

SUMMARY
The H3K4me3 mark in chromatin is closely correlated with actively transcribed genes, although the mechanisms involved in its generation and function are not fully understood. In vitro studies with recombinant chromatin and purified human factors demonstrate a robust SET1 complex (SET1C)-mediated H3K4 trimethylation that is dependent upon p53- and p300-mediated H3 acetylation, a corresponding SET1C-mediated enhancement of p53- and p300-dependent transcription that reflects a primary effect of SET1C through H3K4 trimethylation, and direct SET1C-p53 and SET1C-p300 interactions indicative of a targeted recruitment mechanism. Complementary cell-based assays demonstrate a DNA-damage-induced p53-SET1C interaction, a corresponding enrichment of SET1C and H3K4me3 on a p53 target gene (p21/WAF1), and a corresponding codependency of H3K4 trimethylation and transcription upon p300 and SET1C. These results establish a mechanism in which SET1C and p300 act cooperatively, through direct interactions and coupled histone modifications, to facilitate the function of p53.
INTRODUCTION
The transcription networks that underlie many physiological processes are controlled by sequence-specific DNA-binding transcription factors. These factors act in part through the recruitment of diverse cofactors that generate specific histone modification signatures, some involving histone crosstalk, associated with gene activation or repression events (Suganuma and Workman, 2011; Young, 2011). Modified histones in turn may alter transcription through direct effects on chromatin structure or through recruitment of other effectors (Patel and Wang, 2013). Of special interest here, two modifications closely correlated with gene activation are H3 acetylation and H3K4 methylation (Kouzarides, 2007; Li et al., 2007).
In human cells, H3K4 methylation is catalyzed by the SET1/MLL family (SET1A, SET1B, MLL1, MLL2, MLL3, and MLL4) of histone methyltransferases (HMTs). Like the prototype yeast Set1 (Shilatifard, 2012), the human SET1/MLL proteins exist in multiprotein complexes that possess both unique subunits and common subunits (WDR5, RbBP5, ASH2L, and DPY30) related to those in yeast COMPASS/Set1C (Vermeulen and Timmers, 2010). Biochemical studies have shown that human SET1/MLL complexes possess variable intrinsic abilities to catalyze mono-, di-, and trimethylation of H3K4, although, intriguingly, these complexes appear to have largely nonredundant roles (Eissenberg and Shilatifard, 2010). Notably, however, SET1C appears responsible for the majority of H3K4 trimethylation in mammalian cells (Shilatifard, 2012).
Genome-wide studies have shown that H3K4me3 is localized mainly to promoter regions, whereas H3K4me1 is enriched at enhancers (Heintzman et al., 2007; Zhou et al., 2011), raising the general question of whether these events are mediated by distinct enzymes or by position-specific activities of a common enzyme. Thus, distinct recruitment and/or enzymatic regulatory mechanisms for SET1/MLL complexes might be critical for the position-specific distribution of the distinct methylation states. A variety of alternative (nonmutually exclusive) mechanisms for recruitment (or stabilization) of SET1/MLL complexes have been described (reviewed in Ruthenburg et al., 2007; Schuettengruber et al., 2011; Smith and Shilatifard, 2010), with recruitment by sequence-specific DNA-binding transcription factors (Goo et al., 2003; Vermeulen and Timmers, 2010) being of special interest here. Position-specific H3K4 methylation states might also be regulated by other factors, including other (local) histone modifications. In this regard, early studies in yeast reported that H2B ubiquitylation, which is generally coupled to transcription, is essential for H3K4 trimethylation, but not H3K4 monomethylation, by COMPASS/Set1C (Shilatifard, 2012), whereas more recent studies have shown that ubiquitylated H2B directly stimulates H3K4 trimethylation of chromatin templates by the human and yeast SET1 complexes (Kim et al., 2009, 2013).
As a transcription factor, the tumor suppressor p53 is tightly regulated by complex mechanisms, which include various covalent modifications, in response to distinct cellular stresses (Kruse and Gu, 2009; Vousden and Prives, 2009). Although p53 is well studied with respect to its regulation, the mechanisms by which it differentially activates target genes through interactions with various cofactors, in particular those that effect epigenetic changes in chromatin, are less well understood. However, in relation to the H3K4 methylation mechanisms of interest here, previous studies have demonstrated DNA-damage-induced accumulation of H3K4me3, as well as other histone modifications, on p53 target genes (An et al., 2004; Lee et al., 2009).
To further investigate the factors and mechanisms involved in p53-dependent H3K4 methylation, we have employed cell-free systems reconstituted with purified factors and recombinant chromatin templates in conjunction with cell-based assays of a p53-dependent DNA-damage response. Our results establish a p53-p300/H3ac-SET1C interaction network that leads to synergistic SET1C and p300 functions in H3K4 methylation and transcription activation and, overall, insights into underlying mechanisms.
RESULTS
Human SET1C-Mediated H3K4 Trimethylation on Recombinant Chromatin Is Strongly Enhanced by p53-and p300-Dependent H3 Acetylation in the Absence of H2B Ubiquitylation
SET1C was purified to near homogeneity through Flag-tagged subunits (CFP1, WDR82, or SET1A) unique to SET1 complexes (Figure S1 available online). In an initial analysis of their HMT activities, these complexes exhibited clear H3K4 mono-, di-, and trimethylation activities on recombinant histone octamer substrates (Figure S2A) but very low activities on recombinant chromatin substrates (data not shown). In view of reports of enhanced H3K4 methylation of acetylated H3 peptides by an isolated MLL1 SET domain (Milne et al., 2002; Nightingale et al., 2007), as well as H3K4 methylation in the absence (Wang et al., 2009) or near absence (Vethantham et al., 2012) of H2B ubiquitylation in some physiological situations, we tested the ability of p300-mediated histone acetylation to increase the activity of SET1C on histone octamers with unmodified H2B but found no effects (data not shown).
We next tested possible effects of histone acetylation on H3K4 methylation by SET1C in the more physiological context of a p53- and p300-activated recombinant chromatin template (Figures 1A and S3) according to the protocol (An and Roeder, 2004) in Figure 1A. The histone acetyltransferase (HAT) assay revealed robust p53- and p300-dependent acetylation of H3 (Figure 1B, lane 1 versus lane 2), as reported (An et al., 2004), but failed to reveal any HAT activity in the SET1C preparation (lanes 9 and 10) or any effect of SET1C on p300-dependent histone acetylation (lane 2 versus lane 5). An immunoblot revealed p53- and p300-dependent H3K18 and H3K27 acetylation that was unaffected by SET1C (Figure 1C).
Figure 1. Human SET1C-Mediated H3K4 Trimethylation Is Stimulated by p53- and p300-Dependent H3 Acetylation on Recombinant Chromatin Templates.
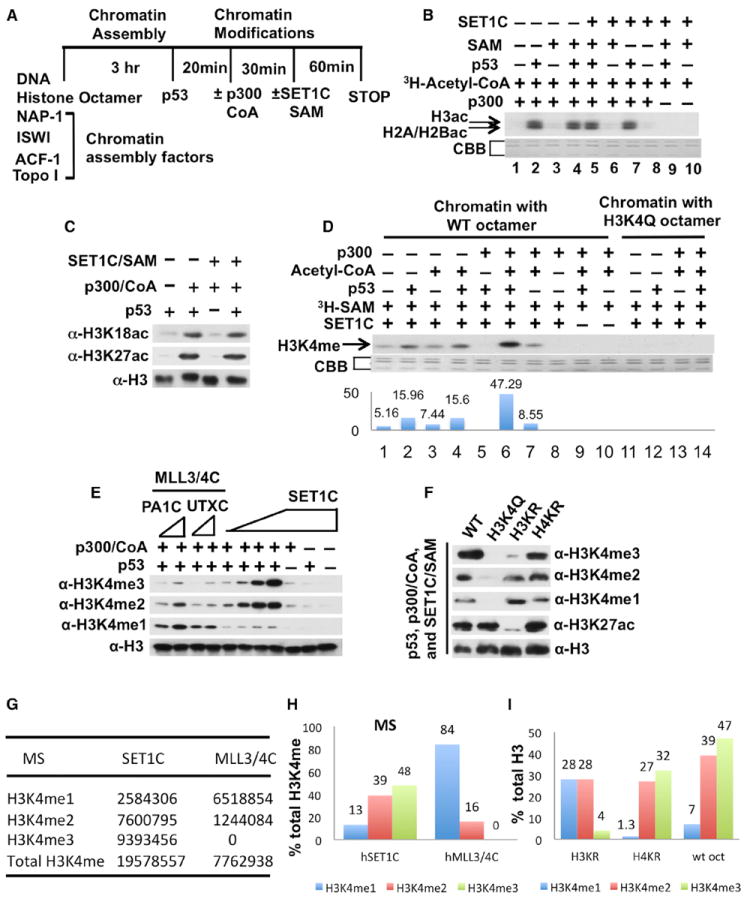
(A) HAT and HMT assay scheme for p300 and SET1C.
(B) HAT assay with indicated factors and 3H-acetyl-CoA (autoradiographic analysis). Lower panels in (B) and (D) show loading controls stained with Coomassie brilliant blue (CBB).
(C) H3 site-specific HAT assays with indicated factors and acetyl-CoA (immunoblot analysis with antibodies indicated on the left).
(D) HMT assays with indicated factors, chromatin substrates, and 3H-SAM (autoradiographic analysis).
(E) H3K4 methylation state-specific HMT assays with indicated factors (immunoblot analysis with antibodies indicated on the left).
(F) HAT and HMT assays with indicated factors and chromatin templates assembled with the mutant H3 and H4 histones indicated at the top and detailed in Figure S3 (immunoblot analysis with antibodies indicated on the right).
(G–I) MS analysis of p53- and p300-dependent methylation products generated by SET1C and hMLL3/4C (f:PA1C) on chromatin with WT histones (as in E; G shows raw data values, and H shows plotted values) or by SET1C on chromatin with WT or mutant histone (H3KR or H4KR) octamers (as in F; I shows plotted values).
See also Figures S1, S2, S3, S4, and S6.
In the HMT assay with recombinant chromatin (Figure 1D), a low level of H3 methylation that was observed with SET1C alone was increased about 3-fold with addition of p53 alone (lane 2 versus lane 1), only marginally with addition of p300/acetyl coenzyme A (acetyl-CoA) alone (lane 7 versus lane 1), and more dramatically (~10-fold) with the joint addition of p53 and p300/acetyl-CoA (lane 1 versus lane 6). These methylation activities were not observed in recombinant chromatin assembled with an H3K4Q mutant (Figure 1D, lanes 11 to 14), indicating that the observed SET1C-mediated H3 methylation is specific to H3K4. In reactions without acetyl-CoA (Figure 1D, lane 5 versus lane 2), the p53-enhanced H3K4 methylation activity of SET1C was inhibited, rather than enhanced, by p300. This result likely reflects direct p300-SET1C interactions that are described below. In a further analysis with methylation state-specific antibodies, and consistent with the results of Figure 1D, SET1C alone generated only low levels of H3K4me2 and H3K4me3 that were minimally stimulated by single additions of p53 or p300 but dramatically enhanced by their joint addition (Figure 1E). In confirmation, a mass spectrometric (MS) analysis of the methylated H3 products from parallel reactions indicated comparably high levels of H3K4me3 and H3K4me2 and a much lower level of H3K4me1 (Figures 1G and 1H).
To confirm a role for histone acetylation per se in the p53- and p300-dependent stimulation of H3K4 methylation by SET1C, we assembled recombinant chromatin templates with mutant H3 (H3KR) or H4 (H4KR) containing lysine-to-arginine mutations at known acetylation sites and, as a control, with the H3K4Q mutant (Figure S3G). Notably, the H3KR substrate showed increased levels of H3K4 monomethylation and nearly normal levels of H3K4 dimethylation but drastically reduced levels of H3K4 trimethylation, whereas the H4KR substrate showed nearly normal levels of H3K4 mono-, di-, and trimethylation (Figure 1F). As a control, H3K27 acetylation was unaffected in the H3K4Q and H4KR chromatin substrates but eliminated in the H3KR chromatin substrate. Based on quantitation of the data in Figure 1F (Figure 1I), and comparisons of the relative levels of mono-, di-, and trimethylation by the immunoblot and MS analyses in control (wild-type [WT] H3) reactions, the major effect of the H3KR mutations is on H3K4 trimethylation. These results clearly establish a critical role for H3 acetylation, but not H4 acetylation, in the p300-dependent enhancement of H3K4 trimethylation by SET1C. In further support of primary effects of p300 through histone acetylation, rather than p53 acetylation (Kruse and Gu, 2009), p53 mutations (Figures S4A and S4B) that effectively eliminated p53 acetylation by p300 had no significant effects on histone acetylation by p300 or p53/p300-dependent H3K4 methylation by SET1C (Figure S4C). Related analyses failed to show any acetylation of SET1C by p300 or methylation of p53 or p300 by SET1C (Figure S4D).
Interestingly, at comparable inputs based on methyltransferase activities on histone octamers and immunoblots of common subunits (Figures S2B and S2C), MLL3/4 complexes (purified through either Flag:PA1 or Flag:UTX subunits) effected predominantly H3K4 monomethylation and (to a lesser extent) dimethylation, but only very low levels of H3K4 trimethylation, in the presence of p53 and p300 (Figure 1E); and the MLL3/4C-mediated H3K4 monomethylation was dependent upon the presence of p53 and p300 (Figure S2D). These results were confirmed by MS analysis of MLL3/4C reaction products relative to the SET1C reaction products (Figures 1G and 1H). A reconstituted core MLL1 complex (Dou et al., 2006) exhibited similar properties under these conditions (data not shown).
H3 Acetylation-Dependent H3K4 Trimethylation Involves Histone Acetylation-Dependent Recruitment of p300, through Its Bromodomain, and p300-Mediated Recruitment of SET1C through Direct Interactions
Previous studies have demonstrated interactions of chromatin-modifying factors, through bromodomains, with acetylated histone residues (Filippakopoulos and Knapp, 2012). Therefore, and given the absence of bromodomains in SET1C subunits, we assessed the possible function of the bromodomain in the ACF1 subunit of the ACF complex, which was used for chromatin assembly and retained in the assay (Figure 1A), and the bromodomain in p300 (Figure 2A). The bromodomain-deleted ACF1 was functionally equivalent to intact ACF1 in our assay (Figure 2B). Most importantly, however, a bromodomain-deleted p300 mutant (p300 Δbromo, Figure S3F) that retained its autoacetylation and p53 acetylation capabilities (Figure 2C) failed to elicit either H3 acetylation or SET1C-mediated H3K4 trimethylation on recombinant chromatin (Figure 2B). As expected, a catalytically inactive p300 mutant (p300 DY, Figure S3F) also failed to support either histone acetylation (Figure 2C) or SET1C-mediated H3K4 trimethylation (data not shown). The loss of histone acetylation by p300 Δbromo is consistent with its reported deficiency in chromatin-based transcription (Santoso and Kadonaga, 2006) and in acetylation of nucleosomal histones (Kraus et al., 1999), whereas the loss of SET1C-mediated H3K4 trimethylation is consistent with our demonstration of histone acetylation-dependent methylation by SET1C. These results led us to further explore the mechanistic aspects of p300 and its resident bromodomain in both H3 acetylation and subsequent H3 methylation.
Figure 2. Role of the p300 Bromodomain in Regulating Histone Acetylation, H3K4 Trimethylation, p300 Recruitment, and SET1C Recruitment on Recombinant Chromatin.
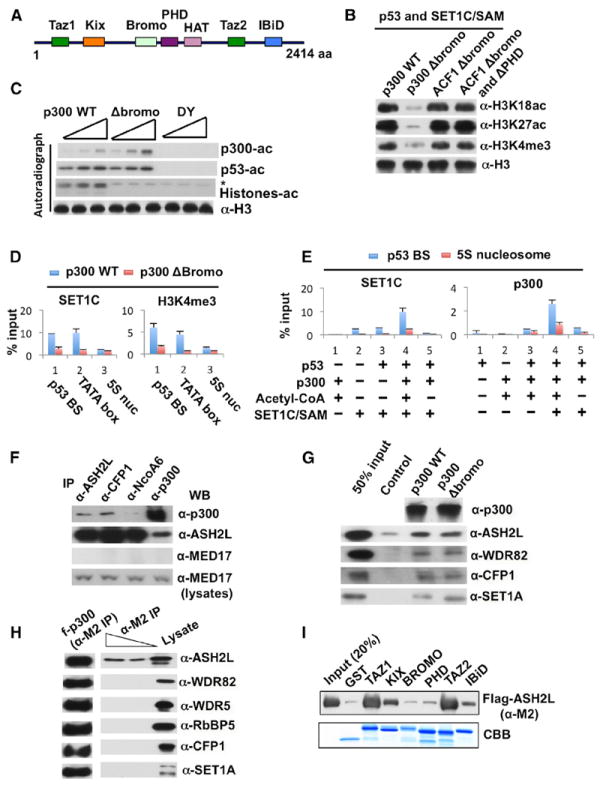
(A) Schematic representation of p300 domains.
(B) HAT and HMT assays as indicated, with either (1) p300 WT or p300 Δbromo with ACF1 WT-assembled chromatin or (2) p300 WT with either ACF1 Δbromodomain- or ACF1 Δbromodomain/ΔPHD-assembled chromatin. Analysis was by immunoblot with indicated antibodies (right). Analyses in (B)–(E) according to Figure 1A.
(C) HAT assays in the presence of p53, 3H-acetyl-CoA, and normalized amounts (Figure S3F) of either p300 WT, p300 Δbromo, or p300 DY. Analysis of HAT activities was by autoradiography (*nonspecific signal), and H3 inputs by immunoblot.
(D) In vitro ChIP assays for SET1C recruitment and H3K4 trimethylation in the presence of p53, indicated p300 proteins, and acetyl-CoA. Analysis by real-time PCR from the indicated locations (Figure S3C) on the chromatin template. Error bars in (D) and (E) denote standard deviations (SD).
(E) In vitro ChIP assays for recruitment of SET1C and p300 in the presence of the indicated components. Analysis at the indicated locations (Figure S3C) was by real-time PCR.
(F) Reciprocal coimmunoprecipitation of p300 and SET1C from nuclear extract by indicated antibodies (top) followed by immunoblots with indicated antibodies (right).
(G) Analysis of purified SET1C binding to recombinant p300 WT or p300 Δbromo (top) monitored by immunoblot with indicated antibodies (right).
(H) Analysis of Flag-p300 binding to individually coexpressed SET1C subunits (monitored by immunoblot with indicated antibodies).
(I) Analysis of purified Flag-ASH2L binding to recombinant GST-p300 domains (monitored by anti-Flag [M2] immunoblot).
See also Figures S1, S3, and S4.
As the recruitment of p300 and SET1C to the promoter is a key aspect of acetylation and methylation mechanisms, we employed in vitro chromatin immunoprecipitation (ChIP) assays to explore their position-specific recruitment to the recombinant chromatin template. As shown in Figure 2D, SET1C and H3K4me3 marks were selectively localized to the adjacent TATA-box and p53-binding-site regions relative to the flanking 5S nucleosome-positioning sequences (Figure S3C). Notably, and in agreement with the methylation data in Figure 2B, SET1C recruitment and accumulation of H3K4me3 were abrogated by deletion of the p300 bromodomain. In additional analyses, a low level of site-specific SET1C binding that was marginally enhanced by p53 was dramatically enhanced by p300 and, indicative of a histone acetylation requirement, dependent upon acetyl-CoA (Figure 2E, left panel). Similarly, p300 recruitment in the presence of acetyl-CoA was dependent upon p53, consistent with previous reports of direct interactions (An et al., 2004), and, somewhat surprisingly, significantly enhanced by the presence of SET1C (Figure 2E, right panel). Notably, the SET1C-enhanced p300 binding (Figure 2E, right) and the p53-dependent/SET1C-independent binding of p300 (data not shown) to the promoter were completely dependent upon the presence of acetyl-CoA—again indicative of a role for histone acetylation in both p300 and SET1C recruitment.
Although these results clearly indicated roles for p300 interactions with p53 and with acetylated histones (through the bromodomain) in p300 recruitment and stabilization, they left open the molecular basis for SET1C recruitment. Although studies described below document a direct p53 interaction as part of the SET1C recruitment mechanism, we also explored the possibility of a direct p300-SET1C interaction in light of the above-mentioned effect of SET1C on p300 binding. In support of this possibility, p300 was coimmunoprecipitated from HeLa nuclear extract by antibodies to ASH2L (common subunit of SET1/MLL complexes) and CFP1 (SET1C-specific) but much more weakly by an antibody to NCoA6 (MLL3/4C-specific), and, reciprocally, ASH2L was coimmunoprecipitated with anti-p300 (Figure 2F). As a control, the MED17 mediator subunit was not coimmunoprecipitated by any of these antibodies. In a more definitive analysis that clearly demonstrates a direct p300-SET1C interaction, a purified SET1C complex was shown to bind directly, and equivalently, to purified p300 WT and p300 Δbromo (Figure 2G). These results also suggest that the bromodomain requirement for SET1C promoter recruitment is indirect—with the bromodomain being required for p300 stabilization and p300, in turn, being required (through direct interactions) for SET1C recruitment. In a further dissection of the p300-SET1C interactions, p300 was found to selectively interact with the ASH2L subunit of the SET1 complex (Figure 2H), and ASH2L was found to interact strongly with the TAZ1 and TAZ2 domains of p300 and weakly with the KIX and IBiD domains (Figures 2A and 2I). These results overall argue strongly for roles for bromodomain-dependent p300-acetylated histone and p300-SET1C interactions in SET1C promoter recruitment and function.
Human SET1C-Mediated H3K4 Trimethylation Markedly Enhances p53- and p300-Dependent Transcription on Recombinant Chromatin
To determine whether SET1C-mediated H3K4 trimethylation directly facilitates p53-dependent transcription, either alone or in conjunction with p300, we tested its effect on transcription of recombinant chromatin according to the protocol in Figure 3A. This assay revealed a robust p53-dependent transcriptional activation in the presence of p300/acetyl-CoA (Figure 3B, lane 4 versus lane 3), as reported previously (An et al., 2004), whereas SET1C/SAM alone failed to elicit any detectable p53-dependent transcription (Figure 3B, lane 1 versus lane 2). Remarkably, however, SET1C/SAM markedly increased the p53-dependent transcription observed with p300/acetyl-CoA (Figure 3B, lanes 2 and 4 versus lane 10). In contrast to the results observed with SET1C, but consistent with its low H3K4 trimethylation activity (Figure 1E), the MLL3/4 complex failed to enhance p53- and p300-dependent transcription (data not shown).
Figure 3. SET1C Facilitates p53- and p300-Dependent Transcription on Recombinant Chromatin through H3K4 Trimethylation.
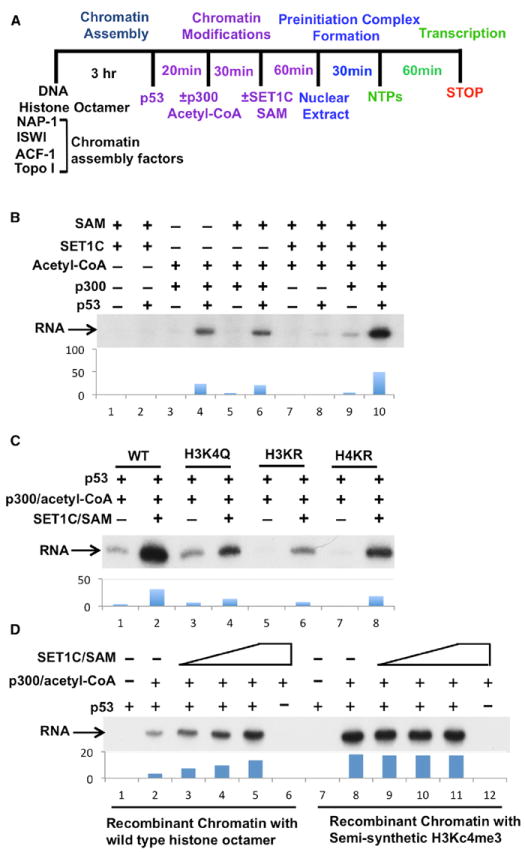
(A) Scheme for recombinant chromatin-based transcription assays.
(B) Stimulation of p53-and p300-dependent transcription by SET1C.
(C) Effects of H3 and H4 mutations on SET1C- and p300-dependent transcription. Chromatin templates were reconstituted with either control (WT) or indicated mutant histones (detailed in Figure S3G).
(D) Stimulation of p53- and p300-dependent transcription by prior incorporation of semisynthetic H3Kc4me3 into the chromatin template. Factor additions as indicated in (B)–(D). Autoradiographs in (B), (C), and (D) were scanned and quantitated by Quantity One Program (Bio-Rad). See also Figures S1 and S3.
To further establish the function of H3K4 trimethylation by SET1C in p53-dependent transcription, we analyzed chromatin templates assembled with the mutant H3KR, H4KR, and H3K4Q histones described above. p53- and p300-dependent transcription was significantly reduced by the lysine-to-arginine mutations in either H3 (H3KR) or H4 (H4KR), consistent with our previous results (An et al., 2004), but not by the H3K4Q mutation (Figure 3C, lanes 3, 5, and 7 versus lane 1). Most importantly, however, the large SET1C enhancement of p53/p300-dependent activity (lane 2 versus lane 1) was largely eliminated by the H3K4Q mutation that precludes H3K4 methylation (lane 4 versus lane 2). This result indicates that the stimulatory effect of SET1C is mediated mainly through H3K4 methylation, whereas the residual SET1C-enhanced activity may reflect other functions of SET1C such as the stabilization of p300 binding observed in Figure 2E. We also observed significant levels of transcription (above the nondetectable baseline levels) from the H3KR and (most notably) H4KR templates (lane 6 versus 5 and 8 versus 7) in the presence of p53, p300, and SET1C, further indicative of SET1C-mediated H3K4 trimethylation functions that partially compensate for the loss of H3 or H4 acetylation-dependent functions.
To further establish a direct role for H3K4 trimethylation in p53- and p300-dependent transcription, we reconstituted a recombinant chromatin template with a semisynthetic methyl-lysine analog (H3Kc4me3) of H3K4me3 (Simon et al., 2007) in place of unmodified H3. Consistent with the observed effect of SET1C-mediated methylation on transcription, preincorporated H3Kc4me3 markedly enhanced p53- and p300-dependent transcription in the absence of SET1C (Figure 3D, lane 8 versus lane 2) but failed to elicit detectable transcription in the absence of p53 or p300 (lanes 7 and 12). Moreover, and further indicative of a predominant function of SET1C through H3K4 trimethylation, the activity observed with the H3Kc4me3 template was comparable to that observed with SET1C (lane 8 versus lane 5) and not further enhanced by addition of SET1C (lanes 8–11). (The small differential likely reflects a lower level of substrate trimethylation in the SET1C reactions [Figure 1H]). Two recent studies also reported stimulatory effects of H3Kc4me3 on activator-mediated transcription from chromatin templates but did not investigate effects of histone acetylation on H3K4 trimethylation or transcription by endogenous SET1C (Lauberth et al., 2013; Lin et al., 2011). Altogether, our biochemical studies establish that SET1C-mediated H3K4 trimethylation plays a critical and causative role in facilitating p53- and p300-dependent transcription.
Direct and Specific Physical Interaction of SET1C with p53
Although the above-described studies established a role for p300-SET1C interactions in p53- and p300-dependent H3K4 trimethylation and transcriptional enhancement by SET1C, they also indicated a modest (but significant) stimulation of SET1C-mediated H3K4 methylation by p53 in the absence of p300 (see also Figure S6 below). To assess whether this reflects a direct p53-SET1C interaction that might complement the p53/p300-enhanced function of SET1C, we initially determined whether endogenous p53 directly associates with SET1C under physiological conditions. As shown in Figure 4A (upper panel), following treatment of HCT116 cells (a human colorectal cancer cell line) with doxorubicin, an anti-CFP1 antibody coimmunoprecipitated p53 in parallel to its induction. Coimmunoprecipitation of hSET1, RbBP5, and ASH2L with anti-CFP1 indicated that the integrity of SET1C was not affected by DNA damage. A reciprocal coimmunoprecipitation with an anti-p53 antibody also revealed a doxorubicin-enhanced association of CFP1, SET1, and ASH2L with p53 (Figure 4A, middle panel). The lysate immunoblots of p53, p21, RbBP5, and β-actin are shown as controls (Figure 4A, lower panel). Quantitation of these data confirms a doxorubicin-induced increase in the p53-SET1C association that parallels the increase in the cellular level of p53 (Figure S5). In a further analysis, p53 was selectively coimmunoprecipitated from p53 WT HCT116 cells by both anti-SET1 and anti-CFP1 antibodies but not by IgG or an anti-MLL1 antibody that coimmunoprecipitated an equivalent level of the common RbBP5 subunit (Figure 4B).
Figure 4. Direct Doxorubicin-Induced Interactions of p53 with SET1C in HCT116 Cells.
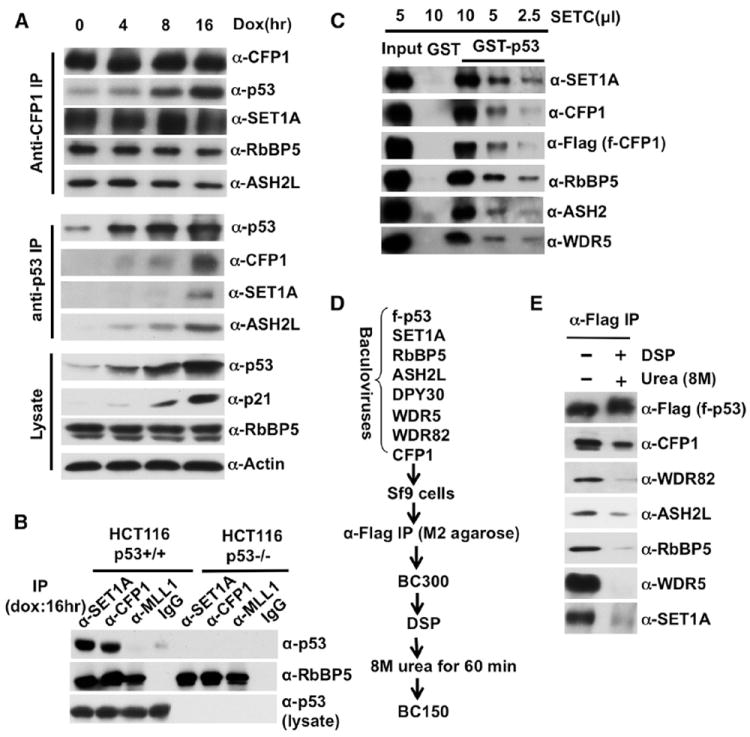
(A) Doxorubicin-induced interaction of p53 with endogenous SET1C. IPs (left) were monitored by immunoblot with indicated antibodies (right).
(B) SET1C-specific interaction with p53. IPs (indicated at the top) from doxorubicin-treated (16 hr) p53 WT and p53 null HCT116 cells were monitored by immunoblot with indicated antibodies (right).
(C) Direct binding of purified SET1C (monitored by antibodies indicated on right) to p53.
(D) Scheme for DSP-mediated crosslinking of p53 to SET1C.
(E) CFP1-mediated intracellular association of p53 with a reconstituted SET1C (details in the Extended Experimental Procedures). See also Figures S1 and S5.
In a more direct analysis with purified proteins, SET1C bound strongly to GST-p53 but not to GST (Figure 4C). Related, analysis of a p53-associated SET1C (reconstituted in Sf9 cells) in a dithiobis[succinimidyl propionate] (DSP)-based crosslinking assay (Figure 4D) revealed a dominant interaction of p53 with the CFP1 subunit of SET1C (Figure 4E). These results clearly establish a direct p53-SET1C interaction, primarily through the SET1C-specific subunit CFP1, that may account for the observed intracellular association of doxorubicin-induced p53 with SET1C.
SET1C and p300 Act Cooperatively to Regulate H3K4 Trimethylation and Transcription of a p53 Target Gene in a DNA-Damage Response
Because H3K4me3 is enriched at the canonical p53 target gene p21/WAF1 in HCT116 cells in response to DNA damage (Kim et al., 2009, 2010), we chose this system for analysis of SET1C function in vivo. In an initial analysis, a small interfering RNA (siRNA)-based knockdown of either CFP1 or WDR82 (both unique to SET1C) significantly reduced both global H3K4me3 (Figure 5A), as expected (Shilatifard, 2012), and, most importantly, the doxorubicin-induced enrichment of H3K4me3 at the p21/WAF1 promoter (Figures 5B and 5C), whereas the global levels of H3K18/27ac were not affected (Figure 5A). Notably, the doxorubicin-induced expression of p21/WAF1 was dramatically reduced, at both protein (Figure 5A) and messenger RNA (mRNA) (Figure 5D) levels, by knockdown of either CFP1 or WDR82.
Figure 5. p300 and SET1C Act Cooperatively to Regulate DNA-Damage-Induced Transcription of the p21/WAF1 Gene by p53.
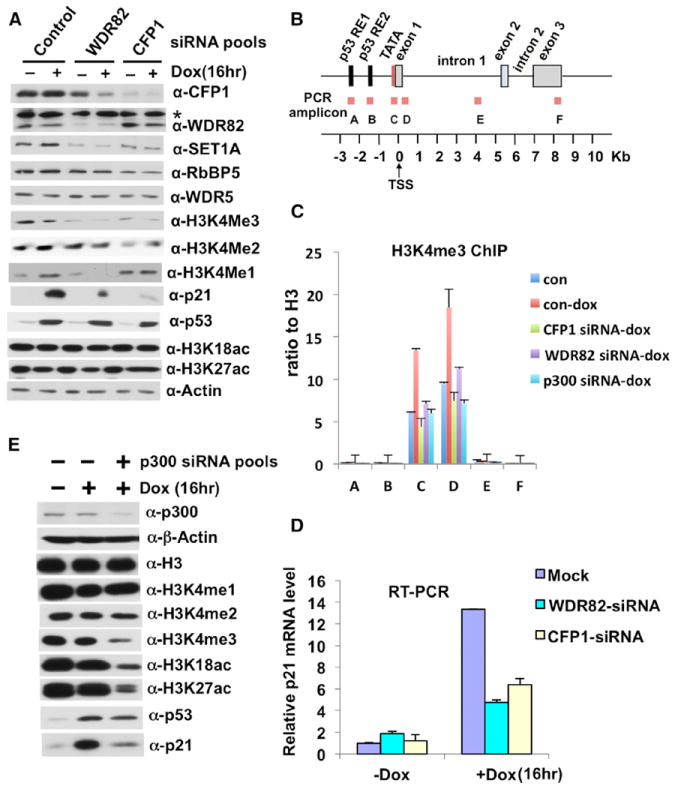
(A) Effects of siRNA-based CFP1 and WDR82 knockdowns on global H3K4 methylation and doxorubicin-induced p53 target gene (p21/WAF1) expression in HCT116 cells. The asterisk indicates a cross-reactive band. In (A) and (E), siRNA pools are indicated at the top, and immunoblot antibodies on the right. The instability of SET1A in the absence of WDR82 was previously noted in yeast and human cells.
(B) Diagram of the p21/WAF1 gene and PCR amplicons used for ChIP analyses.
(C) Effects of siRNA-based p300, CFP1, and WDR82 depletions on doxorubicin-induced (16 hr) enrichment of H3K4me3 on 21/WAF1 in HCT116 p53+/+ cells. ChIP analysis at amplicons A through F as indicated in (B).
(D) Effects of siRNA-based CFP1 and WDR82 depletions on p21/WAF1 expression in doxorubicin-treated HCT116 p53+/+ cells. RNA levels were monitored by real-time RT-PCR. Errors bars in (C) and (D) denote SD from duplicate reactions by real-time PCR.
(E) Effects of p300 knockdown on global H3K18/27 acetylation and H3K4 methylation and on expression of p21/WAF1.
In view of the effects of p300-mediated acetylation on H3K4 methylation in vitro (Figure 1), we also tested the effects of p300 depletion on p21/WAF1 activation events in vivo. In an initial siRNA-based approach (see also below), the knockdown of p300 in doxorubicin-treated HCT116 cells reduced the global levels not only of the H3K18ac and H3K27ac (p300-specific marks) but also of H3K4me3 (Figure 5E). A ChIP analysis also revealed a significant reduction in p21/WAF1 promoter-associated H3K4me3 following p300 knockdown (Figures 5B and 5C), correlating well with the biochemical results (Figures 1D and 1E). Finally, the doxorubicin-induced expression of p21/WAF1 was also significantly reduced upon p300 knockdown (Figure 5E), consistent with previous results (Iyer et al., 2004). The joint requirement of p300 and SET1C (above) for doxorubicin-induced H3K4 methylation and expression of p21/WAF1 indicates a clear functional cooperativity between these factors.
p53-Dependent Recruitment of SET1C and Enrichment of H3K4me3 at the p21/WAF1 Gene during DNA-Damage-Induced Transcription
Our demonstration of a direct physical interaction between p53 and SET1C suggested that p53 may play a direct role, complementary to that involving p300-SET1C interactions, in facilitating recruitment of SET1C to specific regions in the p21/WAF1 gene during a DNA-damage response. To further test this hypothesis, we performed real-time PCR-based ChIP assays, at the gene regions indicated in Figure 6A, at various times after exposure of HCT116 cells to doxorubicin. These analyses revealed (1) significant doxorubicin-induced enrichments of p53, H3K18ac, H3K27ac, and H3K4me1 at the enhancer (Figures 6B–6E) and (2) significant doxorubicin-induced enrichments of Pol II, the SET1C-specific subunit CFP1, and H3K4me3 at the promoter (Figures 6F–6H). The doxorubicin-induced accumulation of p53 and Pol II at p21/WAF1 enhancer and promoter elements, respectively, is consistent with results of a previous study (Kim et al., 2010). Similarly, the differential distribution of H3K4me1 and H3K4me3 to enhancer and promoter regions, respectively, is consistent with previous genome-wide studies in human cells (Heintzman et al., 2007). Thus, these results suggest that p53 is instrumental in facilitating recruitment of SET1C and a corresponding enrichment of H3K4me3 and H3K4me1 at the p21/WAF1 gene upon DNA-damage-induced transcription.
Figure 6. Role of p300 in the DNA-Damage-Induced Accumulation of SET1C and H3K4me3 on the p21/WAF1 Gene.
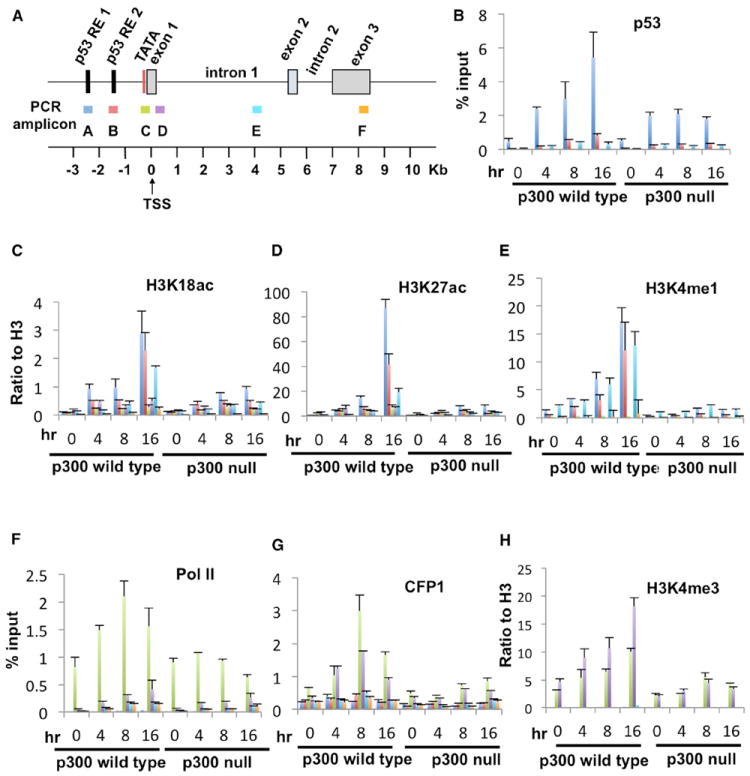
(A) Diagram of p21/WAF1 and PCR amplicons used for ChIP analyses.
(B–H) ChIP assays with the indicated antibodies at the indicated p21/WAF1 loci. Error bars in (B)–(H) denote the SD from duplicate reactions by real-time PCR (all ChIP assays were repeated twice).
Depletion of p300 Leads to Significant Reductions of SET1C and Both H3K4me1 and H3K4me3 at the p21/WAF1 Gene in Response to DNA Damage
In a further analysis of doxorubicin-induced changes on p21/WAF1, and taking advantage of HCT116 p300 WT and null cells (Iyer et al., 2004), comparative ChIP analyses (Figure 6) revealed (1) dramatic reductions of the doxorubicin-induced, enhancer-associated H3K27ac and H3K18ac marks, as well as the H3K4me1 mark, in the p300 null cells relative to p300 WT cells, (2) significant reductions of the doxorubicin-induced enrichments of SET1C and H3K4me3 at the proximal promoter in p300 null cells, and (3) significant, but less dramatic, reductions in the doxorubicin-induced enrichments of Pol II at the promoter and p53 at the enhancers in p300 null cells, the latter result indicating a stabilizing effect of histone acetylation on p53 binding. Most importantly, the loss of both CFP1/SET1C and the H3K4me3 and H3K4me1 marks is strongly correlated with the loss of p300 and related histone acetylation events. Thus, these results provide strong support for the proposal, from the more direct biochemical experiments, that p300-mediated histone acetylation events play a key role in SET1C-mediated H3K4 methylation events that are also important for transcriptional activation of the p21/WAF1 gene by p53.
Independent and Cooperative Effects of p300/H3 Acetylation and H2B Ubiquitylation on SET1C-Mediated H3K4 Trimethylation
Beyond our demonstration of p53 and p300 interactions with SET1C that facilitate its function in H2Bub-independent H3K4 methylation, we examined H2Bub-dependent H3 methylation by SET1C by employing chromatin templates assembled with either WT H2B or a semisynthetic H2Bub analog (Kim et al., 2009, 2013). This analysis (Figure S6) revealed (1) comparable basal (p53-independent) levels of trimethylation activity with p300/acetyl-CoA versus H2Bub (lanes 2 and 5), (2) comparable levels of p53-dependent activity with p300/acetyl-CoA versus H2Bub (lanes 4 and 7), (3) cooperativity between p300/acetyl-CoA and H2Bub in stimulating p53-dependent H3K4 trimethylation by SET1C (lane 8 versus lanes 4 and 7), and (4) a large enhancement of SET1C activity by p53 in the absence of p300/acetyl-CoA on the H2Bub-containing template (lane 7 versus lane 5), an observation that strongly supports a proposed complementary role for the direct p53-SET1C interaction in SET1C recruitment. Most importantly, the quantitatively comparable effects of H2B ubiquitylation and p300/acetyl-CoA on SET1C activity, as well as their cooperativity, suggest that they may be equally important in vivo.
DISCUSSION
A variety of genetic and biochemical studies have provided important examples of crosstalk between different histone modifications (Lee et al., 2010; Suganuma and Workman, 2011). Here, in relation to p53-dependent transcription, we have established strong physical and functional linkages between p300 and SET1C and their corresponding histone modifications. These results, discussed below, have revealed mechanistic insights into the regulation of H3K4 methylation that are likely of broad significance.
Histone Crosstalk: A Cooperativity between p300 and SET1C that Parallels and Complements Cooperativity between Ubiquitylated H2B and SET1C
Although there are a number of prior examples of histone crosstalk (Lee et al., 2010; Suganuma and Workman, 2011), most of these have been inferred either from cell-based assays, where indirect effects or secondary contributions of other endogenous histone modifications are hard to rule out, or from in vitro assays with nonphysiological substrates (e.g., histone peptides or isolated mononucleosomes) and enzymes (e.g., isolated catalytic domains or catalytic subunits of larger complexes). In the present case, our in vitro assays with unmodified recombinant chromatin clearly establish a direct effect of p300-dependent H3 acetylation and subsequent p300 stabilization (discussed below) on H3K4 trimethylation by SET1C. Importantly, our complementary cell-based assays lend further support to the notion of a direct role for p300-dependent H3 acetylation and associated p300 stabilization in SET1C-mediated H3K4 trimethylation (discussed further below).
It is noteworthy that the p53- and p300-dependent H3K4 trimethylation observed in our defined in vitro system occurs independently of any H2B ubiquitylation. Hence, although H2B ubiquitylation is critical for H3K4 trimethylation by COMPASS/Set1C in yeast (Shilatifard, 2012) and can enhance H3K4 trimethylation by both human (Kim et al., 2009) and yeast (Kim et al., 2013) SET1 complexes on unacetylated nucleosomal substrates, our results clearly demonstrate an intrinsic ability of SET1C to effect significant H3K4 trimethylation, in conjunction with p53 and p300, in the absence of H2B ubiquitylation. This result is consistent with H3K4 methylation in the absence of H2B ubiquitylation in Tetrahymena (Wang et al., 2009) and normal levels of H3K4 trimethylation in yeast (Lee et al., 2012) and in differentiated myoblasts (Vethantham et al., 2012) at very low levels of H2Bub. Notably, as p53 and p300 functions may be more closely associated with enhancer/promoter functions and transcription initiation, whereas H2B ubiquitylation appears to be associated with and dependent upon transcription elongation (Kim et al., 2009), our results raise the possibility of partially distinct functions, in initiation and elongation, of the two SET1C regulatory mechanisms. Nonetheless, our demonstration of cooperative effects of p300 and H2Bub on SET1C activity in vitro leaves open the possibility of cooperative intracellular functions in initiation and/or elongation.
The p300 Bromodomain Links Histone Acetylation and H3K4 Trimethylation by Stabilizing p300 for Subsequent SET1C Recruitment through Direct Interactions
Interactions of histone acetyl-lysine residues with the bromodomains of chromatin remodeling and transcription factors are thought to play key roles in the recruitment and/or function of these factors (Filippakopoulos and Knapp, 2012). Here, our in vitro analyses have demonstrated (1) that the p300 bromodomain is important not only for histone acetylation by p300, as previously reported (Kraus et al., 1999), but also for H3K4 trimethylation by SET1C, (2) that the p300 bromodomain-dependent histone acetylation is actually required, along with p53, for the stable association of p300 with the chromatin template, (3) that the stable association of SET1C with the template is dependent upon both p300 and the bromodomain-dependent histone acetylation, and, related, (4) that SET1C, through its ASH2L subunit, interacts directly with p300 through its TAZ domains, thus providing a clear mechanism for the p300-dependent recruitment and function of SET1C. These in vitro results lead to a model (Figure 7) in which the first important feature is an initial p53-mediated recruitment of p300 that is stabilized by primary p300-mediated acetylation events, which in turn lead to a feed-forward mechanism for p300-mediated histone acetylation that could ultimately become independent of p53. A similar situation has been described for the recruitment and function of the yeast SAGA complex (Hassan et al., 2002). The second important feature is the p300-enhanced recruitment of SET1C, which is complementary to the p53-based recruitment and leads directly to H3K4 trimethylation. These results thus add another important mechanism, namely interactions with the coactivator p300, to the growing list of mechanisms (reviewed in Smith and Shilatifard, 2010; Schuettengruber et al., 2011) for recruitment of SET1/MLL complexes.
Figure 7. Model for Cooperative p300 and SET1C Functions, Involving Direct Recruitment by p53 and Coupled Histone Modifications, in Facilitating p53-Dependent Transcription In Vitro.
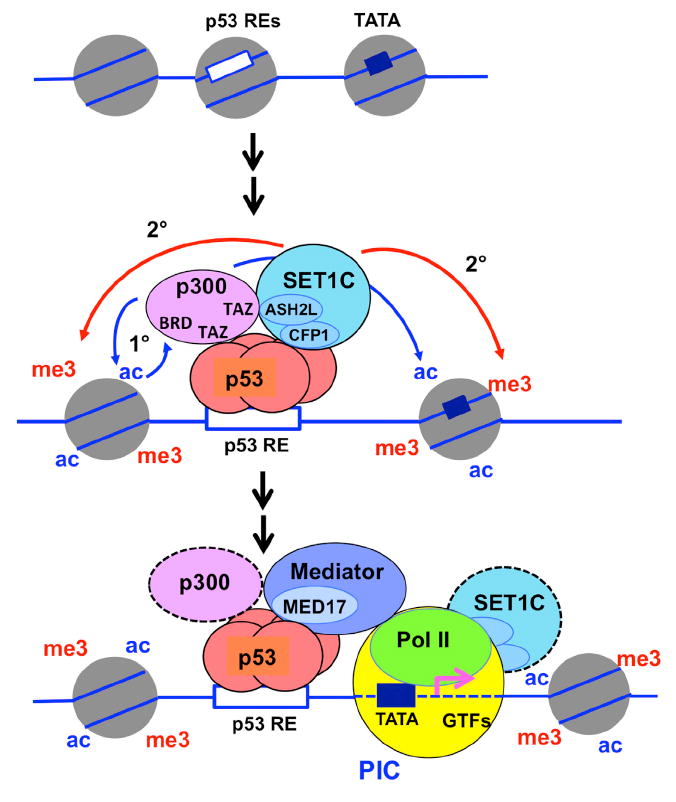
Based on in vitro biochemical analyses, the following model is proposed: (1) site-specific binding of p53, (2) p53-mediated recruitment of p300 with concomitant H3 acetylation and further stabilization of p300 through bromodomain interactions with acetylated H3 residues, (3) p53- mediated recruitment of SET1C, which in turn is stabilized by direct interactions with p300, and subsequent H3K4 trimethylation at the promoter by SET1C, and (4) functions of the histone acetyl and H3K4me3 marks in the recruitment and function of downstream effector molecules (not indicated), leading to the formation and function of initiation and elongation complexes.
See text for details and relevance to DNA-damage-induced activation of p53 target genes in cells. See also Figure S7.
Dominant Function of SET1C, Relative to MLL Complexes, in H3K4 Trimethylation Is Related to p53- and p300-Dependent Transcription
The existence of a family of SET1C-related complexes in mammalian cells (see Introduction) raises significant questions regarding the complexes’ potential gene- and cell-specific functions, as well as their intrinsic capabilities for mono-, di-, and trimethylation. Here, we demonstrate that a highly purified SET1C, in a p53- and p300-dependent manner, exhibits a high degree of H3K4 tri- and dimethylation relative to monomethylation, whereas related MLL1 and MLL3/4 complexes show the opposite behavior. Consistent with this observation, and a major role for SET1C in p53 target-gene activation, depletion of SET1C-specific subunits (WDR82 or CFP1) significantly reduced H3K4me3 both on the p53-activated p21/WAF1 gene and, as expected (Shilatifard, 2012), at a global level. Consistent with our results, a recent study showed a global decrease of H3K4me3 upon depletion of p300/CBP in mouse embryonic fibroblasts (MEFs) but did not establish either the responsible methyltransferase or a direct effect of acetylation on methylation (Jin et al., 2011).
Consistent with the existence of a family of related SET1/MLL enzymes and corresponding complexes in mammalian cells, there is increasing evidence for selective (nonredundant) functions for these enzymes (Eissenberg and Shilatifard, 2010; Vermeulen and Timmers, 2010). However, it remains to be determined whether these enzymes may function cooperatively on most genes—for example, at promoters versus enhancers, at different steps (initiation versus elongation) in the transcription cycle, or in effecting different states of H3K4 methylation. Thus, whereas our present results clearly establish a major role for SET1C in p53- and p300-dependent H3K4 trimethylation in vitro and in vivo, they do not exclude a secondary role for a SET1C-related MLL complex in p53 function. In this regard, recent studies have demonstrated (1) roles for both SET1A and MLL1 in activation of specific estrogen receptor target genes (Jeong et al., 2011), (2) a role for the Drosophila homolog (Trr) of MLL3/4 in the genome-wide maintenance of H3K4 monomethylation on enhancers (Herz et al., 2012), and, of most relevance here, (3) roles for MLL3 (Lee et al., 2009) and MLL4 (Guo et al., 2012) in p53 function in cell-based assays—including localization of MLL3/4C to the p21/WAF1 enhancer in MEF cells (Lee et al., 2009) as discussed below.
Direct Role for p53, as well as p300, in Mediating SET1C Recruitment and Function
Although studies in yeast have emphasized transcription-dependent recruitment mechanisms (Shilatifard, 2012), clear evidence exists for alternative or complementary mechanisms in mammalian cells (reviewed in Smith and Shilatifard, 2010; Schuettengruber et al., 2011). Here, our demonstration of a low but significant p53-dependent H3K4 trimethylation by SET1C on recombinant chromatin templates in the absence of p300 and, especially, a direct interaction between p53 and SET1C argues strongly for a direct recruitment of SET1C by enhancer/promoter-bound p53 that parallels the previously reported p300 recruitment by p53 (An et al., 2004). Although these biochemical studies provide the best evidence for this mechanism, it is strongly supported by cell-based assays that show a DNA-damage-induced intracellular association of p53 and SET1C extrinsic to the chromatin template. Importantly, the ChIP assays in p300 WT versus p300 null HCT116 cells, which show that the loss of p300 and accompanying H3 acetylation has a much greater effect on SET1C recruitment and H3K4 methylation than on p53 binding, also suggest a role for p300-dependent histone acetylation in SET1C recruitment or stabilization. These results are consistent with the in vitro ChIP data showing that SET1C recruitment is highly dependent on both p53 and p300. Thus, as discussed earlier, we favor a mechanism in which SET1C is initially recruited (targeted) by p53 but subsequently stabilized by its interaction with p300 (Figure 7). This type of two-step mechanism has also been proposed for the recruitment and stabilization of other chromatin-modifying factors (Ruthenburg et al., 2007; Suganuma and Workman, 2011). In further support of the proposed mechanism for SET1C recruitment by p53, several studies have reported interactions of DNA-binding transcription factors with SET1C-related MLL complexes (Goo et al., 2003; reviewed in Vermeulen and Timmers, 2010) but, unlike the present study, did not establish direct roles for specific histone modifications in facilitating the recruitment or function of these complexes. In relation to our demonstration that p300 interacts with SET1C through its ASH2L subunit, which is common to all SET1/MLL complexes, it is interesting that ASH2L also plays an important role in H2Bub-mediated H3K4 methylation by the MLL1 complex in vitro (Wu et al., 2013).
Causal Role for SET1C-Mediated H3K4 Trimethylation in p53-Dependent Transcription
Although H3K4 trimethylation has been correlated with actively transcribed or poised genes in organisms ranging from yeast to human, its depletion has little effect on global gene expression in yeast, where it appears to be mainly a consequence rather than a cause of transcription (Shilatifard, 2012), or on transcription of H3K4me3-marked pluripotency genes in embryonic stem cells (ESCs) (Jiang et al., 2011). On the other hand, demonstrations of effects of H3K4me3 depletion on expression of specific genes (Goo et al., 2003; Jiang et al., 2011; Shilatifard, 2012) and of H3K4me3 recognition by various transcription factors and chromatin-remodeling factors (Ruthenburg et al., 2007; Vermeulen and Timmers, 2010) have indicated functions (some potentially indirect) on transcription of specific genes. In this report, a causal role for SET1C-mediated H3K4 trimethylation in p53-dependent transcription is clearly established by (1) the enhancement of p53- and p300-dependent transcription by a purified SET1C complex in a cell-free system reconstituted with a recombinant chromatin template containing a single promoter/enhancer, (2) the loss of this SET1C-enhanced transcription on a chromatin template reconstituted with an H3K4Q mutant, and (3) the quantitative recapitulation of this SET1C-enhanced transcription (in the absence of SET1C) by prior incorporation of H3Kc4me3 into the chromatin template. Our demonstration of SET1C-dependent transcription of the endogenous p53- and p300-activated p21/WAF1 gene in HTC116 cells further supports this conclusion. Moreover, in contrast to the less definitive cell-based analysis of endogenous genes, the in vitro assays also indicate that robust SET1C-enhanced H3K4 methylation does not depend, intrinsically, upon histone modifications other than those generated by p300. However, this does not preclude positive effects of other histone modifications, including H2Bub (discussed above) and the recently described H3R2 symmetric dimethylation (Migliori et al., 2012), on H3K4 methylation.
H3K4 Methylation and H3 Acetylation Events in Relation to Enhancer-Promoter Interactions and a Simplified Model
Our in vitro analyses with a model template lead to a simple model (Figure 7) in which enhancer/promoter-bound p53, through direct interactions, recruits p300 and SET1C to effect linked H3 acetylation and H3K4 trimethylation events through additional p300-SET1C interactions (as discussed earlier) at the contiguous enhancer-core promoter region. Interactions of these histone marks with other effectors (Ruthenburg et al., 2007; Vermeulen and Timmers, 2010) may then stimulate later steps in transcription. Importantly, our in vivo analyses are consistent with the in vitro results with respect to corresponding p300 and SET1C requirements, colocalization of SET1C and H3K4me3, and colocalization of H3K4me1 and H3K27ac marks. However, they present a more complicated situation related to the promoter-distal localization of the enhancer in the natural p21/WAF1 gene. Thus, although we presume from the various in vitro results that enhancer-bound p53 and p300 are involved in the DNA-damage-induced recruitment of SET1C to the p21/WAF1 gene in vivo, the DNA-damage-induced SET1C and H3K4me3 marks localize primarily to the promoter, whereas the induced H3K4me1 and H3K18ac/H3K27ac marks localize mainly to the enhancer. These results raise the possibility of dynamic enhancer-promoter interactions, potentially involving chromatin looping (Young, 2011), that result in primary transient interactions of SET1C at the enhancer with transfer and subsequent stabilization and function at the promoter in the steady-state situation (Figure S7). In apparent support of this model, a recent study has documented accumulation of SET1C and H3K4me3 at p300-associated enhancer elements upon mutation of the CFP1 CXXC domain that, through CpG interactions, normally restricts stable SET1C binding to promoters (Clouaire et al., 2012). Related, the exact origin of the p21/WAF1 enhancer-localized H3K4me1 mark remains unclear. One possibility is acetylated H3-dependent monomethylation by SET1C, with processivity restricted by other local factors. Another possibility, as discussed earlier, is monomethylation by a SET1C-related MLL complex such as MLL3/4 (Figure S7). Noteworthy in this regard are (1) our demonstration that p53- and p300-dependent H3K4 methylation of a chromatin template by the MLL3/4 complex results primarily in monomethylation, (2) our demonstration that p21/WAF1 enhancer monomethylation in doxorubicin-induced HTC116 cells, like the MLL3/4-mediated H3K4 monomethylation in vitro, is also dependent upon p300 (and potentially p300-ASH2L/MLLC interactions), and, as mentioned above, (3) a prior localization of MLL3/4C to the p21 enhancer in doxorubicin-treated MEFs (Lee et al., 2009). Thus, despite current uncertainty about the mechanisms involved in H3K4 monomethylation at the p21/WAF1 and other enhancers, our results add major insights into mechanisms that underlie p300-dependent H3K4 trimethylation by SET1C at the p21/WAF1 promoter and that may be relevant to H3 acetylation and H3K4 monomethylation at the enhancer.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
HCT116 cells were cultured, treated with doxorubicin, and transfected with siRNAs as described (Kim et al., 2009). Stable cell lines expressing Flagtagged SET1C subunits were established by standard methods detailed in the Extended Experimental Procedures.
Recombinant Proteins and SET1/MLL Complexes
Recombinant histones, chromatin-assembly factors, p53, and p300 were prepared as described (An et al., 2004). Methyl-lysine analogs in H3 were installed as described (Simon et al., 2007). Human SET1/MLL complexes were affinity purified as detailed in the Extended Experimental Procedures.
Recombinant Chromatin Assembly and HAT, HMT, and Transcription Assays
All assays were performed as previously described (An et al., 2004; Kim et al., 2009), and as outlined in Figure 3A, with modifications detailed in the Extended Experimental Procedures.
Protein Binding, Coimmunoprecipitation, ChIP, and RT-PCR Assays
Assays were performed as previously described (Kim et al., 2009), with modifications and relevant antibodies detailed in the Extended Experimental Procedures. The p53-SET1C crosslinking analysis with DSP is also detailed in the Extended Experimental Procedures.
Mass Spectrometry
H3K4 methylated products were analyzed by standard liquid chromatography-tandem MS (LC-MS/MS) as detailed in the Extended Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Ming Yu for advice on ChIP assays; laboratory members for discussions; Sohail Malik for critical reading of the manuscript; Wei Gu (Columbia University) for p53 7KR/8KR mutant plasmids; W. Lee Kraus (UT Southwestern) and James T. Kadonaga (UCSD) for the mutant p300 plasmids; Peter B. Becker (Adolf-Butenandt-Institute, Germany) for the ACF1 Δbromodomain plasmid; Christina Hughes (The Rockefeller University), Winship Herr (University of Lausanne), and David G. Skalnik (Indiana University) for antibodies; and Carlos Caldas (University of Cambridge) for p300 WT and null HCT116 cell lines. This work was supported by grants from the NIH (CA129325), the Leukemia and Lymphoma Society, the Ellison Medical Foundation, and the Starr Cancer Consortium to R.G.R.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Experimental Procedures, seven figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2013.06.027.
References
- An W, Roeder RG. Reconstitution and transcriptional analysis of chromatin in vitro. Methods Enzymol. 2004;377:460–474. doi: 10.1016/S0076-6879(03)77030-X. [DOI] [PubMed] [Google Scholar]
- An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Clouaire T, Webb S, Skene P, Illingworth R, Kerr A, Andrews R, Lee JH, Skalnik D, Bird A. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012;26:1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- Eissenberg JC, Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010;339:240–249. doi: 10.1016/j.ydbio.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett. 2012;586:2692–2704. doi: 10.1016/j.febslet.2012.04.045. [DOI] [PubMed] [Google Scholar]
- Goo YH, Sohn YC, Kim DH, Kim SW, Kang MJ, Jung DJ, Kwak E, Barlev NA, Berger SL, Chow VT, et al. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol Cell Biol. 2003;23:140–149. doi: 10.1128/MCB.23.1.140-149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Chang CC, Wortham M, Chen LH, Kernagis DN, Qin X, Cho YW, Chi JT, Grant GA, McLendon RE, et al. Global identification of MLL2-targeted loci reveals MLL2’s role in diverse signaling pathways. Proc Natl Acad Sci USA. 2012;109:17603–17608. doi: 10.1073/pnas.1208807109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan AH, Prochasson P, Neely KE, Galasinski SC, Chandy M, Carrozza MJ, Workman JL. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell. 2002;111:369–379. doi: 10.1016/s0092-8674(02)01005-x. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garruss AS, Liang K, Takahashi YH, Mickey K, Voets O, Verrijzer CP, Shilatifard A. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 2012;26:2604–2620. doi: 10.1101/gad.201327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NG, Chin SF, Ozdag H, Daigo Y, Hu DE, Cariati M, Brindle K, Aparicio S, Caldas C. p300 regulates p53-dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proc Natl Acad Sci USA. 2004;101:7386–7391. doi: 10.1073/pnas.0401002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong KW, Kim K, Situ AJ, Ulmer TS, An W, Stallcup MR. Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat Struct Mol Biol. 2011;18:1358–1365. doi: 10.1038/nsmb.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Shukla A, Wang X, Chen WY, Bernstein BE, Roeder RG. Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell. 2011;144:513–525. doi: 10.1016/j.cell.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY, Ge K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30:249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Guermah M, McGinty RK, Lee JS, Tang Z, Milne TA, Shilatifard A, Muir TW, Roeder RG. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell. 2009;137:459–471. doi: 10.1016/j.cell.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Guermah M, Roeder RG. The human PAF1 complex acts in chromatin transcription elongation both independently and cooperatively with SII/TFIIS. Cell. 2010;140:491–503. doi: 10.1016/j.cell.2009.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim JA, McGinty RK, Nguyen UT, Muir TW, Allis CD, Roeder RG. The n-SET domain of Set1 regulates H2B ubiquitylation-dependent H3K4 methylation. Mol Cell. 2013;49:1121–1133. doi: 10.1016/j.molcel.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kraus WL, Manning ET, Kadonaga JT. Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol Cell Biol. 1999;19:8123–8135. doi: 10.1128/mcb.19.12.8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, Roeder RG. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152:1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim DH, Lee S, Yang QH, Lee DK, Lee SK, Roeder RG, Lee JW. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc Natl Acad Sci USA. 2009;106:8513–8518. doi: 10.1073/pnas.0902873106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Garrett AS, Yen K, Takahashi YH, Hu D, Jackson J, Seidel C, Pugh BF, Shilatifard A. Codependency of H2B monoubiquitination and nucleosome reassembly on Chd1. Genes Dev. 2012;26:914–919. doi: 10.1101/gad.186841.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell. 2010;142:682–685. doi: 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Lehmann LW, Bonora G, Sridharan R, Vashisht AA, Tran N, Plath K, Wohlschlegel JA, Carey M. Mediator coordinates PIC assembly with recruitment of CHD1. Genes Dev. 2011;25:2198–2209. doi: 10.1101/gad.17554711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliori V, Müller J, Phalke S, Low D, Bezzi M, Mok WC, Sahu SK, Gunaratne J, Capasso P, Bassi C, et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol. 2012;19:136–144. doi: 10.1038/nsmb.2209. [DOI] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- Nightingale KP, Gendreizig S, White DA, Bradbury C, Hollfelder F, Turner BM. Cross-talk between histone modifications in response to histone deacetylase inhibitors: MLL4 links histone H3 acetylation and histone H3K4 methylation. J Biol Chem. 2007;282:4408–4416. doi: 10.1074/jbc.M606773200. [DOI] [PubMed] [Google Scholar]
- Patel DJ, Wang Z. Readout of epigenetic modifications. Annu Rev Biochem. 2013;82:81–118. doi: 10.1146/annurev-biochem-072711-165700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Santoso B, Kadonaga JT. Reconstitution of chromatin transcription with purified components reveals a chromatin-specific repressive activity of p300. Nat Struct Mol Biol. 2006;13:131–139. doi: 10.1038/nsmb1048. [DOI] [PubMed] [Google Scholar]
- Schuettengruber B, Martinez AM, Iovino N, Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol. 2011;12:799–814. doi: 10.1038/nrm3230. [DOI] [PubMed] [Google Scholar]
- Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, Shilatifard A. The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol Cell. 2010;40:689–701. doi: 10.1016/j.molcel.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu Rev Biochem. 2011;80:473–499. doi: 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- Vermeulen M, Timmers HT. Grasping trimethylation of histone H3 at lysine 4. Epigenomics. 2010;2:395–406. doi: 10.2217/epi.10.11. [DOI] [PubMed] [Google Scholar]
- Vethantham V, Yang Y, Bowman C, Asp P, Lee JH, Skalnik DG, Dynlacht BD. Dynamic loss of H2B ubiquitylation without corresponding changes in H3K4 trimethylation during myogenic differentiation. Mol Cell Biol. 2012;32:1044–1055. doi: 10.1128/MCB.06026-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the light: the Growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wang Z, Cui B, Gorovsky MA. Histone H2B ubiquitylation is not required for histone H3 methylation at lysine 4 in tetrahymena. J Biol Chem. 2009;284:34870–34879. doi: 10.1074/jbc.M109.046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Lee SY, Zhou B, Nguyen UT, Muir TW, Tan S, Dou Y. ASH2L regulates ubiquitylation signaling to MLL: trans-regulation of H3 K4 methylation in higher eukaryotes. Mol Cell. 2013;49:1108–1120. doi: 10.1016/j.molcel.2013.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.