

Abstract
Epigenetic modifications are implicated in the maintenance and regulation of transcriptional memory by marking genes that were previously transcribed to facilitate transmission of these expression patterns through cell division. During germline specification and maintenance, extensive epigenetic modifications are acquired. Yet somehow at fertilization, the fusion of the highly differentiated sperm and egg results in formation of the totipotent zygote. This massive change in cell fate implies that the selective erasure and maintenance of epigenetic modifications at fertilization may be critical for the re-establishment of totipotency. In this review, we discuss recent studies that provide insight into the extensive epigenetic reprogramming that occurs around fertilization and the mechanisms that may be involved in the re-establishment of totipotency in the embryo.
INTRODUCTION
In 1893, August Weismann was among the first to appreciate that germ cells are specialized cells and that only these cells can give rise to offspring of the subsequent generation. Importantly, from the observation of several organisms, Weismann also concluded that the germ lineage occurs along ‘germ-tracks’ (the germline lineage) that undergo a high degree of differentiation to become the highly specialized sperm and eggs cells. Indeed, Weismann mused that ‘I can see no advantage in objecting to describe a cell of the germ-track as a somatic cell.’ [1]. Yet as Weismann also observed, following the completion of the germ-track, the egg and sperm must come together at fertilization to produce a totipotent zygote. To reconcile these facts, Weismann proposed that the germ lineage contains a special germ plasm that passively allows the germ-track to proceed through these ‘somatic events’ while retaining the ability to return to totipotency following fertilization [1]. Today, evidence from a number of model systems is beginning to provide molecular evidence of Weismann’s vision.
Since all tissues must be specified from the same set of genes, the process of tissue differentiation is inherently epigenetic. Over the past several years, we have begun to understand epigenetic gene regulation and how epigenetic phenomena are used to control differentiation during development. This understanding has led to an opportunity to elucidate how the germ lineage is specified and maintained at the epigenetic level and how this specification may have to be reversed after fertilization to restore totipotency. In this review, we will not attempt to exhaustively present all that is known about epigenetics in the germline cycle [2]. Rather, we will highlight recent epigenetic germline studies in both invertebrates and vertebrates that hint that extensive epigenetic reprogramming occurs around fertilization. This reprogramming is likely critical to maintain totipotency from one generation to the next.
EVIDENCE FOR EPIGENETIC REPROGRAMMING AT FERTILIZATION
Cloning experiments performed in various vertebrates provide evidence for extensive epigenetic reprogramming at fertilization. Beginning with the Xenopus laevis cloning experiments performed by John Gurdon in the late 1950’s along with the cloning of Dolly the sheep in 1996, it has been possible to transform a somatic cell nucleus into a cloned animal by transplanting it into enucleated oocyte (Figure 1A) [3, 4]. The donor chromosomes from the somatic nucleus inherently contain all of the genetic material necessary to produce all cells of the body, and yet these chromosomes have been epigenetically modified so they produce only the proteins that are necessary to specify and maintain the somatic cell type. Because of this differentiated state, it was once thought that it is impossible to reprogram a somatic nucleus to become totipotent. However, the successful cloning of Dolly and subsequently other animals through somatic cell nuclear transfer (SCNT) suggests, as Weismann correctly proposed, that the oocyte cytoplasm natively contains the necessary factors to perform this reprogramming. Recently, somatic reprogramming has been further revolutionized by Yamanaka and colleagues [5], who demonstrated that somatic reprogramming can be triggered by just four transcription factors (Oct4, Sox2, Nanog and c-Myc) (Figure 1A). Three of these four transcription factors are pluripotency factors thought to play a role in reprogramming the embryo back to pluripotency after fertilization. The success of the induced pluripotent stem cell process (iPS) confirms that natural reprogramming occurs through defined genetic pathways, and yet the mechanisms involved in natural reprogramming and in the induction of pluripotent stem cells remain largely unknown. In addition, cloning through SCNT and iPS is highly inefficient compared to the normal process and often results in severe abnormalities, including kidney and liver defects as well as placental overgrowth [6]. These difficulties hint at the extent of epigenetic reprogramming that must occur naturally. Remarkably, when cloned animals survive and have offspring, the distinct abnormalities associated with the type of donor nuclei used are no longer observed [6]. This indicates that passage through the natural germline reprogramming process may be sufficient to revert the abnormal epigenetic state.
Figure 1:
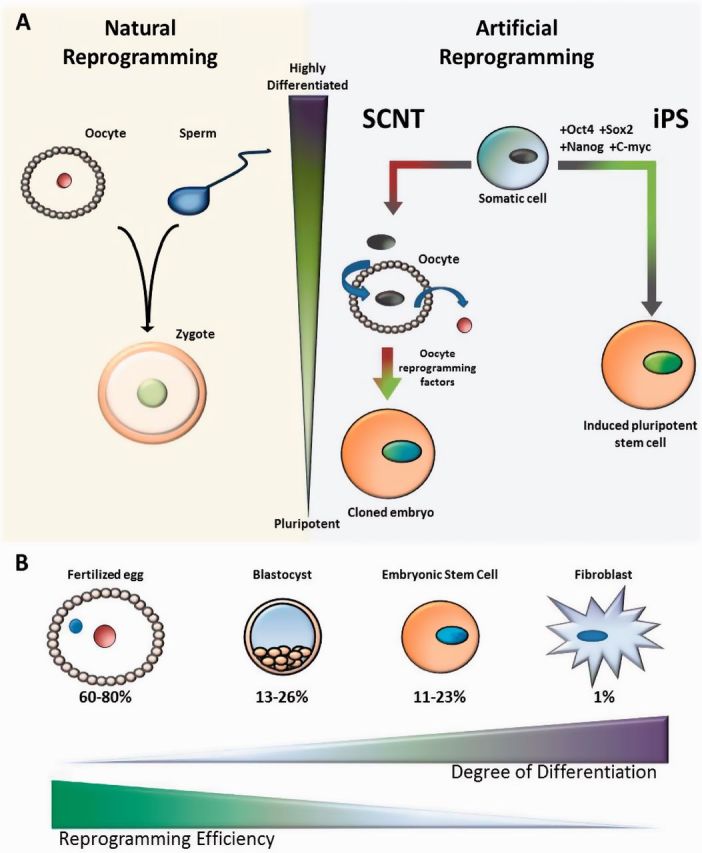
Natural reprogramming versus artificial reprogramming. (A) The natural reprogramming that occurs when the highly differentiated sperm and egg come together at fertilization to generate the totipotent embryo is analogous to what occurs during SCNT and the iPS process. (B) The reprogramming efficiency of artificial reprogramming processes decreases with the increasing differentiation of the donor cell type.
A clear illustration of the requirement for epigenetic resetting in somatic reprogramming can be seen by comparing the efficiency of generating a frog or mouse following SCNT from different donor cell types. In Xenopus, SCNT has been performed using embryonic donor cells with an efficiency of 36% versus an efficiency of 1.5% from differentiated cell types [7]. Similarly in mouse, the efficiencies obtained from cloning from a fertilized egg (60–80%), a blastomere (13–26%), an embryonic stem (ES) cell (11–23%) or a fibroblast cell (1%) decrease with increasing degrees of differentiation (Figure 1B) [8]. These data suggest that increasing epigenetic information acquired during differentiation is increasingly more difficult to reprogram. Yet, even in highly differentiated cases, it is possible to generate viable adult animals. This success, despite the limitations of the artificial process, proves that the oocyte is capable of a high degree of epigenetic reprogramming and implies that extensive epigenetic reprogramming takes place during normal reproduction.
REPROGRAMMING EPIGENETIC MEMORY
Can the inefficiencies and failures of these processes teach us about the types of epigenetic reprogramming that must occur naturally? In addition to the frequent failure to obtain cloned animals, often cloned Xenopus embryos fail to turn on appropriate embryonic genes and inappropriately express genes from the cell type they were cloned from [9, 10]. It is apparent from these difficulties that the donor nuclei have been epigenetically programmed toward specific tissue fates and that this program is not always sufficiently reset during the cloning process. The nature of the epigenetic program in these differentiated donor nuclei is poorly understood. Thus, the mechanisms of reprogramming remain largely unknown. However, over the past several years, parts of the epigenetic program have begun to be elucidated.
Nucleosomes are composed of histones that can be chemically modified and these modifications of the amino-terminal tails are often correlated with the transcription status of the genes that are packaged around them. In addition, certain histone modifications are typically associated with particular histone variants, which contain small numbers of amino acid substitutions compared to canonical histones and can substitute for their corresponding canonical histones within the nucleosome. For example, one of the most well-characterized histone modifications is methylation of lysine 4 on histone H3 (H3K4me). Histone 3 lysine 4 can be mono-, di- or tri-methylated and these varying levels of methylation may have slightly different functions [11]. However, almost all H3K4me is associated with the histone variant H3.3 and is found at active genes [12, 13]. This finding initially led to the hypothesis that H3K4me plays a role in gene activation, but recent work suggests that H3K4me may play a slightly different role in transcription. Accumulating evidence suggests that the acquisition of H3K4me is associated with RNA polymerase II elongation (Figure 2). For example, experiments in yeast have shown that the H3K4 methyltransferase, Set-1 (the yeast ortholog of the mammalian MLL), is in a complex with RNA polymerase II and is recruited to active genes by this interaction [14]. In addition, in Drosophila melanogaster S2 cells, high-resolution mapping of H3K4me2 and RNA polymerase II has demonstrated that their distribution closely matches genome wide [15]. These findings have led to a new model where H3K4me acts as an epigenetic memory to maintain transcription patterns during tissue differentiation rather than in de novo transcriptional activation (Figure 2).
Figure 2:

A model for H3K4me2 epigenetic memory. H3K4me2 is acquired co-transcriptionally as RNA polymerase II elongates. H3K4me2 can be faithfully propagated as a cell divides and may act as an epigenetic transcriptional memory, but may have to be reprogrammed to allow for changes in cell fate.
Recently, this model has been more directly tested in Dictyoselium discoideum cells [16]. Employing a live-cell RNA imaging technique, Muramoto et al. [16] directly examined the expression of genes during inheritance from mother to daughter. These experiments found that expression levels were more faithfully maintained in a cell lineage than in cells that are not lineage related. Furthermore, this epigenetic memory of active transcription was dependent upon H3K4 as well as the H3K4 methyltransferase Set-1.
Could H3K4me comprise part of the epigenetic signal that must be reset during SCNT and natural reproduction? The Gurdon Lab found that the inappropriate expression of endodermal genes was detected in Xenopus embryos derived from the transfer of differentiated endodermal nuclei. This inappropriate epigenetic memory was dependent upon lysine 4 of the histone variant H3.3 [17]. If H3K4me2 functions in the maintenance of transcriptional patterns, then methylation on lysine 4 of histone H3.3 may have to be reprogrammed during SCNT and natural reproduction.
The best evidence that covalent epigenetic modifications can act as an epigenetic memory comes from the trithorax group (trxG) and Polycomb group (PcG) of genes. The trxG and PcG genes were originally identified in Drosophila as genes that are required to maintain the expression pattern of Hox genes after the initial set of transcription factors, which establish their expression, are no longer present (Figure 3A). These findings suggested that trxG and PcG act to maintain transcriptional memory [18]. In the absence of trxG and PcG proteins, flies exhibit homeotic transformations [18].
Figure 3:
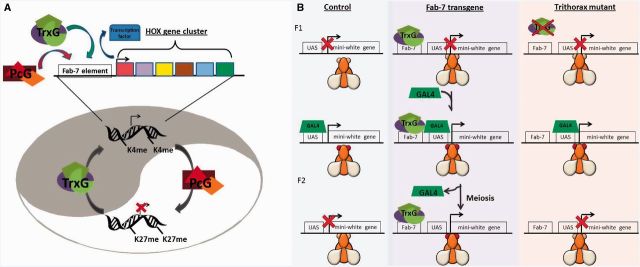
Trithorax and Polycomb group protein complexes maintain epigenetic cellular memory. (A) Trithorax, the H3K4 methyltransferase, and Polycomb, the H3K27 methyltransferase act antagonistically to maintain active (trxG) or repressed (PcG) chromatin at the Hox cluster in Drosophila. These heritable chromatin states form a cellular memory of transcription that aids in the specification of proper segments. (B) Cavalli and Paro [19] demonstrated in flies that trithorax binding to the Fab-7 element from the Hox cluster confers epigenetic transcriptional stability. In control flies, a pulse of GAL4 activates the mini-white transgene resulting in red eyes. When the transgene contains the Fab-7 element, this effect is meiotically stable. The meiotic stability is eliminated in trithorax mutants indicating that it is likely dependent upon H3K4me.
The founding member of the trxG, trithorax, encodes an H3K4 methyltransferase [19]. This suggests that the role of trithorax in tissue specification may be accomplished through H3K4me. Further evidence can be seen in a transgenic experiment performed in Drosophila. trxG and PcG genes act through cis-DNA response elements. One such element, Fab-7 was engineered on a transgene in Drosophila to surround a UAS-lacZ reporter so that the presence of the Fab-7 elements prevented a heat shock-inducible GAL4 driver from inducing the flanking mini-white gene. When activated by a pulse of GAL4, the expression of the flanking mini-white reporter is mitotically and even meiotically stable, in the absence of GAL4. This stability is dependent upon trithorax, the H3K4 methyltransferase, further suggesting that H3K4me can serve as an epigenetic transcriptional memory (Figure 3B) [20, 21].
PLURIPOTENCY IN ES CELLS
The maintenance of pluripotency in ES cells may be regulated by bivalent domains, consisting of H3K4me3 (active) and H3K27me3 (repressive) coexisting at developmentally regulated promoters [22]. At these bivalent promoters, H3K27me3 strongly correlates with the binding of PRC2 and PRC1, encoded by PcG genes, as well as the binding of the pluripotency factors Oct4, Sox2 and Nanog [23, 24]. The EZ subunit of PRC2 encodes an H3K27 methyltransferase, while functions as part of the PRC1 complex to bind H3K27me3 and represses transcription [18]. In addition, the repression of many PRC1 target genes requires Oct4 [25]. This suggests that the pluripotency factors coordinate H3K27me3 to transcriptionally represses developmental control genes and maintain a pluripotent state [23, 26]. At the same time H3K4me3, maintained by the trxG genes, keeps these control regions poised for activation upon differentiation. When ES cells differentiate, the bivalent domains resolve into either H3K4me3 or H3K27me3 domains exclusively [22]. Thus, the bivalent state in ES cells is thought to keep genes poised for rapid conversion to either an ‘on’ or ‘off’ state.
Although it is not clear what triggers resolution of bivalent domains, some data provide insight into possible modes of regulation. For example, the H3K4me jumonji-class demethylase Rbp2 can interact with the H3K27 methyltransferase complex PRC2 and may function in converting bivalent domains into active domains [27]. Conversely, the H3K27me demethylase Utx associates with trithorax and may participate in converting bivalent domains into repressed domains [28]. The conversion of these bivalent domains into either repressed or activated domains may be necessary for tissue specification.
INHERITANCE THROUGH GAMETES
Although much is known about how pluripotency is maintained in ES cells, how pluripotency is re-established in the embryo from highly differentiated gametes remains largely a mystery. In order to gain insight into this process, we must compare the epigenetic status of the gametes to the reprogrammed pluripotent state of the embryo and ES cells. A recent study in mouse demonstrated that mouse oocytes have a lack of H3K4me3 at methylated CpG islands [26]. However, beyond this relatively little is known about the genome-wide patterns of other histone modifications in oocytes.
In contrast, much more is currently known about the patterns of epigenetic modifications in sperm. In mature vertebrate sperm, much of the genome is repackaged from histones to protamines. Because of this replacement, it was originally thought that vertebrate sperm were lacking epigenetic information contained on histone tails. However, a recent study suggests that this is not the case. In mature human sperm, 4% of the genome remains wrapped around canonical histones, and this chromatin is highly enriched at the promoters of genes that function in embryonic development. Remarkably, many of the promoters that contain canonical histones have both H3K4me2/3 and H3K27me3 and these bivalent promoters significantly overlap with the promoters that are bivalent in ES cells [23]. A highly similar bivalent chromatin state is also found in mature mouse sperm [22]. These findings are especially significant considering that these embryonic development genes with bivalent chromatin are not expressed during spermatogenesis. The presence of bivalent chromatin domains at embryonic development genes in sperm suggests an enticing model where the pluripotent state of the embryo may already be established in sperm and may be faithfully propagated through early development to poise genes for embryonic development (Figure 4). Nevertheless, since the presence of bivalent chromatin has not yet been verified in early human or mouse embryos, proof of the maintenance and propagation of these domains requires further investigation.
Figure 4:

Summary of reprogramming events that occur at fertilization. At fertilization, the highly differentiated gametes undergo a dramatic change in cell fate to form the totipotent zygote. During this process, certain epigenetic information is stably propagated while other epigenetic information is reprogrammed. Bivalent domains (H3K4me3 and H3K27me3) acquired through trithorax (MLL) and Polycomb at developmental transcription factor loci are stably propagated from sperm to the embryo in multiple organisms. H3K4me2 at spermatogenesis genes is erased at fertilization by LSD1 in C. elegans. H3K36me3 at germline genes is stably maintained by MES-4 in the C. elegans embryo. Global DNA methylation is erased by Tet3 after fertilization and then returns later in embryogenesis in mice. During this erasure, DNA methylation at Oct4 and Nanog is erased but is maintained at critical CpG residues at imprinted loci due to the protein Stella. Certain retrotransposons, such as IAP elements, are resistant to the global demethylation at fertilization in mice.
In particular, evidence from zebrafish casts some doubt on the model. Similar to what has been observed in human and mouse, bivalent chromatin domains have also been observed in zebrafish sperm [24]. This suggests that the possibility that the use of bivalent chromatin domains to poise embryonic expression may be a highly conserved mechanism. However, when genome-wide chromatin was assayed in the zebrafish embryo to confirm the propagation of bivalent domains, conflicting results were found. Initially, it was observed that bivalent domains are only acquired following zygotic genome activation (ZGA) [25]. This result suggests that bivalent domains may be re-acquired transcriptionally rather than faithfully maintained from sperm. However in a subsequent study, bivalent chromatin domains were observed before ZGA [27]. Based on these two results, it is possible that the bivalent chromatin domains are reduced but not fully erased. This could explain why they were not detected in the original study [25]. If this is the case, then the maintenance of reduced bivalent chromatin domains could act as a seed to re-establish larger domains following ZGA. Intriguingly, this type of ‘signposting’ model is very reminiscent of what occurs at DNA-methylated domains following fertilization in mice.
INHERITANCE AND REPROGRAMMING OF DNA METHYLATION
DNA methylation is found at CpG residues in mammals and can be stably maintained by Dnmt1, which selectively methylates hemi-methylated DNA during DNA replication [28]. Most of the CpG methylation in the mammalian genome is present at repeated sequences, such as retrotransposons and their remnants, where it is thought to play a role in stably repressing these sequences [29]. However, some CpG methylation is also found at developmentally regulated genes. For example, imprinted genes, which are maternally or paternally expressed dependent upon parent-of-origin, are often associated with CpG islands, termed imprinted control regions (ICRs), which are either maternally or paternally methylated (Figure 4) [30].
In oocytes, CpG methylation occurs mainly in CpG island-containing promoters while the most of the genome remains hypomethylated, a pattern that closely resembles the methylation state of the pre-implantation embryo [26, 31]. This suggests that oocyte methylation could have a function in the early embryo. The establishment and maintenance of this DNA methylation is dependent on the activity of Dnmt3a and Dnmt1 [32, 33]. Interestingly, both of these enzymes have CpG island-containing promoters that are hypermethylated in the oocyte, so it possible that DNA methylation is reinforced by a feedback loop. Conversely, in mammalian sperm it has been shown that the methylation distribution in the genome closely resembles the pattern seen in ES cells, with many developmentally regulated genes hypomethylated compared to somatic tissues. These hypomethylated genes correlate strongly with those genes bound by the pluripotency factors Oct4, Sox2 and Nanog. Interestingly, however, the pluripotency genes Oct4 and Nanog themselves are hypermethylated in sperm but become unmethylated and expressed in ES cells [23, 34, 35]. This suggests that these master pluripotency genes may need to be reprogrammed in the embryo to restore pluripotency.
The reprogramming of DNA methylation at Oct4 and Nanog is thought to occur at fertilization during the genome-wide wave of DNA demethylation that occurs actively in the paternal genome and passively in the maternal genome [36, 37]. The mammalian genome contains three Tet family proteins that are thought to function in DNA demethylation through a hydroxymethylation intermediate (Figure 4) [38]. The maternal loss of Tet3 results in the failure to DNA demethylate the paternal genome along with a corresponding increase in the incidence of embryonic lethality and a decrease in the fertility of mutant females [39]. This suggests that the active demethylation of the paternal genome is catalyzed by maternal Tet3. Furthermore, in Tet3 mutants, Oct4 and Nanog fail to be demethylated and an Oct4-EGFP transgene fails to be expressed from a paternally inherited transgene [39]. These results suggest that Oct4 and Nanog are targets of the Tet3 active paternal demethylation and that this demethylation likely plays a critical role in restoring pluripotency in the embryo after fertilization. Consistent with this model, there is a decrease in the efficiency of reprogramming of DNA methylation and a decrease in Oct4 expression when SCNT is performed using Tet3 mutant oocytes [39]. There is also an increase in the efficiency of iPS when DNA methyltransferase activity is inhibited [40].
Remarkably, ICRs, which must remain methylated to maintain parent-of-origin expression, resist demethylation during the genome-wide DNA demethylation that occurs after fertilization in mammalian embryos. During this demethylation, ICRs do lose some DNA methylation. However, critical CpG residues remain methylated and this methylation is thought to seed the re-acquisition of larger methylated domains at ICRs following demethylation [30]. This mechanism is very reminiscent of what may be occurring at bivalent chromatin domains in the zebrasfish embryo. Some retrotransposon sequences such as intra-cisternal A-type particle (IAP) elements, which also must remain repressed, are similarly resistant to this DNA demethylation (Figure 4) [31, 41].
The maintenance of DNA methylation at critical ICR CpG residues is dependent upon Dnmt1, expressed specifically from a maternal promoter termed Dnmt1o. In the absence of Dnmt1o, embryos die late in gestation due to the failure to maintain allele-specific DNA methylation at ICRs [42]. In addition, the resistance of ICRs to demethylation is dependent upon the protein Stella/PGC7 (Figure 4). PGC7 binds specifically to H3K9me2-containing chromatin in the mouse maternal pronucleus [43]. In the absence of maternal provided PGC7, the imprinted genes Peg1, Peg5, Peg10, H19 and Rasgrf1 become inappropriately demethylated [44]. Thus, it seems clear that mammals have evolved a complex set of regulatory mechanisms that allow DNA methylation at certain loci to be inherited through fertilization to the next generation while DNA methylation at other loci is reprogrammed.
INHERITANCE AND REPROGRAMMING OF HISTONE METHYLATION
In addition to demethylating key pluripotency genes to reactivate the embryonic program, it is likely that other epigenetic information must be removed at fertilization to prevent the germline program from being inappropriately propagated in the embryo. For example, along with bivalent domains which may contain critical information for embryogenesis, mature sperm also have H3K4me2/3 in genes that functioned previously in the germline during spermatogenesis [23]. The acquisition of this histone information in sperm is easier to reconcile than the acquisition of bivalent chromatin, because these genes presumably acquired H3K4me2/3 in their chromatin during transcription. Like bivalent domains, these histone modification domains could potentially be stably inherited in the embryo and this ‘epigenetic baggage’ could potentially result in the inappropriate expression of spermatogenesis genes in the embryo. However, recent data suggest that mechanisms may exist to prevent this. The amine-oxidase class demethylase LSD1/KDM1 can specifically demethylate H3K4me2 [45]. Loss of function mutations in LSD1 ortholog in both Drosophila and Caenorhabditis elegans result in sterility, and in C. elegans this sterility is correlated with increasing H3K4me2 and the increasing expression of spermatogenesis genes [46–49]. These results suggest that the role of LSD1 in the germline is to demethylate H3K4me2 at spermatogenesis genes and prevent this epigenetic baggage from heritably persisting in the embryo of the next generation. This finding fits nicely with the findings from human and mouse sperm, where H3K4me2 is observed in spermatogenesis genes [22, 23]. In flies, worms and mice, LSD1 is maternally deposited in the oocyte [46, 47, 50]. Thus, it is intriguing to propose that the H3K4me2 in mature vertebrate sperm at spermatogenesis genes may not be transmitted to the embryo due to the maternal demethylation activity of LSD1 acting at or around fertilization (Figure 4).
Although LSD1 appears to function in reprogramming the germline program at fertilization, there is also evidence in C. elegans that heritable epigenetic mechanisms exist which may facilitate re-initiation of the germline program in the subsequent generation. The co-transcriptional acquisition of H3K36 methylation by Set-2 in yeast is proposed to block spurious intragenic transcription [51]. However, the function of H3K36 methylation in metazoans is not well understood. In C. elegans, H3K36 methylation is carried out by a Set-2-related protein, methyltransferase 1 (MET-1) and a second methyltransferase, maternal effect sterile (MES)-4 [52, 53]. Genome-wide MES-4 mapping in the early embryo revealed that MES-4 associates with germline-specific genes that were previously expressed in the maternal germline and maintains H3K36 methylation at these loci in the absence of transcription [54, 55]. Upon specification of the germline precursors Z2 and Z3, MES-4 transitions from global maintenance to exclusive association with Z2/Z3 [54, 55]. It has been proposed that this H3K36 methylation maintenance by MES-4 serves as an epigenetic memory of transcriptional patterns by marking genes that were previously transcribed in the maternal germline for reactivation in the embryonic germline [54, 55]. Consistent with this hypothesis, maternal loss of MES-4 results in the loss of the embryonic primordial germ cells Z2/Z3 and complete sterility (Figure 4) [56].
Taken together, the data on histone methylation and DNA methylation provide an emerging picture. They suggest that there is a highly specific epigenetic reprogramming event that takes place at fertilization which helps remove epigenetic baggage, while specifically allowing important epigenetic information to be transmitted through to the embryo.
TRANSGENERATIONAL EFFECTS: EVIDENCE FOR ERRORS IN REPROGRAMMING
Based on the complex series of epigenetic reprogramming events that likely occur to restore totipotency following fertilization, it seems probable that there may be instances where epigenetic reprogramming is incomplete, allowing epigenetic information to be inappropriately transmitted through fertilization from the gametes to the embryo of the next generation. These instances could give rise to epialleles with medical implications. Recently, a number of such instances have been documented in model organisms [57]. For example, when a mouse female pronucleus is transplanted into a recipient egg from a different genetic background, the resulting nucleocytoplasmic hybrids have inappropriate transcription and corresponding DNA methylation defects in certain tissues. Remarkably, >50% of the time, these defects are meiotically heritable through sperm [58]. In male rats, exposure to endocrine disruptors during the period of gonadal sex determination causes increased male infertility with corresponding changes in DNA methylation. This reduction in male fertility is meiotically heritable for at least four generations [59]. In viable yellow mice, transcription originating from an IAP retrotransposon results in ectopic expression of the agouti locus and mice with a yellow coat color. This coat color can be preferentially inherited through the mother and correlates with the DNA methylation status of the IAP retrotransposon, suggesting that the preferential inheritance is due to incomplete reprogramming of the DNA methylation in the IAP element. Interestingly, this preferential inheritance of coat color does not occur paternally [60].
CONCLUSION
In recent years, a lot of data have emerged uncovering the role of epigenetic information in tissue differentiation. Based on this data, it has become increasingly clear that a complex set of regulatory mechanisms likely exist to regulate what epigenetic information is stably propagated from the gametes through fertilization to the embryo. While it is clear that much information is yet to be learned about this process, the studies presented here provide a nascent view of the types of information that may be regulated, the function of this reprogramming and how it may be accomplished mechanistically.
Key Points.
At fertilization, the highly differentiated gametes come together to produce the totipotent zygote.
Epigenetic modifications, such as DNA methylation and histone methylation, may provide a cellular memory that aids in the specification and maintenance of cell fates.
A complex set of regulatory mechanisms likely exist to regulate what epigenetic information is stably propagated from the gametes through fertilization to the embryo.
ACKNOWLEDGEMENTS
The authors would like to thank Bill Kelly for very helpful comments on an earlier draft of this review. The authors would also like to thank the entire Katz lab for providing feedback on the article.
Biographies
Jadiel A. Wasson is a Graduate Student in the program at Emory University conducting thesis work in the laboratory of Dr Katz.
Chelsey C. Ruppersburg is also a Graduate Student in the program at Emory University conducting thesis work in the laboratory of Dr Katz.
David J. Katz is an Assistant Professor in the Department of Cell Biology at Emory University.
References
- 1.Weismann A, Parker WN, Rönnfeldt H. The Germ-Plasm: A Theory of Heredity. xxiii. New York: Charles Scribner's Sons; 1893. p. 477. [Google Scholar]
- 2.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447(7143):425–32. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 3.Campbell KH, McWhir J, Ritchie WA, et al. Sheep cloned by nuclear transfer from a cultured cell line. Nature. 1996;380(6569):64–6. doi: 10.1038/380064a0. [DOI] [PubMed] [Google Scholar]
- 4.Gurdon JB, Elsdale TR, Fischberg M. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature. 1958;182(4627):p, 64–5. doi: 10.1038/182064a0. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 6.Hochedlinger K, Jaenisch R. Nuclear transplantation: lessons from frogs and mice. Curr Opin Cell Biol. 2002;14(6):741–8. doi: 10.1016/s0955-0674(02)00380-0. [DOI] [PubMed] [Google Scholar]
- 7.Gurdon JB. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J Embryol Exp Morphol. 1962;10:622–40. [PubMed] [Google Scholar]
- 8.Hochedlinger K, Jaenisch R. Nuclear reprogramming and pluripotency. Nature. 2006;441(7097):1061–7. doi: 10.1038/nature04955. [DOI] [PubMed] [Google Scholar]
- 9.Ng RK, Gurdon JB. Epigenetic memory of active gene transcription is inherited through somatic cell nuclear transfer. Proc Natl Acad Sci USA. 2005;102(6):1957–62. doi: 10.1073/pnas.0409813102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Byrne JA, Simonsson S, Gurdon JB. From intestine to muscle: nuclear reprogramming through defective cloned embryos. Proc Natl Acad Sci USA. 2002;99(9):6059–63. doi: 10.1073/pnas.082112099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25(1):15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 12.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128(4):707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell. 2002;9(6):1191–200. doi: 10.1016/s1097-2765(02)00542-7. [DOI] [PubMed] [Google Scholar]
- 14.Ng HH, Robert F, Young RA, et al. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell. 2003;11(3):709–19. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 15.Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet. 2005;37(10):1090–7. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- 16.Muramoto T, Muller I, Thomas G, et al. Methylation of H3K4 is required for inheritance of active transcriptional states. Curr Biol. 2010;20(5):397–406. doi: 10.1016/j.cub.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Ng RK, Gurdon JB. Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol. 2008;10(1):102–9. doi: 10.1038/ncb1674. [DOI] [PubMed] [Google Scholar]
- 18.Schuettengruber B, Chourrout D, Vervoort M, et al. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128(4):735–45. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 19.Byrd KN, Shearn A. ASH1, a Drosophila trithorax group protein, is required for methylation of lysine 4 residues on histone H3. Proc Natl Acad Sci USA. 2003;100(20):11535–40. doi: 10.1073/pnas.1933593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cavalli G, Paro R. Epigenetic inheritance of active chromatin after removal of the main transactivator. Science. 1999;286(5441):955–8. doi: 10.1126/science.286.5441.955. [DOI] [PubMed] [Google Scholar]
- 21.Cavalli G, Paro R. The Drosophila Fab-7 chromosomal element conveys epigenetic inheritance during mitosis and meiosis. Cell. 1998;93(4):505–18. doi: 10.1016/s0092-8674(00)81181-2. [DOI] [PubMed] [Google Scholar]
- 22.Brykczynska U, Hisano M, Erkek S, et al. Repressive and active histone methylation mark distinct promoters in human and mouse spermatozoa. Nat Struct Mol Biol. 2010;17(6):679–87. doi: 10.1038/nsmb.1821. [DOI] [PubMed] [Google Scholar]
- 23.Hammoud SS, Nix DA, Zhang H, et al. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460(7254):473–8. doi: 10.1038/nature08162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu SF, Zhang H, Cairns BR. Genes for embryo development are packaged in blocks of multivalent chromatin in zebrafish sperm. Genome Res. 2011;21(4):578–89. doi: 10.1101/gr.113167.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vastenhouw NL, Zhang Y, Woods IG, et al. Chromatin signature of embryonic pluripotency is established during genome activation. Nature. 2010;464(7290):922–6. doi: 10.1038/nature08866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smallwood SA, Tomizawa S, Krueger F, et al. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet. 2011;43(8):811–4. doi: 10.1038/ng.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindeman LC, Andersen IS, Reiner AH, et al. Prepatterning of developmental gene expression by modified histones before zygotic genome activation. Dev Cell. 2011;21(6):993–1004. doi: 10.1016/j.devcel.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 28.Yoder JA, Soman NS, Verdine GL, et al. DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J Mol Biol. 1997;270(3):385–95. doi: 10.1006/jmbi.1997.1125. [DOI] [PubMed] [Google Scholar]
- 29.Ooi SK, O'Donnell AH, Bestor TH. Mammalian cytosine methylation at a glance. J Cell Sci. 2009;122(Pt 16):2787–91. doi: 10.1242/jcs.015123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartolomei MS. Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev. 2009;23(18):2124–33. doi: 10.1101/gad.1841409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith ZD, Chan MM, Mikkelsen TS, et al. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484(7394):339–44. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69(6):915–26. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 33.Bourc'his D, Xu GL, Lin CS, et al. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294(5551):2536–9. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 34.Farthing CR, Ficz G, Ng RK, et al. Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genet. 2008;4(6):e1000116. doi: 10.1371/journal.pgen.1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imamura M, Miura K, Iwabuchi K, et al. Transcriptional repression and DNA hypermethylation of a small set of ES cell marker genes in male germline stem cells. BMC Dev Biol. 2006;6:34. doi: 10.1186/1471-213X-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oswald J, Engemann S, Lane N, et al. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000;10(8):475–8. doi: 10.1016/s0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- 37.Mayer W, Niveleau A, Walter J, et al. Demethylation of the zygotic paternal genome. Nature. 2000;403(6769):501–2. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 38.Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25(23):2436–52. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu TP, Guo F, Yang H, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477(7366):606–10. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 40.Huangfu D, Maehr R, Guo W, et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008;26(7):795–7. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lane N, Dean W, Erhardt S, et al. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis. 2003;35(2):88–93. doi: 10.1002/gene.10168. [DOI] [PubMed] [Google Scholar]
- 42.Howell CY, Bestor TH, Ding F, et al. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell. 2001;104(6):829–38. doi: 10.1016/s0092-8674(01)00280-x. [DOI] [PubMed] [Google Scholar]
- 43.Nakamura T, Liu YJ, Nakashima H, et al. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486(7403):415–9. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- 44.Nakamura T, Arai Y, Umehara H, et al. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol. 2007;9(1):64–71. doi: 10.1038/ncb1519. [DOI] [PubMed] [Google Scholar]
- 45.Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–53. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 46.Rudolph T, Yonezawa M, Lein S, et al. Heterochromatin formation in Drosophila is initiated through active removal of H3K4 methylation by the LSD1 homolog SU(VAR)3-3. Mol Cell. 2007;26(1):103–15. doi: 10.1016/j.molcel.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 47.Katz DJ, Edwards TM, Reinke V, et al. A C. elegans LSD1 demethylase contributes to germline immortality by reprogramming epigenetic memory. Cell. 2009;137(2):308–20. doi: 10.1016/j.cell.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Stefano L, Ji JY, Moon NS, et al. Mutation of Drosophila Lsd1 disrupts H3-K4 methylation, resulting in tissue-specific defects during development. Curr Biol. 2007;17(9):808–12. doi: 10.1016/j.cub.2007.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szabad J, Reuter G, Schroder MB. The effects of two mutations connected with chromatin functions on female germ-line cells of Drosophila. Mol Gen Genet. 1988;211(1):56–62. doi: 10.1007/BF00338393. [DOI] [PubMed] [Google Scholar]
- 50.Kageyama S, Gunji W, Nakasato M, et al. Analysis of transcription factor expression during oogenesis and preimplantation development in mice. Zygote. 2007;15(2):117–28. doi: 10.1017/S096719940700411X. [DOI] [PubMed] [Google Scholar]
- 51.Carrozza MJ, Li B, Florens L, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123(4):581–92. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 52.Bender LB, Suh J, Carroll CR, et al. MES-4: an autosome-associated histone methyltransferase that participates in silencing the X chromosomes in the C. elegans germ line. Development. 2006;133(19):3907–17. doi: 10.1242/dev.02584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andersen EC, Horvitz HR. Two C. elegans histone methyltransferases repress lin-3 EGF transcription to inhibit vulval development. Development. 2007;134(16):2991–9. doi: 10.1242/dev.009373. [DOI] [PubMed] [Google Scholar]
- 54.Furuhashi H, Takasaki T, Rechtsteiner A, et al. Trans-generational epigenetic regulation of C. elegans primordial germ cells. Epigenetics Chromatin. 2010;3(1):15. doi: 10.1186/1756-8935-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rechtsteiner A, Ercan S, Takasaki T, et al. The histone H3K36 methyltransferase MES-4 acts epigenetically to transmit the memory of germline gene expression to progeny. PLoS Genet. 2010;6(9) doi: 10.1371/journal.pgen.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fong Y, Bender L, Wang W, et al. Regulation of the different chromatin states of autosomes and X chromosomes in the germ line of C. elegans. Science. 2002;296(5576):2235–8. doi: 10.1126/science.1070790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Richards EJ. Inherited epigenetic variation–revisiting soft inheritance. Nat Rev Genet. 2006;7(5):395–401. doi: 10.1038/nrg1834. [DOI] [PubMed] [Google Scholar]
- 58.Roemer I, Reik W, Dean W, et al. Epigenetic inheritance in the mouse. Curr Biol. 1997;7(4):277–80. doi: 10.1016/s0960-9822(06)00124-2. [DOI] [PubMed] [Google Scholar]
- 59.Anway MD, Cupp AS, Uzumcu M, et al. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308(5727):1466–9. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morgan HD, Sutherland HG, Martin DI, et al. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23(3):314–8. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]