

Abstract
Cardiomyopathies represent an important cause of cardiovascular morbidity and mortality due to heart failure, arrhythmias, and sudden death. Most forms of hypertrophic cardiomyopathy (HCM) are familial with an autosomal-dominant mode of inheritance. Over the last 20 years, the genetic basis of the disease has been largely unravelled. HCM is considered as a sarcomeropathy involving mutations in sarcomeric proteins, most often β-myosin heavy chain and cardiac myosin-binding protein C. ‘Missense’ mutations, more common in the former, are associated with dysfunctional proteins stably integrated into the sarcomere. ‘Nonsense’ and frameshift mutations, more common in the latter, are associated with low mRNA and protein levels derived from the diseased allele, leading to haploinsufficiency of the remaining healthy allele. The two quality control systems responsible for the removal of the affected mRNAs and proteins are the nonsense-mediated mRNA decay (NMD) and the ubiquitin-proteasome system (UPS), respectively. This review discusses clinical and genetic aspects of HCM and the role of NMD and UPS in the regulation of mutant proteins, evidence for impairment of UPS as a pathogenic factor, as well as potential therapies for HCM.
Keywords: Hypertrophy, Cardiomyopathy, Ubiquitin-proteasome system, Nonsense-mediated mRNA decay
1. Hypertrophic cardiomyopathy: a disease of the sarcomere
1.1. Clinical aspects
Cardiomyopathies are defined as diseases of the myocardium with cardiac dysfunction and are classified into four main categories: hypertrophic (HCM), dilated (DCM), arrhythmogenic right ventricular, and restrictive.1,2 HCM is characterized by left ventricular hypertrophy (LVH), which predominantly involves the interventricular septum, LV outflow tract obstruction, and diastolic dysfunction. HCM is associated with marked myocardial and myofibrillar disarray as well as increased interstitial fibrosis. The disease prevalence has been estimated at approximately 1:500 in young adults.3 HCM is therefore much more common than previously recognized; in fact, it is one of the most common monogenic diseases.
The diagnosis of HCM is usually made incidentally or after the diagnosis of a family member with sudden death. Diagnosis relies on physical examination, two-dimensional echocardiography, electrocardiography (ECG), and family history.4 Echocardiographic criteria include an LV wall thickness ≥15 mm, in the absence of another cardiac or systemic disease that could induce this hypertrophy (e.g. hypertension or aortic stenosis). ECGs have diagnostic value in raising a suspicion of HCM in family members without LVH on echocardiography. Genetic testing in the laboratory can also be performed to rule out ‘phenocopy’ diseases and to identify the specific mutations.5,6 Although endomyocardial biopsy is not required for patients with suspected HCM, it would be relevant in differentiating HCM with other known causes of increased LV wall thickness, e.g. infiltrative disorders such as amyloidosis, and storage disorders such as Fabry disease.7 In addition, myectomy is considered to be the most appropriate surgical treatment for patients with obstructive HCM. Finally, biopsy is relevant for investigating the molecular mechanisms of HCM.
The clinical course of HCM is highly variable and is associated with an increased risk of sudden death. Whereas most patients are asymptomatic, HCM patients may present with symptoms of congestive heart failure with exertional dyspnoea and chest pain but usually preserved systolic function, and atrial fibrillation. Sudden death has been recognized as the most devastating consequence of HCM in young adults and particularly young athletes since the first modern description of the disease.8 The presence of at least one of the following clinical indicators has been shown to be associated with increased risk of sudden death and therefore should require implantable cardioverter–defibrillator placement (for detailed reviews, see 9,10): history of sudden death in first-degree family members, prior cardiac arrest, sustained ventricular tachycardia, bursts of non-sustained ventricular tachycardia on serial Holter ECGs, abnormal blood pressure during exercise, and massive (>30 mm) LVH.9,11 The incidence of sudden death has been evaluated to be ∼1%/year in adult HCM patients12 and as high as 3–5% in HCM patients with multiple risk factors.9 Dyspnoea is the most common symptom in HCM (∼90% of patients), which is mainly the consequence of diastolic dysfunction. However, it could also be the consequence of systolic dysfunction in the minority of HCM patients (∼5–10%) who evolved into a ‘burned-out’ phase characterized by LV dilation and wall thinning often resembling the features of DCM and producing progressive and irreversible heart failure.13 Atrial fibrillation is the most common sustained arrhythmia in HCM justifying aggressive treatments, and paroxysmal episodes of chronic atrial fibrillation occur in 20–25% of HCM patients and are strongly associated with left atrial enlargement and increasing age.9
1.2. Mutations in myosin heavy chain and myosin-binding protein are most common
HCM is familial in the majority of cases and is inherited in an autosomal-dominant pattern with an incomplete penetrance.6,14,15 HCM involves more than 450 different mutations in at least 13 genes encoding proteins of the sarcomere (Table 1; for reviews, see 16–18). Therefore HCM is recognized as a sarcomeropathy. Most HCM patients are heterozygous for the mutation, but in 3–5% of cases, patients carry two mutations (in the same or different genes). This is generally associated with a severe phenotype and bad prognosis suggesting a gene-dosage effect.19 The two most frequently mutated genes (∼82% of the genotyped patients) are MYH7 and MYBPC3, encoding β-myosin heavy chain (β-MHC) and cardiac myosin-binding protein C (cMyBP-C), respectively.19 Both proteins are major components of the sarcomere thick filament. Myosin is the molecular motor that transduces energy from the hydrolysis of ATP into directed movement and drives sarcomere shortening and muscle contraction. cMyBP-C is expressed only in the heart of mammals and has both structural and regulatory roles in cardiac muscle20,21 (reviewed in 22). Recent data using cMyBP-C-deficient mice demonstrated that cMyBP-C reduces Ca2+ sensitivity of the myofilament at low Ca2+ concentrations and sarcomere length23 and thereby likely allows complete relaxation in diastole.24,25
Table 1.
Sarcomeric genes and mutations involved in familial HCM
| Gene name | Symbol | Number of mutations | Percentage of total |
|---|---|---|---|
| β-MHC | MYH7 | 212 | 47 |
| cMyBP-C | MYBPC3 | 165 | 37 |
| Cardiac troponin T | TNNT2 | 33 | 7 |
| α-Tropomyosin | TPM1 | 12 | 3 |
| Regulatory myosin light chain | MYL2 | 10 | 2 |
| Cardiac actin | ACTC1 | 7 | 1.5 |
| Essential myosin light chain | MYL3 | 5 | 1.1 |
| Muscle LIM protein | CSRP3 | 3 | <1 |
| Titin | TTN | 2 | <1 |
| Telethonin (T-cap) | TCAP | 2 | <1 |
| Cardiac troponin C | TNNC1 | 1 | <1 |
| α-MHC | MYH6 | 1 | <1 |
HCM exhibits wide phenotypic heterogeneity among affected subjects, characterized by a variable degree of hypertrophy and prognosis. Part of this is explained by locus heterogeneity. For instance, MYBPC3 mutations are usually associated with a delayed onset, a lower penetrance, a milder degree of hypertrophy, and better survival compared with MYH7.26–28 Moreover, genetic studies have revealed that ∼25% of individuals carrying the mutation of first-degree relatives with typical HCM are clinically unaffected.29,30 This suggests the existence of modifier genes, which modulate the phenotypic expression of the disease. Associations have been found with polymorphisms in genes for the angiotensin I-converting enzyme and AT1 and AT2 receptors.31,32 Recently, a polymorphism in the calmodulin III promoter has been shown to contribute to the phenotype in HCM patients carrying a mutation in either MYH7 or MYBPC3.33
2. Quality control systems for mutant mRNAs and proteins
About 200 different mutations were identified in MYH7 and are almost exclusively missense (Table 1). Some of them have been analysed experimentally and were shown to result in stable mutant proteins in human samples34–36 and normal incorporation into the sarcomere after gene transfer in cardiac myocytes.37 These data suggest that missense proteins are present at normal levels in the sarcomere and act as ‘poison polypeptides’ through a dominant-negative effect.
In contrast, 70% of MYBPC3 mutations are nonsense or frameshift.17,30 Nonsense mutations, by definition, introduce a premature termination codon (PTC) at the place of mutations (e.g. by changing CGA to TGA), whereas frameshift mutations result from an insertion or deletion and create a PTC downstream. Both are expected to produce C-terminal truncated cMyBP-Cs. The mechanisms by which MYBPC3 mutations lead to HCM remain elusive and are in the focus of this review. Truncated cMyBP-Cs have been consistently undetectable by western blot in myocardial tissue from patients carrying a frameshift mutation,38–40 after gene transfer in cardiac myocytes37 or in transgenic mice.41 Thus, the prevailing data suggest that mutant mRNAs and/or proteins are unstable and quantitatively degraded. Nonsense and frameshift mutations would therefore act as ‘null alleles’, causing cMyBP-C haploinsufficiency in heterozygous patients. An insufficient amount of normal full-length cMyBP-C could produce an imbalance in the stoichiometry of the thick filament components and alter sarcomeric structure and function. Indeed, heterozygous cMyBP-C-deficient mice carrying only one functional allele exhibit marked activation of the JNK and p38 MAPK pathway and induction of apoptosis,42 followed by asymmetric septal hypertrophy.43 Two major quality control systems, nonsense-mediated mRNA decay (NMD) and ubiquitin-proteasome system (UPS), contribute to low levels of mutant proteins.
2.1. Nonsense-mediated mRNA decay
The NMD is an evolutionary conserved surveillance pathway in all eukaryotes examined to date. NMD targets PTC-containing transcripts for rapid degradation, thereby protecting the organism from the deleterious dominant-negative or gain-of-function effects of resulting C-terminal truncated proteins (Figure 1) (for details, see 44–46). The general rule is that NMD occurs when a PTC is located more than 50–55 nucleotides upstream of the last exon–exon junction within the mRNA, and NMD requires at least one intron and components of translation.47
Figure 1.

The NMD. (A) A PTC located more than 50–55 nucleotides (nt) upstream of the last exon–exon junction within the mRNA (green region) elicits NMD, whereas mRNAs with PTCs downstream of this boundary (red region) escape NMD. (B) Mechanism of the action of NMD. During pre-mRNA splicing, a protein complex called exon junction complex (EJC) is deposited 20–24 nucleotides upstream of every exon–exon junction and functions hereby as a marker to dictate whether a stop codon is premature or not. The EJC containing the up-frameshift (UPF) 3 protein remains bound to the mRNA when exported to the cytoplasm. Here, a second NMD core protein, UPF2, binds to UPF3. In normal mRNAs, the EJCs are then displaced by the ribosome during the pioneer round of translation, and translation stops when the ribosome reaches the normal stop codon (B, top). In contrast, in PTC-bearing mRNAs, the ribosome is blocked at the PTC, and the EJC downstream of the PTC remains associated with the mRNA, because it cannot be displaced by the ribosome (B, bottom). This results in attachment of the so-called SURF complex, which comprises SMG-1 (suppressor with morphogenetic effect on genitalia), UPF1, and the eukaryotic release factors (eRF) 1 and eRF3, to the ribosome. Binding of UPF2 to UPF1 leads to its phosphorylation by SMG-1, which in turn drives dissociation of eRF1 and eRF3 and binding of SMG-7. Ultimately, the mRNA is degraded by different pathways including decapping or deadenylation. Figure adapted from Garneau et al.46
The role of NMD in genetic disease has been well described for Duchenne muscular dystrophy (DMD) and cystic fibrosis (for review, see 48), but has just begun to be recognized in cardiac genetic diseases. Lamin A/C mutations associated with DCM result in much lower level of PTC-bearing transcripts,49,50 which were stabilized by the translation inhibitor cycloheximide, supporting involvement of NMD.50 Similar data were obtained for nonsense mutations in ether-a-go-go-related gene in human long-QT syndrome.51 As described above, frameshift MYBPC3 mutations in human HCM were also associated with lower amount of nonsense than wild-type mRNA, suggesting involvement of NMD.39,40 We recently demonstrated that NMD specifically regulates the level of PTC-bearing, but not of other cMyBP-C transcripts in a mouse model of HCM.52
2.2. The UPS
Like NMD for nonsense mRNAs, quality control for aberrant proteins exists. Most cytosolic, nuclear, and myofibrillar proteins are degraded via the UPS, whereas membrane proteins and organelles are degraded by lysosomes via autophagy (for review, see 53). A major function of the UPS is to prevent accumulation of damaged, misfolded, and mutant proteins, but the system is also involved in intracellular signalling, transcriptional control, or regulation of cell death.53 The UPS functions as an ATP-dependent proteolytic system that requires polyubiquitination via lysine 48 residues of the target protein prior to its degradation by the 26S proteasome (Figure 2).54 Polyubiquitination involves the concerted action of 3 enzymes: E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin ligase), the latter affording substrate specificity. The eukaryotic 26S proteasome is a large, multicatalytic protein complex composed of the 19S regulatory complex and the 20S proteasome (for reviews, see 54,55). The damaged, misfolded, or mutant proteins are degraded by three major peptidase activities (chymotrypsin-like, trypsin-like, and caspase-like) residing on the inner surface of the β-rings of the 20S proteasome (for a detailed review in the same issue, see 56).
Figure 2.
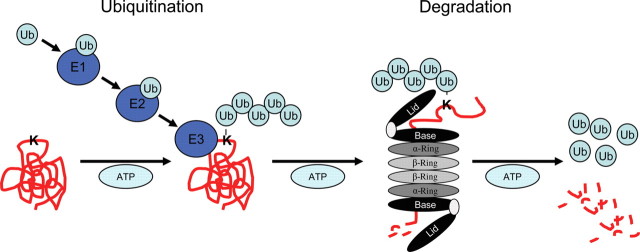
The UPS. The UPS functions as an ATP-dependent proteolytic system that requires polyubiquitination via lysine 48 residues of the target protein prior to its degradation by the 26S proteasome. Polyubiquitination involves the concerted action of three enzymes: E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin ligase), the latter affording substrate specificity. The eukaryotic 26S proteasome is a large, multicatalytic protein complex composed of the 19S regulatory complex and the 20S proteasome. The damaged, misfolded, or mutant proteins are degraded by three major peptidase activities (chymotrypsin-like, trypsin-like, and caspase-like).
The role of the UPS in regulating levels of mutant proteins in HCM has been first suspected by the analysis of myocardial tissue of HCM patients with MYBPC3 frameshift mutations, in which the C-terminal truncated cMyBP-C proteins were never detected despite the presence of (low levels) of nonsense mRNAs.38–40 This suggests that, in addition to NMD, the translation rate of the pathological mRNAs is low or protein degradation rate is high. Support for the latter hypothesis comes from studies overexpressing human truncated cMyBP-Cs in cardiomyocytes or transgenic mice. Despite normal levels of nonsense mRNA, protein levels of truncated cMyBP-C were very low,37,41,57 and blockade of the UPS markedly increased protein levels.57 Recent data suggest that the muscle-specific E3 ubiquitin ligase atrogin-1 is involved in this process.58 The signals that cause quantitative degradation of truncated proteins by the UPS are not clear. Absence of biomolecular interaction between truncated cMyBP-C and human β-MHC59 could promote degradation of truncated cMyBP-C. More likely, truncation and/or the incorporation of new ‘nonsense’ amino acids after the frameshift causes misfolding, and this constitutes a signal for UPS targeting. In line with this idea, a missense cMyBP-C was recently shown to be degraded by the UPS, potentially due to the presence of a new lysine residue in the mutant, which could allow its polyubiquitination.60 Importantly, our recent data obtained with a mouse model of HCM provide evidence that the UPS also regulates the level of aberrant mutant cMyBP-C in vivo.52
Taken together, the data obtained with cMyBP-C mutations associated with HCM suggest that NMD and UPS act as two consecutive quality control systems to effectively eliminate mutant proteins. The mutated allele is thus a functional null allele and the disease is likely caused by a relative lack of wild-type cMyBP-C in the sarcomere, i.e. by the mechanism of haploinsufficiency. In the following sections, we will discuss whether involvement of the UPS per se may constitute a pathogenic mechanism in HCM and whether interfering with NMD or UPS could represent a valuable therapeutic strategy.
3. Alterations of the UPS in cardiac disease
3.1. Impaired UPS in familial HCM and DCM
Apart from the involvement of UPS in degrading mutant proteins in HCM, recent investigations suggest that UPS impairment might play a role in the pathophysiology of HCM and desmin-related cardiomyopathy (DRM). DRM is an autosomal-dominant disorder characterized by marked accumulation of desmin in skeletal and cardiac muscle.61 αB-crystallin (CryAB) is the most abundant heat shock protein in the heart and stabilizes desmin. A missense R120G CryAB mutation leads to DRM.62 Transgenic mice expressing a CryABR120G mutant develop HCM, which gradually progresses to heart failure.63 Perinuclear aggregates of desmin and preamyloid oligomer in cardiomyocytes64 suggest that CryAB-DRM is a subclass of the aggresomal and amyloid-related neurodegenerative disorders such as Alzheimer's and Parkinson's diseases.64 Using GFPdgn reporter mice, in which modified GFP is used as a UPS substrate, aggregates and marked UPS impairment were detected in CryABR120G mice.65 Similar UPS impairment was found in another DRM mouse model associated with a desmin mutation.66,67 The molecular mechanism by which aggregates impair the UPS is not clear. It has been proposed that aggregated proteins directly inhibit the 26S proteasome by ‘chocking’ the proteases. Alternatively, protein aggregates may indirectly interfere with UPS function by inactivating or depleting UPS components.
Adenoviral expression of truncated cMyBP-C in cardiomyocytes resulted in the formation of ubiquitin-positive aggregates of truncated cMyBP-C and impairment of the UPS as evidenced with a GFP-reporter system.57 Overexpression of missense cMyBP-C in COS cells also reduced proteasome activity and increased apoptosis.60 Adrenergic stress-induced hypertrophy revealed a decrease in proteasome activity in heterozygous cMyBP-C mutant mice carrying both mutant and wild-type alleles, but not in heterozygous cMyBP-C null mice carrying only a functional allele.68 The mechanism by which the UPS was impaired under the different conditions has not been elucidated. However, one could speculate that cMyBP-C mutants, similar to misfolded desmin, chock the proteasome and/or compete with other degradation-prone proteins for UPS removal. This could result in the accumulation of factors regulated by the UPS such as transcription factors involved in the hypertrophic response (e.g. SRF, GATA4) or apoptosis (e.g. p53). Viewed from this angle, UPS impairment could contribute to the pathogenesis of HCM.
3.2. Altered UPS in cardiac hypertrophy and heart failure
Is UPS impairment a common feature of LV hypertrophy? A series of investigations indeed reported UPS abnormalities in various cardiac diseases (for review, see 54). Accumulation of ubiquitinated proteins appears to be almost a stereotypic finding in cardiac hypertrophy. But it becomes increasingly clear that increased steady-state levels of ubiquitinated proteins do not necessarily indicate impairment of the UPS. Instead, they are often associated with increase in proteasomal activity (measured with fluorogenic substrates) and/or ubiquitinating or deubiquitinating enzymes. Thus, it is important to keep in mind that steady-state levels of ubiquitinated proteins reflect the balance of several variables: the production rate of misfolded proteins, their ubiquitination, deubiquitination, and finally degradation.
In a mouse model of pressure overload-induced LV hypertrophy, increased levels of ubiquitinated proteins were associated with depressed proteasome activities and induction of apoptosis. These alterations preceded the onset of cardiac dysfunction,69 suggesting that UPS dysfunction could be a causative factor. On the other hand, in a rat model of pressure overload-induced cardiac hypertrophy, ubiquitin B, the E2-conjugating enzyme UbcH2, the E3 ubiquitin ligases atrogin-1 and MuRF1, and the proteasomal subunit PSMB4 transcripts were all increased after 7 days.70 In a feline model of pressure overload-induced cardiac hypertrophy, transient accumulation of ubiquitinated proteins was identified as well as increased levels of numerous E3 ubiquitin ligases.71 Increased steady-state levels of ubiquitinated proteins associated with greater proteasome activities were also found in genetically engineered mouse models with HCM or DCM.65,72 Interestingly, increased chymotrypsin-like activities followed the development of LV hypertrophy in homozygous cMyBP-C-targeted mutant mice72 and the development of heart failure in transgenic CryABR120G mutant mice.65 In terminally failing human hearts, marked accumulation of ubiquitinated proteins was associated with the upregulation of several UPS components.73 Moreover, a well-known UPS target, the proapoptotic p53, was recently found to be increased in human DCM.74 This was associated with increased levels of the p53-specific E3 ubiquitin ligase MDM2 and increased chymotrypsin-like activity, but also increased levels of the deubiquitinating enzyme HAUSP.74 The interpretation of this counterintuitive finding was that the increase in HAUSP overrides the increase in MDM2, pointing to the complexity of the system.
Taken together, these data show that measurements of single components of the UPS are not sufficient to conclude about the state of the system and that the role of the UPS in cardiac disease is just beginning to be unravelled. A few statements can be made with some certainty: (i) the steady-state level of ubiquitinated proteins integrates numerous factors and is not directly indicative of the degradation capacity of the proteasome in vivo; (ii) an increase in an E3 ubiquitin ligase does not allow the conclusion that its substrates are decreased; (iii) impairment of the UPS in vivo, i.e. a mismatch between the level of UPS-targeted proteins and proteasomal capacity, can be determined by UPS-reporter systems; (iv) accumulation of ubiquitinated proteins occurs regularly during hypertrophy and other states of remodelling; and (v) increased proteasome activities during hypertrophy and ‘cardiac stress’ are likely mechanisms to cope with increased appearance of misfolded proteins (e.g. by energy deficit, oxidative stress, overload of sarcomeres). When the latter exceeds the degradation capacity of the UPS, misfolded proteins may accumulate and form aggregates as described in DRM. In addition, any imbalance of ubiquitinating and deubiquitinating enzymes and proteasomal activity could contribute to cardiac pathology by dysregulation of specific factors such as p53, SRF, GATA4, or the inducible cAMP early repressor (ICER), as suggested previously.75 Much more work is needed to fully understand the relative contribution of these processes and the key factors.
4. Targeting nonsense mRNA or the UPS for HCM therapy?
Different approaches have been used and are summarized in Table 2.
Table 2.
Targeting nonsense mRNAs or the UPS for therapy
| Therapy | Disease | Drug | System | Response | Reference |
|---|---|---|---|---|---|
| PTC therapy | Cystic fibrosis, DMD | Aminoglycoside (gentamicin) | Mouse models, human patients | Expression of full-length protein | 78,79; for review, see 48 |
| NMD therapy | Cystic fibrosis, Ullrich's disease | Wortmannin, siRNA UPF1/UPF2 (components of NMD) | Cell lines, human cells from patients | Restoration of cell phenotype | 77; for review, see 48 |
| RNA-based therapy | Genetic diseases | Antisense oligonucleotides (exon skipping, trans-splicing, etc.) | Cell lines, mdx mice | Restoration of the reading frame | 83 |
| UPS therapy | LVH | Proteasome inhibition | Cells, mice, rats | Inhibition/regression of LVH | 84–86 |
| DMD | Proteasome inhibition | mdx mice | Restoration of dystrophin and reduction of muscle damage | 92 | |
| Cancer | E3 ubiquitin ligase inhibition | In vitro | Destruction of p53-expressing cells | 90 |
PTC, premature termination codon; NMD, nonsense-mediated mRNA decay; UPS, ubiquitin-proteasome system; DMD, Duchenne muscular dystrophy; LVH, left ventricular hypertrophy.
4.1. Targeting nonsense mRNA?
Nearly 30% of mutations identified in human genetic diseases result in PTC-bearing transcripts that are degraded by NMD. The biological sense of this system probably lies in the elimination at the transcript level of non-functional proteins that can have a dominant-negative effect; the downside is haploinsufficiency. Selective inhibition of NMD may provide a strategy to rescue the phenotype in those cases, in which the mutant protein shows no dominant-negative effect and has a near-normal function. The experimental NMD inhibitor wortmannin inhibits the PI3-kinase-related protein SMG-1 that phosphorylates UPF1 during NMD.76 Owing to its cytotoxic effects, wortmannin is unacceptable as a therapeutic agent in humans, but siRNA-mediated knockdown of SMG-1 or UPF1 restored the level of PTC-bearing collagen VI-2 mRNA and rescued the phenotype in fibroblasts of a patient with Ullrich's disease.77
An alternative are drugs that target the detection of PTCs. They have been tested in animal models and patients with cystic fibrosis and DMD (for review, see 48). By enforcing reading through PTCs, these therapies can induce the synthesis of full-length proteins in case of nonsense mutations. However, PTC readthrough is unlikely of value in case of frameshift mutations resulting in PTC, because all the amino acids after the frameshift are expected to be non-functional. Different PTC readthrough mechanisms are known. For example, aminoglycosides affect the decoding site of ribosomes and promote the incorporation of an amino acid at a PTC.48 The aminoglycoside gentamicin has been already used in human patients to suppress PTC-bearing transcripts associated with cystic fibrosis78 and DMD.79 However, responses to gentamicin varied widely from one study to the other. Several factors may account for this variability including the identity and surrounding sequences of the PTC, the amount of PTC-bearing transcripts, and the duration of treatment (see 48 for details). The usefulness of aminoglycosides is also limited because of severe side effects, including kidney damage, hearing loss, and tinnitus.80,81 Recently, a new non-aminoglycoside, orally bioavailable compound, PTC124, has been developed for the sole purpose of inducing selective ribosomal PTC readthrough.82 In contrast to aminoclycosides, PTC24 has not evoked kidney failure or deafness in phase I of safety trials.82
Alternative RNA-based therapies, developed over the last decade, present a unique potential for native mRNA modification within the endogenous regulatory environment (for review, see 83). The active targeting molecules are usually short antisense oligoribonucleotides that can block cryptic splicing, or exclude/include one or more exons to restore the reading frame. These RNA-targeting therapeutics could be well suited to treat HCM.
4.2. Targeting the UPS?
As discussed above, impairment of UPS function appears to be common in cardiac disease and may be even causally related to the progression to heart failure. Global inhibition of the UPS is therefore expected to worsen rather than to rescue the phenotype. However, partial inhibition of the UPS could theoretically lead to preferential accumulation of factors such as hypoxia-inducible factor 1α, the NFκB-inhibitor IκB, and the hypertrophy-repressor ICER that could have beneficial consequences and may underlie positive results seen in animal models. For example, low concentrations of MG132 prevented hypertrophy in neonatal rat cardiomyocytes, and low doses of the FDA-approved proteasome inhibitor Velcade® effectively reduced spontaneous cardiac hypertrophy in hypertensive rats.84 Two additional studies have demonstrated that proteasome inhibition can induce regression of established cardiac hypertrophy. In the first model of isoprenaline-induced cardiac hypertrophy, the proteasome inhibitor PS-519 induced significant regression of hypertrophy.85 In a mouse model of pressure overload, the proteasome inhibitor epoxomicin inhibited cardiac hypertrophy, reduced apoptosis, and stabilized ejection fraction.86 Of note however, cardiomyopathic side effects have been reported in patients using proteasome inhibitors for longer periods of time,87–89 raising concerns about their utility for therapy.
Therapies designed to regulate specific E3 ubiquitin ligases have not been developed for the heart but in cancer. The E3 ubiquitin ligase MDM2 is commonly overexpressed in cancer and regulates p53 levels, and small-molecule inhibitors of MDM2 have been recently shown to induce cancer cell death by stabilizing p53.90 Targeting the E3 ubiquitin ligases MDM2, atrogin-1, or MuRF1 in cardiac hypertrophy is likely not a suitable strategy, because the latter has been shown to be cardioprotective, and inhibiting MDM2 would enhance cardiac apoptosis (see recent reviews 54,91). But targeting the E3 ubiquitin ligases for HIF1α, IκB, or ICER could have beneficial consequences.
In cases of HCM, where the remaining PTC-bearing transcripts are detected,38–40 partial inhibition of the proteasome or selective targeting of the responsible E3 ligase could be a treatment of choice if the truncated protein is functional. Such approach has not yet been tested in cardiac disease, but inhibition of the proteasome in vivo restored the expression of truncated dystrophin at the sarcolemma and reduced muscle membrane damage in skeletal muscles of mdx mice.92
5. Conclusion
NMD and UPS are two quality control systems directed towards eliminating potentially harmful abnormal proteins. In HCM associated with MYBPC3 mutations, elimination of aberrant mRNA and proteins resulting from nonsense or frameshift mutations causes a functional null allele and haploinsufficiency. In cases where the aberrant protein is expected to be functional and non-toxic, interference with NMD or underlying RNA-editing mechanisms could (partially) correct the defect. Whether deficient production of cMyBP-C is sufficient to explain the phenotype of HCM or whether incomplete elimination of mutant proteins and/or impairment of the UPS play an additional pathogenic role is still not entirely clear. Both increases and decreases in UPS function are regularly observed in animal models of HCM, cardiac hypertrophy, and heart failure, suggesting that regulation of the UPS belongs to the important adaptations in cardiac disease. Pharmacological proteasome inhibitors were associated with antihypertrophic effects and improved function in animals models, which is probably not the consequence of a global decrease in UPS capacity, but rather a preferential interference with the degradation of antihypertrophic or otherwise beneficial factors particularly sensitive to UPS inhibition. Better understanding of these processes may allow the development of specific inhibitors devoid of the risks of general UPS inhibition and establish the UPS as a promising target in the therapy of cardiac diseases.
Conflict of interest: none declared.
Funding
This work was supported by the sixth Framework Program of the European Union (Marie Curie EXT-014051), the Deutsche Forschungsgemeinschaft (FOR-604/2, CA 618/1-2), and the American Heart Association (Scientist Development Grant to M.S.W.). S.S. is a postdoctoral fellow supported by the DFG-FOR-604/2 grant.
References
- 1.Richardson P, McKenna W, Bristow M, Maish B, Mautner B, O’Connell J, et al. Report of the 1995 World Health Organisation/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996;93:841–842. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 2.Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 3.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation. 1995;92:785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 4.Charron P, Dubourg O, Desnos M, Bouhour JB, Isnard R, Hagège A, et al. Diagnostic value of electrocardiography and echocardiography for familial hypertrophic cardiomyopathy in genotyped children. Eur Heart J. 1998;19:1377–1382. doi: 10.1053/euhj.1998.1049. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez JE, McCudden CR, Willis MS. Familial hypertrophic cardiomyopathy: basic concepts and future molecular diagnostics. Clin Biochem. 2009;42:755–765. doi: 10.1016/j.clinbiochem.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 6.Charron P, Heron D, Gargiulo M, Richard P, Dubourg O, Desnos M, et al. Genetic testing and genetic counselling in hypertrophic cardiomyopathy: the French experience. J Med Genet. 2002;39:741–746. doi: 10.1136/jmg.39.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soor GS, Luk A, Ahn E, Abraham JR, Woo A, Ralph-Edwards A, et al. Hypertrophic cardiomyopathy: current understanding and treatment objectives. J Clin Pathol. 2009;62:226–235. doi: 10.1136/jcp.2008.061655. [DOI] [PubMed] [Google Scholar]
- 8.Teare D. Asymetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1–8. doi: 10.1136/hrt.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 10.Maron BJ, Spirito P, Shen WK, Haas TS, Formisano F, Link MS, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405–412. doi: 10.1001/jama.298.4.405. [DOI] [PubMed] [Google Scholar]
- 11.Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342:1778–1785. doi: 10.1056/NEJM200006153422403. [DOI] [PubMed] [Google Scholar]
- 12.Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet. 2004;363:1881–1891. doi: 10.1016/S0140-6736(04)16358-7. [DOI] [PubMed] [Google Scholar]
- 13.Biagini E, Coccolo F, Ferlito M, Perugini E, Rocchi G, Bacchi-Reggiani L, et al. Dilated-hypokinetic evolution of hypertrophic cardiomyopathy: prevalence, incidence, risk factors, and prognostic implications in pediatric and adult patients. J Am Coll Cardiol. 2005;46:1543–1550. doi: 10.1016/j.jacc.2005.04.062. [DOI] [PubMed] [Google Scholar]
- 14.Charron P, Carrier L, Dubourg O, Tesson F, Desnos M, Richard P, et al. Penetrance of familial hypertrophic cardiomyopathy. Genet Couns. 1997;8:107–114. [PubMed] [Google Scholar]
- 15.Charron P, Dubourg O, Desnos M, Isnard R, Hagège A, Millaire A, et al. Diagnostic value of electrocardiography and echocardiography for familial hypertrophic cardiomyopathy in a genotyped adult population. Circulation. 1997;96:214–219. doi: 10.1161/01.cir.96.1.214. [DOI] [PubMed] [Google Scholar]
- 16.Keller DI, Carrier L, Schwartz K. Genetics of familial hypertrophic cardiomyopathies and arrhythmias. Swiss Med Wkly. 2002;132:401–407. doi: 10.4414/smw.2002.10037. [DOI] [PubMed] [Google Scholar]
- 17.Richard P, Villard E, Charron P, Isnard R. The genetic bases of cardiomyopathies. J Am Coll Cardiol. 2006;48:A79–A89. [Google Scholar]
- 18.Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol. 2008;19:104–110. doi: 10.1111/j.1540-8167.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 19.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations and implications for molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 20.Fougerousse F, Delezoide AL, Fiszman MY, Schwartz K, Beckmann JS, Carrier L. Cardiac myosin binding protein C gene is specifically expressed in heart during murine and human development. Circ Res. 1998;82:130–133. doi: 10.1161/01.res.82.1.130. [DOI] [PubMed] [Google Scholar]
- 21.Lecarpentier Y, Vignier N, Oliviero P, Guellich A, Carrier L, Coirault C. Cardiac myosin-binding protein C modulates the tuning of the molecular motor in the heart. Biophys J. 2008;95:720–728. doi: 10.1529/biophysj.107.127787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrier L. Cardiac myosin-binding protein C in the heart. Arch Mal Coeur Vaiss. 2007;100:238–243. [PubMed] [Google Scholar]
- 23.Cazorla O, Szilagyi S, Vignier N, Salazar G, Kramer E, Vassort G, et al. Length and protein kinase A modulations of myocytes in cardiac myosin binding protein C-deficient mice. Cardiovasc Res. 2006;69:370–380. doi: 10.1016/j.cardiores.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Pohlmann L, Kroger I, Vignier N, Schlossarek S, Kramer E, Coirault C, et al. Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ Res. 2007;101:928–938. doi: 10.1161/CIRCRESAHA.107.158774. [DOI] [PubMed] [Google Scholar]
- 25.Colson BA, Bekyarova T, Fitzsimons DP, Irving TC, Moss RL. Radial displacement of myosin cross-bridges in mouse myocardium due to ablation of myosin binding protein-C. J Mol Biol. 2007;367:36–41. doi: 10.1016/j.jmb.2006.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charron P, Dubourg O, Desnos M, Isnard R, Hagège A, Bonne G, et al. Genotype-phenotype correlations in familial hypertrophic cardiomyopathy: a comparison between mutations in the cardiac protein-C and the b-myosin heavy chain genes. Eur Heart J. 1998;19:139–145. doi: 10.1053/euhj.1997.0575. [DOI] [PubMed] [Google Scholar]
- 27.Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248–1257. doi: 10.1056/NEJM199804303381802. [DOI] [PubMed] [Google Scholar]
- 28.Yu B, French JA, Carrier L, Jeremy RW, McTaggart DR, Nicholson MR, et al. Molecular pathology of familial hypertrophic cardiomyopathy caused by mutations in the cardiac myosin binding protein C gene. J Med Genet. 1998;35:205–210. doi: 10.1136/jmg.35.3.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dausse E, Komajda M, Dubourg O, Fetler L, Dufour C, Carrier L, et al. Familial hypertrophic cardiomyopathy: microsatellite haplotyping and identification of a hot-spot for mutations in the b-myosin heavy chain gene. J Clin Invest. 1993;92:2807–2813. doi: 10.1172/JCI116900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrier L, Bonne G, Bährend E, Yu B, Richard P, Niel F, et al. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ Res. 1997;80:427–434. [PubMed] [Google Scholar]
- 31.Osterop AP, Kofflard MJ, Sandkuijl LA, ten Cate FJ, Krams R, Schalekamp MA, et al. AT1 receptor A/C1166 polymorphism contributes to cardiac hypertrophy in subjects with hypertrophic cardiomyopathy. Hypertension. 1998;32:825–830. doi: 10.1161/01.hyp.32.5.825. [DOI] [PubMed] [Google Scholar]
- 32.Deinum J, van Gool JM, Kofflard MJ, ten Cate FJ, Danser AH. Angiotensin II type 2 receptors and cardiac hypertrophy in women with hypertrophic cardiomyopathy. Hypertension. 2001;38:1278–1281. doi: 10.1161/hy1101.096114. [DOI] [PubMed] [Google Scholar]
- 33.Friedrich F, Bausero P, Sun Y, Treszl A, Krämer E, Juhr D, et al. A new polymorphism in human calmodulin III promoter is a potential modifier gene for familial hypertrophic cardiomyopathy. Eur Heart J. 2009;30:1648–1655. doi: 10.1093/eurheartj/ehp153. [DOI] [PubMed] [Google Scholar]
- 34.Cuda G, Fananapazir L, Epstein ND, Sellers JR. The in vitro motility activity of b-cardiac myosin depends on the nature of the b-myosin heavy chain gene mutation in hypertrophic cardiomyopathy. J Muscle Res Cell Motil. 1997;18:275–283. doi: 10.1023/a:1018613907574. [DOI] [PubMed] [Google Scholar]
- 35.Bottinelli R, Coviello DA, Redwood CS, Pellegrino MA, Maron BJ, Spirito P, et al. A mutant tropomyosin that causes hypertrophic cardiomyopathy is expressed in vivo and associated with an increased calcium sensitivity. Circ Res. 1998;82:106–115. doi: 10.1161/01.res.82.1.106. [DOI] [PubMed] [Google Scholar]
- 36.Keller DI, Coirault C, Rau T, Cheav T, Weyand M, Amann K, et al. Human homozygous R403W mutant cardiac myosin presents disproportionate enhancement of mechanical and enzymatic properties. J Mol Cell Cardiol. 2004;36:355–362. doi: 10.1016/j.yjmcc.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 37.Flavigny J, Souchet M, Sébillon P, Berrebi-Bertrand I, Hainque B, Mallet A, et al. COOH-terminal truncated cardiac myosin-binding protein C mutants resulting from familial hypertrophic cardiomyopathy mutations exhibit altered expression and/or incorporation in fetal rat cardiomyocytes. J Mol Biol. 1999;294:443–456. doi: 10.1006/jmbi.1999.3276. [DOI] [PubMed] [Google Scholar]
- 38.Rottbauer W, Gautel M, Zehelein J, Labeit S, Franz WM, Fischer C, et al. Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy. Characterization of cardiac transcript and protein. J Clin Invest. 1997;100:475–482. doi: 10.1172/JCI119555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moolman JA, Reith S, Uhl K, Bailey S, Gautel M, Jeschke B, et al. A newly created splice donor site in exon 25 of the MyBP-C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation. 2000;101:1396–1402. doi: 10.1161/01.cir.101.12.1396. [DOI] [PubMed] [Google Scholar]
- 40.van Dijk SJ, Dooijes D, Dos Remedios C, Michels M, Lamers JM, Winegrad S, et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 41.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. In vivo modeling of myosin binding protein C familial hypertrophic cardiomyopathy. Circ Res. 1999;85:841–847. doi: 10.1161/01.res.85.9.841. [DOI] [PubMed] [Google Scholar]
- 42.Eijssen LM, van den Bosch BJ, Vignier N, Lindsey PJ, van den Burg CM, Carrier L, et al. Altered myocardial gene expression reveals possible maladaptive processes in heterozygous and homozygous cardiac myosin-binding protein C knockout mice. Genomics. 2008;91:52–60. doi: 10.1016/j.ygeno.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 43.Carrier L, Knoell R, Vignier N, Keller DI, Bausero P, Prudhon B, et al. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc Res. 2004;63:293–304. doi: 10.1016/j.cardiores.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 44.Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 45.Maquat LE. Nonsense-mediated mRNA decay in mammals. J Cell Sci. 2005;118:1773–1776. doi: 10.1242/jcs.01701. [DOI] [PubMed] [Google Scholar]
- 46.Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J, Sun X, Qian Y, LaDuca JP, Maquat LE. At least one intron is required for the nonsense-mediated decay of triosephosphate isomerase mRNA: a possible link between nuclear splicing and cytoplasmic translation. Mol Cell Biol. 1998;18:5272–5283. doi: 10.1128/mcb.18.9.5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Linde L, Kerem B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008;24:552–563. doi: 10.1016/j.tig.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 49.Geiger SK, Bar H, Ehlermann P, Walde S, Rutschow D, Zeller R, et al. Incomplete nonsense-mediated decay of mutant lamin A/C mRNA provokes dilated cardiomyopathy and ventricular tachycardia. J Mol Med. 2008;86:281–289. doi: 10.1007/s00109-007-0275-1. [DOI] [PubMed] [Google Scholar]
- 50.Muchir A, Massart C, van Engelen BG, Lammens M, Bonne G, Worman HJ. Proteasome-mediated degradation of integral inner nuclear membrane protein emerin in fibroblasts lacking A-type lamins. Biochem Biophys Res Commun. 2006;351:1011–1017. doi: 10.1016/j.bbrc.2006.10.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gong Q, Zhang L, Vincent GM, Horne BD, Zhou Z. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome. Circulation. 2007;116:17–24. doi: 10.1161/CIRCULATIONAHA.107.708818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vignier N, Schlossarek S, Fraysse B, Mearini G, Kraemer E, Pointu H, et al. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cMyBP-C mutant levels in cardiomyopathic mice. Circ Res. 2009;105:239–248. doi: 10.1161/CIRCRESAHA.109.201251. [DOI] [PubMed] [Google Scholar]
- 53.Zolk O, Schenke C, Sarikas A. The ubiquitin-proteasome system: focus on the heart. Cardiovasc Res. 2006;70:410–421. doi: 10.1016/j.cardiores.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 54.Mearini G, Schlossarek S, Willis MS, Carrier L. The ubiquitin-proteasome system in cardiac dysfunction. Biochim Biophys Acta. 2008;1782:749–763. doi: 10.1016/j.bbadis.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 55.Ciechanover A. The ubiquitin proteolytic system: from a vague idea through basic mechanisms, and onto human diseases and drug targeting. Neurology. 2006;66:S7–S19. doi: 10.1212/01.wnl.0000192261.02023.b8. [DOI] [PubMed] [Google Scholar]
- 56.Hedhli N, Depre C. Proteasome inhibitors and cardiac cell growth. Cardiovasc Res. 2010;85:321–329. doi: 10.1093/cvr/cvp226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sarikas A, Carrier L, Schenke C, Doll D, Flavigny J, Lindenberg KS, et al. Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res. 2005;66:33–44. doi: 10.1016/j.cardiores.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 58.Mearini G, Gedicke G, Schlossarek S, Witt CC, Krämer E, Cao P, et al. Atrogin-1 and MuRF1 regulate cardiac MyBP-C levels via different mechanisms. Cardiovasc Res. 2010;85:357–366. doi: 10.1093/cvr/cvp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flavigny J, Robert P, Camelin J, Schwartz K, Carrier L, Berebbi-Bertrand I. Biomolecular interactions between human recombinant b-MyHC and cMyBP-Cs implicated in familial hypertrophic cardiomyopathy. Cardiovasc Res. 2003;60:388–396. doi: 10.1016/j.cardiores.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 60.Bahrudin U, Morisaki H, Morisaki T, Ninomiya H, Higaki K, Nanba E, et al. Ubiquitin-proteasome system impairment caused by a missense cardiac myosin-binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J Mol Biol. 2008;384:896–907. doi: 10.1016/j.jmb.2008.09.070. [DOI] [PubMed] [Google Scholar]
- 61.Rappaport L, Contard F, Samuel JL, Delcayre C, Marotte F, Tome F, et al. Storage of phosphorylated desmin in a familial myopathy. FEBS Lett. 1988;231:421–425. doi: 10.1016/0014-5793(88)80863-9. [DOI] [PubMed] [Google Scholar]
- 62.Vicart P, Caron A, Guicheney P, Li Z, Prevost MC, Faure A, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 63.Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, et al. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 64.Sanbe A, Osinska H, Villa C, Gulick J, Klevitsky R, Glabe CG, et al. Reversal of amyloid-induced heart disease in desmin-related cardiomyopathy. Proc Natl Acad Sci USA. 2005;102:13592–13597. doi: 10.1073/pnas.0503324102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen Q, Liu JB, Horak KM, Zheng H, Kumarapeli AR, Li J, et al. Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ Res. 2005;97:1018–1026. doi: 10.1161/01.RES.0000189262.92896.0b. [DOI] [PubMed] [Google Scholar]
- 66.Liu J, Chen Q, Huang W, Horak KM, Zheng H, Mestril R, et al. Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J. 2006;20:362–364. doi: 10.1096/fj.05-4869fje. [DOI] [PubMed] [Google Scholar]
- 67.Liu J, Tang M, Mestril R, Wang X. Aberrant protein aggregation is essential for a mutant desmin to impair the proteolytic function of the ubiquitin-proteasome system in cardiomyocytes. J Mol Cell Cardiol. 2006;40:451–454. doi: 10.1016/j.yjmcc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 68.Schuermann F, Schlossarek S, Mearini G, Geertz B, Vignier N, Eschenhagen T, et al. Adrenergic stress unmasks exaggerated septal hypertrophy and proteasome impairment in heterozygous cMyBP-C mutant mice. (Abstract) Circulation. 2008;118:S521–S521. [Google Scholar]
- 69.Tsukamoto O, Minamino T, Okada K, Shintani Y, Takashima S, Kato H, et al. Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem Biophys Res Commun. 2006;340:1125–1133. doi: 10.1016/j.bbrc.2005.12.120. [DOI] [PubMed] [Google Scholar]
- 70.Razeghi P, Baskin KK, Sharma S, Young ME, Stepkowski S, Essop MF, et al. Atrophy, hypertrophy, and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem Biophys Res Commun. 2006;342:361–364. doi: 10.1016/j.bbrc.2006.01.163. [DOI] [PubMed] [Google Scholar]
- 71.Balasubramanian S, Mani S, Shiraishi H, Johnston RK, Yamane K, Willey CD, et al. Enhanced ubiquitination of cytoskeletal proteins in pressure overloaded myocardium is accompanied by changes in specific E3 ligases. J Mol Cell Cardiol. 2006;41:669–679. doi: 10.1016/j.yjmcc.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 72.Sultan K, Schlossarek S, Englmann D, Vignier N, Eschenhagen T, Carrier L. Alterations of the ubiquitin-proteasome system in cardiomyopathic cMyBP-C mice. (Abstract) J Mol Cell Cardiol. 2007;42:S167. [Google Scholar]
- 73.Weekes J, Morrison K, Mullen A, Wait R, Barton P, Dunn MJ. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003;3:208–216. doi: 10.1002/pmic.200390029. [DOI] [PubMed] [Google Scholar]
- 74.Birks EJ, Latif N, Enesa K, Folkvang T, Luong le A, Sarathchandra P, et al. Elevated p53 expression is associated with dysregulation of the ubiquitin-proteasome system in dilated cardiomyopathy. Cardiovasc Res. 2008;79:472–480. doi: 10.1093/cvr/cvn083. [DOI] [PubMed] [Google Scholar]
- 75.Hedhli N, Wang L, Wang Q, Rashed E, Tian Y, Sui X, et al. Proteasome activation during cardiac hypertrophy by the chaperone H11 Kinase/Hsp22. Cardiovasc Res. 2008;77:497–505. doi: 10.1093/cvr/cvm054. [DOI] [PubMed] [Google Scholar]
- 76.Rehwinkel J, Raes J, Izaurralde E. Nonsense-mediated mRNA decay: target genes and functional diversification of effectors. Trends Biochem Sci. 2006;31:639–646. doi: 10.1016/j.tibs.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 77.Usuki F, Yamashita A, Kashima I, Higuchi I, Osame M, Ohno S. Specific inhibition of nonsense-mediated mRNA decay components, SMG-1 or Upf1, rescues the phenotype of Ullrich disease fibroblasts. Mol Ther. 2006;14:351–360. doi: 10.1016/j.ymthe.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 78.Kerem E. Pharmacologic therapy for stop mutations: how much CFTR activity is enough? Curr Opin Pulm Med. 2004;10:547–552. doi: 10.1097/01.mcp.0000141247.22078.46. [DOI] [PubMed] [Google Scholar]
- 79.Politano L, Nigro G, Nigro V, Piluso G, Papparella S, Paciello O, et al. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003;22:15–21. [PubMed] [Google Scholar]
- 80.Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. 1999;43:1003–1012. doi: 10.1128/aac.43.5.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fischel-Ghodsian N. Genetic factors in aminoglycoside toxicity. Pharmacogenomics. 2005;6:27–36. doi: 10.1517/14622416.6.1.27. [DOI] [PubMed] [Google Scholar]
- 82.Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol. 2007;47:430–444. doi: 10.1177/0091270006297140. [DOI] [PubMed] [Google Scholar]
- 83.Wood M, Yin H, McClorey G. Modulating the expression of disease genes with RNA-based therapy. PLoS Genet. 2007;3:e109. doi: 10.1371/journal.pgen.0030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meiners S, Dreger H, Fechner M, Bieler S, Rother W, Gunther C, et al. Suppression of cardiomyocyte hypertrophy by inhibition of the ubiquitin-proteasome system. Hypertension. 2008;51:302–308. doi: 10.1161/HYPERTENSIONAHA.107.097816. [DOI] [PubMed] [Google Scholar]
- 85.Stansfield WE, Tang RH, Moss NC, Baldwin AS, Willis MS, Selzman CH. Proteasome inhibition promotes regression of left ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2008;294:H645–H650. doi: 10.1152/ajpheart.00196.2007. [DOI] [PubMed] [Google Scholar]
- 86.Hedhli N, Lizano P, Hong C, Fritzky LF, Dhar SK, Liu H, et al. Proteasome inhibition decreases cardiac remodeling after initiation of pressure overload. Am J Physiol Heart Circ Physiol. 2008;295:H1385–H1393. doi: 10.1152/ajpheart.00532.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hacihanefioglu A, Tarkun P, Gonullu E. Acute severe cardiac failure in a myeloma patient due to proteasome inhibitor bortezomib. Int J Hematol. 2008;88:219–222. doi: 10.1007/s12185-008-0139-7. [DOI] [PubMed] [Google Scholar]
- 88.Orciuolo E, Buda G, Cecconi N, Galimberti S, Versari D, Cervetti G, et al. Unexpected cardiotoxicity in haematological bortezomib treated patients. Br J Haematol. 2007;138:396–397. doi: 10.1111/j.1365-2141.2007.06659.x. [DOI] [PubMed] [Google Scholar]
- 89.Voortman J, Giaccone G. Severe reversible cardiac failure after bortezomib treatment combined with chemotherapy in a non-small cell lung cancer patient: a case report. BMC Cancer. 2006;6:129. doi: 10.1186/1471-2407-6-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weissman AM, Yang Y, Kitagaki J, Sasiela CA, Beutler JA, O’Keefe BR. Inhibiting Hdm2 and ubiquitin-activating enzyme: targeting the ubiquitin conjugating system in cancer. Ernst Schering Found Symp Proc. 2008:171–190. doi: 10.1007/2789_2008_108. [DOI] [PubMed] [Google Scholar]
- 91.Willis MS, Schisler JC, Portbury AL, Patterson C. Build it up-tear it down: protein quality control in the cardiac sarcomere. Cardiovasc Res. 2009;81:439–448. doi: 10.1093/cvr/cvn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bonuccelli G, Sotgia F, Schubert W, Park DS, Frank PG, Woodman SE, et al. Proteasome inhibitor (MG-132) treatment of mdx mice rescues the expression and membrane localization of dystrophin and dystrophin-associated proteins. Am J Pathol. 2003;163:1663–1675. doi: 10.1016/S0002-9440(10)63523-7. [DOI] [PMC free article] [PubMed] [Google Scholar]