

Abstract
Objectives
Telavancin is approved in Europe for the treatment of nosocomial pneumonia caused by methicillin-resistant Staphylococcus aureus when other alternatives are not suitable. The approved European prescribing information contraindicates the use of telavancin in patients with severe renal impairment (creatinine clearance <30 mL/min, including patients on haemodialysis) and pre-existing acute renal failure owing to the higher observed mortality in these patients. Data from the ATTAIN studies were reanalysed, excluding patients with these contraindicating conditions at baseline. (At the time of submission of this article, the European marketing authorization of telavancin for the treatment of nosocomial pneumonia was suspended pending evidence of a new European Medicines Agency-approved supplier. Clinigen Healthcare Ltd, Theravance's commercialization partner for telavancin in Europe, is in the process of seeking approval of a new manufacturing source.)
Methods
A post hoc analysis of data from two Phase 3 ATTAIN trials of telavancin for the treatment of Gram-positive nosocomial pneumonia assessing clinical outcomes and safety.
Results
The all-treated population for this analysis represented 84.2% (1266/1503) of the ATTAIN all-treated population. The cure rates in the clinically evaluable population were similar in the telavancin (82.5%, 231/280) and vancomycin (81.3%, 243/299) groups [treatment difference (95% CI): 1.3% (−5.0% to 7.6%)], and were consistent with the overall ATTAIN study results. The cure rate was higher in the telavancin than the vancomycin treatment group in microbiologically evaluable patients with only Gram-positive pathogens isolated at baseline [85.0% (130/153) versus 75.2% (109/145), respectively; treatment difference (95% CI): 9.7% (0.6%–18.8%)]. The incidences of adverse events were similar between treatment groups and consistent with the overall findings of the ATTAIN study.
Conclusions
This analysis demonstrated that in the subset of patients without severe renal impairment or pre-existing acute renal failure, clinical and safety outcomes were similar in the telavancin and vancomycin treatment groups.
Keywords: hospital-acquired bacterial pneumonia, HABP; methicillin-resistant Staphylococcus aureus; telavancin; vancomycin; ventilator-associated bacterial pneumonia, VABP
Introduction
Telavancin is a lipoglycopeptide antimicrobial agent synthetically derived from vancomycin.1 Telavancin has bactericidal activity against a range of Gram-positive pathogens, mediated by a dual mechanism of action involving the inhibition of bacterial cell wall synthesis (a mechanism shared with vancomycin) and disruption of bacterial membrane function.1–3
Telavancin is approved in the USA and Canada for the treatment of complicated skin and skin structure infections (more recently termed acute bacterial skin and skin structure infections4) caused by susceptible Gram-positive pathogens, and in the USA for the treatment of hospital-acquired bacterial pneumonia (HABP) and ventilator-associated bacterial pneumonia (VABP) caused by susceptible isolates of Staphylococcus aureus when other alternatives are not suitable.5
The efficacy of telavancin for the treatment of nosocomial pneumonia was demonstrated by the Phase 3 ATTAIN programme (Assessment of Telavancin for Treatment of Hospital-Acquired Pneumonia; ClinicalTrials.gov registration numbers: NCT00107952 and NCT00124020).6 In 2011, telavancin was approved in Europe for the treatment of nosocomial pneumonia, including VABP, known or suspected to be caused by methicillin-resistant S. aureus when other alternatives are not suitable.
Prescribing information for Europe states that telavancin is contraindicated in patients with severe renal impairment (creatinine clearance <30 mL/min), including patients on haemodialysis, and in patients with pre-existing acute renal failure (not specifically defined).7 These contraindications are based on an observed increased risk of all-cause mortality in such patients while receiving telavancin in the ATTAIN studies (G. R. Corey, M. H. Kollef, A. F. Shorr, E. Rubinstein, M. E. Stryjewski, A. Hopkins and S. L. Barriere, unpublished results).
We report here the findings of a post hoc analysis of pooled data from the ATTAIN studies, investigating the clinical outcomes, safety and tolerability of telavancin for the treatment of nosocomial pneumonia when patients with the aforementioned contraindicating conditions are excluded from the analysis. Furthermore, consistent with the draft guidance issued by the US FDA for trials of treatments for nosocomial pneumonia,8 all-cause mortality through 28 days after randomization was analysed and a composite endpoint of clinical cure and 28 day survival was investigated.
(At the time of submission of this article, the European marketing authorization of telavancin for the treatment of nosocomial pneumonia was suspended pending evidence of a new European Medicines Agency-approved supplier. Clinigen Healthcare Ltd, Theravance's commercialization partner for telavancin in Europe, is in the process of seeking approval of a new manufacturing source.)
Methods
The ATTAIN studies
The ATTAIN studies were two identical multicentre, randomized, double-blind, comparator-controlled, parallel-group Phase 3 trials that enrolled patients between January 2005 and June 2007 and were conducted at 203 (study 0015) and 253 (study 0019) centres in 43 countries. The Institutional Review Board at each study site approved the protocol, and all patients or their authorized representatives provided written informed consent. The methodology of the ATTAIN studies has previously been reported in detail6 and is summarized in brief here, together with the specific details of the current analysis.
Patients6
Full details of the inclusion/exclusion criteria have previously been described in detail.6 Briefly, adult male and non-pregnant female patients were eligible for enrolment if they had pneumonia that had been acquired after 48 h in an inpatient acute or chronic care facility, or that developed within 7 days after their discharge. Patients were required to have at least two signs or symptoms of pneumonia, or a respiratory pathogen isolated from the respiratory tract or blood samples. In addition, patients were required to have at least two signs of systemic inflammatory response, new or progressive infiltrates, consolidation, with or without pleural effusion (chest X-ray or CT scan) and an adequate respiratory specimen for culture.
Patients were excluded if they had received potentially effective systemic antibiotic therapy for Gram-positive pneumonia for >24 h (unless clinical failure or in vitro resistance was documented), only Gram-negative pathogens obtained from baseline microbiological samples by Gram-stain or culture, an absolute neutrophil count <500 cells/mm3, a QTc interval >500 ms, uncompensated heart failure or pulmonary disease that precluded evaluation.
This post hoc sub-analysis evaluated the subset of ATTAIN patients excluding those with severe renal impairment (creatinine clearance <30 mL/min, including patients on haemodialysis) and patients with acute renal failure at baseline, in accordance with the European prescribing information for telavancin.7
Study treatments6
Patients were randomized (1 : 1) to receive either 10 mg/kg telavancin intravenously (iv) every 24 h or 1 g of vancomycin iv every 12 h for 7–21 days. The dose of vancomycin could be modified per site-specific guidelines on the basis of weight, renal function or monitoring of vancomycin levels as long as the study blinding was not compromised. The dose of telavancin was adjusted to 7.5 mg/kg every 24 h in patients with a creatinine clearance of 30–50 mL/min and to 10 mg/kg every 48 h in patients with a creatinine clearance of <30 mL/min. For patients with infections that were suspected or known to involve methicillin-susceptible S. aureus, vancomycin could be replaced with an antistaphylococcal penicillin. Patients with mixed Gram-positive/Gram-negative infections could also receive concomitant aztreonam, or piperacillin/tazobactam if resistance to aztreonam was suspected or documented.
Efficacy and safety assessments6
Clinical assessments were performed at baseline, daily throughout the study treatment, at the end of therapy (EOT) and at the follow-up (FU)/test of cure (TOC) visit conducted 7–14 days after EOT. Patient deaths were monitored up to the FU/TOC visit or were retrospectively assessed up to 28 days after EOT if no FU/TOC visit occurred. Additional data were collected after completion of the study to determine the vital status of the patients (alive/dead) at 28 days post-randomization.
Clinical cure was defined as an improvement or lack of progression of the baseline radiographic findings at EOT and a resolution of the signs and symptoms of pneumonia at FU/TOC. Failure was defined as: persistence or progression of signs and symptoms, or progression of radiological signs of pneumonia at EOT; termination of study medications due to ‘lack of efficacy’ and initiation within two calendar days of a different potentially effective antistaphylococcal medication; death on or after day 3 attributable to primary infection; or relapsed infection at FU/TOC after termination of the study medications. An indeterminate response was defined as an inability to determine the outcome.
Respiratory specimens and two blood samples were obtained for Gram-staining and culture; isolated pathogens were submitted to a central laboratory for confirmation of identification and susceptibility testing. Laboratory assessments were performed every 3 days up to EOT. Adverse events (AEs), vital signs and ECGs were evaluated during treatment and up to FU/TOC.
Patient populations
The ATTAIN patient populations were as follows.6
The all-treated (AT) population included all patients who received at least one dose of study medication. The modified AT (MAT) population consisted of AT patients who had a respiratory pathogen isolated from baseline respiratory samples or blood cultures. The clinically evaluable (CE) population consisted of AT population patients who were protocol-adherent, or who died on or after study day 3 (if death was attributable to the pneumonia under study). The microbiologically evaluable (ME) group consisted of CE patients who had a Gram-positive respiratory pathogen isolated from baseline respiratory samples or blood cultures.
The target AT, target MAT, target CE and target ME populations for this post hoc analysis were as described above, but excluded patients with severe renal impairment (creatinine clearance <30 mL/min, including patients on haemodialysis) and patients with pre-existing acute renal failure at baseline, in accordance with the European prescribing information for telavancin.7
Statistical analyses
This was a post hoc analysis of data from the ATTAIN studies. Consistent with the overall study, the primary endpoint for efficacy was the clinical response at FU/TOC in the AT and CE populations. In addition, a composite endpoint of clinical cure and survival 28 days after EOT was also investigated. The clinical response and composite endpoints were examined in the subgroup of patients with Gram-positive pathogens only isolated at baseline (the MAT and ME populations). Data from the two studies were combined to obtain more precise estimates of treatment effect in the analyses. Both studies were conducted contemporaneously under identical protocols. Two-sided 95% CIs were calculated on the difference in clinical cure rates at FU/TOC.
Baseline characteristics and AE data were summarized using descriptive statistics.
Results
A total of 1266 patients were included in this revised analysis (the target AT population), representing 84.2% of the overall ATTAIN AT population (n = 1503; Figure 1). Of these patients, 579 patients (45.7%) comprised the target CE population and 423 (33.4%) comprised the target ME population (Figure 1).
Figure 1.

Study disposition and analysis groups. aEach reason for exclusion is noted; the number of exclusions may therefore exceed the number of patients if a patient fulfilled more than one exclusion criteria. bCreatinine clearance <30 mL/min, including patients on haemodialysis, and those with or without acute renal failure. cPatients with acute renal failure, but without severe renal impairment. dThe target population excluded patients with severe renal impairment (creatinine clearance <30 mL/min, including patients on haemodialysis) and patients with acute renal failure, in accordance with the European prescribing information for telavancin.7 eOther reasons were: TOC visit outside the window (n = 14); did not comply with inclusion (n = 17) or exclusion (n = 11) criteria; S. aureus in blood culture during the study (n = 13); prior vancomycin or teicoplanin use (n = 4); received <80% of the intended dose (n = 2); and did not receive the intended drug (n = 2).
Baseline characteristics
Baseline characteristics were generally similar between the two treatment groups and were also similar between the target AT population and the overall AT population (Table 1). As expected, the proportion of patients with high APACHE II (Acute Physiology and Chronic Health Evaluation II) scores (≥20) was lower in the target AT population compared with the overall ATTAIN population (19.3% versus 23.8%). The mean (±SD) APACHE II scores in the target AT population were 14.1 (±5.6) in the telavancin group and 14.8 (±6.0) in the vancomycin group. In the vancomycin group, there were 20 patients (3.1%) who received an alternative antistaphylococcal penicillin in place of vancomycin.
Table 1.
Baseline characteristics, AT target populationa (efficacy analysesb)
| Characteristic | AT population, n (%) |
AT target population, n (%) |
||
|---|---|---|---|---|
| telavancin (N = 749) | vancomycin (N = 754) | telavancin (N = 625) | vancomycin (N = 641) | |
| Age, years | ||||
| ≥65 | 397 (53.0) | 408 (54.1) | 312 (49.9) | 315 (49.1) |
| ≥75 | 230 (30.7) | 233 (30.9) | 172 (27.5) | 176 (27.5) |
| APACHE II score | ||||
| mean (SD) | 15.0 (6.1) | 15.5 (6.2) | 14.1 (5.6) | 14.8 (6.0) |
| ≥20 | 167 (22.3) | 191 (25.3) | 107 (17.1) | 137 (21.4) |
| Creatinine clearancec | ||||
| >80 mL/min | 317 (42.3) | 322 (42.7) | 315 (50.4) | 320 (49.9) |
| >50 and ≤80 mL/min | 178 (23.8) | 173 (22.9) | 173 (27.7) | 171 (26.7) |
| ≥30 and ≤50 mL/min | 141 (18.8) | 147 (19.5) | 121 (19.4) | 129 (20.1) |
| <30 mL/mind | 94 (12.6) | 88 (11.7) | 0 | 0 |
| missing | 19 (2.5) | 24 (3.2) | 16 (2.6) | 21 (3.3) |
| Acute renal failured | 73 (9.7) | 64 (8.5) | 0 | 0 |
| Multilobar pneumonia | 473 (63.2) | 460 (61.0) | 394 (63.0) | 386 (60.2) |
| Ventilated at baseline | 345 (46.1) | 346 (45.9) | 276 (44.2) | 284 (44.3) |
| Vasopressor use | 46 (6.1) | 83 (11.0) | 27 (4.3) | 58 (9.0) |
| Mixed infectione | 144 (19.2) | 126 (16.7) | 124 (19.8) | 112 (17.5) |
aThe target population excluded patients with severe renal impairment (creatinine clearance <30 mL/min, including patients on haemodialysis) and patients with acute renal failure, in accordance with the European prescribing information for telavancin.7
bTwo patients in study 0019 randomized to receive vancomycin actually received telavancin. These two patients were included in the vancomycin group for the efficacy analysis (AT population), but were included in the telavancin group for the safety analysis. Neither patient was included in the CE population.
cResult from local laboratories was included when the central laboratory result was unavailable (in contrast to data previously reported6).
dExclusion criteria for the target population.
eGram-positive and Gram-negative pathogens isolated from baseline samples.
Efficacy
Clinical cure rates and clinical cure plus 28 day survival rates were similar for telavancin and vancomycin in both the target AT and CE populations (Figures 2 and 3). In both of these populations, 28 day survival rates were also similar (Figure 4).
Figure 2.

Clinical cure rates and between-treatment differences in the target population. Data are presented as point estimates of between-treatment differences (telavancin–vancomycin) and 95% CIs. The target population excluded patients with severe renal impairment (creatinine clearance <30 mL/min, including patients on haemodialysis) and patients with acute renal failure, in accordance with the European prescribing information for telavancin.7
Figure 3.
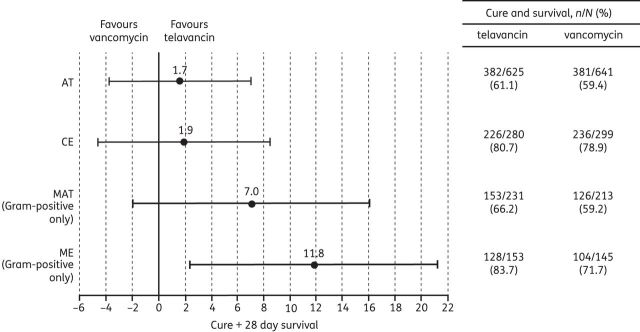
Composite endpoint of clinical cure and 28 day survival rates and between-treatment differences in the target population. Data are presented as point estimates of between-treatment differences (telavancin–vancomycin) and 95% CIs. The target population excluded patients with severe renal impairment (creatinine clearance <30 mL/min, including patients on haemodialysis) and patients with acute renal failure, in accordance with the European prescribing information for telavancin.7
Figure 4.
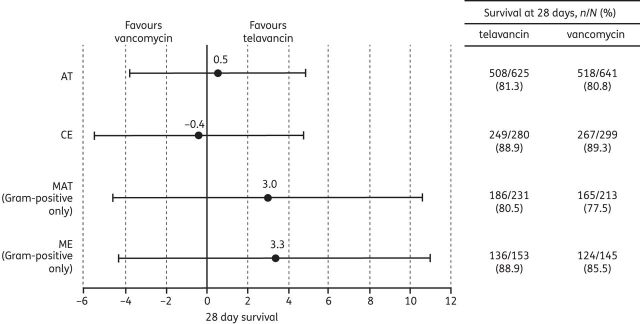
28 day survival rates and between-treatment differences in the target population. Data are presented as point estimates of between-treatment differences (telavancin–vancomycin) and 95% CIs. The target population excluded patients with severe renal impairment (creatinine clearance <30 mL/min, including patients on haemodialysis) and patients with acute renal failure, in accordance with the European prescribing information for telavancin.7
Cure rates in ME patients with only Gram-positive pathogens isolated were higher in the telavancin group [85.0% versus 75.2%; treatment difference (95% CI): 9.7% (0.6%–18.8%)]. Similarly, ME patients with only Gram-positive pathogens achieved the combined endpoint (cure plus 28 day survival) more often in the telavancin group [83.7% versus 71.7%; treatment difference (95% CI): 11.8% (2.4%–21.3%)]. However, 28 day survival in the overall ME patient population did not differ between the two treatment groups (Figure 4).
Safety and tolerability in the target population
The type and frequency of AEs were similar in telavancin- and vancomycin-treated patients (Table 2). Serious AEs (SAEs) were experienced by 171 (27.3%) patients in the telavancin group and 147 (23.0%) patients in the vancomycin group. The most frequent SAEs in the telavancin group were septic shock (3.7%), acute renal failure (2.6%) and respiratory failure (2.4%); and in the vancomycin group were septic shock (3.0%), respiratory failure (2.8%), multi-organ failure (1.7%) and pneumonia (1.7%).
Table 2.
AEs and laboratory abnormalities, AT target populationa (safety analysesb)
| Safety parameter |
n (%) |
|
|---|---|---|
| telavancin (N = 627) | vancomycin (N = 639) | |
| Any AE | 500 (79.7) | 508 (79.5) |
| Any SAE | 171 (27.3) | 147 (23.0) |
| Discontinuation due to AE | 48 (7.7) | 32 (5.0) |
| AE ≥5% in either treatment group | ||
| diarrhoea | 74 (11.8) | 78 (12.2) |
| constipation | 63 (10.0) | 58 (9.1) |
| anaemia | 50 (8.0) | 67 (10.5) |
| hypokalaemia | 50 (8.0) | 65 (10.2) |
| hypotension | 41 (6.5) | 42 (6.6) |
| nausea | 35 (5.6) | 22 (3.4) |
| decubitus ulcer | 29 (4.6) | 38 (5.9) |
| insomnia | 29 (4.6) | 36 (5.6) |
| peripheral oedema | 29 (4.6) | 32 (5.0) |
aThe target population excluded patients with severe renal impairment (creatinine clearance <30 mL/min, including patients on haemodialysis) and patients with acute renal failure, in accordance with the European prescribing information for telavancin.7
bTwo patients in study 0019 randomized to receive vancomycin actually received telavancin. These two patients were included in the vancomycin group for the efficacy analysis (AT population), but were included in the telavancin group for the safety analysis. Neither patient was included in the CE population.
A similar number of patients in the telavancin and vancomycin groups discontinued study medication due to an AE (7.7% versus 5.0%, respectively).
Deaths up to 28 days occurred in 101 (16.1%) patients in the telavancin group and 108 (16.9%) patients in the vancomycin group.
Renal AEs (acute renal failure, chronic renal failure, renal insufficiency, renal impairment and blood creatinine increase) occurred in 55 (8.8%) patients in the telavancin group and 43 (6.7%) patients in the vancomycin group (Table 3). Of these events, the majority had resolved or were resolving by the last study visit (63.6% and 53.5% in the telavancin and vancomycin groups, respectively). Increases in serum creatinine levels (>50% increase from baseline) were experienced by 140 (22.3%) patients in the telavancin group and 104 (16.3%) patients in the vancomycin group.
Table 3.
Renal AEs, AT target populationa (safety analysesb)
| Parameter |
n (%) |
|
|---|---|---|
| telavancin (N = 627) | vancomycin (N = 639) | |
| Renal AEsc | 55 (8.8) | 43 (6.7) |
| renal AEs that had not recovered or were not improving by the last study visitd | 20 (36.4) | 20 (46.5) |
| renal AEs that had recovered or were improving by the last study visitd | 35 (63.6) | 23 (53.5) |
| Renal SAEs | 21 (3.3) | 12 (1.9) |
| Renal AEs leading to discontinuation | 13 (2.1) | 7 (1.1) |
| Renal AEs leading to death | 1 (0.2) | 1 (0.2) |
aThe target population excluded patients with severe renal impairment (creatinine clearance <30 mL/min, including patients on haemodialysis) and patients with acute renal failure, in accordance with the European prescribing information for telavancin.7
bTwo patients in study 0019 randomized to receive vancomycin actually received telavancin. These two patients were included in the vancomycin group for the efficacy analysis (AT population), but were included in the telavancin group for the safety analysis. Neither patient was included in the CE population.
cAcute renal failure, chronic renal failure, renal insufficiency, renal impairment and blood creatinine increase (>50% from baseline and with a maximum value >1.5 mg/dL regardless of the initial value).
dPercentages calculated based on those patients who reported any renal AE.
Discussion
In the subset of ATTAIN study patients without severe renal impairment (creatinine clearance <30 mL/min, including patients on haemodialysis, or with acute renal failure at baseline), the cure rates for telavancin were similar to those for vancomycin. The cure rates for the telavancin and vancomycin treatment groups in the target AT population (62.6% and 61.2%, respectively) and target CE population (82.5% and 81.3%, respectively) were similar to those for the overall ATTAIN AT population (58.9% and 59.5%, respectively) and ATTAIN CE population (82.4% and 80.7%, respectively).6
For the composite endpoint of clinical cure plus 28 day survival, rates were comparable in the telavancin and vancomycin groups in the target AT and target CE populations. Clinical cure and clinical cure plus 28 day survival rates favoured telavancin over vancomycin in patients from whom only Gram-positive pathogens were recovered at baseline.
The 28 day mortality rate in the telavancin and vancomycin target treatment groups was similar (16.1% and 16.9%, respectively) to the overall ATTAIN programme (20.0% and 18.6%, respectively).
Subsequent to the initiation and completion of the ATTAIN trials, the US regulatory focus shifted to all-cause mortality as a potential primary endpoint for nosocomial pneumonia. Vital status information (through to at least day 49, i.e. up to 21 treatment days plus 28 post-treatment days) was collected for all patients. Results from the additional patient follow-up provide a near-complete dataset for the analysis of mortality and showed slightly higher rates for both treatment groups (target AT: telavancin 18.8% versus vancomycin 19.1%). Consistent with the findings of the overall ATTAIN studies, renal AEs, including potentially significant increases in serum creatinine, were more frequent in telavancin-treated patients. No other major differences in clinical or safety outcomes were noted.
This analysis has several limitations. Primarily, this post hoc subgroup analysis of the ATTAIN studies was not pre-specified, and, in addition, 54% of patients in the target AT group were excluded from the target CE analysis group. However, the analysis population is representative of the patients for whom the approved product is intended, and remains representative of the overall population of patients with nosocomial pneumonia. Notably, similar proportions of telavancin- and vancomycin-treated patients with severe renal impairment at baseline were excluded to arrive at the target population (16.6% and 15.0%, respectively). Our analysis shows that 28 day survival rates did not differ between the telavancin and vancomycin groups. However, the ATTAIN trials were not designed with mortality as an endpoint, and thus we chose to incorporate the endpoint of clinical cure plus 28 day survival into this analysis, although this endpoint should be considered as exploratory. Other limitations are shared with the ATTAIN studies6 and include a low usage of semi-invasive diagnostic procedures to determine the aetiology of the pneumonia, and potential controversies regarding the use of vancomycin as a comparator agent and the vancomycin dosages used (new dosage guidance having been published after the ATTAIN trials were conducted9).
This post hoc study shows that, in the ME group with only Gram-positive pathogens at baseline, telavancin has greater efficacy compared with vancomycin in terms of both cure rates and the composite endpoint of clinical cure plus 28 day survival. From a clinical perspective, our data show that clinicians now have an additional antistaphylococcal agent to treat HABP and VABP in patients with normal baseline renal function. The secondary renal effects of telavancin are well known and result from renal tubular toxicity, particularly in patients at risk. Patients without these risk factors are at no greater risk of renal adverse effects when receiving telavancin than they would be with vancomycin. Furthermore, animal studies have shown that this type of toxicity is reversible.5,7
In conclusion, this post hoc sub-analysis of the Phase 3 ATTAIN trials, excluding patients with pre-existing severe renal impairment, demonstrated clinical and safety outcomes similar to those of the overall study population,6 with better outcomes among telavancin-treated patients who had only Gram-positive pathogens recovered from baseline cultures. The patients included in this analysis comprised a large proportion of those who participated in the ATTAIN studies, one of the largest programmes to date of patients with nosocomial pneumonia.6 As such, these findings are supportive of telavancin for the treatment of nosocomial pneumonia in patients without severe renal impairment or acute renal failure at baseline, in accordance with the European prescribing information.7
Funding
The ATTAIN studies were supported jointly by Theravance, Inc. and Astellas Scientific and Medical Affairs, Inc. Medical writing assistance was funded jointly by Theravance, Inc. and Astellas Pharma Europe, Ltd.
Transparency declarations
A. T. has no conflicts of interest to declare. E. R. has served on advisory boards for Astellas, Atox Bio, Bayer, BiondVax, Pfizer and Theravance, has served as a consultant for Roche, and has received payment for lectures/speakers bureaus for Astellas, Bayer and Pfizer. G. R. C. has participated in advisory boards for Cempra, Cerexa, Inimex, Pfizer and Trius, has served as a consultant for Cempra, Cerexa, Dr Reddy's Lab, Inimex, PolyMedix, Pfizer, PRA, Theravance and Trius, has received grants from Innocoll, The Medicines Company and Theravance, and has received other financial support (including reimbursement for travel expenses and manuscript preparation) from Theravance. M. E. S. has served as a consultant for Astellas, Cempra, Cerexa, Furiex, Nabriva, PRA, The Medicines Company, Theravance and Trius, has received grants from Duke University (NIH), and has received other financial support (including reimbursement for travel expenses and manuscript preparation) from Cempra and Theravance. S. L. B. is an employee of, and holds equity securities in, Theravance.
Medical writing assistance was provided by Emily Howard of Envision Scientific Solutions.
All authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors.
S. L. B. had full access to the data and acts as guarantor.
Author contributions
All authors were involved in the concept and design of the analysis. E. R., G. R. C. and M. E. S. were involved in the collection of data, and all authors were involved in the subsequent analysis and interpretation of the data. All authors were involved in drafting the manuscript and were responsible for revising the manuscript for important intellectual content. All authors approved the final version of the manuscript.
Acknowledgements
We would like to acknowledge the contributions of Tracy Taylor (formerly of Astellas Pharma Europe, Ltd), Bernhardt Zeiher (Astellas Pharma US, Inc.) and Rodney Croos-Dabrera (Astellas Pharma US, Inc.), who were involved in the data analyses and development of the manuscript concept.
References
- 1.Saravolatz LD, Stein GE, Johnson LB. Telavancin: a novel lipoglycopeptide. Clin Infect Dis. 2009;49:1908–14. doi: 10.1086/648438. [DOI] [PubMed] [Google Scholar]
- 2.Higgins DL, Chang R, Debabov DV, et al. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2005;49:1127–34. doi: 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lunde CS, Hartouni SR, Janc JW, et al. Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor lipid II. Antimicrob Agents Chemother. 2009;53:3375–83. doi: 10.1128/AAC.01710-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.US Department of Health and Human Services, Food and Drug Admini-stration, Center for Drug Evaluation and Research (CDER) Acute Bacterial Skin and Skin Structure Infections: Developing Drugs for Treatment, Revision 1 (Draft Guidance for Industry) 2010. (August) http://www.fda.gov/downloads/Drugs/.../Guidances/ucm071185.pdf. (8 July 2013, date last accessed)
- 5.Theravance, Inc. VIBATIV® (Telavancin) For Injection, U.S. Prescribing Information. 2012. http://www.vibativ.com/docs/VIBATIV_PI_Final.pdf. (8 July 2013, date last accessed)
- 6.Rubinstein E, Lalani T, Corey GR, et al. Telavancin versus vancomycin for hospital-acquired pneumonia due to gram-positive pathogens. Clin Infect Dis. 2011;52:31–40. doi: 10.1093/cid/ciq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theravance, Inc. VIBATIV® (Telavancin) For Injection, Europe Prescribing Information. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001240/WC500115364.pdf. (9 September 2013, date last accessed)
- 8.US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) Hospital-Acquired Bacterial Pneumonia and Ventilator-Associated Bacterial Pneumonia: Developing Drugs for Treatment, Revision 1 (Draft Guidance for Industry) 2010. (November) http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM234907.pdf. (8 July 2013, date last accessed)
- 9.Rybak MJ, Lomaestro BM, Rotschafer JC, et al. Vancomycin therapeutic guidelines: a summary of consensus recommendations from the Infectious Diseases Society of America, the American Society of Health-System Pharmacists, and the Society of Infectious Diseases Pharmacists. Clin Infect Dis. 2009;49:325–7. doi: 10.1086/600877. [DOI] [PubMed] [Google Scholar]