

Abstract
In this paper we compare and contrast three approaches for labelling polymers with functional groups via ring-opening metathesis polymerization (ROMP). We explored the incorporation of functionality via initiation, termination and propagation employing an array of novel initiators, termination agents and monomers. The goal was to allow the generation of selectively labelled and well-defined polymers that would in turn lead to the formation of labelled nanomaterials. Norbornene analogues, prepared as functionalized monomers for ROMP, included fluorescent dyes (rhodamine, fluorescein, EDANS, and coumarin), quenchers (DABCYL), conjugatable moieties (NHS esters, pentafluorophenyl esters), and protected amines. In addition, a set of symmetrical olefins for terminally labelling polymers, and for the generation of initiators in situ is described.
Introduction
In the development of labelled polymers, and polymeric nanoparticles, one desires synthetic approaches that allow the most direct route to the incorporation of functional moieties with minimal post-polymerization modifications, and/or post-particle-formation conjugations1–17. The most desirable route is via the direct incorporation of functional groups during the polymerization process itself (i.e. as monomers, initiators or termination agents), preventing the need for subsequent low yielding and difficult to characterize graft-to reactions on macromolecules or at particle surfaces. Moreover, post-polymerization modifications and reactions on particle surfaces are difficult to control and yield unpredictably labelled materials. The problem is compounded for particles where chemical functionalities can be difficult to elucidate on nano- and microscale surfaces, and/or exceptionally difficult to reproduce. In our work, we have chosen to focus on a functional group tolerant living polymerization method precisely because of the multiple options available for directly incorporating complex functional groups18–21. The goal is to avoid the need for as many reactions performed post-polymerization as possible given the substrate scope of the chosen initiator. Where this is not possible, in the last resort, one desires a polymer or particle decorated with functional groups enabling standard, high yielding conjugation reactions. This is a common goal for those interested in functional nanoparticles capable of expressing some functionality on their shell and/or their core. Therefore, our aim in this paper is to elucidate the capability of ring-opening metathesis polymerization (ROMP) with respect to the incorporation of several classes of functional moiety, representing a range of generally desirable properties. ROMP was chosen as it is an important and useful polymerization method for the generation of well-defined polymers of low polydispersity and highly functionalized architecture 3, 16, 17, 22. Multiple initiators are commercially available23–26 and can exhibit good stability in ambient conditions making them generally accessible25–29. These properties make ROMP particularly amenable to producing specialized functional polymers for synthetic, biomedical and nanomaterials applications, especially where complex copolymers generated via direct polymerization of functional groups are desirable3, 16, 17, 20, 21, 30–34. There are three opportunities to introduce functionality into polymers via ROMP, (1) the use of an initiator containing a functional alkylidene (Figure 1 - ii), (2) the use of strained olefin-based monomers containing various functionalities (Figure 1 - i, iii), and (3) the use of functionalized termination (or chain transfer) agents (Figure 1 - iv)35. The most popular and easily deployed method for preparing functional polymers is through the use of monomers that either contain the desired functionality or allow for its incorporation via a post-polymerization modification16, 17, 35. In addition, the use of functionalized termination agents that allow end-labelling of polymers has garnered increasing attention30, 36–43. By contrast, specially functionalized initiators are underutilized5, 26, 44–46, most likely because changes to the initiator structure can result in changes to the initiation and propagation rates and hence overall polymer quality and predictability in synthesis. Herein, each of these approaches are assessed and utilized for the preparation of micellar nanoparticles assembled from fluorescently labelled amphiphilic block copolymers.
Fig. 1.

General scheme for the synthesis of functionalized polymers via ROMP. Monomers (i, iii), initiators (ii) and termination agents (iv) containing functional groups can be used to synthesize labeled amphiphilic polymers that assemble into labeled micelles.
RESULTS AND DISCUSSION
Polymer labelling via ROM/ROMP
Labelling studies were initially conducted by employing the most convenient approach; namely, the incorporation of dye-modified monomers by doping them in small quantities together with nonfunctional monomers. This procedure has been used via ROMP, for introducing small quantities of functional groups into polymers as tags, without dominating polymer structure47. To produce end-functionalized polymers, with incorporation of minimal quantities of dye label, we sought to begin polymerization reactions with a 1:1 ratio of monomer to initiator (M:I), followed by addition of a second monomer (in appropriate stoichiometric excess with respect to initiator) for chain extension (Figure 2). We term this procedure, ROM/ROMP, for ring-opening metathesis followed by ROMP. This procedure was planned, as postulated by Sampson and coworkers, as a facile approach to polymer tagging47. To examine the viability of this method, norbornene monomers 1 (N-hydroxysuccinimide (NHS) ester) and 2 (a Boc-protected amine) were utilized as they contain functional groups amenable to subsequent conjugation reactions. The first step was to verify the ability of these monomers to undergo ROMP as determined by 1H NMR. The disappearance of the norbornene olefin at ~6.3 ppm was monitored at various time points after addition of the catalyst (M:I = 20:1). Both monomers 1 and 2 show complete conversion to polymer after 4 minutes (Supporting Information). For the labelling experiment, 1 was mixed with either initiator I or II and allowed to stir for 20 min to insure complete conversion to the ring opened form (Figure 2, step i). Subsequently, the reaction was quenched by addition of an excess of ethyl vinyl ether (Figure 2 step ii). The ring-opened product corresponding to a single monomer was isolated by column chromatography and characterized by NMR and mass spectrometry (Table 1 entries 1 and 2). The olefin region of the proton NMR shows the presence of multiple isomers consistent with the splitting pattern expected (Supporting Information) and an observed 1:1 ratio of backbone olefin protons to phenyl protons. When norbornene monomer 2 was subjected to the same experimental conditions, similar results were obtained, as shown in Table 1 entries 3 and 4. As an additional confirmation of our characterization of the single ring-opened product, we subjected monomers 1 and 2 to conditions favouring the cross-metathesis reaction. This consisted of 5 mol % initiator I or II with respect to monomer, and 10 equivalents of styrene. The isolated products gave NMR and mass spectrometry data that are consistent with those obtained for material isolated after quenching a 1:1 reaction of monomer to initiator with ethyl vinyl ether (Supporting Information).
Fig. 2.

General scheme for end-functionalized polymer synthesis via ROM/ROMP. (i) One equivalent of monomer is added to the Ru(IV) initiator. (ii) The ring-opened product is isolable following addition of ethyl vinyl ether. (iii) End-functionalized polymers result from the addition of m equivalents of a second monomer, followed by termination with ethyl vinyl ether (iv).
Table 1.
Results for the single opening of monomers 1 and 2
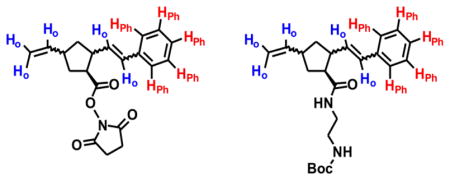
| ||||
|---|---|---|---|---|
| Entry | Mon | Initiator | Mass | Ho/HPh |
| 1 | 1 | I | 357.00 [M+NH4]+ | 1.18 |
| 2 | 1 | II | 356.93 [M+NH4]+ | 0.9 |
| 3 | 2 | I | 384.93 [M+H]+ | 0.9 |
| 4 | 2 | II | 384.81 [M+H]+ | 1.05 |
Having successfully demonstrated the ability to generate the single ring-opened species, we next focused on demonstrating that these reaction conditions could be employed to synthesize end-functionalized polymers by ROM/ROMP. Monomers 1 and 2 were again used as test cases for this strategy. A 1:1 reaction mixture of monomer to initiator was employed to generate a living species (Figure 2 - i) followed by the addition of monomer 3 (m equivalents) and subsequent quenching of the polymerization by addition of excess ethyl vinyl ether (Figure 2 steps iii and iv). This procedure gave polymers (e.g. 12 and 16, Table 2) with low PDI and expected molecular weights as characterized by 1H-NMR, MALDI-MS (where possible), and size-exclusion chromatography using a multiangle light scattering detector (SEC-MALS). To explore the generality of this tagging approach via ROM/ROMP, we studied norbornene monomers with functionalities generally considered useful in subsequent conjugation reactions, including maleimide (4), hydroxylamine precursor (6) and protected amine (11). Furthermore, several dyes for labelling polymers were used, consisting of coumarin (5), DABCYL (7), fluorescein (8), and EDANS (9). Finally, the simple hydrophilic moiety (10) was used, which is subsequently useful in micellar nanoparticle formation (vide infra). In each case, polymerizable norbornene moieties were used as derived from commercially available cis-5-norbornene-exo-2,3-dicarboxylic anhydride or 5-norbornene-2-carboxylic acid (resolved prior to use to give exo-5-norbornene-2-carboxylic acid)48. For ROM/ROMP of each functional monomer, 3 was used as the monomer included for chain extension. Conveniently, 1H-NMR made it possible to determine the ratio of either the phenyl or benzyl protons from 3 with respect to distinct proton signals for the single ring-opened monomers (unless otherwise noted in Table 2). In this manner, the number of each monomer incorporated could be estimated by applying that ratio to Mw values obtained from SEC-MALS.
Table 2.
Terminally functionalized polymers synthesized by ROM/ROMP of combinations of three different monomers indicated here as “Mon1” with “Mon2”, and/or “Mon3”.
| Entry | Polymer | Initiator | Mon1 (ratio to initiator) | Mon2 (ratio to initiator) | Mon3 (ratio to initiator) | Ratio by 1H NMR (Mon1: Mon2: Mon3)a,b | Theoret. Mn (g/mol) | Obs. Mw (g/mol)c | Obs. Mn (g/mol)d | PDIc | % Areae | % Yieldf |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 12 | II | 1 (1:1) | 3 (20:1) | - | nd | 5417 | 6353 | - | 1.01 | 100 | 84 |
| B | 13 | II | 1 (1:1) | 3 (40:1) | - | nd | 10497 | 11440 | - | 1.00 | 100 | 99 |
| C | 14 | II | 1 (1:1) | 3 (60:1) | - | nd | 15577 | 18020 | - | 1.00 | 100 | 90 |
| D | 15 | II | 1 (1:1) | 3 (6:1) | - | nd | 1861 | 2310 | - | 1.04 | 100 | 71 |
| E | 16 | II | 2 (1:1) | 3 (10:1) | - | (1.0: 16.0: 0) | 2921 | 3750 | 4268 | 1.02 | 100 | 67 |
| F | 17 | I | 2 (1:1) | 3 (10:1) | - | (1.0: 12.0: 0) | 2921 | 4838 | 4516 | 1.04 | 99 | 75 |
| G | 18 | I | 2 (1:1) | 3 (10:1) | 5 (1:1) | (1.0: 12.0: 2.3) | 3203 | 5734 | - | 1.06 | 99 | 95 |
| H | 19 | I | 2 (1:1) | 3 (10:1) | 2 (1:1) | (1.0: 12.0: 1.2) | 3201 | 4961 | - | 1.04 | 99 | 70 |
| I | 20 | I | 2 (1:1) | 3 (30:1) | - | (1.0: 36.0) | 7960 | 9571 | - | 1.04 | 100 | 76 |
| J | 21 | I | 4 (1:1) | 3 (10:1) | - | nd | 2844 | 6127 | 4265 | 1.14 | 97 | 88 |
| K | 22 | II | 4 (1:1) | 3 (10:1) | - | nd | 2844 | 6884 | - | 1.02 | 75 | 88 |
| L | 23 | II | 5 (1:1) | 3 (10:1) | - | (1.2: 11.0) | 2923 | 3133 | 3251 | 1.04 | 100 | 83 |
| M | 24 | II | 5 (1:1) | 3 (15:1) | - | (1.0: 11.0) | 4192 | 4000 | 4264 | 1.02 | 100 | 75 |
| N | 25 | II | 5 (1:1) | 3 (15:1) | 5 (1:1) | (1.0: 11.0: 2.7) | 4475 | 4100 | 4517 | 1.08 | 100 | 85 |
| O | 26 | I | 6 (1:1) | 3 (30:1) | 1 (1:1) | (n.d.:38: 0.7) | 8268 | 9906 | - | 1.10 | 100 | 74 |
| P | 27 | I | 6 (1:1) | 3 (25:1) | 5 (1:1) | n.d. | 7049 | 10320 | - | 1.09 | 97 | 99 |
| Q | 28 | II | 6 (1:1) | 3 (25:1) | 5 (1:1) | (n.d.: 25: 1) | 7049 | 6875 | - | 1.01 | 100 | 97 |
| R | 29 | I | 6 (1:1) | 3 (25:1) | 6 (1:1) | (n.d.: 8.0: 1.0) | 7120 | 7850 | - | 1.07 | 95 | 82 |
| S | 30 | II | 6 (1:1) | 3 (25:1) | 6 (1:1) | (n.d.: 22: 1.0) | 7119 | 6309 | - | 1.00 | 100 | 90 |
| T | 31 | II | 7 (1:1) | 3 (10:1) | - | (1.0: 13.0) | 3015 | 2770 | - | 1.05 | 71 | 72 |
| U | 32 | II | 8 (1:1) | 3 (20:1) | - | (1.0: 24.0) | 5915 | 5715 | 5023 | 1.04 | 100 | 74 |
Determined from the ratio of Mon1 to Mon2 protons, Mon3 to Mon 2 protons or Mon1 to Mon3 protons.
Delay time increased to 10 sec to ensure complete relaxation.
Determined by SEC-MALS.
Determined by MALDI mass spectrometry where possible.
Area of largest peak in SEC-MALS trace.
Isolated yield. (n.d. = not determinable).
Entries A–D in Table 2 show polymers of different length synthesized using NHS monomer 1 as the short label or tag. Due to overlapping signals in the NMR spectra the ratio of NHS to Ph groups could not be obtained. Entries E–I correspond to polymers synthesized using protected amine 2. Polymers 17–19 (entries F–H) were synthesized from a single reaction mixture by splitting the solution into three equal portions and either terminating the reaction with ethyl vinyl ether (EVE) or adding monomer 2 or 5 followed by EVE. The ratio of monomers incorporated in these systems could be determined using 1H-NMR by integration of the phenyl or benzyl protons to the protons of the Boc protecting group (for 2) or one of the aromatic protons of coumarin (for 5) (See Supplemental Information). Entry I corresponds to a system where we increased the number of equivalents of monomer 3 to generate a larger polymer. Entries J and K correspond to polymers made starting with maleimide 4 using initiator I or II. Low signal intensity precluded the determination of monomer ratios by NMR for these systems. Entries L–N correspond to polymers synthesized starting with coumarin monomer 5, with entries M and N being synthesized from a single reaction mixture by splitting the solution into two equal portions and either terminating the reaction with ethyl vinyl ether (EVE) or adding coumarin monomer 5 followed by EVE to give the doubly end-labelled polymer. The ratio of monomers incorporated in these systems was determined using 1H-NMR by integration of the benzyl protons of 3 to one of the aromatic protons of 5. In the case of polymer 25 the integral for the benzyl protons was set to the same value as observed in polymer 24. The change in integration of the coumarin proton signal was then determined to give the number of monomers incorporated at the terminal end of the polymer. Entries O–S corresponds to polymers synthesized starting with hydroxylamine precursor 6 and either initiator I or II. In all cases doubly end-labelled polymers were synthesized including telechelic systems incorporating conjugatable monomers 1, 2, and 6. The ratio of monomers incorporated in these systems was determined using 1H-NMR. Entry T corresponds to a polymer synthesized starting with DABCYL monomer 7. The ratio of monomers incorporated in this polymer was determined using 1H-NMR by integration of the phenyl protons of 3 to one of the aromatic protons of 7. Finally, entry U corresponds to a polymer synthesized starting with fluorescein monomer 8. The ratio of monomers incorporated in this polymer was determined using 1H-NMR by integration of the phenyl protons of 3 to the protons of the pivilac protecting group of 8.
In addition to the low PDI measured for each polymer, the observed Mw showed expected deviation from theoretically calculated values based on monomer to initiator ratios, within the range commonly observed for ROMP. Mn was measured where possible by MALDI-TOF mass spectrometry and was in good agreement with Mw and Mn determined by SEC-MALS (e.g. Figure 4). The MALDI-TOF spectra also allowed us to assess the effectiveness of this single monomer opening strategy as the incorporation of a single monomer would result in a single mass distribution curve with peak spacing equal to the molecular weight of 3 (253.11 g/mol). The presence of multiple distributions in the spectra with spacing equal to the molecular weight of 5 (282.09 g/mol) indicates the incorporation of more than one functional monomer distributed throughout the population. The number of signals between two adjacent high intensity peaks corresponds to the number of different functional monomers incorporated (Figure 4). As expected the distribution of functional monomers incorporated in the polymers is dependent on the monomer itself, most likely due to differences in the rate of initiation. The number of incorporated monomers ranges from 1 to 5 with varying distributions depending on the polymer (polymer 23 contains 66% of the singly tagged species) (Supporting information). This combination of data supports the conclusion that it is of course not possible to truly tag each polymer with a single monomer via this method. Rather, it provides a facile method for including close to one equivalent of functional moiety dispersed throughout the population.
Fig. 4.

MALDI MS of polymer 23 showing the polydispersity and revealing the efficiency of the single monomer tagging strategy (3, Mw = 253.11 g/mol, 5 Mw = 282.09 g/mol).
Exceptions to the generally well-defined polymers shown in Table 2 were observed for polymers using maleimide monomer 4 as well as dye monomer 7. These polymerization reactions gave small amounts of high molecular weight products in addition to polymers of the desired molecular weight (see Supporting information). In addition, the higher polydispersities arising from utilizing these monomers in ROM/ROMP is consistent with our observations for corresponding homopolymers of 4 (Supporting information).
The spectrophotometric properties of dye monomers such as 5 were used as a handle for characterizing the degree of incorporation into the population. For example, monitoring the UV absorption at 325 nm allows one to observe the coumarin moiety present in polymers 18, 23–25, 27, and 28. To utilize this feature in the analysis of end-functionalized polymers, a telechelic polymer containing a protected amine group at one end, and a coumarin moiety at the other (18), was synthesized by splitting off an aliquot of 17 prior to quenching, for addition to 5. The SEC traces showing light scattering (LS), refractive index (RI) (Figure 5A) and UV intensities (Figure 5B) are given for polymers 17 and 18. The plot of intensity vs. time for light scattering and differential refractive index (Figure 5C) shows similar traces for 17 and 18, however there is a dramatic increase in the absorption at 325 nm for entry 18 (Figure 5B), consistent with the incorporation of coumarin monomer 5. Based on Mw values derived from SEC-MALS analysis and UV absorption, the number of coumarin moieties in the polymer could be calculated. For example, using UV absorption at 325 nm for entries G, L–N, P, and Q (Table 2) the number of coumarin moieties incorporated into the polymers was calculated to be 3.60 ± 0.13, 1.02 ± 0.02, 1.09 ± 0.08, 1.81 ± 0.18, 1.02, and 1.00, respectively (Supporting Information).
Fig. 5.

SEC traces and fluorescence spectra for polymers 17, 18, 12, and 32. (A) SEC with scattering intensity and refractive index for 17 and 18. (B) SEC with UV absorbance for 17 and 18. (C) SEC with scattering intensity and refractive index for 12 and 12 + 7-hydroxycoumarin. (D) UV traces of polymer 12 before and after conjugation with 4-hydroxycoumarin. (E) SEC-MALS of polymer 32 and (F) fluorescence spectra of polymer 32 before and after removal of the pivilac protecting groups by treatment with base.
In addition to the polymerization of coumarin monomer 5 for tagging polymers, we assessed the efficiency of post-polymerization modification of polymers containing NHS ester functionalities. To achieve this polymer 12 was treated with 5 equivalents of 7-hydroxycoumarin and 10 equivalents DIPEA in CH2Cl2 for 18 hrs. After purification via repeated precipitations from dichloromethane by addition to cold methanol, the polymer was analyzed by SEC-MALS (Figure 5C). An increase in UV absorption at 325 nm is again noted for the polymer treated with coumarin when compared to the original polymer (Figure 5D). A coupling efficiency of only 13% was obtained using this post-polymerization modification reaction. This highlights the problem with relying on post polymerization modifications to install functionality as rigorous optimization of reaction conditions and or conjugatable groups may be required to obtain a high level of conjugation. In addition to our coumarin studies, protected fluorescein monomer 8 was used to synthesize polymer 32 (Table 2, with the SEC-MALS trace for polymer 32 given in Figure 5E). Deprotection was facilitated by treatment of the polymer with a solution of ammonium hydroxide in DMF. Upon addition of base, the colour of the solution turned yellow to the eye, signifying the presence of the fluorescein moiety. Fluorescence spectra of 32 before and after deprotection are shown in Figure 5F and indicate a large increase in fluorescence after treatment with base confirming the incorporation of the fluorescein monomer. Using UV absorption at 510 nm, the number of fluorescein units incorporated in polymer 32 was calculated to be 1.11 ± 0.15 (Supporting Information).
Notably, polymer, 32, demonstrated that labels could be incorporated on the end of the chain in small quantities. Therefore, we next aimed to prepare two polymers that were identical except for the dye used as the end-label. Specifically, we aimed to determine if amphiphilic block copolymers made in this fashion could form labelled micelles incorporating different dyes, but with equivalent morphologies in their final state. This would imply that one could use this tagging strategy to routinely label polymers and particles with different groups, without greatly influencing their overall properties with respect to particle formation. To examine this, we prepared a set of amphiphilic polymers containing either a tag of EDANS or DABCYL (Table 3, Figure 6). A block copolymer of structure, 391-b-106 was prepared, and then split into two equal parts while the polymer was still living. To these two solutions was added one equivalent of 7 or 9. The resulting polymers 33 and 34 were formulated separately into two particles, P1 and P2 via dialysis from DMSO into water49. In addition, they were mixed together in a 1:1 ratio in DMSO and slowly transitioned from organic solvent into water to form a fluorescently quenched particle (P3). The newly formed micellar aggregates were characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM) (Table 3, Figure 6). As revealed by TEM, P1, P2, and P3 each exhibit a mixed phase of spherical and cylindrical micelles, but were dominated by cylindrical micelles of similar dimensions. The spectral properties of fluorescently labelled aggregates P1–P3 were analyzed by exciting each of the structures at 335 nm. As shown in Figure 6D, the characteristic EDANS emission maximum is observed at 450 nm for P1. However, P3, containing both donor (EDANS) and quencher (DABCYL) shows a significant decrease in fluorescence consistent with these dyes now interacting within the Förster radius50. Indeed, it can be concluded that the EDANS and DABCYL in P3 must be within 33 Å of one another51. Most notably, the consistency in overall morphology is extremely promising with respect to deploying this tagging approach without subsequently perturbing the formation of polymeric nanoparticles.
Table 3.
Summary of EDANS and DABCYL labelled ROMP polymers and particle characterization.
| Polymer (Particle)a | Mon1 (DP)b | Mon2 (DP)b | Mon3 (DP)b | Mn (g/mol) | Mw/Mn | Dhc (nm) | PDId |
|---|---|---|---|---|---|---|---|
| 33 (P1) | 3 (91) | 10 (6) | 9 (2) | 10540 | 1.038 | 255 | 0.308 |
| 34 (P2) | 3 (91) | 10 (6) | 7 (4) | 10480 | 1.103 | 164 | 0.265 |
| 33 + 34 (P3) | -e | - | - | - | - | 295 | 0.285 |
Subsequent particles formed from amphiphilic polymer 33, 34, or 33 + 34 is shown in parenthesis (i.e. P#).
Degree of polymerization (DP) of Monx.
Hydrodynamic diameter Dh (uncorrected) of micelles formed from polymers 33, 34, or 33 + 34 as determined by dynamic light scattering (DLS).
Polydispersity (PDI) as determined by DLS.
See polymers 33 and 34 for polymer characterization.
Fig. 6.

Transmission electron micrographs (TEM) and fluorescent spectra of P1, P2, and P3. A) TEM of P1, B) TEM of P2, C) TEM of P3, (1% uranyl acetate stain) and D) Fluorescent emission spectrum of P1 (54.9 μM), P2 (12.5 μM), and P3 (54.9 μM EDANS, 12.5 μM DABCYL) with an excitation of 335 nm.
Polymer labelling via termination of ROMP
While we have successfully shown in the previous section, that a 1:1 ratio of monomer to initiator is effective for incorporating tags of functional monomers on to the ends of polymers, it is desirable to have a more robust approach where one can more reliably incorporate a single functional unit onto each polymer. To facilitate this, we synthesized a number of termination agents (35–43) that incorporate dyes, conjugatable groups, and hydrophilic groups (Figure 7). These termination agents (TAs) were synthesized from cis-1,4-dichloro-2-butene by nucleophilic substitution reactions (Supporting Information). To develop these novel agents for use by us and others, the efficiency of termination was determined using a 1H-NMR assay. We postulated that the 1H-NMR signal corresponding to the alkylidene moiety would be useful for monitoring the termination (cross metathesis) process (Figure 8). As an initial test of this idea, we chose to evaluate the pentafluorophenol (PFP) termination agent (PFP-TA) 36. 1H-NMR spectra were recorded for the free initiator (Figure 8A-i, 8B-i) prior to the addition of monomer 11, and of the living polymer 20 min after the addition of 11 (Figure 8A-ii, 8B-ii) The living polymer was then split into two pots, with either a large excess of ethyl vinyl ether added to quench the reaction and generate ruthenium species iii (Figure 8A-iii, 8B-iii) or 2 equivalents of 36 added to the reaction to generate species iv (Figure 8A-iv). Spectra were recorded 30 min and 60 min after addition of 36 (Figure 8B-iv). A large excess of ethyl vinyl ether was added to the PFP-TA sample to generate ruthenium species iii (Figure 8A-iii′), and after 20 min a spectrum recorded (Figure 8B-iii′). As shown in Figure 8B, the chemical shift of the alkylidene proton of each species has a distinct chemical shift and could be used to monitor the efficiency of termination. Addition of 36 results in a change in the chemical shift of the alkylidene proton from 18.5 ppm to 19.0 ppm (Figure 8B-ii and 8B-iv respectively). After 60 minutes, complete conversion of living polymer (ii) to terminated polymer (iv) was observed. Addition of ethyl vinyl ether results in the conversion of complex (iv) to (iii′) which is obtained directly by addition of ethyl vinyl ether to living polymer ii (Figure 8A-iii). The efficiencies for all terminations agents were determined to be quantitative via this method except for 41 (DABCYL) (Figure 8C). This was attributed to the relatively poor solubility of 41 which begins to precipitate from solution after 30 minutes. Increasing the length of time for termination and changing the solvent to DMF did not increase the efficiency of termination.
Fig. 7.
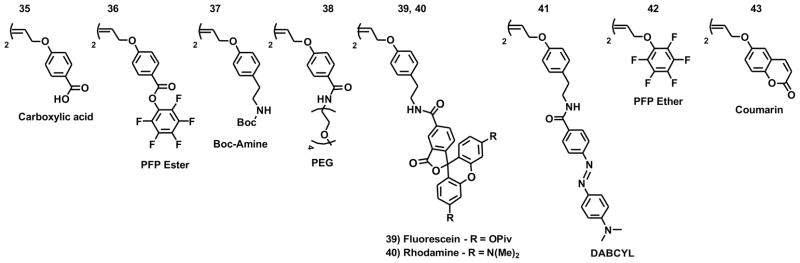
Structures of the symmetrical termination agents (TA) synthesized.
Fig. 8.

Termination agent efficiency for 35–43. A) Scheme outlining termination agent efficiency experiment. B) 1H NMR spectra of the alkylidine proton of the molecules shown in A for TA 36. i) Catalyst, ii) Living polymer, iii) Terminated catalyst with ethyl vinyl ether, iv catalyst terminated with functional termination agent, iii′) After quenching the reaction with ethyl vinyl ether. c) 1H NMR spectra of the alkylidine proton on polymers treated with TAs 35–43.Bottom (black) spectra corresponds to the living polymer, Top spectra (red) corresponds to polymers after termination with TA’s 35–43.
As a demonstration of the utility of the tagging with functional termination agents, we synthesized a set of amphiphilic block copolymers end-labelled with dyes and formulated them into micellar nanoparticles. An amphiphilic block copolymer 341-b-1123 was synthesized using initiator II, and split into 4 portions while living, followed by termination with 39, 40, 41, or ethyl vinyl ether. Similar to the study performed as described for amphiphilic polymers 33 and 34 generated via the ROM/ROMP process, we chose dye TA combinations known to operate either as a quencher (DABCYL, 41) or as a FRET acceptor (rhodamine, 40) of the donor, fluorescein (39). Polymers 44–47 were characterized by SEC-MALS (Figure 9A) displaying a major peak at approx. 22 min for all four polymers. Polymers 46 and 47 display a detectable peak due to a higher molecular weight species (shoulder at earlier retention times) prevalent in the light scattering data. We postulate this is due to some aggregation or only due to small quantities of material as the RI signal is weak (Figure 9B) at 20 min retention times in comparison with the major peak observed at 22 min. Indeed, percent by mass characterization by SEC-MALS indicates the peak at 22 min contains >99% of the material with a low polydispersity between 1.01 – 1.09 (Table 4).
Fig. 9.
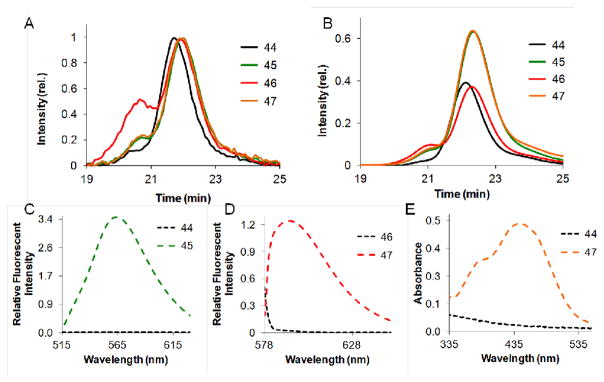
SEC and spectral characterization for polymers 44–47. A) SEC trace and scattering intensity of polymers 44–47. B) SEC trace and refractive index for polymers 44–47. C) Fluorescent emission spectra of polymers 44 and 45 with an excitation of 470 nm. D) Fluorescent emission spectra of polymers 45 and 47 with an excitation of 543 nm. E) UV-VIS of polymers 44 and 47.
Table 4.
Summary of end-group (EG) functionalized polymers synthesized by ROMP of 3 with 10 and terminated with TAs 39–41
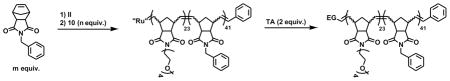
| ||||
|---|---|---|---|---|
| Polymer | TA | Degree of polymerization of 3 and 13 in the polymer as determined by SEC-MALS | Mn (g/mol)b | Mw/Mnb,c |
| 44 | EVEa | 41, 23 | 18460 | 1.01 |
| 45 | 39 | 41, 23 | 18730 | 1.03 |
| 46 | 40 | 41, 23 | 21550 | 1.09 |
| 47 | 41 | 41, 23 | 18660 | 1.04 |
Polymer terminated with ethyl vinyl ether (EVE).
Determined by SEC-MALS.
Determined from the low molecular weight species only (< 99% of sample).
To further confirm the incorporation of the dye-labelled termination agents, fluorescence and UV-VIS spectra of polymers 44–47 were taken (Figure 9C–E). The emission spectra of fluorescein were compared to polymer 44 (terminated with EVE) by exciting both polymers 44 and 45 at 470 nm in DMF and monitoring the fluorescence intensity at 563 nm. As shown in Figure 9C, there is no detectable peak at 563 nm for polymer 44 as expected, with a significant increase with fluorescein incorporated. Similarly, 46 was excited at 543 nm and the fluorescence intensity monitored at 592 nm, showing observable fluorescence with rhodamine present, compared to 44. Lastly, the DABCYL labelled polymer 47, was examined via UV spectroscopy and compared to 44. As expected, no significant absorbance was visible at 444 nm for polymer 44, however polymer 47 containing the DABCYL TA, shows a characteristic absorbance at 444 nm in DMF (Supporting Information).
Following this initial characterization, dye-labelled block copolymers 44–47 were formulated into particles via slow transition into water by dropwise addition of water to a 1 mg/mL solution of polymer in DMSO followed by dialysis against water49. These dye combinations were chosen because fluorescein is a donor for both rhodamine and DABCYL and can be used to study the assembly properties of resulting nanoparticles made up of given ratios of donor to acceptor carrying polymers11, 52–59. Initially, each of the individual polymers were dialyzed from DMSO into water (to generate P4 – P7) and were analyzed by DLS and TEM (Figure 10A–D and Table 5). To account for any possible self-quenching (arising from fluorescein), a mixed particle (P8) containing the unlabeled 44 and fluorescein-labelled 45 was prepared (Figure 10E). Mixed dye particles P9 and P10 were prepared by dialysis by diluting the acceptor into the donor in a co-solvent (DMSO) such that the amount of acceptor was higher than that of the donor (Supporting Information). It was determined by UV spectroscopy that P9 contained a 1:9 ratio of donor-to-acceptor and P10 contained a 1:1.5 ratio of fluorescein-to-DABCYL. All particles P4 – P10 were characterized by TEM and DLS (Table 5 and Figure 10A–G). Each of these particles is spherical in morphology, although some are spherical micelles, while others are larger, and are likely vesicular (e.g. multilamellar vesicles). This set of nanoparticles were then analyzed by fluorescence spectroscopy to characterize the fluorescence properties of the singly labelled particles and mixed nanoparticles. First, a fluorescence emission spectrum was measured for P5–P7 while exciting at 470 nm to determine if indeed no significant fluorescence intensity was visible without fluorescein (Figure 10H). Figure 10I shows the result of exciting P8 at 470 nm, exhibiting a characteristic fluorescein maximum intensity emission at 522 nm. In mixed particles P9 and P10 a decrease in the overall fluorescein emission intensity is apparent due to the presence of acceptor. A decrease in the fluorescein emission intensity and an increase in rhodamine fluorescence at 569 nm are observed for P9, indicating that the donor and acceptor must be within the Förster radius. In the case of P10, it illustrates the principle by completely quenching the fluorescein signal by DABCYL, again indicating that the fluorescein and quencher are in communication.
Fig. 10.

TEM and fluorescent data of nanoparticles made of A) P4, B) P5, C) P6, D) P7, E) P8 F) P9, G) P10 (1% uranyl acetate stain), H) fluorescence emission spectra of P5 (4.15 μM), P6 (36.7 μM), and P7 (6.19 μM), λex = 470 nm, and I) fluorescence emission spectra of P8 (4.15 μM), P9 (4.15 μM fluorescein, 37.7 μM rhodamine), and P10 (4.15 μM fluorescein, 37.7 μM DABCYL), λex = 470 nm
Table 5.
Summary of particles synthesized from polymers 44–47
| Particle | Compositiona | EG(s)b | Dhc (nm) | PDId |
|---|---|---|---|---|
| P4 | 44 | EVE | 146.6 | 0.02489 |
| P5 | 45 | Fluorescein | 314.4 | 0.01418 |
| P6 | 46 | Rhodamine | 312.6 | 0.05280 |
| P7 | 47 | DABCYL | 111.5 | 0.08099 |
| P8 | 44 + 45 | EVE, Fluorescein | 442.1 | 0.01482 |
| P9e | 45 + 46 | Fluorescein, Rhodamine | 498.8 | 0.02155 |
| P10 | 45 + 47 | Fluorescein, DABCYL | 204.2 | 0.03774 |
| P11f | 45 + 46 | Fluorescein, Rhodamine | 450.3 | 0.1109 |
Polymer(s) used in the formulation of the particles P4–P11.
End group of the polymer after termination.
Hydrodynamic diameter (uncorrected)as determined by DLS.
Polydispersity index (PDI) as determined by DLS.
Particle used for CMC measurements.
Particle used for fluorescence lifetime measurements.
Although we previously have used this type of donor/acceptor interaction in determining the upper limit of the critical aggregation concentration (CAC)58, we were unable to do so for these systems as the detection limit of fluorescence is reached before a loss in FRET-signal intensity is observed (Supporting Information). Other common methods used to determine this concentration, including the use of solvochromatic dyes such as pyrene to observe successful micellarization60–65 have not yet met with considerable success in our hands for these ROMP-based micelles, possibly due to their high stability.
In addition to determining particle stability, we sought to utilize the direct labelling strategy to elucidate structural features of the particles. The donor–acceptor (D–A) distance distribution was determined by analyzing the time-domain intensity decay of the donor (Figure 11A). Therefore, the data was fit as a summation of donor decays for all accessible D–A distances (Figure 11A). The mean distance between fluorophores, r, was determined by fluorescence lifetime and plotted as a probability function (Figure 11B–C) for both P10 and P11. For P10 (particle radius from TEM = 50 nm), the mean distance between donor and acceptor is, r = 6.57 ± 0.14 nm, which was then converted to a decay lifetime τDA = 2.66 ± 0.23 ns (Supporting Information). In the case of P11 (particle radius from TEM = 75 nm), the mean distance between donor and acceptor is, r = 6.55 ± 0.08 nm, and τDA = 2.23 ± 0.13 ns. This change in lifetime indicates that the fluorophores are interacting with one another, in positions distributed over the surface of the particles within the Förster radius.
Fig. 11.

Time-domain fluorescence lifetime analysis of P10 and P11 and distance distribution of dyes on nanoparticle surfaces. (A) Fluorescence lifetime of P5, P10, and P11 giving τD = 3.01 ± 0.22 ns for unquenched fluorescein, P5. (B–C) A range of distances, determined from lifetime data, between donor and acceptor in P10 and P11 are plotted and expressed as a probability function P(r). (B) The mean distance between fluorescein and rhodamine in P11, r = 6.55 ± 0.08 nm with decay lifetime τDA = 2.23 ± 0.13 ns. (C) The mean distance between fluorescein and DABCYL in P10, r = 6.57 ± 0.14 nm, giving τDA = 2.66 ± 0.23 ns. (D) Postulated schematic of the relationship in space between donor and acceptor (r) for a bilayer (vesicular/multilamellar) or spherical arrangement left and right respectively.
Polymer labelling via initiation of ROMP
As noted in the introduction, the most underutilized strategy for labelling polymers via ROMP has been the use of novel initiators5, 26, 44–46. We take inspiration here from a method demonstrated by Grubbs and co-workers that allows the generation of multiple polymer batches from a single catalyst through a recycling process66. The strategy relies on the difference in reactivity between the olefin termination agent and the olefin of the strained norbornene67. Initially, the initiator and a chain transfer reagent were stirred together for 20 min to generate the cross-metathesis product (i.e. new functional initiator). Utilizing this approach, addition of a norbornene monomer followed by quenching with ethyl vinyl ether results in polymers with a single functional group installed via the initiator rather than by termination of the reaction (Figure 12). This strategy allows for the single incorporation of two different functional groups at each end of the polymer if a functional TA is used (telechelic polymers). Here, the fluorescein termination agent 39 was used to demonstrate the viability of this method. TA 39 (2 equiv.) was stirred with initiator II for 20 minutes followed by addition of monomer 3 (25 equiv.). After 3 minutes the reaction was split in two and quenched with either ethyl vinyl ether or the Boc-protected amine TA 37 (10 equiv.). The latter was quenched with ethyl vinyl ether after 30 minutes to ensure complete termination of the reaction and quenching of the initiator. The polymers were purified by repeated precipitation from cold methanol until pure as indicated by thin-layer chromatography on silica. 1H NMR of the two resulting polymers shows the presence of both the fluorescein and Boc-amine moieties in addition to the phenyl moiety of monomer 3. Integration of the t-Butyl group for Pivalic protected fluorescein to the phenyl protons of monomer 3 and the, O-t-Butyl for the Boc-amine gives a ratio of 1: 30: 0 for the EVE terminated polymer and a ratio of 0.8:30: 1 for Boc-protected amine terminated polymer. The decrease in the number of Pivalic groups compared to the number of phenyl groups demonstrates the limitation of integration as complete monomer conversion was observed before splitting the reaction mixture for termination as this ratio should be constant in both polymers. However, molecular weights determined by SEC-MALS are consistent with the ratio determined by NMR (see supporting information). We conclude that this approach is easily deployed for the end-labelling of polymers via ROMP for monomers that polymerize rapidly as this minimizes the time that the growing polymer is exposed to the still reactive CTA. Indeed, it provides a clean approach for the generation of telechelic systems, and should be useful where synthetic limitations require the incorporation of functional groups at the beginning of polymerizations. This, for example, can occur where a given monomer is insoluble or problematic during polymerization or characterization.
Fig. 12.

A) Synthesis of telechelic polymer 48 via in situ generation of a functional initiator using TA 39. B) NMR data: Top spectra, polymer after purification showing signals corresponding to both functional groups. Bottom spectra, polymer quenched with ethyl vinyl ether in step 2. C) Fluorescence data of polymer 48 in DMF after treatment with NH4OH to remove the protecting groups.
Conclusions
In this paper we have described three approaches for the incorporation of functional groups into polynorbornenes via ROMP. These involve dosing in small quantities of modified norbornene for the polymerization of dyes, the use of modified termination agents for efficient end-labelling, or the use of in situ generated initiators. To demonstrate these strategies, we have prepared several new monomers and TAs; both conjugatable and fluorescent. Furthermore, we have formulated a variety of the resulting polymers into labelled nanoparticles. In summary, with an array of strategies in hand for preparing well-defined, labelled polymers, materials can be synthesized that carry a diverse array of functionality. Here we have described this in the context of fluorescence, however, ongoing studies in our laboratories involve the incorporation of peptides, nucleic acids and other types of contrast agents. These studies include investigations of the utility of such systems in targeting/imaging strategies in vivo. We contend that it is absolutely critical to the success and progress of relatively complex nanoparticle systems that we have the ability to easily prepare and characterize them. Finally, our goal here was not to conduct an exhaustive, comprehensive study of all possible functional groups of interest, but rather to demonstrate and contrast easily utilized approaches to achieving polymer labelling via ROMP.
Supplementary Material
Fig. 3.
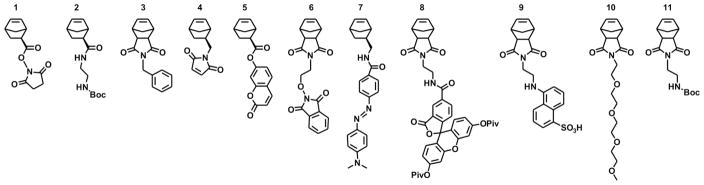
Structures of norbornene-based monomers used in this study.
Acknowledgments
The authors acknowledge support for this program from AFOSR through a PECASE (FA9550-11-1-0105) and a BRI grant (FA9550-12-1-0435). In addition, we acknowledge support from ARO (W911NF-11-1-0264). Furthermore, we thank NIH (NIBIB) for their generous support (1R01EB011633) and via a NIH Director’s New Innovator Award (1DP2OD008724). N.C.G. acknowledges the Henry & Camille Dreyfus Foundation for a New Faculty Award.
Footnotes
Electronic Supplementary Information (ESI) available: [Synthetic procedures and characterization are available]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Aoshima S, Kanaoka S. Chemical Reviews. 2009;109:5245–5287. doi: 10.1021/cr900225g. [DOI] [PubMed] [Google Scholar]
- 2.Beija M, Charreyre MT, Martinho JMG. Progress in Polymer Science. 2011;36:568–602. [Google Scholar]
- 3.Bielawski CW, Grubbs RH. Prog Polym Sci. 2007;32:1–29. [Google Scholar]
- 4.Broyer RM, Grover GN, Maynard HD. Chem Commun. 2011;47:2212–2226. doi: 10.1039/c0cc04062b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castle TC, Hutchings LR, Khosravi E. Macromolecules. 2004;37:2035–2040. [Google Scholar]
- 6.Guo R, Wang X, Guo C, Dong A, Zhang J. Macromolecular Chemistry and Physics. 2012;213:1851–1862. [Google Scholar]
- 7.Lele BS, Murata H, Matyjaszewski K, Russell AJ. Biomacromolecules. 2005;6:3380–3387. doi: 10.1021/bm050428w. [DOI] [PubMed] [Google Scholar]
- 8.Li M, Li H, De P, Sumerlin BS. Macromolecular Rapid Communications. 2011;32:354–359. doi: 10.1002/marc.201000619. [DOI] [PubMed] [Google Scholar]
- 9.Moad G, Rizzardo E, Thang SH. Australian Journal of Chemistry. 2012;65:985–1076. [Google Scholar]
- 10.Nicolas J, Mantovani G, Haddleton DM. Macromolecular Rapid Communications. 2007;28:1083–1111. [Google Scholar]
- 11.Prazeres TJV, Beija M, Charreyre MT, Farinha JPS, Martinho JMG. Polymer. 2010;51:355–367. [Google Scholar]
- 12.Schaefer M, Hanik N, Kilbinger AFM. Macromolecules. 2012;45:6807–6818. [Google Scholar]
- 13.Siegwart DJ, Oh JK, Matyjaszewski K. Progress in Polymer Science. 2012;37:18–37. doi: 10.1016/j.progpolymsci.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sumerlin BS. ACS Macro Letters. 2011;1:141–145. doi: 10.1021/mz200176g. [DOI] [PubMed] [Google Scholar]
- 15.Tasdelen MA, Kahveci MU, Yagci Y. Progress in Polymer Science. 2011;36:455–567. [Google Scholar]
- 16.Leitgeb A, Wappel J, Slugovc C. Polymer. 2010;51:2927–2946. [Google Scholar]
- 17.Slugovc C. Macromol Rapid Commun. 2004;25:1283–1297. [Google Scholar]
- 18.Scholl M, Ding S, Lee CW, Grubbs RH. Organic Letters. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 19.Trnka TM, Grubbs RH. Accounts of Chemical Research. 2000;34:18–29. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]
- 20.Hahn ME, Randolph LM, Adamiak L, Thompson MP, Gianneschi NC. Chem Commun. 2013;49:2873–2875. doi: 10.1039/c3cc40472b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kammeyer JK, Blum AP, Adamiak L, Hahn ME, Gianneschi NC. Polymer Chemistry. 2013;4:3929–3933. doi: 10.1039/C3PY00526G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grubbs RH. Tetrahedron. 2004;60:7117–7140. [Google Scholar]
- 23.Harrity JPA, La DS, Cefalo DR, Visser MS, Hoveyda AH. Journal of the American Chemical Society. 1998;120:2343–2351. [Google Scholar]
- 24.Schrock RR, Murdzek JS, Bazan GC, Robbins J, DiMare M, O’Regan M. Journal of the American Chemical Society. 1990;112:3875–3886. [Google Scholar]
- 25.Schwab P, France MB, Ziller JW, Grubbs RH. Angewandte Chemie International Edition. 1995;34:2039–2041. [Google Scholar]
- 26.Schwab P, Grubbs RH, Ziller JW. Journal of the American Chemical Society. 1996;118:100–110. [Google Scholar]
- 27.Frenzel U, Nuyken O. Journal of Polymer Science Part A: Polymer Chemistry. 2002;40:2895–2916. [Google Scholar]
- 28.Sanford MS, Love JA, Grubbs RH. Organometallics. 2001;20:5314–5318. [Google Scholar]
- 29.Sanford MS, Love JA, Grubbs RH. Journal of the American Chemical Society. 2001;123:6543–6554. doi: 10.1021/ja010624k. [DOI] [PubMed] [Google Scholar]
- 30.Hilf S, Kilbinger AFM. Nat Chem. 2009;1:537–546. doi: 10.1038/nchem.347. [DOI] [PubMed] [Google Scholar]
- 31.Khosravi E, Castle TC, Kujawa M, Leejarkpai J, Hutchings LR, Hine PJ. In: New Smart Materials via Metal Mediated Macromolecular Engineering. Khosravi E, Yagci Y, Savelyev Y, editors. Springer; Netherlands: 2009. pp. 223–236. [Google Scholar]
- 32.Kiessling LL, Mangold SL. In: Polymer Science: A Comprehensive Reference. Krzysztof M Editors-in-Chief, Martin M, editors. Elsevier; Amsterdam: 2012. pp. 695–717. [Google Scholar]
- 33.Nomura K, Abdellatif MM. Polymer. 2010;51:1861–1881. [Google Scholar]
- 34.Smith D, Pentzer EB, Nguyen ST. Polymer Reviews. 2007;47:419–459. [Google Scholar]
- 35.Kilbinger AFM. Functional Polymers by Post-Polymerization Modification. Wiley-VCH Verlag GmbH & Co; KGaA: 2012. pp. 153–171. [Google Scholar]
- 36.Madkour AE, Koch AHR, Lienkamp K, Tew GN. Macromolecules. 2010;43:4557–4561. doi: 10.1021/ma100330u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen B, Metera K, Sleiman HF. Macromolecules. 2005;38:1084–1090. [Google Scholar]
- 38.Kolonko EM, Kiessling LL. Journal of the American Chemical Society. 2008;130:5626–5627. doi: 10.1021/ja8001716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mangold SL, Carpenter RT, Kiessling LL. Organic Letters. 2008;10:2997–3000. doi: 10.1021/ol800932w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matson JB, Grubbs RH. Macromolecules. 2010;43:213–221. doi: 10.1021/ma9019366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Owen RM, Gestwicki JE, Young T, Kiessling LL. Organic Letters. 2002;4:2293–2296. doi: 10.1021/ol0259239. [DOI] [PubMed] [Google Scholar]
- 42.Chien M-P, Thompson MP, Barback CV, Ku T-H, Hall DJ, Gianneschi NC. Advanced Materials. 2013:n/a–n/a. doi: 10.1002/adma.201300823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rush AM, Thompson MP, Tatro ET, Gianneschi NC. ACS Nano. 2013;7:1379–1387. doi: 10.1021/nn305030g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ambade AV, Burd C, Higley MN, Nair KP, Weck M. Chemistry – A European Journal. 2009;15:11904–11911. doi: 10.1002/chem.200801647. [DOI] [PubMed] [Google Scholar]
- 45.Burtscher D, Saf R, Slugovc C. Journal of Polymer Science Part A: Polymer Chemistry. 2006;44:6136–6145. [Google Scholar]
- 46.Ambade AV, Yang SK, Weck M. Angew Chem Int Edit. 2009;48:2894–2898. doi: 10.1002/anie.200805116. [DOI] [PubMed] [Google Scholar]
- 47.Roberts KS, Sampson NS. Organic Letters. 2004;6:3253–3255. doi: 10.1021/ol048935y. [DOI] [PubMed] [Google Scholar]
- 48.Pontrello JK, Allen MJ, Underbakke ES, Kiessling LL. Journal of the American Chemical Society. 2005;127:14536–14537. doi: 10.1021/ja053931p. [DOI] [PubMed] [Google Scholar]
- 49.Zhang L, Eisenberg A. Polymers for Advanced Technologies. 1998;9:677–699. [Google Scholar]
- 50.Förster T. Annalen der Physik. 1948;437:55–75. [Google Scholar]
- 51.Matayoshi ED, Wang GT, Krafft GA, Erickson J. Science. 1990;247:954–958. doi: 10.1126/science.2106161. [DOI] [PubMed] [Google Scholar]
- 52.Clegg RM. In: Methods in Enzymology. David JED, Lilley MJ, editors. Vol. 211. Academic Press; 1992. pp. 353–388. [DOI] [PubMed] [Google Scholar]
- 53.Stryer L. Annual Review of Biochemistry. 1978;47:819–846. doi: 10.1146/annurev.bi.47.070178.004131. [DOI] [PubMed] [Google Scholar]
- 54.Lakowicz JR. Principles of Fluorescence Spectroscopy. Springer Science+Business Media, LLC; 1983. [Google Scholar]
- 55.Selvin PR. Nat Struct Mol Biol. 2000;7:730–734. doi: 10.1038/78948. [DOI] [PubMed] [Google Scholar]
- 56.Farinha JPS, Martinho JMG. The Journal of Physical Chemistry C. 2008;112:10591–10601. [Google Scholar]
- 57.Schillén K, Yekta A, Ni S, Farinha JPS, Winnik MA. The Journal of Physical Chemistry B. 1999;103:9090–9103. [Google Scholar]
- 58.Chien MP, Thompson MP, Lin EC, Gianneschi NC. Chemical Science. 2012;3:2690–2694. doi: 10.1039/C2SC20165H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Farinha JPS, Schillén K, Winnik MA. The Journal of Physical Chemistry B. 1999;103:2487–2495. [Google Scholar]
- 60.Acharya KR, Bhattacharyya SC, Moulik SP. Journal of Photochemistry and Photobiology A: Chemistry. 1999;122:47–52. [Google Scholar]
- 61.Hait SK, Majhi PR, Blume A, Moulik SP. The Journal of Physical Chemistry B. 2003;107:3650–3658. [Google Scholar]
- 62.Majhi PR, Mukherjee K, Moulik SP, Sen S, Sahu NP. Langmuir. 1999;15:6624–6630. [Google Scholar]
- 63.Turro NJ, Kuo PL. Langmuir. 1985;1:170–172. doi: 10.1021/la00063a015. [DOI] [PubMed] [Google Scholar]
- 64.Kalyanasundaram K, Thomas JK. Journal of the American Chemical Society. 1977;99:2039–2044. [Google Scholar]
- 65.Aguiar J, Carpena P, Molina-Bolívar JA, Carnero Ruiz C. Journal of Colloid and Interface Science. 2003;258:116–122. [Google Scholar]
- 66.Matson JB, Virgil SC, Grubbs RH. Journal of the American Chemical Society. 2009;131:3355–3362. doi: 10.1021/ja809081h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hanik N, Kilbinger AFM. Journal of Polymer Science Part A: Polymer Chemistry. 2013;51:4183–4190. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.