQ (5 μg/ml) in the presence or absence of the SM164 (SM) (100 nm), zVAD (12.5 μm), and Nec-1 (30 μm). E, peritoneal macrophages from 20 F2 mice were stimulated for 4 h with LPS (100 ng/ml). CYLD mRNA levels were measured by qPCR. Samples from each mouse were sorted by genotype at D8Mit51. Error bars are mean ± S.E. (B and D) or S.D. (C). Data from two (D) or three (B and C) independent experiments were pooled, with statistical significance determined by two-tailed Student's t test. *, p < 0.05; ***, p < 0.0001; ns, not significant.
To establish a list of candidate genes within the locus on chromosome 8, we further narrowed the genomic area of interest using additional microsatellite markers. We considered candidate genes that were expressed in macrophages and prioritized the examination of genes with a known connection to cell death or innate immunity. The linked 5.1-Mb region, flanked by the microsatellite markers D8Mit238 and D8Mit81, contains the candidate genes, Nod2 and Cyld, adjacent genes separated by 8.5 kilobases. Nod2 encodes a pattern recognition receptor that senses bacterium-derived muramyl dipeptide (31) and has been described to have functions in autophagy and inflammasome activation (32). Nod2 knockout mice on a B6 background showed no change in susceptibility to TLR-induced necroptosis, suggesting that Nod2 is not the gene conferring this trait (Fig. 3B). The Nod2 knockout results, together with the mapping data, allowed us to focus on the deubiquitinase Cyld as the main gene candidate. Importantly, other critical mediators of necroptosis, such as Ripk1, Ripk3, and Mlkl, are not located within the regions of linkage on chromosomes 1 or 8, allowing us to dismiss intrinsic, cis-acting differences in these genes from conferring resistance to necroptosis in MOLF because we did not map to the loci in which these genes reside.
Necroptosis activated by the TLR7 agonist imiquimod relies on TNFR1 for activation (Fig. 1B), making it possible that a difference in autocrine TNF production by each strain of macrophages underlies the differences in activation of necroptosis. To rule out this possibility, we measured levels of TNF production from peritoneal macrophages of each strain after imiquimod stimulation. In both the presence and absence of zVAD, MOLF macrophages produced TNF levels similar to B6 macrophages (Fig. 3C), suggesting that a different degree of autocrine TNF signal is not the likely cause of a difference in susceptibility to necroptosis between the strains.
To further investigate at which point in the pathway MOLF necroptosis signaling differs from B6, we treated activated cells with a Smac mimetic, SM164 (33), that degrades the E3 ubiquitin ligases cellular inhibitor of apoptosis 1 and 2 (cIAP1/2), which ligate Lys-63-linked polyubiquitin chains to RIP1 (10). Depletion of cIAP1/2 with SM164 sensitizes cells to necroptosis (28, 34). In macrophages, SM164 treatment abrogated (LPS+Z) or substantially reduced (IMQ+Z) the difference between strains in resistance to necroptosis (Fig. 3D). These data demonstrate that MOLF peritoneal macrophages can become sensitized to necroptosis by disrupting enzymes that control RIP1 ubiquitination. Taken together with the dependence of the trait on RIP1 kinase activity, as evidenced by the loss of a difference between the strains by blocking this activity with Nec-1, these data strongly suggest that the gene conferring resistance in MOLF exerts its effect at or upstream of the level of RIP1 and cIAP1/2.
The deubiquitinase CYLD, the gene for which is located within the genomic region linked to MOLF resistance to necroptosis, is known to promote necroptosis downstream of TNFR1 (20) and exerts its effect directly upon RIP1 (18). On the basis of the data showing that depletion cIAP1/2, which presumably reduces RIP1 ubiquitination, promotes susceptibility to TLR-induced necroptosis in MOLF, we hypothesized that low levels of CYLD in MOLF macrophages would prevent deubiquitination of RIP1 and result in less necroptosis activation. In a panel of F2 mice (an intercross of B6×MOLF F1 mice), we show that CYLD mRNA levels are significantly lower after 4 h of LPS stimulation in F2 mice containing a homozygous MOLF Cyld allele (in close proximity to D8Mit51) when compared with mice homozygous for the B6 allele (Fig. 3E). Although contribution of other genes may impact CYLD expression, these data show that a cis-acting element in the MOLF Cyld locus is sufficient to cause decreased expression of this deubiquitinase.
CYLD Promotes TLR-induced Necroptosis
To investigate whether CYLD knockdown in peritoneal macrophages can prevent TLR-induced necroptosis, we used small interfering RNA to deplete CYLD protein levels in B6 peritoneal macrophages. A double-stranded siRNA targeting CYLD or a negative control siRNA were transfected into B6 peritoneal macrophages, followed by activation with LPS or imiquimod in the presence of zVAD. Knockdown of CYLD was sufficient to reduce susceptibility to necroptosis (Fig. 4A). Primary peritoneal macrophages are difficult to efficiently transfect and have a short life span outside of the mouse. However, compared with negative control siRNA, we achieved nearly 40% knockdown at the protein level (Fig. 4B) and 75% knockdown at the RNA level (Fig. 4C) with CYLD-targeting siRNA. This partial knockdown explains the significant, but not complete, rescue of cell viability in B6 peritoneal macrophages. The observation that the difference between strains in susceptibility to necroptosis can be diminished by the inhibition of cIAP1/2 that ubiquitinate RIP1 (Fig. 3D) further supports the hypothesis that an enzyme that modifies ubiquitin, such as CYLD, underlies the phenotype in MOLF macrophages.
FIGURE 4.

CYLD promotes TLR-induced necroptosis. A, CYLD siRNA and control (NC) siRNA were transfected into peritoneal macrophages, followed by stimulation with LPS or IMQ and zVAD (Z). Knockdown efficiency was assessed by Western blotting for CYLD relative to GAPDH in unstimulated cells (B) and by qPCR after 4 h of LPS stimulation (C). D, RAW264.7 macrophages were infected with lentivirus containing full-length (FL) CYLD or an empty vector and stimulated with IMQ or LPS and zVAD (50 μm) ± Nec-1 (N) (30 μm). E, overexpression level of FL-CYLD measured by qPCR relative to empty vector. F, TNFR−/− BMDM infected with lentivirus containing short hairpin RNA targeting GFP or CYLD stimulated with LPS (100 ng/ml) or polyI:C (200 μg/ml) with zVAD (25 μm). G, Western blotting for CYLD protein expression for samples in F. For all experiments, cell viability was measured at 18 h relative to cells kept in medium with vehicle (DMSO). Data shown are the mean ± S.E. pooled from six (A), two (D), or three (F) pooled, independent experiments. Statistical significance was determined by two-tailed Student's t test. *, p < 0.05; ***, p < 0.0001.
To demonstrate that CYLD overexpression promotes TLR-mediated necroptosis in a cell line derived from peritoneal macrophages, we expressed a lentiviral vector containing CYLD in RAW264.7 macrophages and observed an increase in TLR-induced necroptosis in cells overexpressing CYLD at mRNA levels 2.5-fold higher than controls (Fig. 4, D and E). Although it is possible that CYLD overexpression bypasses the requirement for a TLR signal, we observe a significant decrease in cell viability in LPS+zVAD- and IMQ+zVAD-treated cells relative to untreated controls, suggesting that CYLD promotes TLR-induced cell death.
The role of CYLD in promoting necroptosis has been elucidated exclusively with respect to TNFR1-mediated signaling (18), whereas its role in TLR-mediated necroptosis is unknown, despite controversial reports on negative regulation of TLR-mediated signaling by CYLD (35, 36). We (Fig. 1B) and others (27) have shown that MyD88-dependent TLRs require autocrine TNF signaling for initiation of necroptosis, whereas engagement of the TRIF adaptor by TLR4 and TLR3 overcomes such a requirement. To investigate whether CYLD positively regulates TRIF-dependent (TLR3/TLR4) mediated necroptosis, we performed knockdown of CYLD with short hairpin RNA introduced through lentiviral infection in TNFR1-deficient BMDM. Stimulation with LPS or polyI:C and zVAD resulted in cell death in the control siGFP-infected cells, whereas CYLD knockdown was able to substantially, albeit not completely, protect these cells from necroptosis (Fig. 3G). The efficiency of CYLD knockdown at the protein level was assessed by Western blot analysis (Fig. 3H).
Without TNFR1, autocrine TNF/TNFR1 signaling is absent, and cell death is activated only through intrinsic signaling through TLR3 or TLR4. Here we demonstrate that CYLD promotes TLR3- and TLR4-mediated necroptosis directly, implicating CYLD-mediated deubiquitination of RIP1 as a critical regulatory step in TLR-induced necroptosis in addition to its known role in TNF-mediated cell death. However, because we do not see a complete rescue of cell viability in CYLD knockdown cells, we cannot rule out a role for other deubiquitinases in addition to CYLD to drive TRIF-dependent necroptosis.
MOLF Macrophages Acutely Down-regulate CYLD
To further investigate how the MOLF Cyld locus protects from TLR-induced necroptosis in peritoneal macrophages, we sequenced cDNA in the protein-coding region but found no coding polymorphisms. Next, we compared CYLD expression in B6 and MOLF peritoneal macrophages, reasoning that, because CYLD is a tightly regulated gene, non-coding differences affecting mRNA or protein expression, mRNA splicing, or stability might be responsible for the phenotype (37). Two isoforms of CYLD generated by alternative splicing have been described in spleens of B6 mice, a full-length isoform and a splice isoform lacking exons 7 and 8 (38). The splice isoform is generated by alternative splicing of exon 6 to exon 9, which produces an mRNA lacking exons 7 and 8 but does not introduce a frameshift in the coding region. Interestingly, the protein domains of CYLD coded from exons 7 and 8 are required for the interaction of CYLD with important binding partners such as NF-κB essential modifier and TRAF2/6 (38, 39).
To assess whether alternative splicing between the full-length and exon7/8 splice isoform of CYLD differentially occurs in B6 and MOLF mice, we performed 5′ rapid amplification of cDNA ends on unstimulated and LPS-stimulated peritoneal macrophages. B6 macrophages expressed mRNA from the full-length and splice isoforms in unstimulated and LPS-stimulated cells, whereas MOLF macrophages expressed the full-length isoform in unstimulated cells and lose expression of this isoform after LPS stimulation, instead producing the splice isoform (Fig. 5A). However, the splice isoform was not detected by Western blotting in either strain, and this isoform does not appear to be expressed at the protein level in macrophages. Alternative splicing of CYLD mRNA could be an interesting aspect of regulation between the strains, even in the absence of splice isoform protein expression. However, it is beyond the scope of this report. Here we focus on characterizing the down-regulation of the full-length, functional isoform of CYLD in MOLF peritoneal macrophages.
FIGURE 5.

CYLD is down-regulated by TLR stimulation in MOLF peritoneal macrophages. A–D, RNA extracted from B6 and MOLF peritoneal macrophages. A, CYLD-specific PCR and sequencing of clones obtained from 5′ rapid amplification of cDNA ends shows full-length (FL) and splice (SP) CYLD isoforms in unstimulated (UN) or 4-h LPS-stimulated (100 ng/ml) cells. B, ribonuclease protection assay using CYLD-specific (top row) and GAPDH-specific (bottom row) probes on RNA samples from macrophages stimulated for 1, 2, and 4 h with LPS (100 ng/ml). Shown is the relative amount (Rel Amt) of CYLD/GAPDH in each sample as determined by densitometry. C, quantitative RT-PCR was performed using a full-length, CYLD-specific TaqMan probe after LPS stimulation (4 h). Values are presented as relative units (RU) normalized to GAPDH. D, Northern blot on RNA samples of macrophages stimulated for 4 h with LPS (100 ng/ml) or IMQ (5 μg/ml) using a CYLD-specific probe. 28 S rRNA ethidium bromide staining was the loading control. E, B6 and MOLF peritoneal macrophages unstimulated or stimulated with LPS with zVAD (LPS+Z) for 6 h. Lysates were analyzed by Western blot analysis for CYLD protein levels with GAPDH as a loading control. Densitometry measured the fold change of the CYLD/GAPDH ratio relative to unstimulated levels within each mouse strain. F, LPS-stimulated peritoneal macrophages were cross-linked and lysed, and DNA was sheared by sonication. RNA polymerase II ChIP was performed. Quantitative real-time PCR was performed at locations in the Cyld promoter indicated in reference to the TSS. Shown is the mean ± S.E. (C and F) of pooled data from three independent experiments (C) or from one representative experiment (F) (of two independent experiments). Statistical significance was determined by Student's t test. *, p < 0.05; **, p < 0.001.
Accordingly, we used a ribonuclease protection assay to monitor the presence of CYLD mRNA in B6 and MOLF peritoneal macrophages using a full-length CYLD-specific probe. After stimulation with a time course of LPS, the amount of CYLD mRNA showed a modest increase relative to GAPDH expression (Fig. 5B). Remarkably, LPS stimulation of MOLF macrophages caused a depletion of CYLD mRNA levels after 2 h and left less than 20% of the starting amount after 4 h.
To quantitatively assess differential expression of CYLD transcripts in peritoneal macrophages, we used an isoform-specific TaqMan qPCR probe spanning exons 7 and 8 and found that MOLF macrophages down-regulated CYLD after LPS stimulation, which was in striking contrast with B6 cells that significantly up-regulated CYLD (Fig. 5C). Combined with data from the RNase protection assay, we established that MOLF macrophages acutely down-regulate the mRNA that encodes the full-length isoform of CYLD upon stimulation with LPS. To confirm the size and levels of endogenous CYLD mRNA after stimulation with IMQ or LPS, we used Northern blot hybridization. Unstimulated B6 and MOLF macrophages showed a similar expression of the full-length isoform of CYLD. MOLF macrophages down-regulated CYLD mRNA levels after 4 h of stimulation with either agonist (Fig. 5D). This, to our knowledge, is the first report of TLR activation acutely down-regulating CYLD mRNA.
To confirm that the down-regulation of the full-length CYLD isoform observed at the mRNA level also occurs at the protein level, we performed SDS-PAGE and Western blotting for CYLD in B6 and MOLF macrophages. Peritoneal macrophages were stimulated for 6 h with LPS in the presence of zVAD, which was used to approximate the conditions used in the necroptosis assays (Fig. 2). The addition of zVAD isolates our analysis to changes in CYLD protein levels independent of CYLD cleavage by caspase 8 (18). In support of the mRNA analysis, CYLD was down-regulated nearly 2-fold in MOLF peritoneal macrophages after activation with LPS and zVAD, whereas there was a more than 5-fold increase in CYLD protein levels in B6 macrophages (Fig. 5E). Taken together, analysis of mRNA levels and protein expression of CYLD revealed TLR-induced, acute down-regulation of CYLD in MOLF macrophages.
Stimulus-dependent down-regulation of CYLD mRNA in MOLF macrophages is a novel mechanism of regulation of CYLD in MOLF macrophages that has not been described previously. The nearly 2-fold decrease of CYLD in MOLF may not reflect the full impact of lower CYLD activity in MOLF. Following NF-κB activation, CYLD is phosphorylated, and its enzymatic activity decreases (40). More rapid synthesis of de novo CYLD by B6 macrophages to replace phosphorylated CYLD that has lost its deubiquitinase activity may underlie the increased sensitivity to necroptosis in these cells compared with MOLF macrophages. Further analysis of CYLD mRNA expression at protein turnover is required for full assessment of the mechanism of CYLD down-regulation.
Differential Transcriptional Regulation of CYLD in Wild-derived Mice
Because sequencing did not reveal a genetic lesion in close proximity to Cyld, we investigated whether the down-regulation observed in MOLF macrophages is a result of a difference in stability mediated by the 3′ UTR or a change in promoter activity. No mutations in the 3′UTR were uncovered in MOLF, making a cis-acting difference between the strains that is regulated through the 3′ UTR unlikely. The broad region on chromosome 1 linked to MOLF resistance contains miR-181b, which has been shown to negatively regulate CYLD (41). However, we found no difference in expression levels of this microRNA by real-time PCR. Additionally, sequencing of the CYLD promoter in MOLF and B6 up to 2kB upstream of the transcription start site (TSS) found no mutations between the strains.
Because CYLD regulation occurs at the RNA level, we further investigated the activity of the promoter region using ChIP with RNA Pol II. Through interaction with a number of transcription factors at the promoter region of a gene, RNA Pol II forms a preinitiation complex at the TSS (42). However, promoter pausing of RNA Pol II soon after transcriptional initiation is a normal regulatory step in some, if not most, genes (43). Thus, we examined whether the Cyld locus differs in RNA Pol II recruitment between B6 and MOLF LPS-stimulated peritoneal macrophages by ChIP with RNA Pol II antibody (Fig. 5F). Although no significant differences in RNA Pol II accumulation were noted at the proximal (-300-bp) or distal (-700-bp) promoter regions, a modest increase in RNA Pol II was noted in B6 macrophages at the TSS, and a significant increase was observed at intron 1 (+300 bp) downstream of the TSS. RNA Pol II pausing is mostly noted at regions immediately downstream of the TSS, and these data suggest that RNA Pol II is stalled or paused in B6 macrophages to a greater extent than in MOLF macrophages. RNA Pol II pausing is often associated with expressed genes and is rarely, if ever, associated with unexpressed genes (44). Genome-wide ChIP sequencing data in primary murine B cells confirms that this region is subject to similar regulation in other cell types.3 Taken together, these results strongly suggest that a difference in transcription at the Cyld locus associated with a loss of RNA Pol II pausing may be the underlying reason for the acute down-regulation in CYLD following TLR stimulation in MOLF macrophages, leading to less CYLD deubiquitinase activity on RIP1.
CYLD Expression and Susceptibility to Necroptosis Are Restored in MOLF BMDM
To demonstrate that re-expressing CYLD in MOLF peritoneal macrophages can reverse the resistance to necroptosis, we attempted to express CYLD in this cell type by lentiviral infection and transfection. Disappointingly, MOLF peritoneal macrophages in culture were not amenable to this manipulation because the prolonged duration of culture time required to exogenously express CYLD led to cytotoxicity before we were able to assay these terminally differentiated cells.
We then planned to examine an experimental macrophage model that is more easily manipulated by lentiviral infection to address whether re-expression of MOLF CYLD is sufficient to induce susceptibility to TLR-induced necroptosis. We examined responses in B6 and MOLF BMDM, which are technically easier to manipulate than peritoneal macrophages. Surprisingly, MOLF BMDM were susceptible to necroptosis that was induced by LPS or IMQ in the presence of zVAD (Fig. 6A). Furthermore, the full-length isoform of CYLD was up-regulated in MOLF BMDM to the same extent as B6 BMDM (Fig. 6B). Because an expression difference rather than a coding mutation underlies the difference in CYLD regulation between the B6 and MOLF mouse strains, we can reconcile the difference between the two sources of macrophages on the basis of how effectively CYLD mRNA is transcribed after activation of TLRs. In peritoneal macrophages, the absence RNA Pol II pausing or stalling at the Cyld promoter leads to a down-regulation of CYLD mRNA and protein levels following TLR stimulation. In MOLF BMDM, factors required for in vitro differentiation activate MAPK signaling (45) and may allow cells to bypass the aforementioned regulatory mechanism, leading to up-regulation of CYLD and susceptibility to necroptosis. We recently described a similar event in the context of mannose receptor (MRC1) expression, which is required for CpG-induced TLR9 signaling in MOLF macrophages (46). In peritoneal macrophages, MRC1 mRNA is not expressed, and MOLF cells cannot respond to CpG. However, differentiation into BMDM in vitro permits up-regulation of MRC1 and partially restores CpG responses in MOLF.
FIGURE 6.
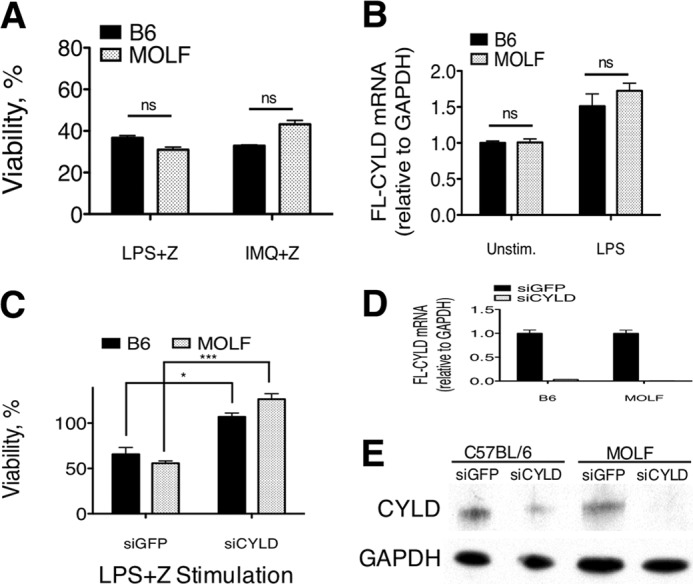
CYLD expression and susceptibility to necroptosis are restored in MOLF BMDM. A, B6 and MOLF BMDM stimulated with LPS (100 ng/ml) or IMQ (5 μg/ml) and zVAD (Z) (25 μm) for 18 h. B, quantitative real-time PCR performed on cDNA reverse-transcribed total RNA from unstimulated (Unstim) and LPS-stimulated B6 and MOLF BMDM. The full-length (FL) CYLD/GAPDH ratio is presented as relative amounts in comparison to B6 unstimulated cells. C, BMDM infected with lentivirus containing siGFP or siCYLD hairpin stimulated with LPS (100 ng/ml) and zVAD (25 μm) for 18 h. D, relative expression of FL-CYLD as measured by qPCR in B6 and MOLF BMDM stimulated for 4 h with LPS. E, CYLD protein expression in B6 and MOLF BMDM measured by Western blot analysis in unstimulated cells. GAPDH was used as a loading control. A and C, cell viability was measured relative to cells in medium with vehicle (DMSO). Data are mean ± S.E. from one representative experiment of three independent experiments. Statistical significance was determined by Student's t test. *, p < 0.05; ***, p < 0.0001; ns, not significant.
Next, we demonstrated that CYLD knockdown using a lentiviral shRNA expression system was sufficient to protect BMDM from each strain from TLR-induced necroptosis. Although siGFP controls from each strain were susceptible to LPS-induced necroptosis, CYLD knockdown protected both strains (Fig. 6C). The knockdown efficiency for both strains was over 90% by mRNA quantitation using qPCR (Fig. 6D), and CYLD protein levels were decreased substantially, as assessed by Western blot analysis (Fig. 6E). Although these data do not directly show that an add-back of CYLD in peritoneal macrophages sensitizes MOLF cells to necroptosis, they demonstrate that, in settings where MOLF macrophages express CYLD, the necroptosis pathway becomes active. Additionally, CYLD knockdown prevents necroptosis in both strains, arguing that down-regulation of this gene is sufficient to cause resistance to necroptosis in both strains of mice.
DISCUSSION
The goal of this study was to investigate whether the genetic diversity of wild-derived inbred mouse strains could reveal novel mechanisms of regulation of necroptosis driven by RIP1. Despite extensive investigation of RIP1 kinase as a regulator of inflammatory signaling and cell death, little is known about its direct targets (47). We approached this question by establishing a genetic screen dependent on the kinase activity of RIP1 and comparing two mouse strains that have been separated by approximately one million years of evolution. MOLF macrophages exhibited resistance to TLR-induced necroptosis compared with B6 macrophages. This difference in resistance was attenuated by the addition of Nec-1, implying that, in B6 peritoneal macrophages, RIP1 kinase activity makes a greater contribution toward necroptosis than in MOLF. To uncover genes that underlie this phenotypic difference, we used linkage analysis in second-generation backcross (N2) progeny. This trait proved to be a complex genetic trait linked significantly to two loci. According to our model of necroptosis resistance in MOLF macrophages, these loci exert their effect on the trait at the level of or upstream of RIP1 and contain components that render RIP1 more pronecrotic in B6 than in MOLF. Alternatively, gene candidates from these loci could be targets of RIP1 kinase that contribute to necroptosis in B6 but not in MOLF mice.
We identified Cyld as one of the genes conferring resistance in MOLF and demonstrated a novel, acute down-regulation of the full-length isoform of CYLD that leads to protection of MOLF macrophages from undergoing TLR-induced necroptosis. Identification of CYLD provided proof of principle of our approach because the identified gene fit into the model of exerting its effect at the level of or upstream of RIP1, and CYLD has been shown to be a necroptosis-promoting protein (18, 20). Additionally, we demonstrated in this study that CYLD is required for TLR-induced necroptosis that occurs without autocrine TNF signaling. The second locus linked to MOLF resistance in the area of the microsatellite marker D1Mit102 also shows significant linkage. However, the linked genomic region is too large to investigate at the level of gene candidates. We plan to produce more meioses to increase the number of recombinations in this region and narrow the size of this locus via high-resolution mapping.
On the basis of our mapping and biochemical analysis data, we propose that differential regulation of RIP1 in MOLF and B6 macrophages underlies their differences in phenotype. According to our model, activation of MOLF macrophages with TLR ligands leads to acute down-regulation of CYLD at the mRNA and protein levels, leading to lower CYLD deubiquitinase activity in MOLF peritoneal macrophages, allowing RIP1 to persist in its ubiquitinated state and polarizing MOLF cells away from necroptotic signaling. In contrast, B6 macrophages exhibit a higher CYLD activity because of its up-regulation, promoting deubiquitination of RIP1, which commits cells to necroptotic death.
With respect to the biology of CYLD, our study revealed a novel regulation of CYLD at the RNA level following TLR stimulation in MOLF peritoneal macrophages. Our findings add to previous work showing that CYLD down-regulation occurs during infection with Sendai virus in the presence of TNF in 293EBNA cells (48), linking the capability of MOLF macrophages to down-regulate CYLD in a signal-dependent fashion with that of human cells, albeit under different conditions. Although CYLD expression has been described widely to be decreased in multiple cancers (49–54), its stimulus-dependent regulation is less clear. Different studies have identified NF-κB (55) and serum response factor (56) as positive regulators of CYLD expression. However, the capability to up-regulate CYLD appears to be agonist-specific (57). Our study in wild-derived mice suggests that further investigation to determine the importance acute down-regulation of CYLD in cell death, proinflammatory signaling, or interferon responses is warranted using mouse strains of diverse genetic backgrounds and human cells in disease models.
Additionally, this work provides insight into how CYLD is regulated at the transcriptional level involving RNA Pol II pausing or stalling. Our data suggest that, in addition to its posttranslational cleavage by caspase 8 (18), CYLD transcriptional down-regulation can prevent the induction of necroptosis. This is perhaps the first time that a regulatory step involving RNA Pol II pausing has been linked to a genetic difference in mouse strains that results in different biological responses to pathogenic signaling. Although we were unable to determine a specific mutation within 2 kilobases of the TSS that explains the lack of RNA Pol II recruitment to the (+300) region in MOLF Cyld, regions of the genome proximal or distal from this location may be mutated so that they affect binding of other factors, like enhancer elements, that could alter the chromatin structure in the Cyld locus (58). Despite this drawback, the utilization of the genetically divergent wild-derived mouse model allowed us to uncover this novel, naturally occurring regulatory mechanism. Further characterization of the genes that confer resistance to necroptosis in MOLF on chromosome 1, elucidation of how MOLF utilizes RIP1 kinase activity for cytokine production, and fine-resolution mapping of the loci conferring this trait may provide insight into signaling components that can be investigated to more completely understand this pathway in humans. Overall, our study revealed an alternative regulation of genes required for necroptosis at the transcriptional level. The outcome of this regulation is a differential predisposition to necroptotic cell death between B6 and MOLF peritoneal macrophages. Furthermore, the ability of Necrostatin 1 to attenuate TNF production by MOLF macrophages suggests that RIP1 kinase activity contributes to the induction of cytokines and potentially other genes through a novel pathway that awaits elucidation. The contribution of RIP1 kinase activity to cytokine production is not entirely surprising, given that it is implicated in a number of spontaneous inflammation models (59, 60) driven by RIP1-dependent inflammatory events independent of cell death that represent pathologic scenarios driven by RIP1 kinase activity. Our data support that MOLF mice can be used to study death-independent models of RIP1 kinase activity promoting inflammation. Because they have a more significant component of the RIP1 kinase-driven cytokine response, they are more likely than B6 mice to reveal striking phenotypic differences that can be used to identify regulators of this pathway using genetic analysis.
Acknowledgment
We thank Bridget Larkin for reading and editing the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants AI056234 and AI090419 (to A. P.). This work was also supported by Government of the Russian Federation (Article 220) Grants GK 11.G34.31.0052, and RFFI 13-04-40267-H13 (to A. P.), by the Eshe Fund, and by the Keck Foundation.
A. Roy, Tufts University School of Medicine, personal communication.
- TLR
- Toll-like receptor
- TNFR
- TNF receptor
- Pol II
- polymerase II
- Pen-Strep
- penicillin-streptomycin
- BMDM
- bone marrow-derived macrophage(s)
- CYLD
- cylindromatosis
- DMSO
- dimethyl sulfoxide
- qPCR
- quantitative PCR
- RIP
- receptor-interacting protein
- IMQ
- imiquimod
- Nec-1
- Necrostatin 1
- TSS
- transcription start site
- zVAD
- Z-Val-Ala-Asp-fluoromethylketone
- TRIF
- TIR-domain-containing adapter-inducing interferon beta
- B6
- C57BL/6
- MOLF
- MOLF/EiJ.
REFERENCES
- 1. Takeuchi O., Akira S. (2010) Pattern recognition receptors and inflammation. Cell 140, 805–820 [DOI] [PubMed] [Google Scholar]
- 2. Duprez L., Wirawan E., Vanden Berghe T., Vandenabeele P. (2009) Major cell death pathways at a glance. Microbes Infect. 11, 1050–1062 [DOI] [PubMed] [Google Scholar]
- 3. Duprez L., Takahashi N., Van Hauwermeiren F., Vandendriessche B., Goossens V., Vanden Berghe T., Declercq W., Libert C., Cauwels A., Vandenabeele P. (2011) RIP Kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35, 908–918 [DOI] [PubMed] [Google Scholar]
- 4. Upton J. W., Kaiser W. J., Mocarski E. S. (2010) Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7, 302–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cho Y. S., Challa S., Moquin D., Genga R., Ray T. D., Guildford M., Chan F. K. (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iwai K. (2012) Diverse ubiquitin signaling in NF-κB activation. Trends Cell Biol. 22, 355–364 [DOI] [PubMed] [Google Scholar]
- 7. Rajput A., Kovalenko A., Bogdanov K., Yang S.-H., Kang T.-B., Kim J.-C., Du J., Wallach D. (2011) RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity 34, 340–351 [DOI] [PubMed] [Google Scholar]
- 8. Oberst A., Dillon C. P., Weinlich R., McCormick L. L., Fitzgerald P., Pop C., Hakem R., Salvesen G. S., Green D. R. (2011) Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin Y., Devin A., Rodriguez Y., Liu Z. (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wertz I. E., Dixit V. M. (2010) Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ. 17, 14–24 [DOI] [PubMed] [Google Scholar]
- 11. Ofengeim D., Yuan J. (2013) Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 14, 727–736 [DOI] [PubMed] [Google Scholar]
- 12. Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G. (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 [DOI] [PubMed] [Google Scholar]
- 13. Sun L., Wang H., Wang Z., He S., Chen S., Liao D., Wang L., Yan J., Liu W., Lei X., Wang X. (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 [DOI] [PubMed] [Google Scholar]
- 14. Wu J., Huang Z., Ren J., Zhang Z., He P., Li Y., Ma J., Chen W., Zhang Y., Zhou X., Yang Z., Wu S.-Q., Chen L., Han J. (2013) Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 23, 994–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murphy J. M., Czabotar P. E., Hildebrand J. M., Lucet I. S., Zhang J.-G., Alvarez-Diaz S., Lewis R., Lalaoui N., Metcalf D., Webb A. I., Young S. N., Varghese L. N., Tannahill G. M., Hatchell E. C., Majewski I. J., Okamoto T., Dobson R. C., Hilton D. J., Babon J. J., Nicola N. A, Strasser A., Silke J., Alexander W. S. (2013) The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453 [DOI] [PubMed] [Google Scholar]
- 16. Komander D., Reyes-Turcu F., Licchesi J. D., Odenwaelder P., Wilkinson K. D., Barford D. (2009) Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 10, 466–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Degterev A., Hitomi J., Germscheid M., Ch'en I. L., Korkina O., Teng X., Abbott D., Cuny G. D., Yuan C., Wagner G., Hedrick S. M., Gerber S. A., Lugovskoy A., Yuan J. (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O'Donnell M. A., Perez-Jimenez E., Oberst A., Ng A., Massoumi R., Xavier R., Green D. R., Ting A. T. (2011) Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 13, 1437–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moquin D. M., McQuade T., Chan F. K. (2013) CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE 8, e76841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hitomi J., Christofferson D. E., Ng A., Yao J., Degterev A., Xavier R. J., Yuan J. (2008) Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He S., Liang Y., Shao F., Wang X. (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. 108, 20054–20059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Günther C., Martini E., Wittkopf N., Amann K., Weigmann B., Neumann H., Waldner M. J., Hedrick S. M., Tenzer S., Neurath M. F., Becker C. (2011) Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guénet J.-L., Bonhomme F. (2003) Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet. 19, 24–31 [DOI] [PubMed] [Google Scholar]
- 24. Conner J. R., Smirnova I. I., Poltorak A. (2008) Forward genetic analysis of Toll-like receptor responses in wild-derived mice reveals a novel antiinflammatory role for IRAK1BP1. J. Exp. Med. 205, 305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang H., Wang J. R., Didion J. P., Buus R. J., Bell T. A., Welsh C. E., Bonhomme F., Yu A. H., Nachman M. W., Pialek J., Tucker P., Boursot P., McMillan L., Churchill G. A., de Villena F. P. (2011) Subspecific origin and haplotype diversity in the laboratory mouse. Nat. Genet. 43, 648–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flannery S. M., Keating S. E., Szymak J., Bowie A. G. (2011) Human interleukin-1 receptor-associated kinase-2 is essential for Toll-like receptor-mediated transcriptional and post-transcriptional regulation of tumor necrosis factor α. J. Biol. Chem. 286, 23688–23697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaiser W. J., Sridharan H., Huang C., Mandal P., Upton J. W., Gough P. J., Sehon C. A, Marquis R. W., Bertin J., Mocarski E. S. (2013) Toll-like receptor 3-mediated necrosis via TRIF, RIP3 and MLKL. J. Biol. Chem. 288, 31268–31279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feoktistova M., Geserick P., Kellert B., Dimitrova D. P., Langlais C., Hupe M., Cain K., MacFarlane M., Häcker G., Leverkus M. (2011) cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Conner J. R., Smirnova I. I., Poltorak A. (2009) A mutation in Irak2c identifies IRAK-2 as a central component of the TLR regulatory network of wild-derived mice. J. Exp. Med. 206, 1615–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Conner J. R., Smirnova I. I., Moseman A. P., Poltorak A. (2010) IRAK1BP1 inhibits inflammation by promoting nuclear translocation of NFκB p50. Proc. Natl. Acad. Sci. 107, 11477–11482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kobayashi K. S., Chamaillard M., Ogura Y., Henegariu O., Inohara N., Nuñez G., Flavell R. A. (2005) Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734 [DOI] [PubMed] [Google Scholar]
- 32. Strober W., Watanabe T. (2011) NOD2, an intracellular innate immune sensor involved in host defense and Crohn's disease. Mucosal Immunol. 4, 484–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu J., Bai L., Sun H., Nikolovska-Coleska Z., McEachern D., Qiu S., Miller R. S., Yi H., Shangary S., Sun Y., Meagher J. L., Stuckey J. A., Wang S. (2008) SM-164: A novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res. 68, 9384–9393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McComb S., Cheung H. H., Korneluk R. G., Wang S., Krishnan L., Sad S. (2012) cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. 19, 1791–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reiley W. W., Zhang M., Jin W., Losiewicz M., Donohue K. B., Norbury C. C., Sun S.-C. (2006) Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat. Immunol. 7, 411–417 [DOI] [PubMed] [Google Scholar]
- 36. Zhang J., Stirling B., Temmerman S. T., Ma C. A., Fuss I. J., Derry J. M., Jain A. (2006) Impaired regulation of NF-κB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J. Clin. Invest. 116, 3042–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Massoumi R. (2010) Ubiquitin chain cleavage: CYLD at work. Trends Biochem. Sci. 35, 392–399 [DOI] [PubMed] [Google Scholar]
- 38. Hövelmeyer N., Wunderlich F. T., Massoumi R., Jakobsen C. G., Song J., Wörns M. A., Merkwirth C., Kovalenko A., Aumailley M., Strand D., Brüning J. C., Galle P. R., Wallach D., Fässler R., Waisman A. (2007) Regulation of B cell homeostasis and activation by the tumor suppressor gene CYLD. J. Exp. Med. 204, 2615–2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Massoumi R., Chmielarska K., Hennecke K., Pfeifer A., Fässler R. (2006) CYLD Inhibits Tumor Cell Proliferation By Blocking Bcl-3-dependent NF-κB Signaling. Cell 125, 665–677 [DOI] [PubMed] [Google Scholar]
- 40. Hutti J. E., Shen R. R., Abbott D. W., Zhou A. Y., Sprott K. M., Asara J. M., Hahn W. C., Cantley L. C. (2009) Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKK[var ϵ] promotes cell transformation. Mol. Cell 34, 461–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iliopoulos D., Jaeger S. A., Hirsch H. A., Bulyk M. L., Struhl K. (2010) STAT3 Activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 39, 493–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adelman K., Lis J. T. (2012) Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat. Rev. Genet. 13, 720–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kwak H., Fuda N. J., Core L. J., Lis J. T. (2013) Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gilchrist D. A., Fromm G., dos Santos G., Pham L. N., McDaniel I. E., Burkholder A., Fargo D. C., Adelman K. (2012) Regulating the regulators: the pervasive effects of Pol II pausing on stimulus-responsive gene networks. Genes Dev. 26, 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gobert Gosse S., Bourgin C., Liu W. Q., Garbay C., Mouchiroud G. (2005) M-CSF stimulated differentiation requires persistent MEK activity and MAPK phosphorylation independent of Grb2-Sos association and phosphatidylinositol 3-kinase activity. Cell. Signal. 17, 1352–1362 [DOI] [PubMed] [Google Scholar]
- 46. Moseman A. P., Moseman E. A., Schworer S., Smirnova I., Volkova T., von Andrian U., Poltorak A. (2013) Mannose receptor 1 mediates cellular uptake and endosomal delivery of CpG-motif containing oligodeoxynucleotides. J. Immunol. 191, 5615–5624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Festjens N., Vanden Berghe T., Cornelis S., Vandenabeele P. (2007) RIP1, a kinase on the crossroads of a cell's decision to live or die. Cell Death Differ. 14, 400–410 [DOI] [PubMed] [Google Scholar]
- 48. Friedman C. S., O'Donnell M. A., Legarda-Addison D., Ng A., Cárdenas W. B., Yount J. S., Moran T. M., Basler C. F., Komuro A., Horvath C. M., Xavier R., Ting A. T. (2008) The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9, 930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang L., Baiocchi R. A., Pal S., Mosialos G., Caligiuri M., Sif S. (2005) The BRG1- and hBRM-associated factor BAF57 induces apoptosis by stimulating expression of the cylindromatosis tumor suppressor gene. Mol. Cell. Biol. 25, 7953–7965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhong S., Fields C. R., Su N., Pan Y. X., Robertson K. D. (2007) Pharmacologic inhibition of epigenetic modifications, coupled with gene expression profiling, reveals novel targets of aberrant DNA methylation and histone deacetylation in lung cancer. Oncogene 26, 2621–2634 [DOI] [PubMed] [Google Scholar]
- 51. Massoumi R., Kuphal S., Hellerbrand C., Haas B., Wild P., Spruss T., Pfeifer A., Fässler R., Bosserhoff A. K. (2009) Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J. Exp. Med. 206, 221–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kuphal S., Shaw-Hallgren G., Eberl M., Karrer S., Aberger F., Bosserhoff A. K., Massoumi R. (2011) GLI1-dependent transcriptional repression of CYLD in basal cell carcinoma. Oncogene 30, 4523–4530 [DOI] [PubMed] [Google Scholar]
- 53. Liu P., Xu B., Shen W., Zhu H., Wu W., Fu Y., Chen H., Dong H., Zhu Y., Miao K., Xu W., Li J. (2012) Dysregulation of TNFα-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia 26, 1293–1300 [DOI] [PubMed] [Google Scholar]
- 54. Urbanik T., Köhler B. C., Boger R. J., Wörns M. A., Heeger S., Otto G., Hövelmeyer N., Galle P. R., Schuchmann M., Waisman A., Schulze-Bergkamen H. (2011) Down-regulation of CYLD as a trigger for NF-κB activation and a mechanism of apoptotic resistance in hepatocellular carcinoma cells. Int. J. Oncol. 38, 121–131 [PubMed] [Google Scholar]
- 55. Jono H., Lim J. H., Chen L.-F., Xu H., Trompouki E., Pan Z. K., Mosialos G., Li J.-D. (2004) NF-κB is essential for induction of CYLD, the negative regulator of NF-κB. J. Biol. Chem. 279, 36171–36174 [DOI] [PubMed] [Google Scholar]
- 56. Liang G., Ahlqvist K., Pannem R., Posern G., Massoumi R. (2011) Serum response factor controls CYLD expression via MAPK signaling pathway. PLoS ONE 6, e19613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jin W., Chang M., Paul E. M., Babu G., Lee A. J., Reiley W., Wright A., Zhang M., You J., Sun S.-C. (2008) Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J. Clin. Invest. 118, 1858–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Calo E., Wysocka J. (2013) Modification of enhancer chromatin: what, how, and why? Mol. Cell 49, 825–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kang T.-B., Yang S.-H., Toth B., Kovalenko A., Wallach D. (2013) Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38, 27–40 [DOI] [PubMed] [Google Scholar]
- 60. Lukens J. R., Vogel P., Johnson G. R., Kelliher M. A, Iwakura Y., Lamkanfi M., Kanneganti T.-D. (2013) RIP1-driven autoinflammation targets IL-1α independently of inflammasomes and RIP3. Nature 498, 224–227 [DOI] [PMC free article] [PubMed] [Google Scholar]