

Abstract
Objective
The proliferation of vascular smooth muscle cells (VSMC) plays a crucial role in vascular diseases such as atherosclerosis and restenosis after percutaneous coronary intervention. Many studies have shown that estrogen inhibits VSMC proliferation in response to vascular injury in the mouse carotid injury model. However, the mechanisms that mediate these effects remain unclear. We here investigated the mechanisms by which estrogen inhibits VSMC proliferation.
Approach and Results
We established a novel transgenic mouse line, referred to as the disrupting peptide mice (DPM), in which rapid estrogen receptor (ER)-mediated signaling is abolished by overexpression of a peptide that prevents the ER from forming a signaling complex necessary for rapid signaling. Carotid artery VSMC from DPM or littermate wild type (WT) female mice were obtained by the explant method. In VSMC derived from WT mice, estrogen significantly inhibited VSMC proliferation. Phosphorylation levels of Akt and ERK induced by PDGF were significantly inhibited by estrogen pretreatment. Estrogen enhanced complex formation between ERα and protein phosphatase 2 (PP2) A, and enhanced PP2A activity. The blockade of PP2A activity abolished the estrogen-induced anti-proliferative effect on VSMC. In contrast, none of these effects of estrogen observed in the WT VSMC were observed in VSMC derived from DPM. These results support that rapid non-nuclear ER signaling is required for estrogen-induced inhibition of VSMC proliferation, and further that PP2A activation by estrogen mediates estrogen-induced anti-proliferative effects.
Conclusions
These findings support that PP2A activation via rapid non-nuclear ER signaling may be a novel target for therapeutic approaches to inhibit VSMC proliferation, which plays a central role in atherosclerosis and restenosis.
Keywords: cardiovascular diseases, hormones, molecular biology, signal transduction
Introduction
Cardiovascular disease (CVD) is the leading cause of death in the United States (1). Premenopausal women have a lower incidence of death due to CVD than age-matched men in the United States, supporting that the hormone estrogen may have an important protective effect on the vasculature (2). Though many observational studies have demonstrated that exogenous estrogen therapy is associated with lower primary risk of CVD in postmenopausal women (3–5), randomized controlled trials, primarily in older postmenopausal women, have not shown reductions in CVD events, and in some instances, they have demonstrated harm (6).
Numerous animal studies have shown that estrogen significantly protects against the development of atherosclerosis. We and others have repeatedly shown that estrogen inhibits the response to vascular injury in the mouse carotid artery injury model, in which in vivo estrogen treatment dramatically inhibits the proliferation of vascular smooth muscle cells (VSMC) (7–11). Taken together with the clinical data, these findings underscore the complexity of estrogen’s effects on the cardiovascular system, and highlight its ability to exert both potentially harmful and potentially beneficial effects. This observation strongly supports the need to better understand the molecular mechanisms by which estrogen exerts its cardiovascular effects.
There are two different forms of the estrogen receptor (ER), ERα and ERβ (12). We previously reported that in ERβ knock out (KO) mice estrogen was still protective against vascular injury (13), whereas in ERα KO mice, in which ERα is fully deleted, estrogen treatment showed no protective effect on VSMC proliferation after vascular injury (8), supporting that ERα is necessary for estrogen’s inhibitory effect on VSMC proliferation. Importantly, the molecular mechanisms by which estrogen inhibits VSMC proliferation are unknown.
Estrogen, acting via ERs activates two distinct signaling pathways, the genomic pathway and the rapid, non-nuclear pathway (2, 14). The genomic pathway has been well studied in which ligand-bound ER translocates to the nucleus and transcriptionally regulates gene expression (2). More recent studies have shown that estrogen also signals through a rapid, non-nuclear signaling pathway that involves activation of specific protein kinases, but does not directly involve regulation of gene expression (though cross-talk between these pathways has also now been described) (15, 16). Since their discovery, estrogen-induced protection against vascular injury and the inhibition of VSMC growth have been believed to be due to long-term genomic effects, while little has been investigated about the role of the rapid non-nuclear pathway in regulating the response to vascular injury and inhibiting VSMC proliferation, even though this pathway is known to be involved in regulating endothelial function and nitric oxide release (12, 17, 18).
We previously reported that activation of the non-nuclear pathway by estrogen requires binding between the ER and a scaffold protein, striatin, and that a peptide derived from amino acids 176–253 of ERα disrupts the binding between ER and striatin, preventing activation of the non-nuclear signaling pathway, while leaving the classical genomic signaling pathway intact (19). We have established a transgenic mouse line in which this disrupting peptide (disrupting peptide mice, DPM) is overexpressed throughout the body (20). In these DPM, ERα-mediated non-nuclear signaling is disrupted, while the canonical genomic pathway is still intact (20). In this study, we investigated the molecular mechanisms by which estrogen inhibits VSMC proliferation and explored whether non-nuclear ER signaling is involved in estrogen’s antiproliferative effect on VSMC.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Estrogen-induced anti-proliferative effect on VSMC requires ERα-striatin binding
E2 significantly inhibited the serum-induced proliferation of VSMC derived from WT mice but not of VSMC derived from DPM (Fig. 1A). E2 had no effect on VSMC proliferation derived from ERαKO mice, while the E2-induced inhibition was restored by ERα overexpression in VSMC derived from ERαKO mice (Fig. 1B). E2 markedly inhibited the increase in mRNA expression of PCNA, a marker of VSMC proliferation, induced by sBGS or PDGF in WT VSMC, but not in DPM VSMC (Fig. 1C). E2 significantly activated an ERE-driven luciferase reporter plasmid to a similar degree in VSMC from both WT and DPM (Fig. 1D), supporting that the genomic ER pathway is still preserved in VSMC from DPM. These data indicate that E2 directly inhibits WT VSMC proliferation in an ERα-dependent manner, and further that the disruption of ER-striatin binding induced by the disrupting peptide abolishes the anti-proliferative effect.
Figure 1. Rapid ER signaling is required for estrogen-mediated inhibition of VSMC proliferation.

(A), (B) VSMC derived from WT, DPM, and ERαKO(st) mice was treated with vehicle (V) or E2, and cell proliferation was assessed in each time point. VSMC from ERαKO(st) mice overexpressing green fluorescent protein (KO) as control or ERα (KO+ERα) by using adenovirus vector. Representative data of three different culture lines was shown. Each experiment was performed at n=4–6 per each condition. # P<0.05, *P<0.01 vs vehicle. (C) qRT-PCR analysis of PCNA mRNA. VSMC derived from WT and DPM mice was pretreated with vehicle (V) or E2 and then treated with vehicle (control), PDGF (5 ng/ml) or sBGS (3 %) for 24 hrs before mRNA was extracted (n=4). #P<0.05. (D) VSMC from WT and DPM mice were transiently transfected with estrogen response element-driven luciferase reporter plasmid and β-galactosidase expression plasmid. Cells were treated with vehicle (V) or E2 for 24 hr (n=3). #P<0.05.
Estrogen inhibits kinase phosphorylation through activation of PP2A
To examine the mechanisms by which estrogen inhibits VSMC proliferation, we assessed phosphorylation levels of kinases which promote cell growth including Akt and ERK. As expected, PDGF stimulation increased phosphorylation levels of Akt and ERK in WT VSMC (Fig. 2A). Meanwhile E2 pretreatment attenuated PDGF-induced phosphorylation of Akt and ERK in a dose dependent manner (Fig. 2A). E2-mediated inhibition reached statistical significance at 10–100 nmol/L of E2 for phospho-Akt, and at 50–100 nmol/L of E2 for phospho-ERK. We therefore used 100 nmol/L E2 for further experiments. PDGF stimulation increased phosphorylation levels of Akt, its down stream target GSK3 α/β, and ERK, not only in WT VSMC but also in DPM VSMC (Fig. 2B). E2 pretreatment significantly attenuated PDGF-induced kinase phosphorylation in VSMC from WT mice. In contrast, E2 had no significant effect on PDGF-induced kinase phosphorylation in DPM VSMC (Fig. 2B).
Figure 2. E2 inhibits PDGF-induced kinase activation in VSMC derived from WT but not DPM mice.

Western blotting and quantification of phosphorylated kinases in VSMC stimulated with PDGF (5 ng/ml) for 15 min with pretreatment of vehicle (V) or E2 for 30 min. (A) E2 inhibited phosphorylation of Akt and ERK in dose dependency in VSMC derived from WT mice (n=3). Quantification data is normalized to PDGF(−) vehicle-treated control. #P<0.05, *P<0.01. (B) E2-mediated inhibition of phosphorylation of Akt, GSK3α/β and ERK in VSMC derived from WT or DPM mice (n=4). #P<0.05.
To investigate the mechanisms by which E2 inhibits kinase phosphorylation, we examined whether ER in VSMC interacts with phosphatases. Co-immunoprecipitation experiments revealed that ERα forms a complex with PP2Ac, but not with either MKP-1 or PTEN, all of which are known to dephosphorylate Akt and/or ERK as their substrate (Suppl. Fig. 1). We therefore investigated the role of PP2A in estrogen-mediated inhibition of kinase phosphorylation. Immunoprecipitation of PP2Ac showed that E2 clearly increased ERα-PP2Ac complex formation in VSMC from WT mice, whereas E2-induced ERα-PP2A complex formation was significantly attenuated in DPM VSMC (Fig. 3A). Similarly, E2 increased PP2A-striatin complex formation in VSMC from WT mice but not DPM (Fig. 3A), suggesting that E2 promotes ERα-PP2Ac complex formation via the scaffold protein striatin. PP2A activity assays revealed that E2 significantly increased PP2A activity in VSMC from WT mice, but had no significant effect in DPM VSMC (Fig. 3B). The expression levels of PP2Ac protein were not altered by 30 min of E2 treatment (Fig. 3C), suggesting that E2 increases PP2A activity independently from alteration of PP2A expression levels.
Figure 3. E2 enhances ER-PP2A complex formation and increases PP2A phosphatase activity in WT but not in DPM VSMC.

(A) PP2Ac was immunoprecipitated from VSMC treated with vehicle (V) or E2 for 30 min. Immunoblotting with striatin, ERα and PP2Ac and quantification are shown. Normal mouse IgG was used as negative control (n=3). #P<0.05. (B) PP2A activity assay. VSMC was treated with vehicle (V) or E2 for specific time (n=4 for each condition). #P<0.05. (C) Western blot of PP2Ac in VSMC treated with vehicle (V) or E2 for 30 min. Three repeated experiments showing similar results were performed.
PP2A activation is required for estrogen-induced inhibition of kinase phosphorylation and anti-proliferative effect on VSMC
To determine whether PP2A is involved in E2-mediated inhibition of PDGF-induced Akt and ERK phosphorylation, we next examined whether knock-down of PP2A by siRNA attenuated this effect of E2. As shown in Fig. 4A, E2 significantly inhibited Akt and ERK phosphorylation in control siRNA-treated WT VSMC, but it no longer inhibited PDGF-induced these phosphorylation in WT VSMC in which PP2A was knocked down.
Figure 4. PP2A inhibition abolished E2-mediated inhibition of kinase phosphorylation and VSMC proliferation.

(A) Western blot and quantification of PP2Ac, phosphorylated Akt and phosphorylated ERK. siRNA oligo targeting PP2Ac or non-targeting control was introduced into VSMC followed by E2 and PDGF treatment as described in Figure 2 (n=4). #P<0.05, *P<0.01. VSMC derived from WT mice was treated with vehicle (V) or E2 in the presence or absence of OA (50 nmol/L), and cell proliferation was assessed in each condition cultured with 3% sBGS (B) and PDGF (5 ng/ml) (C) (n=4 per each condition). #P<0.05 vs vehicle.
To further investigate the role of PP2A in E2-mediated inhibition of VSMC proliferation, we next examined the effect of PP2A inhibition with a pharmacological PP2A inhibitor, okadaic acid, on E2’s effects on VSMC proliferation. Okadaic acid completely inhibited the E2-induced increase in PP2A activity in WT VSMC (data not shown). The E2-induced anti-proliferative effect observed in sBGS-stimulated VSMC from WT mice was completely blocked by okadaic acid (Fig. 4B). Further, okadaic acid also significantly blocked the E2-induced anti-proliferative effect observed in PDGF-stimulated VSMC from WT mice (Fig. 4C).
Since the kinases Akt and ERK also mediate apoptosis signaling cascades, we also examined the effects of E2 on apoptosis of VSMC. TUNEL staining showed that E2 had no effect on apoptosis in VSMC under quiescent, PDGF-stimulated or serum-stimulated conditions (Suppl. Fig. 2).
PP2A activation is required for estrogen-induced inhibitory effect on VSMC migration
We further examined whether the rapid, non-nuclear signaling pathway is involved in E2-mediated inhibition of PDGF-induced VSMC migration which also contributes to the development of maladaptive response after vascular injury. In vitro scratch wound assays showed that E2 significantly inhibited cell migration promoted by PDGF in VSMC derived from WT mice, and that this inhibitory effect of E2 on VSMC migration was abolished in cells treated with okadaic acid (Fig. 5A). In contrast, E2 had no effect on migration in VSMC derived from ERαKO mice (Fig. 5B) or on VSMC derived from DPM (Fig. 5C).
Figure 5. PP2A activation through rapid ER signaling is required for estrogen-mediated inhibition of VSMC migration.

A single scratch was made in VSMC derived from either WT mice (A), ERαKO (KO) mice (B) or DPM (C). Cells were treated with E2 or vehicle (V) followed by PDGF (5 ng/ml) or control vehicle (cont) stimulation in the presence or absence of okadaic acid (OA, 50 nmol/L). Bright-field images were taken at right after and 24 hr after making scratch, and the number of migrated cells into the scratched field was counted. The data were normalized to vehicle-treated, control group (n=4). #P<0.05.
Discussion
Although we and others have previously shown that estrogen inhibits VSMC proliferation in vitro and in vivo, the molecular mechanisms that mediate this effect remain unknown. We recently reported that estrogen-induced protective effects against carotid vascular injury are abolished in DPM mice in vivo (20), supporting that the rapid ERα signaling pathway is necessary for estrogen’s anti-proliferative effects in vivo. However, the molecular mechanisms that mediate these effects, and the specific role of rapid ERα signaling is unknown.
In the current studies, we used cultured VSMC from WT and DPM mice to explore these issues. We now show that in cultured WT VSMC, ERα and PP2A can form a complex, that E2-treatment activates PP2A and inhibits phosphorylation of growth promoting kinases, and that the activation of PP2A by estrogen is required for the inhibitory effect of estrogen both on kinase phosphorylation and on VSMC proliferation. We show further, that these effects are lost in VSMC derived from the DPM mice in which the rapid, non-nuclear signaling pathway is disrupted by overexpression of a peptide consisting of amino acids 176–253 of ERα, which disrupts formation of the striatin-based signaling complex required for rapid signaling. In addition, E2 significantly inhibited cell migration promoted by PDGF in VSMC derived from WT mice, but not from DPM, and the inhibitory effect of E2 on VSMC migration was abolished by PP2A inhibition. Taken together, these data support that the rapid, non-nuclear signaling pathway of ER is required for the estrogen-mediated anti-proliferative/migratory effects in VSMC (Fig 6).
Figure 6. Proposed mechanism by which estrogen inhibits VSMC proliferation via rapid, non-nuclear ER signaling.
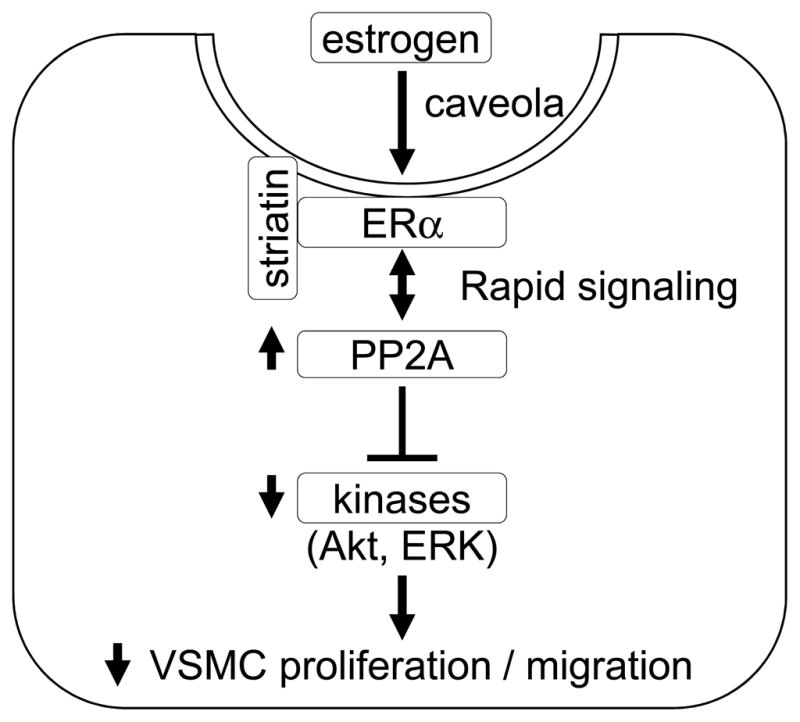
Upon stimulation of estrogen, ERα interacts with and activates PP2A, and modulates activation of kinases induced by growth stimulation, leading to inhibition of VSMC proliferation.
PP2A is a major serine/threonine protein phosphatase that has been highly conserved in all eukaryotes (21). PP2A phosphatase activity is associated with growth inhibition and cell cycle arrest so that PP2A is known to be a key mediator of tumor growth suppression (22–24). Since proliferation of VSMC is also enhanced in large part by protein kinase-dependent pathways, we explored the role of PP2A activation in E2-mediated inhibition of VSMC proliferation. We previously showed that ER binds to PP2Ac which is a catalytic subunit of PP2A complex, and estrogen enhances the interaction between PP2Ac and ERα in endothelial and COS-1 cell lines respectively (25). In that study, inhibition of PP2A activity by OA increased ER-mediated transcriptional activity, which is consistent with our current data showing a trend toward increased transcriptional activity of ERα in VSMC from DPM compared to WT mice, even though the difference was not statistically significant (Fig. 1D). In this study, we further show that in VSMC derived from WT mice, E2 treatment increases the binding between ERα and PP2Ac and increases PP2A activity in VSMC derived from WT mice. These effects of estrogen were diminished or absent in VSMC derived from DPM, supporting that estrogen enhances PP2A activity, possibly by increasing the interaction between ERα and PP2Ac, and that this is dependent on the rapid, non-nuclear pathway. In VSMC derived from DPM, where ERα no longer binds to striatin, E2-induced ERα-PP2A complex formation was significantly attenuated, suggesting that striatin plays a critical role for ERα-PP2Ac complex formation mediated by E2. Furthermore, the inhibition of PP2A activity resulted in loss of estrogen’s ability to inhibit kinase phosphorylation and cell proliferation, suggesting that estrogen-induced antiproliferative effect on VSMC is dependent on PP2A activation.
Estrogen regulates physiology and pathophysiology in both reproductive and non-reproductive target tissues, including the cardiovascular system (12). Estrogen exerts diverse actions in these tissues in part due to the multiple signaling pathways mediated by ER, including both the canonical genomic pathway and the rapid, non-nuclear pathway (12). The canonical genomic pathway of estrogen is itself quite diverse, as a result of differences in expression levels of ERs and ER-interacting proteins, which has produced the concept of “tissue-specific” selective ER modulators (26, 27). Although the molecular mechanisms that mediate aspects of the rapid, non-nuclear pathway of estrogen have been studied in vascular endothelial cells, the role of the rapid signaling in VSMC is poorly explored (12). Although the current findings support that rapid ER signaling is necessary for estrogen-mediated inhibition of VSMC proliferation, they do not address whether rapid signaling by itself is sufficient to inhibit VSMC growth. Similarly, these findings do not rule out a role for genomic signaling in this effect, as it remains possible that both rapid and genomic signaling pathways must be intact to allow for estrogen mediated VSMC growth inhibition. The identification of rapid ER signaling as necessary for estrogen-mediated VSMC inhibition supports the possibility that pathway-selective ER modulators might be potentially promising candidates for specific therapies in cardiovascular diseases such as arthrosclerosis and restenosis after percutaneous coronary interventions. Support for this concept has also recently been provided by the Shaul laboratory who demonstrated that administration of an estrogen dendrimer complex that selectively activates rapid ER signaling, also inhibited the response to vascular injury in a mouse model (28).
Estrogen is known to accelerate proliferation of many cell types, which is clinically relevant to the effects of estrogen on tumors such as breast, ovary and uterus (29, 30). In these cancer cells, estrogen enhances the phosphorylation and activation of the same kinases that are inhibited in VSMC in our study. Since this activation is crucial for the pro-proliferative effects of estrogen in these cells, if we can understand the molecular mechanisms that produce the opposite effect in VSMC, we might be able to gain insight in to how to block the pro-proliferative, cancer promoting effects of estrogen in these other cell types.
The evidence from randomized controlled trials suggests that treatment with estrogen early after menopause may be more effective in reducing the risk of cardiovascular diseases than late treatment with estrogen (6). However, although PP2A might play a role in the different effects of estrogen in early versus late, there is no direct evidence that the function of PP2A in the vasculature is altered after menopause. Further studies will be needed to address this specific point.
Several limitations of the current findings are worthy of mention. First, though the data presented support that E2, acting via ERα activates PP2A catalytic activity, the molecular mechanisms that mediate this effect are unclear. Second, E2 has also been reported to interact with and regulate activity of phosphatases in addition to PP2A, such as MKP-1 and PTEN, (25, 31, 32), and the extent to which effects on other phosphatases might also contribute to regulation of VSMC proliferation has not been tested here. Finally, though our data indicate that estrogen inhibits VSMC proliferation via PP2A activation through the non-nuclear rapid pathway in cultured VSMC, and the relevancy of the rapid pathway in vivo was observed in our previous study using DPM (20), further in vivo studies are needed to assess whether PP2A activation by estrogen is also relevant in vivo.
In conclusion, our results support that the rapid non-nuclear ER signaling is required for estrogen-induced inhibition of VSMC proliferation, and further that PP2A activation by estrogen mediates estrogen-induced anti-proliferative effects in these cells. Taken together, our findings support that rapid non-nuclear ER signaling may be a novel target for therapeutic approaches to inhibit VSMC proliferation, which plays a central role in vascular diseases such as atherosclerosis and restenosis.
Supplementary Material
Significance.
The proliferation of VSMC plays a crucial role in vascular diseases such as atherosclerosis and restenosis after percutaneous coronary intervention (PCI). Although estrogen is known to inhibit VSMC proliferation in response to vascular injury, the mechanisms that mediate these effects remain unclear. We here demonstrate that PP2A activation via non-canonical, rapid non-nuclear ER signaling is required for estrogen-induced inhibition of VSMC proliferation by using a novel transgenic mouse line, in which rapid non-nuclear ER-mediated signaling is abolished. Our findings suggest that PP2A activation via the non-canonical ER signaling may be a novel target for therapeutic approaches to inhibit VSMC proliferation, which plays a central role in atherosclerosis and restenosis.
Acknowledgments
We are grateful to T. Kershaw for her excellent technical assistance.
Sources of Funding
This work is supported by National Institutes of Health grants RO1 HL61298 and T32 HL069770 (RHK), and Japan Society for the Promotion of Science grant Postdoctoral Fellowships for Research Abroad (KU).
Footnotes
Disclosures
none.
References
- 1.Miniño AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep. 2011;59:1–126. [PubMed] [Google Scholar]
- 2.Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science. 2005;308:1583–1587. doi: 10.1126/science.1112062. [DOI] [PubMed] [Google Scholar]
- 3.Falkeborn M, Persson I, Adami HO, Bergström R, Eaker E, Lithell H, Mohsen R, Naessén T. The risk of acute myocardial infarction after oestrogen and oestrogen-progestogen replacement. Br J Obstet Gynaecol. 1992;99:821–828. doi: 10.1111/j.1471-0528.1992.tb14414.x. [DOI] [PubMed] [Google Scholar]
- 4.Sellers TA, Mink PJ, Cerhan JR, Zheng W, Anderson KE, Kushi LH, Folsom AR. The role of hormone replacement therapy in the risk for breast cancer and total mortality in women with a family history of breast cancer. Ann Intern Med. 1997;127:973–980. doi: 10.7326/0003-4819-127-11-199712010-00004. [DOI] [PubMed] [Google Scholar]
- 5.Stampfer MJ, Colditz GA, Willett WC, Manson JE, Rosner B, Speizer FE, Hennekens CH. Postmenopausal estrogen therapy and cardiovascular disease. Ten-year follow-up from the nurses’ health study. N Engl J Med. 1991;325:756–762. doi: 10.1056/NEJM199109123251102. [DOI] [PubMed] [Google Scholar]
- 6.Manson JE, Hsia J, Johnson KC, Rossouw JE, Assaf AR, Lasser NL, Trevisan M, Black HR, Heckbert SR, Detrano R, Strickland OL, Wong ND, Crouse JR, Stein E, Cushman M. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med. 2003;349:523–534. doi: 10.1056/NEJMoa030808. [DOI] [PubMed] [Google Scholar]
- 7.Iafrati MD, Karas RH, Aronovitz M, Kim S, Sullivan TR, Jr, Lubahn DB, O’Donnell TF, Jr, Korach KS, Mendelsohn ME. Estrogen inhibits the vascular injury response in estrogen receptor alpha-deficient mice. Nat Med. 1997;3:545–548. doi: 10.1038/nm0597-545. [DOI] [PubMed] [Google Scholar]
- 8.Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, Mendelsohn ME. Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circ Res. 2002;90:1087–1092. doi: 10.1161/01.res.0000021114.92282.fa. [DOI] [PubMed] [Google Scholar]
- 9.Sullivan TR, Karas RH, Aronovitz M, Faller GT, Ziar JP, Smith JJ, O’Donnell TF, Mendelsohn ME. Estrogen inhibits the response-to-injury in a mouse carotid artery model. J Clin Invest. 1995;96:2482–2488. doi: 10.1172/JCI118307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sivritas D, Becher M, Ebrahimian T, Arfa O, Rapp S, Bohner A, Mueller C, Umemura T, Wassmann S, Nickenig G, Wassmann K. Antiproliferative effect of estrogen in vascular smooth muscle cells is mediated by Kruppel-like factor-4 and manganese superoxide dismutase. Basic Res Cardiol. 2011;106:563–575. doi: 10.1007/s00395-011-0174-z. [DOI] [PubMed] [Google Scholar]
- 11.Yuan Y, Xu J. Loss-of-function deletion of the steroid receptor coactivator-1 gene in mice reduces estrogen effect on the vascular injury response. Arterioscler Thromb Vasc Biol. 2007;27:1521–1527. doi: 10.1161/ATVBAHA.107.144477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–1811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- 13.Karas RH, Hodgin JB, Kwoun M, Krege JH, Aronovitz M, Mackey W, Gustafsson JA, Korach KS, Smithies O, Mendelsohn ME. Estrogen inhibits the vascular injury response in estrogen receptor beta-deficient female mice. Proc Natl Acad Sci US A. 1999;96:15133–15136. doi: 10.1073/pnas.96.26.15133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mendelsohn ME, Karas RH. Rapid progress for non-nuclear estrogen receptor signaling. J Clin Invest. 2010;120:2277–2279. doi: 10.1172/JCI43756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Björnström L, Sjöberg M. Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. Mol Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 16.Le Romancer M, Treilleux I, Leconte N, Robin-Lespinasse Y, Sentis S, Bouchekioua-Bouzaghou K, Goddard S, Gobert-Gosse S, Corbo L. Regulation of Estrogen Rapid Signaling through Arginine Methylation by PRMT1. Mol Cell. 2008;31:212–221. doi: 10.1016/j.molcel.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 17.Ganz P. Vasomotor and vascular effects of hormone replacement therapy. Am J Cardiol. 2002;90:11F–16F. doi: 10.1016/s0002-9149(01)02218-4. [DOI] [PubMed] [Google Scholar]
- 18.Darblade B, Pendaries C, Krust A, Dupont S, Fouque M-J, Rami J, Chambon P, Bayard F, Arnal J-F. Estradiol alters nitric oxide production in the mouse aorta through the alpha-, but not beta-, estrogen receptor. Circ Res. 2002;90:413–419. doi: 10.1161/hh0402.105096. [DOI] [PubMed] [Google Scholar]
- 19.Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha. Proc Natl Acad Sci US A. 2004;101:17126–17131. doi: 10.1073/pnas.0407492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moens SJB, Schnitzler GR, Nickerson M, Guo H, Ueda K, Lu Q, Aronovitz MJ, Nickerson H, Baur WE, Hansen U, Iyer LK, Karas RH. Rapid estrogen receptor signaling is essential for the protective effects of estrogen against vascular injury. Circulation. 2012;126:1993–2004. doi: 10.1161/CIRCULATIONAHA.112.124529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24:186–191. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- 22.Janssens V, Goris J, Van Hoof C. PP2A: the expected tumor suppressor. Curr Opin Genetics Dev. 2005;15:34–41. doi: 10.1016/j.gde.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Junttila MR, Puustinen P, Niemelä M, Ahola R, Arnold H, Böttzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, Lu S-L, Lin S, Chan EKL, Wang X-J, Grènman R, Kast J, Kallunki T, Sears R, Kähäri V-M, Westermarck J. CIP2A Inhibits PP2A in Human Malignancies. Cell. 2007;130:51–62. doi: 10.1016/j.cell.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 24.Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell. 2004;5:127–136. doi: 10.1016/s1535-6108(04)00026-1. [DOI] [PubMed] [Google Scholar]
- 25.Lu Q, Surks HK, Ebling H, Baur WE, Brown D, Pallas DC, Karas RH. Regulation of estrogen receptor alpha-mediated transcription by a direct interaction with protein phosphatase 2A. J Biol Chem. 2003;278:4639–4645. doi: 10.1074/jbc.M210949200. [DOI] [PubMed] [Google Scholar]
- 26.Turgeon JL, McDonnell DP, Martin KA, Wise PM. Hormone Therapy: Physiological Complexity Belies Therapeutic Simplicity. Science. 2004;304:1269–1273. doi: 10.1126/science.1096725. [DOI] [PubMed] [Google Scholar]
- 27.Nilsson S, Koehler KF. Oestrogen receptors and selective oestrogen receptor modulators: molecular and cellular pharmacology. Basic Clin Pharmacol Toxicol. 2005;96:15–25. doi: 10.1111/j.1742-7843.2005.pto960103.x. [DOI] [PubMed] [Google Scholar]
- 28.Chambliss KL, Wu Q, Oltmann S, et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010;120:2319–2330. doi: 10.1172/JCI38291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castoria G, Migliaccio A, Giovannelli P, Auricchio F. Cell proliferation regulated by estradiol receptor: Therapeutic implications. Steroids. 2010;75:524–527. doi: 10.1016/j.steroids.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Acconcia F, Kumar R. Signaling regulation of genomic and nongenomic functions of estrogen receptors. Cancer Lett. 2006;238:1–14. doi: 10.1016/j.canlet.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 31.Takeda-Matsubara Y, Nakagami H, Iwai M, Cui T-X, Shiuchi T, Akishita M, Nahmias C, Ito M, Horiuchi M. Estrogen activates phosphatases and antagonizes growth-promoting effect of angiotensin II. Hypertension. 2002;39:41–45. doi: 10.1161/hy1201.097197. [DOI] [PubMed] [Google Scholar]
- 32.Yang L, Wang Y, Chen P, Hu J, Xiong Y, Feng D, Liu H, Zhang H, Yang H, He J. Na(+)/H(+) exchanger regulatory factor 1 (NHERF1) is required for the estradiol-dependent increase of phosphatase and tensin homolog (PTEN) protein expression. Endocrinology. 2011;152:4537–4549. doi: 10.1210/en.2011-1207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.