

Abstract
Aims
Familial hypertrophic cardiomyopathy (FHC) is frequently caused by cardiac myosin-binding protein C (cMyBP-C) gene mutations, which should result in C-terminal truncated mutants. However, truncated mutants were not detected in myocardial tissue of FHC patients and were rapidly degraded by the ubiquitin-proteasome system (UPS) after gene transfer in cardiac myocytes. Since the diversity and specificity of UPS regulation lie in E3 ubiquitin ligases, we investigated whether the muscle-specific E3 ligases atrogin-1 or muscle ring finger protein-1 (MuRF1) mediate degradation of truncated cMyBP-C.
Methods and results
Human wild-type (WT) and truncated (M7t, resulting from a human mutation) cMyBP-C species were co-immunoprecipitated with atrogin-1 after adenoviral overexpression in cardiac myocytes, and WT-cMyBP-C was identified as an interaction partner of MuRF1 by yeast two-hybrid screens. Overexpression of atrogin-1 in cardiac myocytes decreased the protein level of M7t-cMyBP-C by 80% and left WT-cMyBP-C level unaffected. This was rescued by proteasome inhibition. In contrast, overexpression of MuRF1 in cardiac myocytes not only reduced the protein level of WT- and M7t-cMyBP-C by >60%, but also the level of myosin heavy chains (MHCs) by >40%, which were not rescued by proteasome inhibition. Both exogenous cMyBP-C and endogenous MHC mRNA levels were markedly reduced by MuRF1 overexpression. Similar to cardiac myocytes, MuRF1-overexpressing (TG) mice exhibited 40% lower levels of MHC mRNAs and proteins. Protein levels of cMyBP-C were 29% higher in MuRF1 knockout and 34% lower in TG than in WT, without a corresponding change in mRNA levels.
Conclusion
These data suggest that atrogin-1 specifically targets truncated M7t-cMyBP-C, but not WT-cMyBP-C, for proteasomal degradation and that MuRF1 indirectly reduces cMyBP-C levels by regulating the transcription of MHC.
Keywords: E3 ubiquitin ligase, Sarcomere, Myosin-binding protein C, MuRF1, Atrogin-1
1. Introduction
Familial hypertrophic cardiomyopathy (FHC) is an autosomal-dominant disease characterized by left ventricular (LV) hypertrophy and myocardial disarray.1,2 It is often referred to as a sarcomeropathy since it is associated with mutations in at least 13 genes encoding sarcomeric proteins (reviewed in Richard et al.2). Gene modifiers also contribute to the phenotype.3,4 One of the most frequently mutated FHC genes is MYBPC3 encoding cardiac myosin-binding protein C (cMyBP-C).5 cMyBP-C is a major component of the A-band of the sarcomere and interacts with myosin, actin, and titin (for review, see Carrier6). It contains immunoglobulin- and fibronectin-like domains and it contains at least three phosphorylation sites in the MyBP-C motif. Most of the MYBPC3 mutations are frameshift and should produce C-terminal truncated proteins.5,7,8 However, in myocardial tissue of FHC patients, there was no trace of the truncated proteins.9–12 Previous data have shown that truncated cMyBP-Cs and an E344K cMyBP-C are rapidly and quantitatively degraded by the ubiquitin-proteasome system (UPS) after gene transfer in neonatal rat cardiac myocytes (NRCM), HeLa, or COS cells.13–15
The UPS is the major degradation system of damaged, misfolded, or mutated proteins in eukaryotic cells (for review, see Ciechanover16). The signal for protein degradation is the covalent attachment of ubiquitin, a small 76-amino acid protein, to the target protein, which is then degraded by the 26S proteasome complex. Polyubiquitination involves the concerted action of three enzymes: E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase), the latter affording substrate specificity. Several muscle-specific E3 ubiquitin ligases have been identified (for review, see Mearini et al.17). Among them, atrogin-1/MAFbx and the muscle ring finger proteins (MuRFs) are of particular interest since they are located in the sarcomere. Whereas MuRF1 is mainly located in the M-band but also at the Z-band, atrogin-1 and MuRF3 are restricted to the Z-band.18–22 MuRF1 has also been observed in nuclei.20 The expression of atrogin-1 and MuRF1 is up-regulated in skeletal muscle atrophy23–26 and in experimental heart failure,27,28 but down-regulated in unloading-induced cardiac atrophy in rat.28 Although atrogin-1 targets calcineurin A for proteasomal degradation,22 MuRF1 degrades cardiac troponin I, myosin heavy chain (MHC), and muscle creatine kinase.29–32 Recent data suggest that MuRF1 may also inhibit protein translation.33 Both atrogin-1 and MuRF1 blunt the development of cardiac hypertrophy in mice and/or in NRCM.22,34,35
In the present study, we investigated whether the E3 ubiquitin ligases atrogin-1 or MuRF1 regulate the level of wild-type (WT) or truncated cMyBP-C (M7t), which results from a human FHC mutation and has been shown to be degraded by the UPS in NRCM.14 To address this question, adenoviral gene transfer in NRCM and mice deficient in MuRF1 (MuRF1-KO) or overexpressing MuRF1 (MuRF1-TG) were investigated. We show that atrogin-1 and MuRF1 regulate the level of WT and/or M7t-truncated cMyBP-C via different mechanisms.
2. Methods
An expanded material and methods section is given in the Supplementary material online.
All experiments involving animals were conform to the Guide for the Care and Use of Laboratory Animals published by the National Institute of Health (NIH) (Publication No. 85-23, revised 1985) and were approved by the University of North Carolina Institutional Animal Care Advisory Committee and the Regierungspräsidium Tübingen, Germany.
2.1. MuRF1-KO and MuRF1-TG mice
MuRF1-KO and MuRF1-TG mice have been described previously.33,36 LV mass index was calculated from echocardiography measurements performed with a Vevo 660 ultrasound system (VisualSonics Inc.) equipped with a 30 MHz transducer as described previously.35
2.2. Generation of recombinant adenoviruses
Each adenovirus was constructed in the same way and encodes the protein of interest plus the EGFP in a bicistronic manner under the control of independent CMV promoters (Figure 1A). The adenovirus encoding myc-tagged human full-length cMyBP-C (myc-WT-cMyBP-C), human C-terminal truncated cMyBP-C (myc-M7t-cMyBP-C), and mouse MuRF1 (myc-MuRF1) were generated previously.14,37 The adenovirus encoding 6-myc-tagged mouse atrogin-1 (myc6-atrogin-1) was constructed with the Ad-Easy system.38 In brief, mouse atrogin-1 cDNA24 was PCR-amplified and subcloned into BglII and XbaI sites of pAdTrack-CMV. The pAdTrack vector bearing atrogin-1 was electroporated into Escherichia coli BJ5183-AD-1 (Stratagene) to produce adenoviral DNA through recombination. This DNA was transfected into 293A cells and viral plaques expressing atrogin-1 were selected and amplified in 293A cells using standard adenoviral techniques.
Figure 1.
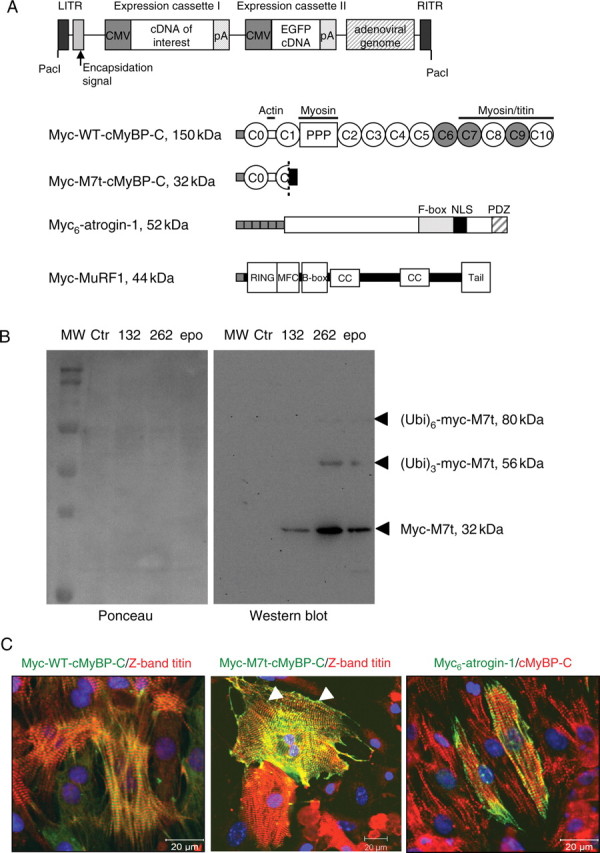
(A) Schematic representation of the adenovirus vectors containing the cDNA of interest and EGFP cDNA under the control of distinct CMV promoters, and schematic structure of the proteins encoded by the adenovirus: human full-length cMyBP-C (myc-WT-cMyBP-C), human C-terminally truncated cMyBP-C (myc-M7t-cMyBP-C), mouse atrogin-1 (myc6-atrogin-1), and mouse MuRF1 (myc-MuRF1). The absence of exon 6 in myc-M7t-cMyBP-C results in a frameshift, loss of the terminal 1055 residues including the MyBP-C motif (PPP), and additional 41 novel amino acids (black box). (B) Effect of proteasome inhibitors on the expression of myc-M7t in NRCM. NRCM were infected with myc-M7t adenovirus in the absence (Ctr) or presence of the proteasome inhibitors MG132 (132; 1 µM), MG262 (262; 100 nM), or epoxomicin (epo; 500 nM) for 24 h. Western blot (right panel) was stained with the anti-myc antibody and left panel shows the corresponding Ponceau. (C) Immunofluorescence staining and confocal microscopy of NRCM 24 h after infection with adenovirus. Myocytes were double-stained with an anti-myc antibody (green) and with the anti-titin (Z1 domain, red) or anti-cMyBP-C antibody (C0C1 domains; red). Arrowheads show aggregates.
2.3. Immunofluorescence analysis
NRCM were fixed and proceeded for immunofluorescence as described previously.39,40 See Supplementary material online for details.
2.4. Western blot analysis
Total cardiac proteins were extracted from mouse ventricles or from myocyte lysates as described previously.40,41 See Supplementary material online for details.
2.5. Chymotrypsin-like activity assay
The chymotrypsin-like activity of the proteasome was assessed in ventricular cytosolic lysates using the synthetic peptide substrate SLLVY linked to the fluorogenic reporter aminomethylcoumarin (Calbiochem) as described previously.42 See Supplementary material online for details.
2.6. Statistical analysis
Data are expressed as mean ± SEM. Statistical analyses were performed using the unpaired Student's t-test with the commercial software GraphPad Prism4. A value of P < 0.05 was considered statistically significant.
3. Results
3.1. Atrogin-1 specifically targets truncated M7t-cMyBP-C for proteasomal degradation in cardiac myocytes
We previously showed that myc-M7t-truncated cMyBP-C is rapidly degraded by the UPS after gene transfer in NRCM.14 Treatment of myc-M7t-infected NRCM with different proteasome inhibitors stabilized myc-M7t levels and revealed higher MW myc-positive bands (Figure 1B), suggesting polyubiquitination and degradation of myc-M7t by the UPS. The localization of exogenous proteins was determined by immunofluorescence (Figure 1C). Myc-WT-cMyBP-C was incorporated into the A-band of the sarcomere as shown by the alternation of myc and Z-band-titin stainings (Figure 1C). In contrast, myc-M7t-cMyBP-C, which lacks the myosin-interacting domains (Figure 1A), was located in the Z-band of the sarcomere as shown by the co-localization of myc and Z-band-titin stainings, and also formed some aggregates (Figure 1C). Atrogin-1 was localized in both the nucleus and the Z-band of the sarcomere, where it showed alternation with cMyBP-C. The localization of both myc-M7t and atrogin-1 at the Z-band suggests that atrogin-1 could interact with M7t-cMyBP-C and mediate M7t-cMyBP-C degradation by the UPS.
To determine whether atrogin-1 interacts with M7t-cMyBP-C, NRCM were co-infected with adenovirus encoding atrogin-1 and M7t- or WT-cMyBP-C in the presence of the proteasome inhibitor MG132, in order to stabilize the amount of protein. Atrogin-1 was efficiently precipitated with the anti-atrogin-1 antibody, and both myc-WT- and myc-M7t-cMyBP-C were co-immunoprecipitated (Figure 2A), suggesting physical interaction between atrogin-1 and cMyBP-C.
Figure 2.

Effect of atrogin-1 on the levels of WT/M7t-cMyBP-C after gene transfer in NRCM. NRCM were co-infected with myc6-atrogin-1 and with either myc-M7t-cMyBP-C or myc-WT-cMyBP-C in the presence or absence of 1 µM MG132 for 24 h. (A) Immunoprecipitation (IP) of the NRCM lysates treated with MG132 was performed with 0.8 µg anti-atrogin-1 antibody (+) or without antibody (−), and western blot (WB) was revealed with anti-atrogin-1 and anti-myc antibodies. P0 and SN correspond to the crude fraction and the supernatant after IP. (B) Western blot of cell lysates (20 µg) was stained with the anti-ubiquitin antibody that recognizes a smear of ubiquitinated proteins (between 50 and 250 kDa), the anti-myc antibody for detection of exogenous myc-M7t-cMyBP-C (32 kDa), myc-WT-cMyBP-C (150 kDa), and myc6-atrogin-1 (52 kDa), and the anti-GFP antibody for loading control (GFP, 29 kDa). (C) Quantification of M7t and WT-cMyBP-C levels normalized to Ponceau in the absence (upper bars) or presence (lower bars) of MG132. Values are expressed as mean ± SEM. ***P < 0.001 vs. absence of atrogin-1, Student's t-test. The number of experiments is indicated in the bars.
We then analysed whether atrogin-1 affects the levels of M7t- and WT-cMyBP-C after infection in NRCM. Treatment with MG132 increased the amount of ubiquitinated proteins in both Ad-M7t- and Ad-WT-infected cells, with or without Ad-atrogin-1, validating the inhibition of the proteasome (Figure 2B). In addition, MG132 significantly increased the amount of both M7t-cMyBP-C and atrogin-1, but not of WT-cMyBP-C. Strikingly, the level of M7t-cMyBP-C was 80% lower with atrogin-1 than without (compare lanes 1 and 3), and this was prevented by proteasome inhibition (Figure 2B and C). In contrast to M7t, the level of WT-cMyBP-C was not altered with atrogin-1. GFP levels did not significantly differ in all conditions. The mRNA levels of WT- and M7t-cMyBP-C were not affected by atrogin-1 (see Supplementary material online, Figure S1A). These data suggest that truncated M7t and atrogin-1 are degraded by the UPS and that atrogin-1 targets M7t-cMyBP-C, but not WT-cMyBP-C for proteasome-mediated degradation.
3.2. MuRF1 indirectly reduces exogenous cMyBP-C levels in cardiac myocytes
Since WT-cMyBP-C is located in the A-band of the sarcomere and therefore around MuRF1, mainly located in the M-band of the sarcomere,20 we then investigated whether MuRF1 regulates the level of WT-cMyBP-C.
Immunoprecipitation with the anti-MuRF1 antibody did not work under our experimental conditions, we thus evaluated whether MuRF1 interacts with cMyBP-C by yeast two-hybrid screens, using an MuRF1 cDNA as a bait to screen an adult heart cDNA library. A total of four interacting clones identified cMyBP-C by sequencing (data not shown). The region of overlap between the different prey clones was found in the C7–C10 domains of cMyBP-C (Figure 3A). This suggests that MuRF1 can regulate the level of WT-cMyBP-C, but likely not of truncated M7t, which does not contain domains C7–C10 (Figure 1A).
Figure 3.

Effect of MuRF1 on the levels of WT/M7t-cMyBP-C after gene transfer in NRCM. (A) Localization of the region of overlap in the four cMyBP-C prey clones identified by yeast two-hybrid screens with MuRF1 bait. (B) NRCM were co-infected with myc-MuRF1 and with either myc-M7t-cMyBP-C or myc-WT-cMyBP-C in the presence or absence of 1 µM MG132 for 24 h. Western blot of cell lysates (15 µg) stained with the anti-ubiquitin antibody, the anti-myc antibody for detection of exogenous myc-M7t-cMyBP-C (32 kDa), myc-WT-cMyBP-C (150 kDa), and myc-MuRF1 (44 kDa), and the anti-GFP antibody for loading control (GFP, 29 kDa). (C) Quantification of M7t- and WT-cMyBP-C levels normalized to Ponceau in the absence (upper bars) or presence (lower bars) of MG132. Values are expressed as mean ± SEM. ***P < 0.001 vs. absence of MuRF1, Student's t-test. The number of experiments is indicated in the bars.
We then investigated whether MuRF1 regulates the level of cMyBP-C after infection of NRCM with adenovirus encoding MuRF1 and WT- or M7t-cMyBP-C in the presence or absence of MG132 (Figure 3B and C). The protein levels of M7t- and WT-cMyBP-C were 86 and 66% lower with MuRF1 than without, respectively (Figure 3B and C). Of note, however, the effect of MuRF1 on the level of both cMyBP-C isoforms was not prevented by proteasome inhibition. Unexpectedly, MuRF1 reduced by >80% the mRNA levels of exogenous M7t- and WT-cMyBP-C (see Supplementary material online, Figure S1B), but not the level of endogenous GAPDH and cMyBP-C (data not shown). MG132 markedly increased the steady-state levels of ubiquitinated proteins in the absence of MuRF1, but only slightly in the presence of MuRF1 (see Supplementary material online, Figure S2). GFP levels did not significantly differ in any condition. We then investigated whether MuRF1 could affect the expression of MHC, which has been shown to be degraded by MuRF1.32,43 Surprisingly, the mRNA levels of α-MHC and β-MHC were markedly lower in the presence of MuRF1 and were associated with lower MHC protein amount (see Supplementary material online, Figure S3). These data suggest that MuRF1 (i) does not directly target exogenous cMyBP-C for proteasomal degradation and (ii) regulates the transcription of MHC. The strong transcriptional regulation by MuRF1 could explain the absence (or minor) effect of MG132 on the accumulation of MHC, cMyBP-C, and ubiquitinated proteins.
3.3. MuRF1 indirectly regulates the level of cMyBP-C in mice
To investigate whether MuRF1 regulates the level of cMyBP-C in the whole animal, we used an MuRF1-KO mouse and an MuRF1-TG mouse that overexpress MuRF1.33,36 MuRF1-KO mice were created by homologous recombination and insertion of a Neo cassette in exon 2 (see Supplementary material online, Figure S4A).33 The genotype of the animals was determined by PCR on genomic tail DNA with primers located around the Neo cassette (see Supplementary material online, Figure S4B). The absence of MuRF1 in the KO was confirmed by nonsense mRNA amplified by RT–PCR with primers located in exons 1 and 3 and by western blot analysis with an antibody directed against the C-terminal part of MuRF1 (see Supplementary material online, Figure S4C). MuRF1-KO mice appeared normal in all aspects and were viable. The heart-weight-to-body-weight ratio did not differ in 24-week-old KO and WT mice (see Supplementary material online, Figure S4D). MuRF1-TG mice were created by overexpression of the mouse MuRF1 cDNA under control of the α-MHC promoter (see Supplementary material online, Figure S4E).36 Overexpression of MuRF1 was confirmed by real-time RT–qPCR and western blot analysis (see Supplementary material online, Figure S4F and G). The LV mass-to-body-weight did not differ in 8- to 12-week-old MuRF1-TG and WT mice (see Supplementary material online, Figure S4H).
We then investigated whether the absence or overexpression of MuRF1 results in alterations of the UPS in general and of cMyBP-C levels in particular. The absence of MuRF1 did not affect the steady-state levels of ubiquitinated proteins (Figure 4A) or the chymotrypsin-like activity of the proteasome (Figure 4B) compared with WT. It was also not associated with a compensatory increase in MuRF2, MuRF3, and atrogin-1 mRNAs (Figure 4C). In MuRF1-TG mice, the steady-state levels of ubiquitinated proteins were slightly higher than in WT (Figure 5A), and the chymotrypsin-like activity of the proteasome (Figure 5B) and mRNA levels of MuRF2, MuRF3 and atrogin-1 were unaltered (Figure 5C).
Figure 4.

Investigation of the UPS in MuRF1-KO mice. (A) Representative western blot stained with the anti-ubiquitin antibody, corresponding Ponceau and steady-state level of ubiquitinated proteins (normalized to Ponceau) in ventricular tissue of 24-week-old WT and MuRF1-KO mice. (B) Chymotrypsin-like activity of the proteasome in WT and KO ventricular tissue. (C) mRNA levels of MuRF2, MuRF3, and atrogin-1 determined by RT–qPCR with specific Taqman probes and normalized to GAPDH. Bars represent the mean ± SEM. The number of animals is indicated in the bars.
Figure 5.

Investigation of the UPS in MuRF1-TG mice. (A) Representative western blot stained with the anti-ubiquitin antibody, corresponding Ponceau and steady-state level of ubiquitinated proteins (normalized to Ponceau) in ventricular tissue from 8- to 12-week-old WT and MuRF1-TG mice. (B) Chymotrypsin-like activity of the proteasome in WT and TG ventricular tissue. (C) mRNA levels of MuRF2, MuRF3, and atrogin-1 determined by RT–qPCR with specific Taqman probes and normalized to GAPDH. Bars represent the mean ± SEM. *P < 0.05 vs. WT, Student's t-test. The number of animals is indicated in the bars.
Importantly, the protein level of cMyBP-C was 29% higher in MuRF1-KO (Figure 6A) and 34% lower in MuRF1-TG (Figure 6B) than in WT littermates. This difference was even more pronounced in neonatal mice, where cMyBP-C levels were 150% higher in MURF1-KO than in WT (see Supplementary material online, Figure S5). Similar levels of cMyBP-C mRNA were found in MuRF1-KO or MuRF1-TG compared with respective WT littermates (Figure 6C), suggesting a post-transcriptional regulation of the expression of cMyBP-C by MuRF1 in vivo. Similar to NRCM, MuRF1 overexpression in transgenic mice resulted in >35% lower levels of MHC (mainly α-MHC) mRNAs and proteins (see Supplementary material online, Figure S6). This supports the view that MuRF1 may target an MHC-specific transcription factor. In contrast to MHC, the protein levels of cTnI, also known to be degraded by MuRF1,29 were not affected in MuRF1-KO or MuRF1-TG mice (see Supplementary material online, Figure S7).
Figure 6.

Determination of the level of cMyBP-C in MuRF1-KO and MuRF1-TG mice. (A) Representative western blot, corresponding Ponceau and quantification of ventricular cMyBP-C levels in 24-week-old WT and MuRF1-KO mice. (B) Representative western blot, corresponding Ponceau and quantification of ventricular cMyBP-C levels in 8- to 12-week-old WT and MuRF1-TG mice. (C) cMyBP-C mRNA levels determined by real-time RT–PCR in ventricular RNA from WT/KO mice and WT/TG mice. Bars represent the mean ± SEM. **P < 0.01, ***P < 0.001 vs. WT, Student's t-test. The number of animals is indicated in the bars.
4. Discussion
We have previously shown that a truncated form of cMyBP-C resulting from a human MYBPC3 mutation (M7t-cMyBP-C) is rapidly and quantitatively degraded by the UPS after gene transfer in cardiac myocytes.14 Since the diversity and specificity of the UPS regulation lie in the E3 ubiquitin ligases, we reasoned that identifying the E3 ubiquitin ligases involved in the degradation of cMyBP-C would represent an important first step to unravel the mechanisms of the UPS-mediated regulation of cMyBP-C. We examined whether the E3 ubiquitin ligases atrogin-1 and MuRF1 affect the level of M7t-cMyBP-C and also WT-cMyBP-C. The major findings of the present study were: (i) atrogin-1 interacted with both cMyBP-C isoforms and targeted truncated M7t-, but not WT-cMyBP-C for UPS-mediated degradation in infected NRCM, (ii) two-hybrid screens identified cMyBP-C as an interacting partner of MuRF1, (iii) MuRF1 overexpression decreased the protein and mRNA levels of both WT- and M7t-cMyBP-C in NRCM, (iv) the absence of MuRF1 was associated with higher cMyBP-C levels in mice, and (v) overexpression of MuRF1 with lower cMyBP-C levels in mice. Our data also provide evidence that MuRF1 regulates the transcription of MHC both in cardiac myocytes and in mice.
The E3 ubiquitin ligases are directly or indirectly involved in the transfer of ubiquitin moieties to the proteins, which will be further degraded by the 26S proteasome (for review see Mearini et al.17). Co-expression of an E3 ubiquitin ligase and its target was expected to result in degradation of the target by the proteasome. Among several known muscle-specific E3 ubiquitin ligases, we focussed on atrogin-1 and MuRF1,19,24 both being located in the sarcomere.20 Our data support the view that atrogin-1 is an E3 ubiquitin ligase for M7-cMyBP-C. First, atrogin-1 and M7t-cMyBP-C are both located in the Z-band of the sarcomere. Thus, the principal prerequisite for interaction is given. Secondly, co-immunoprecipitation assays identified M7t as an interaction partner of atrogin-1. Thirdly, atrogin-1 reduced the level of M7t protein, but not of M7t mRNA, and proteasome inhibition prevented this effect after gene transfer in cells. The effect of overexpressed atrogin-1 appeared to be restricted to the truncated M7t mutant. The lack of effect of atrogin-1 on WT-MyBP-C is apparently not due to an inability to physically interact under the conditions of adenoviral overexpression, but rather the consequence of different localizations in the sarcomere making a direct interaction under normal conditions unlikely. Thus, co-immunoprecipitation of WT-cMyBP-C with atrogin does not prove de facto interaction in living cells.
In contrast to atrogin-1, MuRF1 reduced the levels of both WT- and M7t-cMyBP-C in infected cells and of endogenous cMyBP-C in the whole animal. Although this may suggest that MuRF1 is an E3 ubiquitin ligase for cMyBP-C, several results presented in this study do not support this view. First, the yeast two-hybrid screens identified the C7–C10 domains of cMyBP-C as an MuRF1-interacting region, but these domains are not present in M7t being down-regulated similar to WT-cMyBP-C. Secondly, proteasome inhibition did not rescue the effect of MuRF1 on WT- and M7t-cMyBP-C levels in infected cells, in contrast to the results obtained with atrogin-1. Thirdly, mRNA levels of exogenous cMyBP-C were lower in the MuRF1-overexpressing myocytes. If MuRF1 is not a major E3 ubiquitin ligase for cMyBP-C, by which mechanisms does it regulate cMyBP-C? MuRF1 can affect gene expression via interaction with transcriptional modulators such as GMEB-1.20 This is supported by recent data showing that up-regulation of MuRF1 during muscle atrophy not only reduced the protein levels, but also the mRNA levels of MyBP-C.44 However, our data argue against transcriptional regulation of cMyBP-C by MuRF1, because it did not affect endogenous cMyBP-C mRNA levels neither in NRCM nor in transgenic mice, but only reduced exogenous cMyBP-C mRNAs derived from cDNAs in infected NRCM. MuRF1 could thus indirectly inhibit protein translation by down-regulating factors involved in translation initiation or elongation such as shown for INT-6.33 An alternative mechanism could be that MuRF1 indirectly reduces the protein level of cMyBP-C by regulating the transcription of MHC. Indeed, the levels of MHC mRNAs were markedly lower in MuRF1-overexpressing cells or mice, suggesting that MuRF1 degrades an MHC-specific transcription factor. Knowing that the stoichiometry of the sarcomere proteins is tightly regulated by co-translational and co-assembly mechanisms, lower level of MHC is likely associated with lower amount of cMyBP-C and vice versa. We therefore propose that the unexpected lower level of exogenous cMyBP-C mRNAs in MuRF1-infected NRCM is secondary to the lower transcription of MHC, in order to maintain the stoichiometry of the thick filament's proteins.
Under the in vivo conditions in mice, loss or surplus of MuRF1 was not associated by changes in other MuRFs or global chymotrypsin-like activity, suggesting that MuRF1 does not markedly affect the global UPS function, but rather specifically the turnover of a few specific substrates. In contrast to previous data obtained after gene transfer in cells,29 but in agreement with other in vivo data,36,45 cTnI was not regulated by MuRF1 in mice, suggesting that cTnI is not a major target for MuRF1. Finally, we show that MG132 increased the level of atrogin-1 in NRCM, indicating that the E3 ligase atrogin-1 itself is a target for UPS-mediated degradation. Interestingly, the level of atrogin-1 was 42% lower in MuRF1-TG than in WT littermates (see Supplementary material online, Figure S8) without change in the mRNA levels, suggesting that MuRF1 could also target atrogin-1 for UPS-mediated degradation.
MuRF1 deletion or overexpression in mice did not alter the degree of LV hypertrophy but recent findings showed dilated cardiomyopathy with reduced myocyte cross-sectional area in MuRF1-TG mice.36 The reduced level of both MHC and cMyBP-C could contribute to this phenotype. Similar to our data, previous experiments with another MuRF1-KO mouse model showed no basal phenotype either, but increased sensitivity to TAC-induced cardiac hypertrophy,46 suggesting that the defect becomes apparent only after induction of cardiac stress.
In conclusion, our data suggest that MuRF1 plays an indirect role in the regulation of the expression of cMyBP-C, probably by targeting a transcription factor specific for MHC. Moreover, the present data suggest that atrogin-1 specifically acts as an E3 ubiquitin ligase for truncated M7t-cMyBP-C, resulting from a human MYBPC3 mutation. Further analyses are needed to investigate whether atrogin-1 is critical for the removal of truncated cMyBP-C mutants in mice and to which extent this process may contribute to the pathophysiology of FHC.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the Sixth Framework Program of the European Union (Marie Curie EXT-014051), the Deutsche Forschungsgemeinschaft (FOR-604/1-2, CA 618/1-2 to T.E. and L.C., WI 3278/2-1 to C.C.W.), and the Association Française contre les Myopathies (AFM-9471).
Supplementary Material
Acknowledgements
We thank Nessan Herrgesell, Denise Juhr, and Silke Reischmann (Hamburg) for active involvement in parts of the experiments.
Conflict of interest: none declared.
References
- 1.Richardson P, McKenna W, Bristow M, Maish B, Mautner B, O'Connell J, et al. Report of the 1995 World Health Organisation/International Society and Federation of Cardiology task force on the definition and classification of cardiomyopathies. Circulation. 1996;93:841–842. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 2.Richard P, Villard E, Charron P, Isnard R. The genetic bases of cardiomyopathies. J Am Coll Cardiol. 2006;48:A79–A89. [Google Scholar]
- 3.Friedrich F, Bausero P, Sun Y, Treszl A, Krämer E, Juhr D, et al. A new polymorphism in human calmodulin III promoter is a potential modifier gene for familial hypertrophic cardiomyopathy. Eur Heart J. 2009;30:1648–1655. doi: 10.1093/eurheartj/ehp153. [DOI] [PubMed] [Google Scholar]
- 4.Carrier L, Schlossarek S, Willis MS, Eschenhagen T. Ubiquitin-proteasome system and nonsense-mediated mRNA decay in hypertrophic cardiomyopathy. Cardiovasc Res. 2009;105:239–248. doi: 10.1093/cvr/cvp247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations and implications for molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 6.Carrier L. Cardiac myosin-binding protein C in the heart. Arch Mal Coeur Vaiss. 2007;100:238–243. [PubMed] [Google Scholar]
- 7.Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, et al. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:438–440. doi: 10.1038/ng1295-438. [DOI] [PubMed] [Google Scholar]
- 8.Carrier L, Bonne G, Bährend E, Yu B, Richard P, Niel F, et al. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ Res. 1997;80:427–434. [PubMed] [Google Scholar]
- 9.Rottbauer W, Gautel M, Zehelein J, Labeit S, Franz WM, Fischer C, et al. Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy. Characterization of cardiac transcript and protein. J Clin Invest. 1997;100:475–482. doi: 10.1172/JCI119555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moolman JA, Reith S, Uhl K, Bailey S, Gautel M, Jeschke B, et al. A newly created splice donor site in exon 25 of the MyBP-C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation. 2000;101:1396–1402. doi: 10.1161/01.cir.101.12.1396. [DOI] [PubMed] [Google Scholar]
- 11.van Dijk SJ, Dooijes D, Dos Remedios C, Michels M, Lamers JM, Winegrad S, et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy. haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 12.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, et al. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 13.Flavigny J, Souchet M, Sébillon P, Berrebi-Bertrand I, Hainque B, Mallet A, et al. COOH-terminal truncated cardiac myosin-binding protein C mutants resulting from familial hypertrophic cardiomyopathy mutations exhibit altered expression and/or incorporation in fetal rat cardiomyocytes. J Mol Biol. 1999;294:443–456. doi: 10.1006/jmbi.1999.3276. [DOI] [PubMed] [Google Scholar]
- 14.Sarikas A, Carrier L, Schenke C, Doll D, Flavigny J, Lindenberg KS, et al. Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res. 2005;66:33–44. doi: 10.1016/j.cardiores.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Bahrudin U, Morisaki H, Morisaki T, Ninomiya H, Higaki K, Nanba E, et al. Ubiquitin-proteasome system impairment caused by a missense cardiac myosin-binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J Mol Biol. 2008;384:896–907. doi: 10.1016/j.jmb.2008.09.070. [DOI] [PubMed] [Google Scholar]
- 16.Ciechanover A. The ubiquitin proteolytic system: from a vague idea through basic mechanisms, and onto human diseases and drug targeting. Neurology. 2006;66:S7–S19. doi: 10.1212/01.wnl.0000192261.02023.b8. [DOI] [PubMed] [Google Scholar]
- 17.Mearini G, Schlossarek S, Willis MS, Carrier L. The ubiquitin-proteasome system in cardiac dysfunction. Biochim Biophys Acta. 2008;1782:749–763. doi: 10.1016/j.bbadis.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 18.Spencer JA, Eliazer S, Ilaria RL, Jr, Richardson JA, Olson EN. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein. J Cell Biol. 2000;150:771–784. doi: 10.1083/jcb.150.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol. 2001;306:717–726. doi: 10.1006/jmbi.2001.4448. [DOI] [PubMed] [Google Scholar]
- 20.McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol. 2002;157:125–136. doi: 10.1083/jcb.200108089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pizon V, Iakovenko A, Van Der Ven PF, Kelly R, Fatu C, Furst DO, et al. Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 RING-finger protein. J Cell Sci. 2002;115:4469–4482. doi: 10.1242/jcs.00131. [DOI] [PubMed] [Google Scholar]
- 22.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, et al. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 24.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dehoux MJ, van Beneden RP, Fernandez-Celemin L, Lause PL, Thissen JP. Induction of MafBx and Murf ubiquitin ligase mRNAs in rat skeletal muscle after LPS injection. FEBS Lett. 2003;544:214–217. doi: 10.1016/s0014-5793(03)00505-2. [DOI] [PubMed] [Google Scholar]
- 26.DeRuisseau KC, Kavazis AN, Deering MA, Falk DJ, Van Gammeren D, Yimlamai T, et al. Mechanical ventilation induces alterations of the ubiquitin-proteasome pathway in the diaphragm. J Appl Physiol. 2005;98:1314–1321. doi: 10.1152/japplphysiol.00993.2004. [DOI] [PubMed] [Google Scholar]
- 27.Adams V, Linke A, Wisloff U, Doring C, Erbs S, Krankel N, et al. Myocardial expression of Murf-1 and MAFbx after induction of chronic heart failure: effect on myocardial contractility. Cardiovasc Res. 2007;73:120–129. doi: 10.1016/j.cardiores.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 28.Razeghi P, Baskin KK, Sharma S, Young ME, Stepkowski S, Essop MF, et al. Atrophy, hypertrophy, and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem Biophys Res Commun. 2006;342:361–364. doi: 10.1016/j.bbrc.2006.01.163. [DOI] [PubMed] [Google Scholar]
- 29.Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci USA. 2004;101:18135–18140. doi: 10.1073/pnas.0404341102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koyama S, Hata S, Witt CC, Ono Y, Lerche S, Ojima K, et al. Muscle RING-Finger Protein-1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J Mol Biol. 2008;376:1224–1236. doi: 10.1016/j.jmb.2007.11.049. [DOI] [PubMed] [Google Scholar]
- 31.Witt SH, Granzier H, Witt CC. Labeit S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J Mol Biol. 2005;350:713–722. doi: 10.1016/j.jmb.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 32.Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, et al. The E3 ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007;6:376–385. doi: 10.1016/j.cmet.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 33.Witt CC, Witt SH, Lerche S, Labeit D, Back W, Labeit S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 2008;27:350–360. doi: 10.1038/sj.emboj.7601952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arya R, Kedar V, Hwang JR, McDonough H, Li HH, Taylor J, et al. Muscle ring finger protein-1 inhibits PKC{epsilon} activation and prevents cardiomyocyte hypertrophy. J Cell Biol. 2004;167:1147–1159. doi: 10.1083/jcb.200402033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, et al. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117:3211–3223. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willis MS, Schisler JC, Li L, Rodriguez JE, Hilliard EG, Charles PC, et al. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ Res. 2009;105:80–88. doi: 10.1161/CIRCRESAHA.109.194928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci USA. 2004;101:18135–18140. doi: 10.1073/pnas.0404341102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El-Armouche A, Pohlmann L, Schlossarek S, Starbatty J, Yeh YH, Nattel S, et al. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J Mol Cell Cardiol. 2007;43:223–229. doi: 10.1016/j.yjmcc.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 40.Pohlmann L, Kroger I, Vignier N, Schlossarek S, Kramer E, Coirault C, et al. Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ Res. 2007;101:928–938. doi: 10.1161/CIRCRESAHA.107.158774. [DOI] [PubMed] [Google Scholar]
- 41.Carrier L, Knoell R, Vignier N, Keller DI, Bausero P, Prudhon B, et al. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc Res. 2004;63:293–304. doi: 10.1016/j.cardiores.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 42.Vignier N, Schlossarek S, Fraysse B, Mearini G, Kramer E, Pointu H, et al. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein c mutant levels in cardiomyopathic mice. Circ Res. 2009;105:239–248. doi: 10.1161/CIRCRESAHA.109.201251. [DOI] [PubMed] [Google Scholar]
- 43.Fielitz J, Kim MS, Shelton JM, Latif S, Spencer JA, Glass DJ, et al. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J Clin Invest. 2007;117:2486–2495. doi: 10.1172/JCI32827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, et al. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol. 2009;185:1083–1095. doi: 10.1083/jcb.200901052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willis MS, Rojas M, Li L, Selzman CH, Tang RH, Stansfield WE, et al. Muscle ring finger 1 mediates cardiac atrophy in vivo. Am J Physiol Heart Circ Physiol. 2009;296:H997–H1006. doi: 10.1152/ajpheart.00660.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res. 2007;100:456–459. doi: 10.1161/01.RES.0000259559.48597.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.