

Abstract
Interest in single-cell whole-transcriptome analysis is growing rapidly, especially for profiling rare or heterogeneous populations of cells. We compared commercially available single-cell RNA amplification methods with both microliter and nanoliter volumes, using sequence from bulk total RNA and multiplexed quantitative PCR as benchmarks to systematically evaluate the sensitivity and accuracy of various single-cell RNA-seq approaches. We show that single-cell RNA-seq can be used to perform accurate quantitative transcriptome measurement in individual cells with a relatively small number of sequencing reads and that sequencing large numbers of single cells can recapitulate bulk transcriptome complexity.
High-throughput sequencing of whole transcriptomes, or RNA-seq, has been used extensively to profile gene expression from bulk tissues. There is a growing demand for methods that allow whole-transcriptome profiling of single cells, driven by (i) the need for direct analysis of rare cell types or primary cells for which there may be insufficient material for conventional RNA-seq protocols and (ii) the desire to profile interesting subpopulations of cells from a larger heterogeneous population1,2. It has been shown that the average expression level of a population of cells can be strongly biased by a few cells with high expression and is thus not reflective of a typical individual cell from that population3. Measurements using FISH indicate that levels of specific transcripts can vary as much as 1,000-fold4 between presumably equivalent cells, further illustrating the value of profiling whole transcriptomes at the single-cell level.
Various methods for performing single-cell RNA-seq have been reported5–15, but many questions remain about the throughput and quantitative-versus-qualitative value of single-cell RNA-seq measurements. In particular, performance has mainly been evaluated with respect to sensitivity and precision. Sensitivity is typically measured by counting the number of genes whose expression is detected per cell, and precision is measured by how well the results can be reproduced on replicate samples. However, in order to assess the validity of a measurement, it is also critical to evaluate accuracy, or how close the measurement comes to the true value. Accuracy depends on systematic errors deriving from the data collection method, and it is often estimated by using different measurement techniques on the same sample type.
Here we report quantitative RNA-seq analysis of 102 single-cell human transcriptomes. We assessed the performance of commercially available single-cell RNA amplification methods in both microliter and nanoliter volumes, compared each method to conventional RNA-seq of the same sample using bulk total RNA and evaluated the accuracy of the measurements by independently quantitating expression of 40 genes in the same cell type by multiplexed quantitative PCR (qPCR)16,17. Our results show that it is possible to use single-cell RNA-seq to perform quantitative transcriptome measurements of single cells and that, when such measurements are performed on large numbers of cells, one can recapitulate both the bulk transcriptome complexity and the distributions of gene expression levels found by single-cell qPCR.
RESULTS
Single-cell RNA-seq methods and validation with qPCR
We performed all experiments using cultured HCT116 cells to minimize heterogeneity among single-cell experiments. We made a total of 102 single-cell RNA-seq libraries using two tube-based methods (6 libraries) and one microfluidic method (96 libraries): (i) the SMARTer Ultra Low RNA Kit (Clontech) for cDNA synthesis18 (ii) the TransPlex Kit (Sigma-Aldrich)19 and (iii) SMARTer cDNA synthesis using the C1 microfluidic system (Fluidigm), all followed by Nextera library construction (Illumina) in standard tube format (Fig. 1a and Supplementary Table 1). To obtain a benchmark for comparison, we also made libraries from bulk RNA generated from 1 million HCT116 cells using both SuperScript II reverse transcriptase (Invitrogen) and SMARTer. We sequenced tube-based libraries using Illumina HiSeq, obtaining >26 million raw reads for each. The 96 microfluidics-based libraries were barcoded, and two pooled samples of 48 libraries were each sequenced on a HiSeq lane (for a total of two lanes for all 96 libraries), resulting in an average of 2 million raw reads per library. We also constructed seven tube-based single-cell RNA-seq libraries using Ovation (NuGEN v.1)20, which was followed by library construction with both Nextera and NEBNext (New England BioLabs) (Supplementary Fig. 1).
Figure 1.

Initial validation of single-cell RNA-seq methods. (a) Schematic of the experimental strategy. (b) Reproducibility, as evaluated by the percentage of genes detected in pairs of replicate samples out of the mean total number of genes detected in this pair of samples. (c) Sensitivity, as evaluated by overlap between genes detected by single-cell and bulk RNA-seq measurement. Bulk values listed exclude the overlap values. Percentages are calculated as the number of genes detected in both relative to the number of genes detected in the bulk measurement.
Currently, qPCR is considered the gold standard for validating gene expression studies16,17. Although authors of some previous studies have validated their RNA-seq results by confirming the presence of transcripts of interest using targeted qRT-PCR10,21–23, none has demonstrated quantitative correlation of multiple genes between the two methods. Therefore, we performed single-cell multiplexed qPCR on HCT116 cells to measure expression of 40 genes curated from previous studies, with some genes known to be highly expressed and others known not to be expressed in this cell type1, in order to benchmark the accuracy of RNA-seq against qPCR. A total of 457 single cells were assessed using qPCR, of which 273 were FACS sorted individually into well plates for cDNA synthesis. The other 184 were captured using the C1 microfluidic system, and cDNA synthesis was performed in the microfluidic device (Fig. 1a).
Reproducibility and accuracy of single-cell RNA-seq
We assessed the reproducibility of each single-cell RNA-seq method in two ways. The first was by comparing the number of genes detected in all combinations of replicate sample pairs prepared with the same method to the mean total number of genes detected using that method (Fig. 1b). For bulk samples, this reflects technical variation, whereas for the single-cell samples it combines technical variation with biological variability between cells, which can be substantial. Single-cell reproducibility varied between 57% and 65%, depending on the method used.
The second way was by comparing the levels of External RNA Controls Consortium (ERCC) exogenous spike-in mRNA sequences detected in four randomly selected single-cell samples. These sequences were added to each sample before cell lysis on the C1 platform and were subjected to the same amplification conditions as the cellular mRNA. The spike-ins reflect a diverse range of sequence content and length, have low homology with eukaryotic transcripts and span a large range of concentrations. The pairwise correlation of transcript abundance was >0.9 (Pearson correlation coefficient) for all comparisons among the four samples (Supplementary Fig. 2), indicating a high level of reproducibility among samples generated by the C1 platform.
We determined sensitivity by calculating how many genes were detected by each single-cell method compared to bulk RNA-seq (Fig. 1c), computed as average percentages in a pairwise manner (Online Methods). C1 displayed higher sensitivity than other single-cell methods, detecting ~42% of genes picked up by bulk RNA-seq with no amplification (bulk SuperScript) and ~44% of genes detected by bulk RNA-seq using the same cDNA synthesis kit (bulk SMARTer) (Fig. 1c). Reproducibility on the C1 platform was also high (Fig. 1b), though high sensitivity and reproducibility were not always associated: the Ovation method with Nextera library construction was the most reproducible over several replicates, but it had low sensitivity (Supplementary Fig. 1). The bulk RNA measurements are technical replicates; therefore, the number of genes consistently detected among replicates is high. The variability within single-cell samples is expected to be higher, as each single cell is a biological replicate, and biological noise in gene expression contributes to the variation.
To evaluate accuracy, we compared the expression values of 40 genes generated by single-cell qPCR and RNA-seq (Fig. 2). Because it is challenging to obtain absolute expression values with either method, we calculated gene expression relative to the median expression across all transcripts for each cell (Online Methods). The resulting dimensionless numbers should be independent of the method used to measure them and can be compared directly to determine the accuracy of the measurements. Expression values from qPCR and RNA-seq correlated well for all single-cell methods (r > 0.84), confirming that single-cell RNA-seq methods are able to detect gene expression in a quantitative manner consistent with existing gold standards. The standard error was small relative to the expression level of each gene (Fig. 2).
Figure 2.
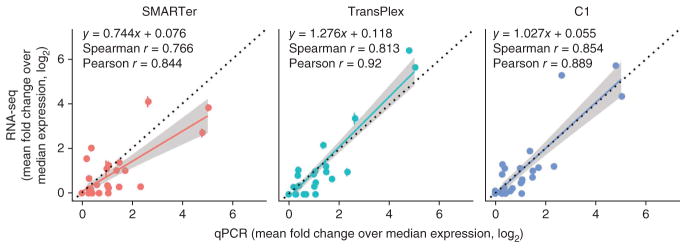
Correlation between single-cell RNA-seq and single-cell multiplexed qPCR for each sample preparation method. Correlation coefficients were computed from log2- transformed values. A linear regression line (color) and the y = x line (black, dotted) are also shown in each panel. Shading represents the 95% confidence interval for each regression line. For RNA-seq data, FPKM values for each of gene of interest were normalized to the median FPKM for each cell and log2 transformed. For qPCR data, threshold cycle (Ct) values for each gene of interest were normalized to the median Ct value for each cell (ΔCt), which also represents a fold change over the median expression. ΔCt values are already in log2 space and were directly plotted. Error bars, standard error (n values as in Fig. 1a).
Interestingly, the correlation coefficients and slopes of the linear regressions varied among methods. The correlation coefficients suggest that each method is able to reproduce expression measured by qPCR to some degree, whereas the slopes indicate some distortion. Notably, performing the sample preparation in nanoliter volumes (C1 system) generated a regression slope that was almost 1, which indicates the highest absolute accuracy. One likely explanation is that reducing the volume of the reaction chamber increases the effective concentration of the reactants and reduces competition for enzymes between template and nonspecific molecules or contaminants, thus minimizing amplification bias24. However, lower bias could also be a result of more uniform reverse transcription or more efficient template switching during reverse-transcription PCR.
Comparison between nanoliter and microliter reactions
We directly compared microliter with nanoliter sample preparation, and found that for both qPCR and RNA-seq, samples prepared in nanoliter volumes yielded fewer false positives. For example, on the basis of previous results with cells of this lineage1, CA1 and AQP8 had sporadic false positive signals in the tube-based qPCR data but were clean in the C1 data (Fig. 3a). Furthermore, RNA-seq on the C1 picked up genes that were not detected very strongly or consistently by RNA-seq in microliter reactions. CD47, a surface marker that is upregulated in cancers to evade phagocytosis25–27, was barely detectable in microliter-volume RNA-seq but was obvious in nanoliter volume (Fig. 3b). The nanoliter-volume data are consistent with functional studies demonstrating the presence of CD47 protein on the surface of HCT116 cells27 (Supplementary Fig. 3). Positive controls GAPDH and ACTB showed a surprisingly wide distribution in microliter-volume samples, spanning several orders of magnitude, whereas C1-generated distributions for these genes were consistent with qPCR data and the distribution of other genes. We also noted that one of the low-expression genes, TERT, was detected by only qPCR (Fig. 3). This result may be due to the relatively low sequencing depth (an average of ~2 million total raw reads per cell) for these libraries.
Figure 3.

Comparison of gene expression distributions for 40 genes between samples prepared in microliter and nanoliter volumes. (a) Frequency distribution of expression values from single-cell qPCR shown as a violin plot for each gene. Expression (vertical axis) is the log2-transformed fold change over median gene expression level for each sample. Width of the violin indicates frequency at that expression level. (b) Frequency distribution of expression from single-cell RNA-seq. Violin plots are presented as in a.
The C1 platform appears to confer several advantages over tube-based preparations, including lower false positives and reduced bias. The narrower distribution of expression values for qPCR indicates it provides higher precision than single-cell RNA-seq, independently of preparation volume. A clustering analysis of gene expression across the measurement approaches is shown in Supplementary Figure 4.
We have also assessed the transcript coverage, length bias and GC bias of these samples by looking at the range and level of correlation between transcript expression (FPKM values, or fragments per kilobase of exon model per million mapped reads) and GC fraction and length (Supplementary Fig. 5). There was no systematic bias for GC content or transcript length in the microfluidically prepared samples as compared to the bulk RNA samples (Supplementary Fig. 5a,b). Coverage over the full length of the transcript was more uniform for single-cell samples prepared using the C1 and the bulk sample prepared using SuperScript. Other single-cell samples prepared in tube format, using SMARTer and TransPlex, showed a large 3′ end bias, with coverage declining rapidly with distance from the 3′ end (Supplementary Fig. 5c). The coverage profile for tube-based SMARTer is similar to that observed by Ramsköld et al.14.
Nanoliter-volume sample preparation reduces bias
To assess bias on the C1 platform, we examined the correlation between the known ERCC spike-in concentrations and their levels measured by RNA-seq (Supplementary Fig. 6a). The spike-in sequences span a large range of concentrations to allow empirical determination of the limit of detection28,29. We found that C1 experiments gave good correlations, even at concentrations below one molecule per chamber; this is possible because the results are averaged over all 96 chambers in the C1 chip. Our data show that ERCC spike-ins at a level of one molecule per chamber have a corresponding FPKM value of approximately 1. As determined by Poisson statistics, at a concentration of one molecule per chamber, the expected fraction of non-empty chambers is ~0.64; empirically, ERCC spike-in transcripts at this concentration were detected at ~0.40 fraction using the C1 platform. This indicates that the ‘quantum efficiency’ of amplification and detection of a single molecule in the C1 is about ~0.63 (Supplementary Note). In comparison, previous single-cell transcriptome analyses reported a detection rate of ~0.25–0.30 (ref. 12).
Combining single-cell data recapitulates bulk RNA profile
We created a synthetic ensemble data set by computationally pooling raw reads from all the single-cell RNA-seq data to mimic a bulk RNA-seq experiment. The correlation between the true bulk gene expression and the single-cell ensemble was remarkably high (Fig. 4a); there was little distortion as demonstrated by the Loess regression curve being virtually linear with a slope close to 1 in this regime, and the Pearson correlation coefficient was 0.870. This analysis confirmed that an ensemble of single cells indeed recapitulates the bulk11. However, it is worth noting that the opposite is generally not true: bulk measurements cannot be used to accurately infer ‘typical’ single-cell expression values, nor can they be used to infer the variation in expression value from cell to cell (T.K., P.D., S.S., M.F.C. and S.R.Q., unpublished data).
Figure 4.
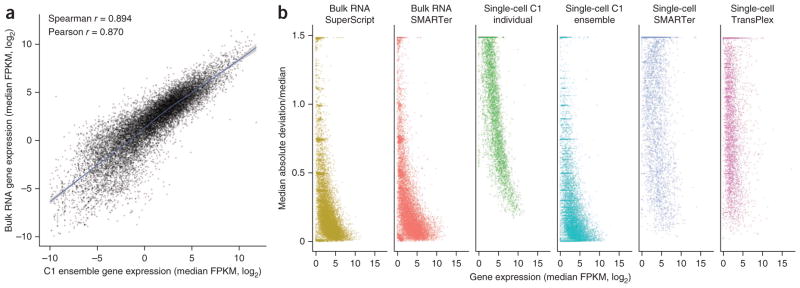
Merging many single-cell transcriptomes quantitatively recreates the bulk measurement. (a) Correlation between the merged single cells (“ensemble”) and the bulk RNA-seq measurement of gene expression. The ensemble was created by computationally pooling all the raw reads obtained from the 96 single-cell transcriptomes generated using the C1 system and then sampling 30 million reads randomly. The bulk and ensemble libraries were depth matched before alignment was performed. For each gene, the log2-transformed median FPKM values from the ensemble and bulk were plotted against each other. (b) Variation in gene expression as a function of gene expression level across sample replicates for each preparation method. Variation (vertical axis) is the median absolute deviation of the FPKM divided by the median FPKM (MAD/M; see Online Methods for the equation). For each gene, the MAD/M is plotted against the log2-transformed median FPKM value for that gene in order to visualize how variation changes with overall transcript abundance. Replicates for single-cell methods are biological replicates, whereas replicates for the bulk and ensemble are technical replicates, as each sample represents a subsampling of a pooled sample.
Next we examined variation among RNA-seq replicates by looking at how dispersion about the median FPKM depends on median gene expression for each method (Fig. 4b). In general, genes with low expression levels exhibited greater variation, and the degree of variability was gene dependent. But as expression level increased, the amount of variation decreased, presumably because genes with high expression are also those that are expressed stably. Low-expression genes that were reliably detected with low variation among replicates were only found in bulk RNA-seq and synthetic ensemble data sets. Interestingly, despite the uniform behavior of ERCC spike-ins (Supplementary Fig. 6b), inter-replicate variation among individual C1 single-cell data sets appeared to monotonically decrease with gene expression level; that is, low-expression genes always had a high intersample variation. This was not seen in the other single-cell methods and is perhaps attributable to the sequencing depth of these samples. In general, the microfluidic single-cell data had a more well-defined relationship, with less scatter, between expression level and variation than the single cells measured in tubes.
Nanoliter sample preparation improves RNA-seq sensitivity
We constructed saturation curves for each preparation method by subsampling the raw reads from each library and determining the number of genes detected (Fig. 5). The number of genes detected with confidence (FPKM > 1) approached saturation at roughly 2 million reads for all methods; in fact, the majority of genes were detected within the first 500,000 reads—and for most methods, >90% of all genes detected at 30 million reads were already detected at a sequencing depth of 2 million (Supplementary Fig. 7a). There was a large difference in the sensitivity of each method, with a wide range of saturation points. The synthetic ensemble experiment matched the bulk experiment generated with the same method (SMARTer): both reached saturation at 2 million sequenced reads at almost identical rates. This again suggests that there is less bias when performing cDNA synthesis in smaller reaction volumes. With less bias, low-abundance transcripts have better representation at lower sequencing depths, and the overall assay sensitivity thus improves. Further confirming this hypothesis is the observation that for individual transcriptomes generated using the microfluidic platform, the average number of genes detected at any sequencing depth is higher than with any other single-cell method (Fig. 5 and Supplementary Figs. 1 and 7).
Figure 5.
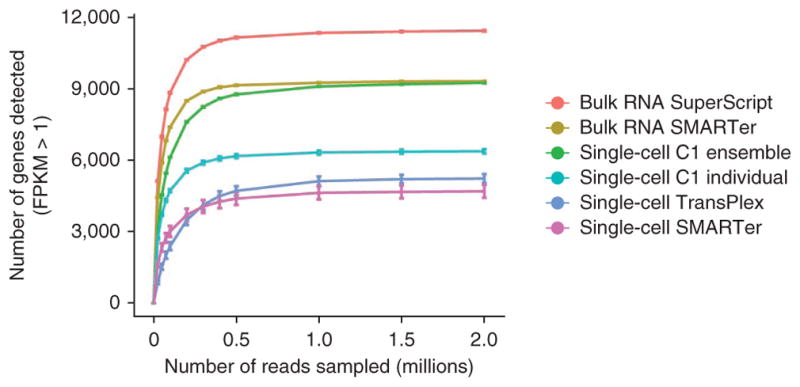
Saturation curves for the different sample preparation methods. Each point on the curve was generated by randomly selecting a number of raw reads from each sample library and then using the same alignment pipeline to call genes with mean FPKM >1. Each point represents four replicate subsamplings. Error bars, standard error.
DISCUSSION
We used microfluidic automation to quantitatively compare the accuracy and precision of single-cell RNA-seq to qPCR. Using two distinct methods, each of which has different biases and sources of error, enabled us to estimate the absolute accuracy of single-cell gene expression. Our study shows that single-cell RNA-seq can generate results that are quantitatively comparable to qPCR, in particular when sample preparation is done in nanoliter-scale reaction volumes, as in a microfluidic device. Bias that is typically introduced during sample preparation is reduced, and correlation further improves. It is not yet clear whether this bias is a fundamental limitation of microliter-volume amplification schemes or whether with further optimization, these approaches will also be able to yield fully accurate transcriptome measurements. We expect that the availability of low-bias, high-throughput single-cell RNA-seq will make studies of primary tissue involving diverse subsets of cell types and hundreds or thousands of individual cells routine.
ONLINE METHODS
Cell growth and sorting
HCT116 is a human colon cancer cell line and can be obtained from the American Tissue-type Culture Collection (ATCC; CCL-247). HCT116 cells were maintained in RPMI-1640 medium, supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 120 μg/ml penicillin, 100 μg/ml streptomycin, 20 mM HEPES and 1 mM sodium pyruvate. Cells were authenticated by HLA typing and were confirmed negative for Mycoplasma contamination using PCR. Cells for single-cell qPCR assays using the Fluidigm Biomark were sorted into wells of 96-well plates containing 5 μl 2× Cells Direct (Invitrogen, Life Technologies) plus 0.4 units/μl Superase In (Ambion, Life Technologies) and pre-amplified with pooled TaqMan probes (AB, Life Technologies) as previously described1. For cDNA synthesis, single HCT116 cells were sorted into the wells of 96-well plates (ABgene PCR plates, Thermo Scientific) containing 5 μl nuclease-free water (IDT) and 0.4 units/μl Superase In (Ambion, Life Technologies). The sealed plates were stored frozen at −80 °C.
Single-cell multiplexed qPCR
For the microliter-volume sample preparations, Biomark qPCR assays were performed as described before1. For the nanoliter-volume sample preparations on the C1 platform, single HCT116 cells were captured on a microfluidic STA chip using the Fluidigm C1, and amplicons were made with pooled TaqMan assays1 and Ambion Cells-to-CT lysis and pre-amplification kit (Life Technologies). A list of the TaqMan assays used is found in Supplementary Table 2.
In order to directly compare with RNA-seq data, gene expression values were calculated relative to the median gene expression across all transcripts for each cell. The Ct values for each gene of interest were normalized to the median Ct for each cell; unexpressed genes with no detectable Ct value were given a value of “NA” for computational purposes.
Bulk RNA-seq library construction
Bulk total RNA was prepared from ~1 million HCT116 cells using the Dynal mRNA Direct kit (Invitrogen) according to the supplier’s recommendations. Aliquots of the bulk prep were stored at −80 °C in nuclease-free water. Replicates of the bulk prep were made into cDNA libraries using two different preparation techniques: (i) SuperScript II RT (Invitrogen) for cDNA synthesis with no amplification, followed by Nextera library construction (limited cycles of amplification to attach sequencing primers), and (ii) SMARTer Ultra Low RNA kit with amplification, followed by Nextera library construction. For each cDNA synthesis method, the maximum recommended amount of RNA was used as input. Bioanalyzer (Agilent) traces were used to confirm library size distribution and quantitation.
Single-cell RNA-seq library construction
For the microliter-volume sample preparations, single cells were sorted into and frozen in standard 96-well plates as described above. Wells were thawed, and those containing single cells were cut from the plate. cDNA synthesis was then performed in an MJ Thermocycler according to the supplier’s protocols. Clean-up reactions were done with Ampure XP beads (Beckman Coulter Genomics). The cDNA synthesis kits compared were NuGEN Ovation RNA Seq System (version 1), Sigma-Aldrich WTA2 TransPlex Complete Whole Transcriptome Amplification Kit and Clontech SMART Ultra Low RNA kit for Illumina Sequencing (Supplementary Table 1). Illumina library construction used either the Epicentre (Illumina) Nextera DNA Sample Prep kit (Illumina-compatible) or New England BioLabs NEBNext DNA library Prep Master Mix Set for Illumina combined with Illumina-compatible adaptors and PCR primers from IDT. Libraries were quantitated by an Agilent Bioanalyzer using the High Sensitivity DNA analysis kit and Kapa Biosystems Illumina library Quantitation kit.
For the nanoliter-volume sample preparations on the C1 platform, single HCT116 cells were captured using the Fluidigm C1 chips using the manufacturers recommended protocol. A concentration of 300,000–350,000 cells per ml was used for chip loading. After cell capture, chips were examined visually to identify empty chambers that were excluded from later analysis. cDNAs were made on-chip with Clontech SMARTer Ultra Low RNA kit for Illumina using manufacturer-provided protocols. External RNA Controls Consortium (ERCC) mRNA spike-ins (Ambion, Life Technologies) were added to the cell lysis mix instead of the manufacturer-provided spike-in controls at a final concentration that was 40,000-fold diluted from the stock. ERCC spike-ins were introduced to each sample within the cell lysis mix microfluidically and underwent the same sample preparation as the cell in each sample. Detailed information on the ERCC transcripts can be found in ref. 28 as well as the supplementary information of ref. 29. Illumina libraries were constructed in 96-well plates using the Illumina Nextera XT DNA Sample Preparation kit according to a protocol supplied by Fluidigm.
DNA sequencing
Sorted single-cell libraries and bulk libraries were sequenced 1 × 50 bases on Illumina HiSeq 2000 with replicate libraries of each type in the same lane. Single-cell C1 libraries were sequenced 2 × 150 bases in two pools of 48 libraries each.
Sequence alignment and analysis
Raw reads were pre-processed with existing sequence grooming tools FastQC (Babraham Institute, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), Cutadapt30 and Prinseq31, before sequence alignment using the Tuxedo Suite (Bowtie32, Bowtie 2 (ref. 33), TopHat34) and SAMtools35. FPKM values used for analyses were generated by TopHat. Where depth matching was done, Seqtk (H. Li, https://github.com/lh3/seqtk/) was used to randomly select raw reads from each library, and the same pre-processing and alignment pipelines were used to obtain FPKM values for the depth-matched samples.
To construct the reproducibility bar graphs, we compared pairs of samples prepared using the same method to determine the number of genes that were detected (FPKM > 0) in both samples of the pair. This was repeated for every combination within each method, and the average number of genes reproducibly detected between pairs was then computed as a percentage of the average of the total number of genes detected in each sample. The percentage calculated this way is plotted in Figure 1b. For the Venn diagrams showing genes found in the single-cell samples that are also detected in the bulk, a similar approach was taken: each pairwise combination of bulk and single-cell samples was compared, and the number of genes lying in the intersection of the two was determined; then for each single-cell method, the average number of genes detected in both samples of the pair was calculated and plotted in the Venn diagram for that single-cell method. Percentages shown in the Venn diagram represents this average divided by the average number of genes detected by the bulk samples.
For comparing qPCR with RNA-seq, fragments per kilobase per million (FPKM) values and threshold cycle (Ct) values were each converted to a differential fold change compared to the median expression level for each sample. For RNA-seq data, FPKM values for each gene of interest were normalized to the median FPKM per cell, whereas for qPCR data the Ct values for each gene of interest were normalized to the median Ct for each cell. R scripts were used to construct violin plots and perform hierarchical clustering.
For generating the ‘synthetic ensemble’ single-cell data set, raw reads from all the single-cell RNA-seq libraries were bioinformatically pooled to mimic a bulk RNA-seq experiment, resulting in an ensemble library of ~186 million reads. For the ‘true’ bulk RNA-seq, 1 million cells were pooled to generate bulk RNA, and from this pool, four samples of 100 ng of RNA each were taken and independently made into four RNA-seq libraries and sequenced. The total number of reads obtained for the true bulk RNA was ~131 million reads from four libraries, with each library contributing ~30 million reads. Subsequently, four samples of 30 million raw reads each were taken from the ‘true’ bulk library and the C1-generated ‘ensemble’ library, and identical read QC and alignment pipelines were applied to each subsampled library. For each gene, mean FPKM values (i.e., the mean FPKM for that gene over the four subsamples from each library) were found for both the true bulk experiment and the ensemble experiment, and these values were plotted to generate the correlogram (Fig. 5a).
To estimate the variation in the measured gene expression as a function of gene expression level across sample replicates, we used the median absolute deviation of the FPKM divided by the median FPKM. The mean absolute deviation, or MAD, is calculated using the following formula:
We chose the median absolute deviation as the measure of statistical dispersion in this case, over using the s.d. (MAD/median rather than coefficient of variation) because the MAD is a robust statistic and is more resilient to outliers than the s.d.36. For single-cell statistics, owing to transcriptional bursting of individual cells, it is very likely that there are outliers in the measured gene expression of a set of single cells; the MAD is less influenced by such deviations.
Saturation plots were generated by randomly selecting the corresponding number of millions of raw reads from each sample library and then using the same alignment pipeline to call FPKM values for each gene. This random subsampling was repeated for each sample replicate a total of four subsampled data sets per point, and the mean number of genes with FPKM greater than 0 or 1 (Supplementary Fig. 7) was plotted. The C1 samples individually were sequenced to a depth of only 2 million reads on average; therefore, no data points beyond 2 million reads were created for those samples.
Supplementary Material
Acknowledgments
The authors would like to acknowledge W. Pan for helping with part of the sample preparation, W. Koh and B. Passarelli for help and discussions regarding bioinformatics pipelines and statistical analysis, and I. de Vlaminck for critical reading of the manuscript. A.R.W. was supported by US National Institutes of Health (NIH) U01HL099999-01 and RC4NS073015. N.F.N. was supported by NIH U01HL099999-01 and U01CA154209. T.K. was supported by P01CA139490. B.T. was supported by U01HL099995-01. F.M.M. was sponsored by the Stanford Institute of Medicine Summer Research Program. P.D. was supported by a scholarship from the Thomas and Stacey Siebel Foundation and by a BD Biosciences Stem Cell Research Grant (Summer 2011).
Footnotes
Accession codes. Gene Expression Omnibus: GSE51254. All analysis was performed using custom R scripts, available for download at http://sourceforge.net/projects/arwu-scrnaseq/files/C1_hiseq_analysis_for_paper_revision.R/download.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
A.R.W., N.F.N., T.K., P.D., M.E.R., M.F.C. and S.R.Q. conceived of the study and designed the experiments. A.R.W., N.F.N., T.K., P.D., M.E.R., B.T., F.M.M., G.L.M. and S.S. performed the experiments. A.R.W., N.F.N., T.K., P.D., M.E.R., B.T., M.F.C. and S.R.Q. analyzed the data and/or provided intellectual guidance in their interpretation. P.D., M.E.R. and M.F.C. provided samples and reagents. A.R.W., N.F.N, B.T. and S.R.Q. wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Dalerba P, et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol. 2011;29:1120–1127. doi: 10.1038/nbt.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalisky T, Blainey P, Quake SR. Genomic analysis at the single-cell level. Annu Rev Genet. 2011;45:431–445. doi: 10.1146/annurev-genet-102209-163607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bengtsson M, Ståhlberg A, Rorsman P, Kubista M. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res. 2005;15:1388–1392. doi: 10.1101/gr.3820805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Islam S, et al. Highly multiplexed and strand-specific single-cell RNA 5′ end sequencing. Nat Protoc. 2012;7:813–828. doi: 10.1038/nprot.2012.022. [DOI] [PubMed] [Google Scholar]
- 6.Islam S, et al. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011;21:1160–1167. doi: 10.1101/gr.110882.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang F, et al. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell. 2010;6:468–478. doi: 10.1016/j.stem.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang F, et al. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat Protoc. 2010;5:516–535. doi: 10.1038/nprot.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 10.Sasagawa Y, et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013;14:R31. doi: 10.1186/gb-2013-14-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shalek AK, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2:666–673. doi: 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 13.Liu CL, Bernstein BE, Schreiber SL. Whole genome amplification by T7-based linear amplification of DNA (TLAD): II. second-strand synthesis and in vitro transcription. CSH Protoc. 2008;2008 doi: 10.1101/pdb.prot5003. pdb.prot5003. [DOI] [PubMed] [Google Scholar]
- 14.Ramsköld D, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30:777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tariq MA, Kim HJ, Jejelowo O, Pourmand N. Whole-transcriptome RNAseq analysis from minute amount of total RNA. Nucleic Acids Res. 2011;39:e120. doi: 10.1093/nar/gkr547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.VanGuilder HD, Vrana KE, Freeman WM. Twenty-five years of quantitative PCR for gene expression analysis. Biotechniques. 2008;44:619–626. doi: 10.2144/000112776. [DOI] [PubMed] [Google Scholar]
- 18.Matz M, et al. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res. 1999;27:1558–1560. doi: 10.1093/nar/27.6.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez-Roca E, et al. Accurate expression profiling of very small cell populations. PLoS ONE. 2010;5:e14418. doi: 10.1371/journal.pone.0014418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurn N, et al. Novel isothermal, linear nucleic acid amplification systems for highly multiplexed applications. Clin Chem. 2005;51:1973–1981. doi: 10.1373/clinchem.2005.053694. [DOI] [PubMed] [Google Scholar]
- 21.Au KF, Jiang H, Lin L, Xing Y, Wong WH. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;38:4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffith M, et al. Alternative expression analysis by RNA sequencing. Nat Methods. 2010;7:843–847. doi: 10.1038/nmeth.1503. [DOI] [PubMed] [Google Scholar]
- 24.Marcy Y, et al. Nanoliter reactors improve multiple displacement amplification of genomes from single cells. PLoS Genet. 2007;3:1702–1708. doi: 10.1371/journal.pgen.0030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaiswal S, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oldenborg PA, Gresham HD, Lindberg FP. Cd47-signal regulatory protein α (Sirpα) regulates Fcγ and complement receptor–mediated phagocytosis. J Exp Med. 2001;193:855–862. doi: 10.1084/jem.193.7.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willingham SB, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA. 2012;109:6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The External RNA Controls Consortium. et al. The External RNA Controls Consortium: a progress report. Nat Methods. 2005;2:731–734. doi: 10.1038/nmeth1005-731. [DOI] [PubMed] [Google Scholar]
- 29.Jiang L, et al. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011;21:1543–1551. doi: 10.1101/gr.121095.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17 http://journal.embnet.org/index.php/embnetjournal/article/view/200/479. [Google Scholar]
- 31.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoaglin DC, Mosteller F, Tukey JW. Understanding Robust and Exploratory Data Analysis. Wiley; New York: 1983. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.