

Abstract
Suppression of gonadal testosterone synthesis represents the standard first line therapy for treatment of metastatic prostate cancer. However, in the majority of patients who develop castration-resistant prostate cancer (CRPC), it is possible to detect persistent activation of the androgen receptor (AR) through androgens produced in the adrenal gland or within the tumor itself. Abiraterone acetate was developed as an irreversible inhibitor of the dual functional cytochrome P450 enzyme CYP17 with activity as a 17α-hydroxylase and 17,20-lyase. CYP17 is necessary for production of nongonadal androgens from cholesterol. Regulatory approval of abiraterone in 2011, based on a phase III trial showing a significant improvement in overall survival (OS) with abiraterone and prednisone versus prednisone, represented proof of principle that targeting AR is essential for improving outcomes in men with CRPC. Inhibition of 17α-hydroxylase by abiraterone results in accumulation of upstream mineralocorticoids due to loss of cortisol-mediated suppression of pituitary adrenocorticotropic hormone (ACTH), providing a rationale for development of CYP17 inhibitors with increased specificity for 17,20-lyase (orteronel, galeterone and VT-464) that can potentially be administered without exogenous corticosteroids. In this article, we review the development of abiraterone and other CYP17 inhibitors; recent studies with abiraterone that inform our understanding of clinical parameters such as drug effects on quality-of-life, potential early predictors of response, and optimal sequencing of abiraterone with respect to other agents; and results of translational studies providing insights into resistance mechanisms to CYP17 inhibitors leading to clinical trials with drug combinations designed to prolong abiraterone benefit or restore abiraterone activity.
Keywords: androgen synthesis, castration-resistant prostate cancer, treatment
INTRODUCTION
Prostate cancer continues to be a significant burden on men's health. Based on 2008 global estimates, prostate cancer was the second most frequently diagnosed cancer of men and it was found to be the 6th leading cause of death in men with close to 258,000 deaths reported worldwide.1,2 In the United States alone, prostate cancer is the second leading cause of cancer mortality with nearly 30,000 lives expected to be lost in 2014.3 Death from prostate cancer is almost always associated with metastatic castration-resistant prostate cancer (CRPC). The past several years have marked a rapid expansion in the therapeutic armamentarium against CRPC. It is helpful to understand the evolution of the treatments aimed at this phase of advanced prostate cancer.
History of androgen deprivation therapy
One of the biggest leaps in prostate cancer treatment occurred at the University of Chicago in the early 1940s. Huggins and Hodges demonstrated that prostate cancer cell growth and spread were dependent on androgen signaling, thus implying that by depriving cancer cells of such endocrine activation the growth of prostate cancer could be hindered.4 Huggins was awarded the Nobel Prize in 1966 for his contribution to revealing the nature of androgen dependent malignancy. Prostate cancer's reliance on androgens would be exploited with little innovation for the next 60 years. Although, the early 2000s saw the rise of taxane-based chemotherapeutics for CPRC,5 androgen deprivation therapy (ADT) remains the foundation of advanced prostate cancer care.6 The development of novel androgen synthesis inhibitors and androgen receptor (AR) antagonists has refocused oncologists’ attention on hormonal manipulation in men with CRPC.7
Androgen receptor signaling
It is well-established that the prostate and seminal vesicles contain 5000–20,000 high affinity AR/cell.8 These ARs are intracellular and are transported both into the nucleus as well as out of the nucleus.8 After AR production, the receptor is neutralized by a multitude of proteins known as chaperones. Phosphorylation or glycosylation of the AR complex may result in activation by way of inhibition or prevention of rebinding of the chaperones. AR activation is also driven by both ligand-dependent and independent binding.9 Normally, the AR's primary role is to regulate normal prostate growth by promoting and inhibiting secretion of various growth factors from stromal cells that act on the epithelial cells of the prostate.10 The precise role of AR activity in the initiation and progression of prostate cancer is an area of active investigation.11 It has been reported that AR activity may assist in the formation of gene fusions between the AR regulated TMPRSS2 gene and the E26 transformation-specific (ETS) related transcription factors that are found in over 50% of prostate cancers.12,13 TMPRSS2-ETS fusions promote prostate cancer cell motility and invasiveness.14 Furthermore, in the setting of loss of the phosphatase and tensin homologue (PTEN) tumor suppressor (which occurs in over 50%) of prostate cancers, AR transcriptional activity appears to be significantly increased by overexpression of the ETS transcription factor in both early and advanced cancer.15,16 Therefore, in at least certain molecular contexts, maintenance of AR signaling may induce genetic rearrangements that in turn prime the cell to be responsive to the AR.
Androgen receptor activation
The primary agonists for AR are the androgens testosterone (T) and dihydrotestosterone (DHT). DHT has a 10-fold higher affinity for the AR and is the primary ligand for the AR in the prostate.17,18 DHT is essential for normal prostate development.19 Androgen binding to the AR induces conformational changes in the AR leading to dimerization and dissociation from nuclear chaperones, with subsequent translocation of the AR into the nucleus. Nuclear AR binds to androgen responsive elements in the DNA with resultant transcriptional activity inducing cellular proliferation.20
Testicular leydig cells produce approximately 97% of circulating T.21 In the noncastrate state, tissue T is taken into the prostate epithelium and converted into DHT by the 5α-reductase, enzymes SRD5A1 and SRD5A2. DHT also synthesized by the adrenal gland, skin and prostate.6 When castration therapy is administered, either surgically or with a gonadotropin-releasing hormone agonist, several groups, going as far back as 1978,22 have demonstrated that DHT can still be detected in locally recurrent23 and metastatic prostate tissue24 at levels that are sufficient to activate the AR.25 The two dominant sources of androgens in men with CRPC are the adrenal androgens dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEA-S)26,27 and de novo synthesis by the prostate gland.24,28,29,30
Irrespective of the site of production of androgens in patients with CRPC, ultimately it is necessary for the 21-carbon pregnenolone to be modified through a series of enzymatic reactions to the 19-carbon DHT (Figure 1).31 The cytochrome P450 enzymes are a superfamily of enzymes that catalyze the oxidation of multiple biosynthetic intermediates and toxins. Cytochrome p450, family 17, subfamily A, polypeptide 1 (CYP17) performs two enzymatic functions essential for cleavage of the bond between carbon 17 and carbon 20 of pregnenolone. The 17α-hydroxylase adds a hydroxyl group at carbon 17 followed by the 17,20-lyase, which cleaves the C17–C20 bond. Deficiency of CYP17 has been identified in children as the cause of congenital adrenal hyperplasia leading to absence of sex steroid and cortisol synthesis.
Figure 1.
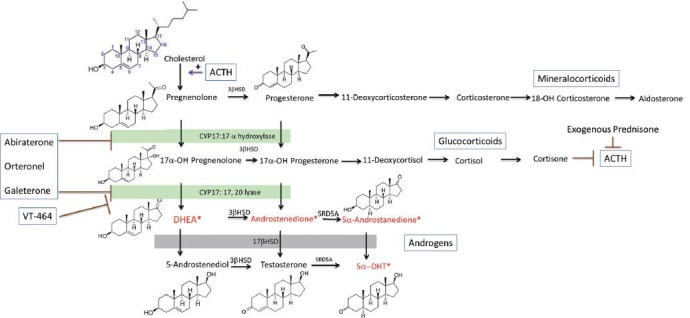
Steroid synthesis pathways. Mineralocorticoid, glucocorticoid, dehydroepiandrosterone, and androstenediol synthesis take place in the adrenal gland. Testosterone is converted to dihydrotestosterone (DHT) in peripheral tissue. Abiraterone inhibits both the 17α-hydroxylase and 17,20-lyase activity of the cytochrome p450 enzyme CYP17. Orteronel and galeterone have increased specificity for 17,20-lyase relative to 17α–hydroxylase. VT-464 has 10-fold in vitro specificity for the 17,20-lyase reaction over 17α-hydroxylase. Androgens in the 5α-androstanedione pathway for production of DHT are noted with*. 3βHSD: 3β-hydroxysteroid dehydrogenase; SRD5A: steroid 5 alpha reductase; DHT: dihydrotestosterone; DHEA: dehydroepiandrosterone.
ABIRATERONE
The essential function of CYP17 in androgen synthesis provided the rationale for development of potent CYP17 inhibitors. Proof of principle regarding activity of T synthesis inhibitors in the treatment of prostate cancer is well-documented in the off-label use of ketoconazole.32 Ketoconazole inhibits multiple CYP enzymes including CYP17; however, it is a comparatively weak inhibitor with significant toxicity including fatigue, hepatotoxicity, nausea, and rash.32 A randomized phase III study in men with CPRC comparing anti-androgen withdrawal to anti-androgen withdrawal plus ketoconazole demonstrated that ketoconazole had modest activity in CRPC. Ketoconazole led to a decrease in serum prostate-specific antigen (PSA) by ≥50% in 27% of patients, while anti-androgen withdrawal caused a PSA response in 11% of patients.33 There was no difference in survival highlighting the need for development of potent CYP17 inhibitors.
Abiraterone was developed by medicinal chemists at the Institute of Cancer Research in London.34 It is structurally similar to pregnenolone, with structural modifications to promote irreversible binding to the CYP17 enzyme, thereby maximizing enzyme inhibition. To increase oral bioavailability, the prodrug, abiraterone acetate was synthesized.31 In vitro studies demonstrated that compared to ketoconazole, abiraterone acetate is 10–30 times more potent as a CYP17 inhibitor.35
Abiraterone phase I trials
The initial phase I report, comprising two single dose studies in castrate and noncastrate men and a 12 day dose escalation study in noncastrate men demonstrated that abiraterone was safe, orally bioavailable and could suppress serum T levels at 500 mg and 800 mg of abiraterone.36 In noncastrate men, a compensatory rise in luteinizing hormone was noted, limiting the ability to maintain complete T suppression in one of three patients treated at a dose of 800 mg, guiding further development of abiraterone specifically in castrate patients. A continuous dosing phase I study in chemotherapy naïve men who had not received prior ketoconazole demonstrated that doses up to 2000 mg were well-tolerated and a maximally tolerated dose was not reached.37 At baseline, castrate but detectable levels of T became undetectable (<1 ng ml−1) by day 8 at all abiraterone dose levels. Measurements of the mineralocorticoid precursor steroids, 11-deoxycorticosterone (DOC) and corticosterone, which are regulated by adrenocorticotropic hormone (ACTH) and are upstream of 17α-hydroxylase in the steroid synthesis pathway, were used as pharmacodyamic markers of CYP17 inhibition. At doses from 250 mg to 750 mg of abiraterone mean levels of DOC and corticosterone increased, while a plateau in DOC and corticosterone levels was observed in patients treated at 750–2000 mg leading to selection of 1000 mg/day as the recommended phase II dose of abiraterone.
In the initial phase I report of abiraterone, 57% of patients experience a decline in PSA of ≥ 50% despite having castrate levels of T at study entry. Objective responses by Response Evaluation Criteria in Solid Tumors (RECIST) criteria were observed in five of eight evaluable subjects. Due to loss of feedback mediated ACTH suppression by cortisol (downstream of 17α-hydroxylase) patients experienced the effects of mineralocorticoid excess including hypertension, hypokalemia and edema, however excess DOC and cortisol prevented adrenocortical insufficiency. Mineralocorticoid excess was reversed by administering dexamethasone, restoring suppression of ACTH, or was managed with the mineralocorticoid receptor antagonist eplerenone. Dexamethasone administration also lead to a PSA response in four of 15 patients progressing while on abiraterone suggesting that promiscuous activation of the AR by steroids upstream of CYP17 mediated resistance in some patients. The terminal half-life of abiraterone was approximately 10 h. Administration of abiraterone with a high fat meal increased absorption by 4.4-fold compared to the fasting state (P = 0.49). Therefore to minimize inter-patient variability in absorption further development of abiraterone was based on fasting administration of medication. In a second phase I trial, allowing patients treated with prior ketoconazole, similar response rates were seen, with PSA responses in 47% and 64% of patients with and without prior ketoconazole treatment, respectively.38
Abiraterone phase II trials
Phase II evaluation of abiraterone 1000 mg per day was performed in the UK and US in both chemotherapy naïve patients and those who had previously been treated with chemotherapy. In the chemotherapy naive population, Attard et al.39 demonstrated a PSA response in 67% of patients with median time to progression of 32 weeks. Similarly, Ryan observed a PSA response rate of 79% with median time to progression of 71 weeks.40 In the former study, dexamethasone 0.5 mg per day was added at progression while the later study administered prednisone 5 mg twice per day in all patients. Post chemotherapy, abiraterone treatment resulted in PSA responses in 51%41 and 36%42 of subjects in two phase II trials. In the former trial 17% of the 42 subjects received prior ketoconazole while in the latter trial 47% of the 58 patients received prior ketoconazole, likely accounting for the difference in the response rate. In studies prospectively incorporating prednisone 5 mg twice per day into the treatment regimen, fatigue was the most common Grade 2 side-effect with symptoms of hyperaldosteronism (hypokalemia, hypertension, and fluid retention) occurring relatively rarely compared with patients treated without prednisone and managed symptomatically with eplerenone. The study by Ryan et al.40 also sought to characterize the frequency of a bone flare response in patients treated with abiraterone. In this study, bone flare was prospectively defined as discordance between a restaging bone scan at 3 months’ time indicating progression while PSA decreased by 50% or more, with follow-up bone scan 3 months later indicating improvement or stable disease. Out of 23 patients with the combination of a positive bone scan at baseline and a 50% of greater decline in PSA with treatment, 11 (48%) of patients had bone flare at 3 months and stable or improved scans at 6 months highlighting the need for confirmatory scans to avoid premature discontinuation of therapy.α
Abiraterone phase III trials
Two randomized phase III trials of abiraterone plus prednisone versus placebo plus prednisone have been reported in patients with metastatic CRPC, COU-AA-301 in patients previously treated with chemotherapy and COU-AA-302 in the prechemotherapy setting. In COU-AA-301, 1195 men were randomized to receive abiraterone 1000 mg per day plus prednisone 5 mg twice per day or placebo plus prednisone 5 mg twice per day in a 2:1 ratio.43,44 Patients with prior ketoconazole and cardiac ejection fraction <50% were excluded. The primary endpoint of the study was OS. Secondary endpoints included PSA response rate (PSA decrease by >50% from baseline), time to PSA progression and radiographic progression free survival (rPFS). The study was unblinded after a planned interim analysis, conducted when 67% of on-study deaths occurred, demonstrated that the study met the prespecified boundary for efficacy. With median follow-up of 12.8 months, median survival improved from 10.9 months to 14.8 months (hazard ratio (HR) = 0.646; P < 0.0001) for patients treated with abiraterone and prednisone.43 Updated survival analysis with median follow-up of 20.2 months and 775 deaths demonstrated an improvement in OS from a median of 11.2 to 15.8 months.44 Secondary endpoints all favored treatment with abiraterone (P < 0.0001). Confirmed PSA response (5.5% vs 29.5%), time to PSA progression (6.6 vs 8.5 months), and response by RECIST (3.3 vs 14.8%) all increased with abiraterone. Side-effects seen in COU-AA-301 were related to increased levels of mineralocorticoids causing fluid retention (31% all grades), hypokalemia (17% all grades), increased transaminases (10% all grades), hypertension (10% all grades), and cardiac events (13% all grades, 3% Grade 3/4). Based on the positive results of this phase III study, abiraterone plus prednisone was approved by the Food and Drug Administration (FDA) in April 2011 for use in men with metastatic CRPC after treatment with chemotherapy.45 Based on the abiraterone side-effect profile, the manufacturer recommends monitoring for hypertension, low potassium, and fluid retention on a monthly basis.46
COU-AA-302 was a randomized, double-bind, placebo controlled phase III study in which 1088 men with progressive (by scans or PSA) asymptomatic or minimally symptomatic metastatic CRPC (mCRPC) were randomized in a 1:1 ratio to either abiraterone 1000 mg per day plus prednisone 5 mg twice per day or to placebo plus prednisone 5 mg twice per day.47 Patients who required narcotic analgesics in the 4 weeks prior to study, had visceral metastatic disease, or had prior chemotherapy or ketoconazole were excluded. The study was designed with a co-primary endpoint of OS and rPFS. An interim evaluation for rPFS was planned on the basis of a blinded review by the central radiologist after 378 progression-free events, providing a statistical power of 91% to detect an HR of 0.67 at a two-tailed level of significance of 0.01. Three interim analyses of OS were planned after 15, 40 and 55% of planned OS events had occurred.
At the time of the second interim analysis, performed with median follow-up of 22.2 months, after 43% of the expected deaths had occurred, median rPFS was 16.5 months with abiraterone plus prednisone and 8.3 months with placebo plus prednisone (HR = 0.53; 95% confidence interval (CI), 0.45–0.62; P < 0.001).47 OS was improved with abiraterone plus prednisone (median not reached, vs 27.2 months for prednisone alone; HR = 0.75; 95% CI, 0.61–0.93; P = 0.01), however the result did not meet the prespecified boundary for statistical significance. Evidence of substantial benefit from abiraterone was seen in all prespecified secondary endpoints including time to PSA progression (11.1 months with abiraterone plus prednisone and 5.6 months with placebo plus prednisone, HR = 0.49), time to initiation of chemotherapy (25.2 vs 16.8 months, HR = 0.58), and time to initiation of narcotic pain medicine. Based on the compelling evidence of benefit from abiraterone plus prednisone administered prior to chemotherapy, the data and safety monitoring committee recommended that the study be unblinded and that patients receiving placebo be treated with abiraterone. Results of the third interim analysis after a median follow-up of 27.1 months demonstrated persistence of the trend toward improved OS of 35.3 months among men treated with abiraterone plus prednisone compared to 30.1 months among men in the placebo plus prednisone control arm (HR = 0.79; 95% CI, 0.66–0.96; P < 0.0151), although these results did not meet the boundary for statistical significance.48
Review by the FDA resulted in an extended indication for abiraterone in men with CRPC prior to chemotherapy, representing the first time a drug has been approved for use in prostate cancer based on an endpoint of rPFS rather than pain or survival.49 In expanding the indication, the FDA cited the demonstrated survival benefit with abiraterone in the postdocetaxel setting combined with a trend toward improved survival in the predocetaxel setting, the large improvement in rPFS, improvement in PFS across all subgroups, benefit seen in clinically meaningful secondary endpoints such as time to initiation of chemotherapy, improvement in patient reported pain outcomes and a favorable toxicity profile. Therefore, while improvement in rPFS was the primary outcome leading to regulatory approval, it was not the sole basis for approval and future trials utilizing rPFS will require additional evidence of clinical benefit in the absence of a definitive link between improvement in rPFS and OS.49
Abiraterone correlative studies
Correlative studies in these phase II trials of abiraterone included evaluation of circulating tumor cells (CTCs) counted with the FDA approved Cell Search System (Veridex, Raritan NJ). CTCs were previously demonstrated to be an independent prognostic factor for survival.50 In the chemotherapy naive population 10 of 17 patients (59%) with unfavorable pretreatment CTC counts of ≥5 CTC converted to the favorable group with < 5 CTCs per 7.5 ml.39 In the post chemotherapy studies CTC conversion from the unfavorable to favorable occurred in 34%42 and 41%41 of patients. These studies provided the basis for developing CTC enumeration as a potential biomarker of response in the phase III trial of abiraterone (see below). In additional to CTC enumeration, CTCs were evaluated for the presence of the TMPRSS2-ETS-related gene (ERG) gene fusions.51,52 While feasibility of detecting gene fusions in CTCs was demonstrated, the value of the TMPRSS2-ERG gene fusion in predicting treatment response remains unclear.53
Correlative studies performed in the COU-AA-301 abiraterone trial have led to the development of biomarkers that may predict prognosis in patients with metastatic CRPC. Measurement of serum androgen levels prior to initiation of abiraterone was performed in patients participating in the COU-AA-301 study.54 Improved survival, regardless of the treatment received, was observed in patients with baseline serum androgen levels above the median compared to patients with serum androgen levels below the median. Patients receiving abiraterone who had T levels above the median level had a 4.2-month improvement in median survival (17.8 vs 13.6 months, HR = 0.54, 95% CI, 0.53–0.77). Likewise in patients receiving prednisone, a significantly improved prognosis was seen when T levels were above the median compared to below the median (15.8 vs 9.3, HR = 0.51, 95% CI, 0.39–0.67). Multivariate analysis demonstrated that T, androstenedione, and DHEA-S above the median were all associated with improved OS. Of note, patients with baseline androgen levels below the median did benefit from abiraterone compared to prednisone, however, patients with the lowest quartile of T at baseline had a particularly poor prognosis with survival of 10.4 months compared to survival of 18.9 months in patients with T in the highest quartile at baseline.
Building upon the potential for CTC quantification to assess patient response to abiraterone, investigators analyzed the ability of CTCs obtained during the course of treatment to provide prognostic information with respect to OS. After 12 weeks on study, patients were risk stratified based on CTC count and lactate dehydrogenase (LDH) into high-risk (CTCs > 5 and LDH > 250 IU l−1), intermediate risk (CTCs ≥ 5 and LDH ≤ 250 IU l−1), and low risk (≤4 and normal LDH levels) groups. Data presented in abstract form suggests that risk stratification using these two biomarkers was able to fulfill the four postulates of the Prentice criteria for surrogacy when assessed at 12 weeks.55,56 Specifically, (1) treatment (abiraterone) has a significant effect on the clinical end point of OS; (2) treatment (abiraterone) has a significant effect on the CTC/LDH biomarker; (3) the CTC/LDH biomarker measured at 12 weeks has a significant impact on the clinical end point (2 year survival for high-risk group = 2%, 2 year survival for low risk group = 46%); (4) the full effect of treatment on the clinical end-point was captured by the biomarker such that at 12 weeks on study survival was determined by the CTC/LDH risk group rather than by the treatment the patients received. In addition to providing clinically useful information to patients and physicians regarding patient risk, this biomarker, if confirmed, could be useful for designing future combination based studies with abiraterone utilizing an intermediate endpoint, or for selecting patients at highest risk for novel therapies and intense molecular assessment of the tumor to elucidate potential characteristics driving tumor resistance.
EFFECTS OF ABIRATERONE ON QUALITY OF LIFE AND SPECIFIC PATIENT POPULATIONS
Mineralocorticoid and cardiovascular effects
CYP17 inhibition with abiraterone is characterized by suppression of androgen and cortisol synthesis. As noted above, the latter is associated with a rise in ACTH that causes elevated mineralocorticoids potentially leading to hypertension, hypokalemia, and fluid overload.57 For patients with CRPC and concurrent cardiovascular comorbidities, these side-effects can pose added risks. The cardiovascular outcomes of abiraterone treatment in 51 patients with mCRPC with cardiovascular comorbidities treated with standard doses of abiraterone and prednisone were assessed.58 Comorbidities included controlled hypertension (41%), cardiac ischemia (14%), stroke (9%), dyslipidemia (18%) and hyperglycemia (30%). All cardiovascular risk factors and comorbidities were controlled using appropriate medical treatments and patients had a risk of ≤ 4% for development of fatal cardiovascular disease according to standard risk assessments. The most common adverse event reported was Grade 1-2 hypertension in 16% of patients. None of the patients had a decrease in left ventricular ejection fraction, nor were there any cardiac events reported during treatment with abiraterone.58 Due to the retrospective nature of this study with limited follow-up, no definitive conclusions can be established; however, the study suggests that abiraterone combined with prednisone therapy can be well-tolerated in patients with well-controlled cardiovascular risk factors.
Body composition effects
A change in body composition, with loss of muscle mass and increased subcutaneous and visceral abdominal fat is a widely recognized as an adverse effect of castration for men with prostate cancer.59 A change in body composition with abiraterone could have important implications for the quality of life of men with advanced prostate cancer, especially men who are asymptomatic or minimally symptomatic.59 Treatment of CRPC with abiraterone results in a further decline in serum T to near undetectable levels.57 Body composition changes in men undergoing maximal androgen suppression with and without exogenous glucocorticoids were assessed in a post-hoc analysis on all patients treated with single-agent abiraterone in clinical trials at the Royal Marsden Foundation Trust, UK. All patients maintained a castrate state throughout study participation by means of ongoing luteinizing-hormone-releasing hormone (LHRH) analog therapy or surgical castration. These trials allowed addition of dexamethasone 0.5 mg daily at the time of PSA progression on abiraterone or for management of abiraterone-related toxicity. Previous studies have shown linear relationships between lumbar skeletal muscle and fat cross-sectional areas measured using multiple-slice computed tomography (CT) and whole-body muscle and fat mass measured using dedicated dual-energy X-ray absorptiometry scans.59 The Royal Marsden researches were able to use CT scans performed for clinical or restaging purposes for analysis of changes in body composition. A total of 55 patients were eligible for body composition analyses. Muscle loss was observed in patients treated with single-agent abiraterone, with loss greatest in patients with a baseline body mass index (BMI) >30 (median change 4.3%). There was no significant change in muscle mass after the addition of dexamethasone. To the authors’ surprise, loss of visceral fat was also observed on single-agent abiraterone, with effect most evident in patients with a baseline BMI > 30 who had a 19.6% loss of visceral fat area. There was also a decrease in BMI on single agent abiraterone, again most significant in patients with a baseline BMI > 30 (−8.1%). In contrast, the addition of dexamethasone led to a striking increase in central fat in all patients regardless of baseline BMI, with a recovery in fat to preabiraterone levels. After the addition of dexamethasone, all groups experienced a marked increase in BMI.59 With the use of abiraterone for prolonged periods of time in chemotherapy naive patients, additional studies are needed to further characterize the metabolic effects of abiraterone.
Skeletal-related events
The natural history of prostate cancer often includes painful bone metastases resulting in a decreased health-related quality of life (HRQoL), increase in skeletal-related events (fractures), and correlating with poor survival data on pain control and skeletal-related events was prospectively collected as part of the randomized, phase 3 COU-AA-301 trial of abiraterone acetate plus prednisone versus placebo plus prednisone after docetaxel chemotherapy, as noted above.60 In this study of 1195 patients, 1068 had bone metastasis and an ECOG performance status of two or less. Pain intensity and interference of pain with daily activities were assessed with the Brief Pain Inventory-Short Form questionnaire. With a median follow-up of 20.2 months, patients with clinically significant pain at baseline who received abiraterone and prednisone compared to patients receiving placebo plus prednisone experienced significantly better palliation (45.0% vs 28.8%); P = 0.0005) and faster palliation (median time to palliation 5.6 months [95% CI 3.7–9.2] vs 13.7 months [5.4–not estimable]; P = 0.0018) of pain intensity. In the overall population, median time to occurrence of first skeletal-related event was significantly longer with abiraterone and prednisone than with prednisone only (25.0 months [95% CI 25.0–not estimable] vs 20.3 months [16.9–not estimable]; P = 0.0001).
Quality of life
Further evidence of a palliative benefit from abiraterone comes from analyses of the prospective data for patient-reported pain and functional status from the phase III COU-AA-302 trial described above.61 Pain was assessed with the Brief Pain Inventory Short Form questionnaire and HRQoL was measured with the Functional Assessment of Cancer Therapy-Prostate (FACT-P) questionnaire. At a median follow-up of 22.2 months, median time to progression of mean pain intensity was longer in patients assigned to abiraterone plus prednisone (26.7 months [95% CI 19.3–not estimable]) than in those assigned to placebo plus prednisone (18.4 months [14.9–not estimable]; HR = 0.82, 95% CI 0.67–1.00; P = 0.0490). Median time to HRQoL deterioration was longer in patients assigned to abiraterone plus prednisone than in those assigned to placebo plus prednisone as assessed by the FACT-P total score (12.7 months [95% CI 11.1–14.0] vs 8.3 months [7.4–10.6]; HR = 0.78, 95% CI 0.66–0.92; P = 0.003),61 further substantiating overall benefit from abiraterone.
INHIBITORS OF ANDROGEN SYNTHESIS IN DEVELOPMENT
Orteronel
CYP17 is a bifunctional enzyme with both 17α-hydroxylase and 17,20-lyase activity. While synthesis of mineralocorticoids and the glucocorticoid corticosterone is not dependent on CYP17, synthesis of glucocorticoids and androgens requires CYP17 activity. 17α-hydroxylase is necessary for synthesis of 17α-hydroxyprogesterone, the precursor of most glucocorticoids, including cortisol. 17,20-lyase is required to trim 21 carbon steroids, including the progestins, to 19-carbon steroids such as the androgens DHEA and androstenedione. Selective inhibition of 17,20-lyase therefore offers the promise of inhibiting androgen synthesis while permitting upstream synthesis of glucocorticoids thereby blunting ACTH feedback that drives mineralocorticoid synthesis when CYP17 is blocked.62
Based on the potential advantages of a CYP17 inhibitor with increased 17,20-lyase lyase specificity, medicinal chemists at Takedea developed nonsteroidal imidazole derivatives with increased 17,20-lyase activity compared to 17α -hydroxylase activity and that would also have high specificity for CYP17,20 over other CYP enzymes (such as CYP 3A4) that are crucial for drug metabolism.63 In addition, nonsteroidal drugs would not be expected to bind to the AR in their own right. Based on considerations of potency and specificity orteronel (also known as TAK-700) was identified as a lead compound for development. In vitro studies demonstrated that orteronel inhibited human 17,20-lyase and 17α-hydroxylase with IC50 of 140 and 760 nmol l−1, respectively. IC50 values for abiraterone were 27 and 30 nmol l−1 respectively and IC50 values for ketoconazole were 110 and 580 nmol l−1. With regard to specificity for CYP17, orteronel had no activity against CYP 11 while abiraterone had modest activity (IC50 = 600 nmol l−1) and ketoconazole had notable off target effects with IC50 = 270 nmol l−1.64
Phase I/II studies
A phase I/II trial of orteronel in patients with in mCRPC demonstrated that orteronel has a good safety profile across several doses.65 The phase I portion of the trial revealed no dose-limiting toxicities. Patients received five different dose levels of orteronel orally twice daily. Decline in PSA was noted in all patients who received 300 mg or more of orteronel. In the phase II portion of the study 4 dose levels were evaluated in cohorts of approximately 24 patients each: (1) 300 mg orteronel twice daily, (2) 400 mg orteronel and 5 mg prednisone both given twice per day, (3) 600 mg orteronel and 5 mg prednisone both given twice per day and (4) 600 mg orteronel daily. After 12 weeks of treatment, PSA decreases of ≥50% from baseline in patients from these four cohorts were 63%, 50%, 41%, and 60%, respectively. Fatigue (76% overall, 12% Grade 3/4), nausea (47% overall), and constipation (38% overall) were the most common adverse events noted. Despite the putative increase in CYP 17,20 specificity, adverse effects associated with mineralocorticoid excess were observed in some patients, with 8% of subjects having Grade 3 hypokalemia.
A second phase I/II study (NCT01084655) was conducted to evaluate orteronel (200 mg or 400 mg given twice daily) in combination with docetaxel given every 3 weeks at 75 mg m−2 and prednisone 5 mg twice pre day, in patients with mCRPC.66 The combination was generally well-tolerated with no dose-limiting or unexpected toxicities reported. PSA decreases of ≥ 50% and 90% occurred in 86% and 36% of patients. The phase II portion of the study using orteronel 400 mg twice daily has been completed and results are pending. Ultimately, it remains to be seen if an improvement in OS can be obtained by combining docetaxel with CYP17 inhibitors or if using these two agents sequentially is preferable.
Phase III studies
Two phase III trials evaluating orteronel in patients with metastatic CRPC have completed enrollment. In ELM PC-5 (NCT01193257) patients who had received prior docetaxel chemotherapy were randomized in a 2:1 ratio to orteronel 400 mg plus prednisone 5 mg, both given twice per day or to placebo plus prednisone 5 mg twice per day. The primary endpoint of this study is OS. Planned enrollment was 1083 subjects. In July 2013, Takeda Pharmaceutical Company announced that it unblinded ELM PC-5 based on the recommendation of the Independent Data Monitoring Committee after a prespecified interim analysis indicated that orteronel plus prednisone would likely not meet the primary endpoint of improved OS compared to the control arm (HR = 0.886, P = 0.18976).67 The interim analysis did show an advantage for orteronel plus prednisone versus placebo plus prednisone for the secondary endpoint, rPFS (HR = 0.775, P = 0.00038). In addition, no safety concerns were noted. Approximately 25% of patients in the study went on to receive abiraterone plus prednisone or the anti-androgen enzalutamide at the time of disease progression as these agents received FDA approval mid-way through ELM-5 accrual, potentially confounding the primary endpoint. Based on the encouraging improvement in PFS, the failure of this trial to meet its primary endpoint did not impact accrual to other ongoing trials with orteronal.
A second phase III trial, ELM PC-4 (NCT01193244) randomized chemotherapy-naive patients with mCRPC to orteronel 400 mg plus prednisone 5 mg, both given twice per day or to placebo plus prednisone 5 mg twice per day. The primary endpoints of the study are OS and rPFS with a planned enrollment of 1454 subjects. Secondary endpoints include PSA decrease of 50% at 12 weeks, changes in CTC count at 24 weeks, and time to pain progression. Accrual has been reached and the results are pending.
Ongoing studies
Several studies are now ongoing or completed evaluating orteronel without concomitant prednisone based on the increased selectivity of orteronel for CYP17, 20 lyase activities over CYP17 hydroxylase. Preliminary results of a phase II trial, in which 39 patients with nonmetastatic CRPC and rising PSA were treated with ortenonel 300 mg twice daily without concomitant steroid administration (NCT01046916) demonstrated that treatment without prednisone was feasible.68 Adverse events included fatigue (62%), diarrhea (38%), and hypertension (38%). The most common adverse events of Grade 3 or higher were hypertension (15%), dyspnea (8%), and fatigue, hypokalemia and pneumonitis (each 5%). One patient was treated with corticosteroid replacement for laboratory findings consistent with a hypoadrenal state. The drug was active with 76% and 32% experiencing a decline in PSA of ≥ 50% and 90%, respectively at 3 months. Median time to PSA progression was 14.8 months. Orteronel administered without prednisone suppressed T by 87%–89% and the adrenal androgen DHEA by 85%–89%.
In SWOG1216 (NCT01809691), subjects with hormone sensitive, newly diagnosed metastatic prostate cancer are randomized to ADT with the LHRH agonist leuprolide plus orteronel 300 mg twice per day or to leuprolide plus the anti-androgen bicalutamide. 1486 subjects will be enrolled to test the hypothesis that superior androgen blockade provided by orteronel compared with bicalutamide will result in a 25% improvement in OS. RTOG1115 is a phase III clinical trial currently accruing patients with high-risk, locally advanced prostate cancer. This study tests dose-escalated radiation therapy (ex. 79.2 Gy using external beam radiation) combined with either standard ADT (LHRH agonist for 24 months plus bicalutamide until the end of radiation) or enhanced ADT (LHRH agonist plus orteronel 300 mg twice per day without concomitant steroids (NCT01546987).
VT-464
VT-464, developed by Viamet Pharmaceuticals is small bioavailable molecule reported as a selective oral CYP17 inhibitor that preferentially inhibits the 17,20-lyase reaction over 17α-hydroxylase.69 In vitro studies of VT-464 demonstrated IC50 = 69 nM for 17,20-lyase and 670 nM for 17α-hydroxylase. In the same assay abiraterone showed IC50 values of 15 nM and 2.5 nM for the 17,20-lyase and 17α-hydroxylase.69 In experiments performed in adult, castrate, male rhesus monkeys the increase in specificity for CYP 17 lyase versus hydroxylase translated into the absence of any change in cortisol levels with administration of VT-464.70 By contrast, abiraterone administration resulted in significant cortisol suppression (P < 0.005) compared with vehicle with resultant increase in steroids upstream from abiraterone.71 In male monkey both VT-464 and abiraterone effectively suppressed T levels by 90% after a single subcutaneous injection.72 VT-464 was active against LnCAP prostate cancer subcutaneous xenografts with the degree of growth inhibition comparable to surgical castration.72 VT-464 is being evaluated in a phase I/II study being performed in the US and Europe in patients with mCRPC and no prior chemotherapy (NCT02012920).
GALETERONE
Galeterone (also known as VN/124-1, TOK-001) developed by Tokai Pharmaceuticals, Cambridge, MA is an oral, steroidal 17,20-lyase inhibitor.73,74 In vitro studies of galeterone activity inhibiting human CYP17 demonstrated a 3.2-fold selectivity for the 17,20-lyase (IC50 = 23 nmol l−1) versus 17α-hydroxylase (IC50 = 73 nmol l−1).75 In this assay abiraterone was a more potent 17,20-lyase (IC50 = 12 nmol l−1) and hydroxylase (IC50 = 7 nmol l−1) inhibitor, but was less selective inhibitor for 17,20-lyase (lyase: hydroxylase selectivity = 0.6). Orteronel was a more potent CYP 17 lyase (IC50 = 64 nmol l−1) and 17α- hydroxylase (IC50 = 348 nmol l−1) inhibitor, but was the most selective inhibitor for 17,20-lyase (lyase: hydroxylase selectivity = 5.4).75 In a cell-based assay, galeterone, abiraterone, and orteronel all inhibited T synthesis ≥ 94% at 1 μmol l−1. However, at this concentration, galeterone produced minimal changes in cortisol (decreased 14%) while abiraterone and orteronel reduced cortisol by 91% and 70%, respectively, potentially leading to a clinical increase in ACTH and resultant symptoms of mineralocorticoid excess.75 Additional studies suggest that galeterone may block translation of the AR through inhibition of cap-dependent translation.74,76 Galeterone also binds to the AR and blocks AR transactivation with a 10-fold tighter binding for wild type AR (galeterone IC50 = 405 nm) compared bicalutamide (IC50 = 4300 nm).77 However, unlike bicalutamide, galeterone had no agonist activity toward mutated AR. In mouse Los Angeles Prostate Cancer-4 (LAPC-4) xenografts galeterone demonstrated a statistically significant reduction in tumor growth compared to abiraterone.78 It is possible that the combined mechanisms of action observed in galeterone may contribute to overcoming resistance mechanisms observed in other agents that exhibit CYP17-inhibitor properties.
ARMOR1 (NCT00959959), a phase I dose escalation study, was conducted in chemotherapy-naive subjects with both metastatic and nonmetastatic CPRC with the goal of evaluating safety and preliminary efficacy of galeterone.79 Forty nine chemotherapy-naive patients with CRPC were treated with one of eight doses ranging from 650 to 2600 mg every day. Across all dose levels, after 12 weeks, 22% of patients achieved PSA reduction of ≥ 50%. Maximum tolerated dose was not reached in this study. Fatigue (37%), aspartate transaminase/alanine transaminase elevation (33%/31%), nausea (29%), diarrhea (27%), and pruritus (25%) constituted the most common reported adverse events. Liver function tests abnormalities occurred in a total of 15 of 49 patients who were generally asymptomatic. Patients with Grade 3 liver abnormalities were successfully re-challenged after a dose interruption without recurrent liver function tests abnormalities. No evidence of adrenal mineralocorticoid excess was noted. Based on ARMOR1 data, galeterone received fast-track designation from FDA for the potential treatment of mCRPC. Due to a significant food effect resulting in increased drug exposure in the fed state, the galeterone was reformulated into a tablet, eliminating the food effect and providing more consistent bioavailability.80
ARMOR2 (NCT01709734), a phase II trial, that is currently enrolling subjects, is designed to evaluate galeterone in 144 patients with progressive CRPC. The primary endpoints of the study are reduction in PSA levels and safety. This trial will be split into two parts. Part 1 is designed to confirm the phase II dose of galeterone and to select a target patient population for further development. Patients will be enrolled into one of three cohorts depending on prior treatment (1) no prior CYP17 inhibitor or enzalutamide, (2) abiraterone refractory prostate cancer, (3) enzalutamide refractory prostate cancer. Part 2 will be an expansion of the dose and patient population selected in Part 1.
OVERCOMING RESISTANCE TO ABIRATERONE
Understanding the mechanisms of resistance to CYP17 inhibitors is essential for optimizing outcomes in patients with CRPC (Figure 2). With the approval of abiraterone, preclinical and clinical data continue to inform our understanding of resistance mechanisms and the design of clinical trials to overcome resistance to abiraterone (Table 1). In the majority of patients progressing during treatment with abiraterone, PSA rise is an early indicator of ultimate clinical progression, occurring 5 months before radiographic progression in the COU-AA-302 study.47 The increasing PSA implies persistence of AR signaling as the gene for PSA is exclusively regulated by the AR.9
Figure 2.
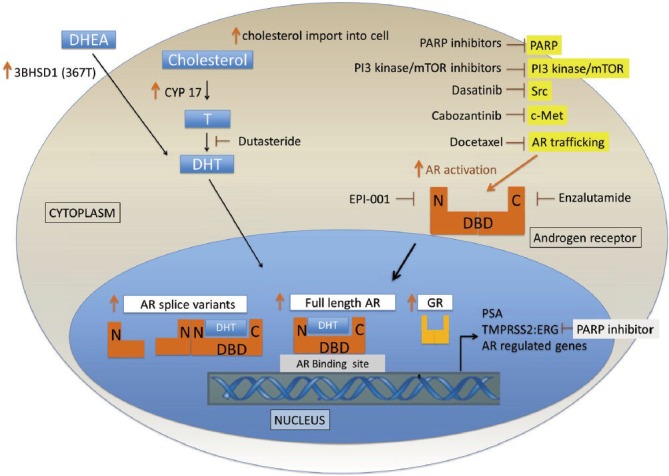
Mechanisms of resistance to CYP17 inhibitors. Androgen receptor (AR) ligand dependent mechanisms of resistance include increase of intratumoral androgen concentrations through increase in CYP17, increase in cholesterol import into the cell, acquired mutation in the androgen synthesis enzyme 3BHSD. Ligand independent activation may occur through induction of AR splice variants, through alterations of co-factors modulating AR function and potentially through activity of the GR. N: N-terminal domain of androgen receptor; C: C-terminal domain of androgen receptor; DBD: DNA binding domain.
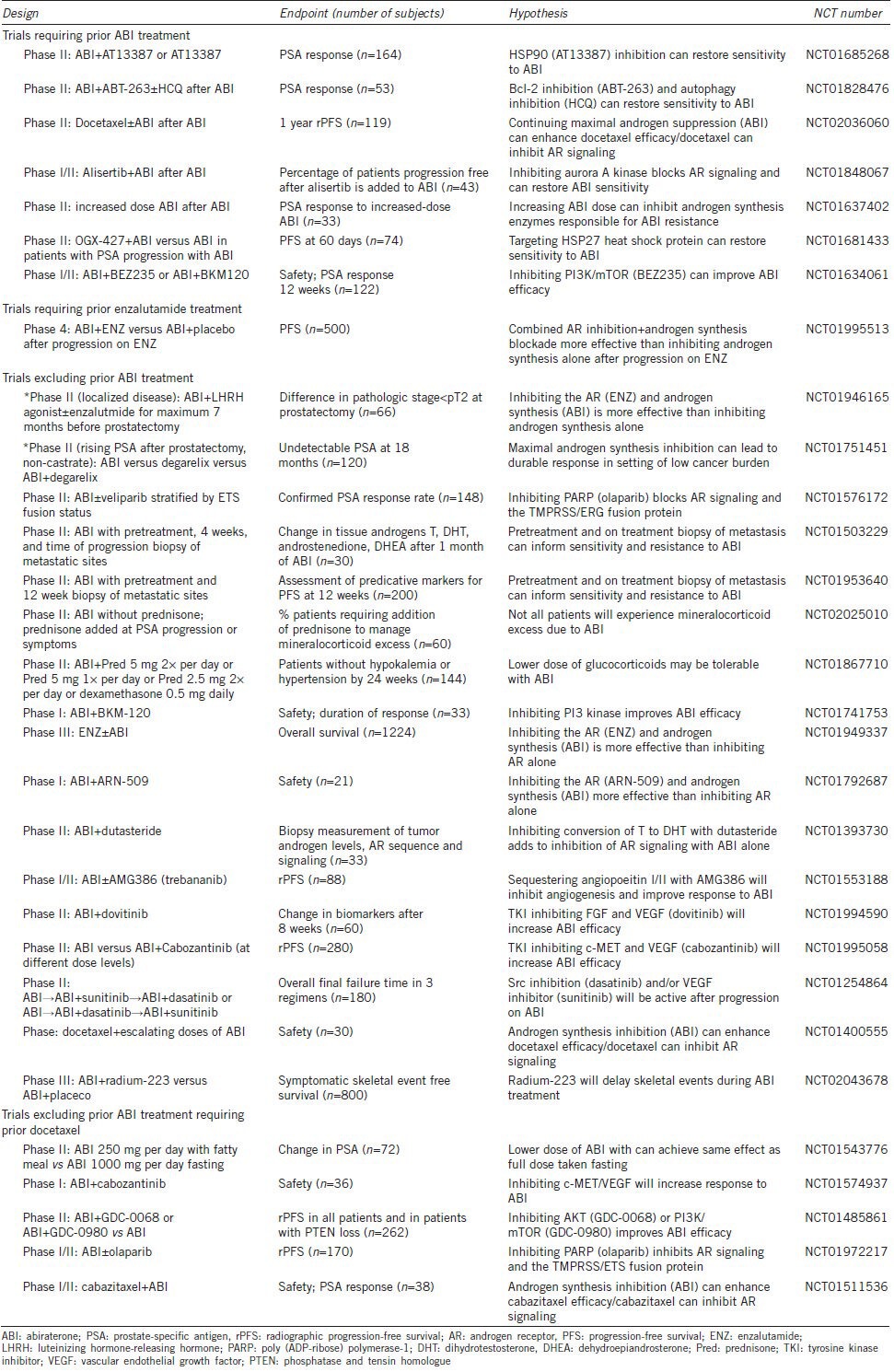
Table 1.
Planned/ongoing clinical trials to optimize use or overcome resistance to Abiraterone in patients with metastatic CRPC (or in other disease states as noted with*)
Ligand dependent mechanisms of resistance
Emerging evidence is converging around several mechanisms that may contribute to persistence of androgen signaling despite treatment with CYP 17 inhibitors.9,81 Broadly speaking, tumors may adapt through the increase of intratumoral androgen concentrations, alteration of the AR itself (through a change in either its structure or number), or through alterations of co-factors modulating AR function.
In support of de novo DHT synthesis from cholesterol as a mechanism of abiraterone resistance, in a xenograft prostate cancer model, treatment with abiraterone lead to decreased AR activity followed by marked increase in CYP17 expression in the relapsing tumors.28 Furthermore, Mostaghel et al.82 demonstrated that treatment of two LuCap prostate cancer xenografts with abiraterone resulted in induction of CYP17 and other genes involved in the synthesis of intratumoral androgens. In addition to de novo synthesis of DHT from cholesterol, tumors may also adapt to CYP17 inhibition through efficient utilization of available androgen precursors that may be present at low levels despite CYP17 inhibition.81,83 Using ultrasensitive techniques, it is possible to detect low levels of androgen metabolites in the urine of patients receiving abiraterone.57 In addition, CRPC cells have adapted mechanisms to efficiently import residual androgens from the circulation.84,85,86
The potential significance of residual androgens in mediating resistance to abiraterone has been highlighted by the discovery of an adaptive mutation in an essential enzyme for DHT synthesis.87 3 beta hydroxysteroid dehydrogenase type 1, (3BHSD1) is a catalyst in the initial rate limiting step for the conversion of DHEA to DHT through an efficient three-step synthetic pathway, the 5-alpha androstanedione pathway, in which DHT is synthesized while bypassing synthesis of T as in intermediate step.25,88 In CRPC cell lines and two autopsy specimens this pathway was the dominant mechanism for DHT production.89 Sharifi et al. identified an acquired somatic mutation in 3BHSD1 (367T) containing a point mutation at nucleotide position 1245 resulting in exchange of asparagine for threonine at amino acid 367.87 This mutation confers resistance of 3BHSD1 to ubiquitination and degradation resulting in increasing metabolic flux from DHEA to DHT, thereby sustaining DHT concentrations in orchiectomized mice. CRPC tumors from 3 of 25 men with germ line homozygous wild type 3BHSD1 had acquired a somatic mutation for 3BHSD1 (367T). In two of eight prostate cancer LAPC4 enografts, treatment with abiraterone resulted in acquisition of the 3BHSD1 (367T) mutation while no mutations were acquired in the absence of abiraterone.
Discovery of the 3BHSD1 (367T) mutation suggests that in at least some cases, increased steroidogenesis causes abiraterone resistance and that targeting multiple steps in the DHT synthesis pathway may be beneficial. To that end, trials targeting steroidogenesis at multiple levels are currently in process. Recent preclinical studies demonstrated that abiraterone can inhibit 3BHSD1, at concentrations of approximately 1 μmol l−1.83 As previously noted, in the initial phase I studies, the recommended abiraterone dose of 1000 mg per day administered in the fasting state, was chosen based on the ability of this dose of abiraterone to maximally de-repress ACTH, which is normally suppressed by cortisol in the absence of abiraterone. However, the ability of abiraterone to inhibit other sterodiogenic enzymes at higher levels was not explored. Furthermore, in the phase I trial doses of up to 2000 mg per day were tolerated without significant toxicity.37 NCT 01637402 is a phase II trial evaluating high dose abiraterone, given at 1000 mg twice daily in patients who have experienced disease progression while being treated with the standard dose of abiraterone. An alternative approach to increasing abiraterone concentration is drug administration with a high fat meal37,38 leading to concentrations in excess of 1 μmol l−1. However, concerns about inter-patient drug level variability, ensuring adequate drug exposure, and patient safety when taking abiraterone with a high fat meal have been noted.90 Currently, a phase II trial is ongoing evaluating the safety and efficacy of administering low dose (250 mg) abiraterone with a fatty meal (NCT01543776). Another approach to inhibiting DHT synthesis was evaluated in a phase II trial combining CYP17 inhibition with 5-alpha reductase inhibition using dutasteride (NCT01393730). The primary end point of this study is a tissue based analysis of the AR in serial biopsies while the secondary end points include assessment of serum levels of T, DHT, and androgen precursors at baseline and at progression as well as time to radiographic and PSA progression.
In addition to T and dihydrotestosterone, other ligands may also activate the AR thereby mediating resistance to inhibitors of CYP17. Richards demonstrated that wild-type AR and mutant AR can be activated by the glucocorticoids typically administered with abiraterone in order to dampen production of ACTH.91 In addition, the mineralocorticoid antagonist eplerenone has the ability to bind the AR.91 Finally, other potential activators of the AR include steroids produced upstream of CYP17 binding to a mutated AR.92,93,94
The interaction between the AR and kinase signaling pathways suggests additional potential targets for overcoming resistance to abiraterone therapy. Src kinase is a drugable target that has been implicated in AR activation,95 and potentially in abiraterone resistance.96 Dasatinib, a Src kinase inhibitor, is currently being evaluated in combination with abiraterone in a phase II trial to overcome resistance to abiraterone (NCT01254864). Poly (ADP-ribose) polymerase-1 (PARP-1) is nuclear enzyme that functions as DNA repair protein and transcriptional regulator. Recently, PARP-1 has been identified as a critical promoter of AR function97 as well as ETS gene-mediated transcription.98 Clinical trials are currently underway evaluating the potential for PARP inhibitors to augment the efficacy of abiraterone by inhibiting AR function and activity of the AR target TMPRSS2: EGR fusion protein, found in the majority of prostate cancers. The PI3kinase-AKT signaling pathway may also play a critical role in AR regulation. In PTEN-negative prostate cancer with resultant activation of the PI3K-AKT-mTOR–signaling pathway, pharmacologic inhibition of mTORC1 (everolimus) or PI3K and mTORC1/2 (BEZ225) resulted in reciprocal increase in AR protein levels and AR signalling.99 Conversely, inhibition of AR led to increased activation of AKT due to down regulation of the AKT phosphatases PHLP99 and INPP4B.100 These studies provide a strong rationale for a clinical trial with dual AR/PI3K pathway inhibition. NCT01634061 is a phase I trial evaluating the safety of abiraterone with prednisone plus the PI3K/mTOR inhibitor BEZ235 or the panPI3 kinase inhibitor BKM-120 in patients previously treated with abiraterone. NCT01485861 is a phase I/II trial evaluating abiraterone in combination with PI3Kinase/mTOR inhibition (GDC-0980) or AKT inhibition (GDC-0068) in patients with prior chemotherapy, but without prior abiraterone therapy.
Alteration in androgen receptor expression
Experimental data suggests that increase in AR number is another, and perhaps the most immediate, method by which the cell compensates for decreased AR ligand as a result of treatment with testicular suppressive therapy and abiraterone. In an experimental model of abiraterone resistance, resistant tumors demonstrated increase in the expression of full length AR as well as an increase in AR variants lacking the c-terminal domain responsible for ligand binding.82 In a similar, reciprocal manner, cancer cells may overcome potent inhibition of the AR by the second generation AR inhibitor enzalutamide through compensatory increases in androgen levels in the blood and bone marrow.101,102 These data suggest that dual inhibition of androgen signaling by combining a CYP17 inhibitor with a potent AR antagonist may overcome resistance to single agent CYP17 inhibitors. Preliminary data suggests that the combined inhibition of androgen synthesis and the AR may be able to overcome the compensatory pathways induced by either therapy alone. Efstathiou et al.103 treated 57 patients with metastatic CRPC with the standard dose of abiraterone (1000 mg per day) plus standard dose of enzalutamide (160 mg per day) and demonstrated that with combined therapy 72% of patients achieved a 50% of greater decrease in PSA. Notably, 48% of patients achieved a 90% or greater PSA decline and 10% of patients achieved undetectable PSA. To determine if the combination of abiraterone plus enzalutamide provides a significant increase in clinical benefit, the Alliance-National Cancer Institute cooperative group is conducting a phase III study of enzalutamide at standard dose versus the combination of enzalutamide plus abiraterone (both at full dose) in patients with previously untreated CRPC with OS as the primary endpoint (NCT01949337).
Ligand independent mechanisms of resistance
In addition to ligand dependent mechanisms of resistance to CYP17 inhibition, resistance may result through ligand independent mechanisms as well. AR splice variants result from loss of the c-terminal androgen binding domain of the receptor.104 Multiple different splice variants have been identified in prostate cancer cell lines and clinical specimens. The clinical significance of any given variant as opposed to variants with a different DNA deletion remains unclear. In an experimental model, treatment with abiraterone lead to the production of the full length AR as well as the AR7 and the ARdel567es splice variants.82 Novel drugs are being developed to target emergent splice variants. EPI-001 is a drug that disrupts activity of the AR N-terminal domain, thereby maintaining activity against AR splice variants with c-terminal ligand-binding domain deletions as well as inhibiting full-length AR.105
In addition to ligand dependent and ligand independent mechanisms of AR activation, induction of glucocorticoid receptor (GR) expression has recently been described as a frequent occurrence in preclinical models of enzalutamide resistance and patients with enzalutamide-resistant prostate cancer.106 After 8 weeks of treatment with enzalutamide, the percentage of GR-positive cells was significantly higher in poor compared to good responders (29% vs 10%, P = 0.02). Through transcriptome analysis the authors demonstrate that GR can bind to > 50% of the AR binding sites in enzalutamide resistant cells, including the AR target genes a kallikrein-3 (PSA) and TMPRSS2. GR activation by dexamethasone was sufficient to confer enzalutamide resistance in prostate cancer cell lines treated with enzalutamide and could be reversed with a glucocorticoid antagonist. At this point, it is unknown if increased GR expression is a relevant mechanism of resistance to androgen synthesis inhibitors such as abiraterone which is approved for use in combination with prednisone; however, this question clearly needs further investigation, particularly as trials exploring the efficacy of combined abiraterone, prednisone, and enzalutamide proceed.
CURRENT CLINICAL CONSIDERATIONS WITH CYP17 INHIBITORS
Currently, abiraterone is approved for use in patients with metastatic CRPC both before and after docetaxel-based chemotherapy. In addition to abiraterone six other agents are now improved for treatment of metastatic CRPC including the AR antagonist enzalutamide101 (currently approved for use after docetaxel chemotherapy, with recent results of the randomized phase 3 trial demonstrating improved OS with enzalutamide given prior to chemotherapy107), the immunotherapy sipuleucel-T108 (approved for patients with asymptomatic or minimally symptomatic CRPC), two taxane-based chemotherapies with a survival benefit (docetaxel109 and cabazitaxel110), mitoxantrone111 (approved for palliation of pain) and the radionucleotide radium-223112 (for patients with symptomatic bone metastases, demonstrating both improvement in pain and improvement in survival). Optimal sequencing of these medications to maximize benefit remains an active area of investigation.
Sequence and combination strategies with sipuleucel-T
For patients with asymptomatic or minimally symptomatic metastatic CRPC, a phase III trial of the autologous cellular immune therapy sipuleucel-T versus placebo demonstrated an improvement in OS with sipuleucel-T (25.8 months vs 21.7 months P = 0.032).108 As this population can also benefit from treatment with abiraterone or enzalutamide studies have been designed to define the immune implications of maximal androgen blockade with these agents. A practical consideration in patients appropriate for treatment with sipuleucel-T and abiraterone plus prednisone, is if prednisone 5 mg twice/day impairs the production or efficacy of sipuleucel-T.
In a randomized phase II study 64 subjects were randomized to treatment with sipuleucel-T plus abiraterone and prednisone starting 1 day after the first sipuleucel-T infusion or at 10 weeks following the first sipuleucel-T infusion. The primary endpoint of this study was to determine if corticosteroids interfered with sipuleucel-T product production, as measured by CD54 upregulation in the product.113 CD54 upregulation has previously been shown to be an effective means to assess the biological activation of antigen presenting cells (APCs) and specifically the potency of sipuleucel-T, an APC manufactured to elicit a prostate tumor specific immune response.114 The authors reported no significant differences in median cumulative APC activation or APC count and similar profiles of prostate antigen-specific humoral and cellular immune responses were generated in patients on both arms (P > 0.05). These results suggest that regardless of whether or not subjects received abiraterone and prednisone concomitant with sipuleucel-T, an equally effective sipuleucel-T could be generated which could then generate an effective immune response in the subject. These results are consistent with the lack of significant effect of corticosteroids on the immune response seen in the context of influenza vaccination.115 Ultimately, while this study provides indirect evidence that no clinical antagonism exists between sipuleucel-T and abiraterone plus prednisone, this study was not designed to reject the possibility of diminished sipuleucel-T efficacy when manufactured and administered to patients receiving low dose glucocorticoids. In this regard, CYP17 inhibitors that are specific for the 17,20-lyase, such as VT-464 and can safely be administered without corticosteroids may obviate the need for further investigation of the interaction between corticosteroids and immune therapy.
Additional studies are also ongoing to determine if maximal inhibition of the androgen axis can augment the immune response and synergize with immune therapies such as sipuleucel-T. A randomized phase II study evaluating the optimal sequencing of sipuleucel-T and ADT in noncastrate, biochemically recurrent prostate cancer was recently reported.116 Patients received sipuleucel-T followed by ADT starting 2 weeks after the final dose of sipuleucel-T or ADT for 12 weeks prior to sipuleucel-T. Both arms received 1 year total of ADT. There were 34 patients in each arm. This study demonstrated a trend toward a stronger T-cell immune response to prostate antigen (as measured by ELISPOT assay) when ADT preceded treatment with sipuleucel-T (Arm 2 vs Arm 1–40.5 vs 12.8 spots; P = 0.086). In addition, higher levels of serum cytokines in Arm 2 vs Arm 1 (P < 0.05) and increased T cell activation was when ADT preceded sipuleucel-T. This study suggests that ADT may induce cell death and antigen release thereby augmenting an immune response, at least in noncastrate subjects. However, in men with CRPC, a phase II study of the viral immune therapy Prostvac came to the opposite conclusion.117 Median survival was 6.2 years in patients with nonmetastatic CRPC treated with vaccine prior to treatment with a second line hormonal therapy (nilutamide) vs 3.7 years in men who started on nilutamide prior to vaccine (P = 0.045). Future studies in patients with metastatic CRPC to further elucidate the interaction between immune stimulation and maximal androgen signaling inhibition are planned such as a phase II trial of enzalutamide and concurrent sipuleucel-T versus enzalutamide starting 10 weeks after the first infusion of sipuleucel-T (NCT01981122).
Potential cross-resistance with taxanes
Optimal sequencing of abiraterone must also take into account data suggesting that there is cross resistance between these androgen pathway inhibitors and taxane-based chemotherapies. Emerging evidence suggests that taxanes such as docetaxel and cabazitaxel inhibit AR mediated gene expression.118,119 Intact microtubule function is required for translocation of the AR to the nucleus, with taxanes causing accumulation of cytoplasmic AR.120,121 Taxanes also increase accumulation of the FOXO1 transcription factor in the nucleus where FOXO1 functions as a potent inhibitor of AR transcriptional activity.122 In vitro studies of docetaxel, cabazitaxel, and enzalutamide in cells lines with or without abiraterone resistance demonstrate impaired efficacy of the taxanes and enzalutamide in the abiraterone resistant cells.123 Similar cross resistance with taxanes and abiraterone was seen in enzalutamide resistant cells. Clinically, a retrospective analysis of docetaxel treatment after abiraterone treatment as part of a single institution phase II study suggested that docetaxel postabiraterone was less effective than docetaxel preabiraterone.124 Patients receiving docetaxel after abiraterone had median OS of 12.5 months and a PSA response rate (>50% decline) of 26%. In the TAX-327 docetaxel registration trial, conducted before the advent of abiraterone, OS was 19 months for treatment with docetaxel with PSA response of 48%. Likewise, PSA response to abiraterone before docetaxel in the COU-AA-302 study was 62% vs 30% after in the COU-AA-301 trial after docetaxel. At this point, randomized studies of abiraterone followed by docetaxel or docetaxel followed by abiraterone with an OS survival endpoint to conclusively address optimal sequencing have not been conducted. Such a trial, prospectively incorporating clinical and molecular factors is essential for determining if the current paradigm of using the relatively well-tolerated androgen signaling inhibitors prior to chemotherapy in patients with CRPC should be applied to all patients regardless of clinical factors that may suggest a poor outcome.
Predictors of cross-resistance with androgen receptor antagonists
Finally, predictors of cross resistance between androgen synthesis inhibitors (i.e. abiraterone) and AR antagonists (i.e. enzalutamide) must be considered in future trials. The relatively limited activity of abiraterone after treatment with enzalutamide, or the converse is beginning to be defined in retrospective studies. In a report of 38 patients receiving abiraterone after treatment with docetaxel and enzalutamide in the enzalutamide post docetaxel-A Study Evaluating the Efficacy and Safety of the Investigational Drug MDV3100 (AFFIRM) study, a PSA decrease of > 50% was seen in three patients (8%) with PFS = 2.7 months. By comparison, in the same cohort, of 16 patients who received placebo in the AFFIRM trial followed by abiraterone, PSA decline of > 50% occurred in 29% of patients with PFS = 6.5 months.125 Of 17 patients with insensitivity to enzalutamide, one (6%) had > 50% decrease in PSA with abiraterone. In a second 30 patient cohort treated with abiraterone after progression with enzalutamide and docetaxel similarly poor abiraterone activity was seen was PSA decline > 50% in 3% of patients (compared to 29% PSA response rate in the COU-AA-301 study). Of nine patients with minimal or no PSA decline with enzalutamide, two (22%) had a > 30% decline with abiraterone.126
Modest benefit has likewise been noted when enzalutamide is administered after abiraterone and docetaxel. In 39 patients treated with docetaxel and abiraterone followed by enzalutamide at progression, PSA decline > 50% was seen in 5/39 (13%) of patients. Median duration of treatment was 2.9 months. In 22 patients with <50% PSA decline with abiraterone, two patients (9%) had a confirmed >30% PSA decrease with enzalutamide.127 In a second study of 35 patients, enzalutamide after abiraterone and docetaxel caused PSA decline >50% in 10/35 (29%) patients. Three of 19 (16%) patients with <50% PSA decrease with abiraterone had a > 50% PSA decrease with enzalutamide.128
Taken together, these four early reports of limited efficacy when two androgen pathway inhibitors are used in sequence, serve as a cautionary reminder of the need to consider all treatment options, including chemotherapy or radium-223, in patients with progression with a CYP17 inhibitor, particularly in patients with symptomatic or extensive metastatic disease. Future studies will need to be designed to assess pathways of AR inhibition escape for individual patients potentially using tumor biopsies or CTC analysis to gather data needed to make rational therapeutic choices.129
SUMMARY
The development of abiraterone, a potent inhibitor of androgen synthesis, through blockade of the CYP17 enzyme has rapidly redefined the therapeutic landscape for the treatment of prostate cancer. Detailed analysis of the phase III trials leading to abiraterone approval has provided important insights into optimal use of this medication though development of biomarkers predictive of clinical response, and clinical indicators of benefit and or potential side-effects. Second generation inhibitors of androgen synthesis hold the promise of overcoming the need for long-term corticosteroid administration when taking abiraterone. Despite the remarkable activity of CYP17 inhibitors in patients with CRPC, they are not ultimately a curative therapy in this setting and additional translational studies to clarify mechanism of resistance are urgently needed. Additional challenges include defining the optimal sequence of CYP17 inhibitors and other AR pathway inhibitors, and developing combination therapies that can overcome resistance in a tolerable manner. Incorporation of CYP17 into earlier disease states such as the treatment of rising PSA after prostatectomy may also help to push more patients over the threshold of cure (NCT01751451). Prioritization of clinical trials and obtaining resources to answer fundament translational questions remain ongoing challenges. Ultimately, development of CYP17 inhibitors represents a triumph of data driven science over our assumptions about the definition of castrate resistance, opening up new vistas for treatment of the prostate cancer.
COMPETING INTERESTS
All authors declare no competing interests.
ACKNOWLEDGMENTS
This work is supported by a grant from the National Cancer Institute (P30CA072720).
REFERENCES
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Center MM, Jemal A, Lortet-Tieulent J, Ward E, Ferlay J, et al. International variation in prostate cancer incidence and mortality rates. Eur Urol. 2012;61:1079–92. doi: 10.1016/j.eururo.2012.02.054. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 4.Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941;1:293–7. [Google Scholar]
- 5.Singer EA, Srinivasan R. Intravenous therapies for castration-resistant prostate cancer: toxicities and adverse events. Urol Oncol. 2012;30:S15–9. doi: 10.1016/j.urolonc.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6:76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cookson MS, Roth BJ, Dahm P, Engstrom C, Freedland SJ, et al. Castration-resistant prostate cancer: AUA Guideline. J Urol. 2013;190:429–38. doi: 10.1016/j.juro.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Fang S, Liao S. Androgen receptors. Steroid- and tissue-specific retention of a 17 beta-hydroxy-5 alpha-androstan-3-one-protein complex by the cell nuclei of ventral prostate. J Biol Chem. 1971;246:16–24. [PubMed] [Google Scholar]
- 9.Nelson PS. Molecular states underlying androgen receptor activation: a framework for therapeutics targeting androgen signaling in prostate cancer. J Clin Oncol. 2012;30:644–6. doi: 10.1200/JCO.2011.39.1300. [DOI] [PubMed] [Google Scholar]
- 10.Cunha GR, Cooke PS, Kurita T. Role of stromal-epithelial interactions in hormonal responses. Arch Histol Cytol. 2004;67:417–34. doi: 10.1679/aohc.67.417. [DOI] [PubMed] [Google Scholar]
- 11.Schrecengost R, Knudsen KE. Molecular pathogenesis and progression of prostate cancer. Semin Oncol. 2013;40:244–58. doi: 10.1053/j.seminoncol.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 13.Lin C, Yang L, Tanasa B, Hutt K, Ju BG, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–83. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tian TV, Tomavo N, Huot L, Flourens A, Bonnelye E, et al. Identification of novel TMPRSS2: ERG mechanisms in prostate cancer metastasis: involvement of MMP9 and PLXNA2. Oncogene. 2013 May 27; doi: 10.1038/onc.2013.176. doi: 10.1038/onc.2013.176. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 15.Demichelis F, Attard G. A step toward functionally characterized prostate cancer molecular subtypes. Nat Med. 2013;19:966–7. doi: 10.1038/nm.3285. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med. 2013;19:1023–9. doi: 10.1038/nm.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grino PB, Griffin JE, Wilson JD. Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology. 1990;126:1165–72. doi: 10.1210/endo-126-2-1165. [DOI] [PubMed] [Google Scholar]
- 18.Askew EB, Gampe RT, Jr, Stanley TB, Faggart JL, Wilson EM. Modulation of androgen receptor activation function 2 by testosterone and dihydrotestosterone. J Biol Chem. 2007;282:25801–16. doi: 10.1074/jbc.M703268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imperato-McGinley J, Guerrero L, Gautier T, Peterson RE. Steroid 5alpha-reductase deficiency in man: an inherited form of male pseudohermaphroditism. Science. 1974;186:1213–5. doi: 10.1126/science.186.4170.1213. [DOI] [PubMed] [Google Scholar]
- 20.Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007;67:5033–41. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 21.Labrie F, Cusan L, Gomez JL, Martel C, Bérubé R, et al. Comparable amounts of sex steroids are made outside the gonads in men and women: strong lesson for hormone therapy of prostate and breast cancer. J Steroid Biochem Mol Biol. 2009;113:52–6. doi: 10.1016/j.jsbmb.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Geller J, Albert J, Loza D, Geller S, Stoeltzing W, et al. DHT concentrations in human prostate cancer tissue. J Clin Endocrinol Metab. 1978;46:440–4. doi: 10.1210/jcem-46-3-440. [DOI] [PubMed] [Google Scholar]
- 23.Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–7. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- 24.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharifi N. Minireview: androgen metabolism in castration-resistant prostate cancer. Mol Endocrinol. 2013;27:708–14. doi: 10.1210/me.2013-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharifi N, McPhaul MJ, Auchus RJ. “Getting from here to there” – Mechanisms and limitations to the activation of the androgen receptor in castration-resistant prostate cancer. J Investig Med. 2010;58:938–44. doi: 10.231/JIM.0b013e3181ff6bb8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 28.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503–13. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–15. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 30.Mitsiades N, Sung CC, Schultz N, Danila DC, He B, et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012;72:6142–52. doi: 10.1158/0008-5472.CAN-12-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Attard G, Belldegrun AS, de Bono JS. Selective blockade of androgenic steroid synthesis by novel lyase inhibitors as a therapeutic strategy for treating metastatic prostate cancer. BJU Int. 2005;96:1241–6. doi: 10.1111/j.1464-410X.2005.05821.x. [DOI] [PubMed] [Google Scholar]
- 32.Small EJ, Baron AD, Fippin L, Apodaca D. Ketoconazole retains activity in advanced prostate cancer patients with progression despite flutamide withdrawal. J Urol. 1997;157:1204–7. [PubMed] [Google Scholar]
- 33.Small EJ, Halabi S, Dawson NA, Stadler WM, Rini BI, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: a phase III trial (CALGB 9583) J Clin Oncol. 2004;22:1025–33. doi: 10.1200/JCO.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 34.Potter GA, Barrie SE, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017 alpha (17 alpha-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. J Med Chem. 1995;38:2463–71. doi: 10.1021/jm00013a022. [DOI] [PubMed] [Google Scholar]
- 35.Chan FC, Potter GA, Barrie SE, Haynes BP, Rowlands MG, et al. 3- and 4-pyridylalkyl adamantanecarboxylates: inhibitors of human cytochrome P450 (17 alpha) (17 alpha-hydroxylase/C17,20-lyase). Potential nonsteroidal agents for the treatment of prostatic cancer. J Med Chem. 1996;39:3319–23. doi: 10.1021/jm950749y. [DOI] [PubMed] [Google Scholar]
- 36.O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, et al. Hormonal impact of the 17alpha-hydroxylase/C (17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer. 2004;90:2317–25. doi: 10.1038/sj.bjc.6601879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 38.Ryan CJ, Smith MR, Fong L, Rosenberg JE, Kantoff P, et al. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol. 2010;28:1481–8. doi: 10.1200/JCO.2009.24.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–8. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryan CJ, Shah S, Efstathiou E, Smith MR, Taplin ME, et al. Phase II study of abiraterone acetate in chemotherapy-naive metastatic castration-resistant prostate cancer displaying bone flare discordant with serologic response. Clin Cancer Res. 2011;17:4854–61. doi: 10.1158/1078-0432.CCR-11-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reid AH, Attard G, Danila DC, Oommen NB, Olmos D, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28:1489–95. doi: 10.1200/JCO.2009.24.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Danila DC, Morris MJ, de Bono JS, Ryan CJ, Denmeade SR, et al. Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol. 2010;28:1496–501. doi: 10.1200/JCO.2009.25.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fizazi K, Scher HI, Molina A, Logothetis CJ, Chi KN, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012;13:983–92. doi: 10.1016/S1470-2045(12)70379-0. [DOI] [PubMed] [Google Scholar]
- 45.Ning YM, Kluetz P, Liu K. Center for drug evaluation and research: clinical review of NDA 202379. Zytiga™ (abiraterone acetate) for metastatic castration-resistant prostate cancer after prior chemotherapy. 2011 [Google Scholar]
- 46.Cai C, Balk SP. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer. 2011;18:R175–82. doi: 10.1530/ERC-10-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rathkopf DE, Smith MR, De Bono JS, Logothetis C, Shore N, et al. Long-term safety and efficacy analysis of abiraterone acetate (AA) plus prednisone (P) in metastatic castration-resistant prostate cancer (mCRPC) without prior chemotherapy (COU-AA-302) ASCO Meeting Abstr. 2013;31:5009. [Google Scholar]
- 49.Kluetz PG, Ning YM, Maher VE, Zhang L, Tang S, et al. Abiraterone acetate in combination with prednisone for the treatment of patients with metastatic castration-resistant prostate cancer: U.S. Food and Drug Administration drug approval summary. Clin Cancer Res. 2013;19:6650–6. doi: 10.1158/1078-0432.CCR-13-2134. [DOI] [PubMed] [Google Scholar]
- 50.de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14:6302–9. doi: 10.1158/1078-0432.CCR-08-0872. [DOI] [PubMed] [Google Scholar]
- 51.Danila DC, Fleisher M, Scher HI. Circulating tumor cells as biomarkers in prostate cancer. Clin Cancer Res. 2011;17:3903–12. doi: 10.1158/1078-0432.CCR-10-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Attard G, Swennenhuis JF, Olmos D, Reid AH, Vickers E, et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009;69:2912–8. doi: 10.1158/0008-5472.CAN-08-3667. [DOI] [PubMed] [Google Scholar]
- 53.Danila DC, Anand A, Sung CC, Heller G, Leversha MA, et al. TMPRSS2-ERG status in circulating tumor cells as a predictive biomarker of sensitivity in castration-resistant prostate cancer patients treated with abiraterone acetate. Eur Urol. 2011;60:897–904. doi: 10.1016/j.eururo.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryan CJ, Molina A, Li J, Kheoh T, Small EJ, et al. Serum androgens as prognostic biomarkers in castration-resistant prostate cancer: results from an analysis of a randomized phase III trial. J Clin Oncol. 2013;31:2791–8. doi: 10.1200/JCO.2012.45.4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scher H, Heller G, Molina A, Attard G, McCormack R, et al. Amsterdam: presented at: European Cancer Congress; 2013. Evaluation of a composite biomarker panel including circulating tumor cell enumeration as a surrogate for survival in metastatic castration-resistant prostate cancer. [Google Scholar]
- 56.Janssen Diagnostics LPR. New study shows circulating tumor cell enumeration-as part of composite biomarker panel-may serve as a surrogate for efficacy response in metastatic castration-resistant prostate cancer. 2013 [Google Scholar]
- 57.Attard G, Reid AH, Auchus RJ, Hughes BA, Cassidy AM, et al. Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J Clin Endocrinol Metab. 2012;97:507–16. doi: 10.1210/jc.2011-2189. [DOI] [PubMed] [Google Scholar]
- 58.Procopio G, Grassi P, Testa I, Verzoni E, Torri V, et al. Safety of abiraterone acetate in castration-resistant prostate cancer patients with concomitant cardiovascular risk factors. Am J Clin Oncol. 2013 Sep 21; doi: 10.1097/COC.0b013e3182a790ce. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 59.Pezaro C, Mukherji D, Tunariu N, Cassidy AM, Omlin A, et al. Sarcopenia and change in body composition following maximal androgen suppression with abiraterone in men with castration-resistant prostate cancer. Br J Cancer. 2013;109:325–31. doi: 10.1038/bjc.2013.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Logothetis CJ, Basch E, Molina A, Fizazi K, North SA, et al. Effect of abiraterone acetate and prednisone compared with placebo and prednisone on pain control and skeletal-related events in patients with metastatic castration-resistant prostate cancer: exploratory analysis of data from the COU-AA-301 randomised trial. Lancet Oncol. 2012;13:1210–7. doi: 10.1016/S1470-2045(12)70473-4. [DOI] [PubMed] [Google Scholar]
- 61.Basch E, Autio K, Ryan CJ, Mulders P, Shore N, et al. Abiraterone acetate plus prednisone versus prednisone alone in chemotherapy-naive men with metastatic castration-resistant prostate cancer: patient-reported outcome results of a randomised phase 3 trial. Lancet Oncol. 2013;14:1193–9. doi: 10.1016/S1470-2045(13)70424-8. [DOI] [PubMed] [Google Scholar]
- 62.Yamaoka M, Hara T, Kusaka M. Overcoming persistent dependency on androgen signaling after progression to castration-resistant prostate cancer. Clin Cancer Res. 2010;16:4319–24. doi: 10.1158/1078-0432.CCR-10-0255. [DOI] [PubMed] [Google Scholar]
- 63.Kaku T, Hitaka T, Ojida A, Matsunaga N, Adachi M, et al. Discovery of orteronel (TAK-700), a naphthylmethylimidazole derivative, as a highly selective 17,20-lyase inhibitor with potential utility in the treatment of prostate cancer. Bioorg Med Chem. 2011;19:6383–99. doi: 10.1016/j.bmc.2011.08.066. [DOI] [PubMed] [Google Scholar]
- 64.Yamaoka M, Hara T, Hitaka T, Kaku T, Takeuchi T, et al. Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J Steroid Biochem Mol Biol. 2012;129:115–28. doi: 10.1016/j.jsbmb.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 65.Agus DB, Stadler WM, Shevrin DH, Hart L, MacVicar GR, et al. Safety, efficacy, and pharmacodynamics of the investigational agent orteronel (TAK-700) in metastatic castration-resistant prostate cancer (mCRPC): updated data from a phase I/II study. ASCO Meeting Abstr. 2012;30:98. [Google Scholar]
- 66.Petrylak D, Gandhi JG, Clark WR, Heath EI, Lin J, et al. A phase I/II study of safety and efficacy of orteronel (TAK-700), an oral, investigational, nonsteroidal 17,20-lyase inhibitor, with docetaxel and prednisone (DP) in metastatic castration-resistant prostate cancer (mCRPC): updated phase II results. ASCO Meeting Abstr. 2013;31:59. [Google Scholar]
- 67.Dreicer R, Jones R, Oudard S, Efstathiou E, Saad F, et al. Results from a phase 3, randomized, double-blind, multicenter, placebo-controlled trial of orteronel (Tak-700) plus prednisone in patients with metastatic castration-resistant prostate cancer (mCRPC) that has progressed during or following docetaxel-based therapy (Elm-Pc 5 trial) ASCO Meeting Abstracts. 2014;32(4 suppl):7. [Google Scholar]
- 68.George DJ, Corn PG, Michaelson MD, Hammers HJ, Alumkal JJ, et al. Safety and activity of the investigational agent orteronel (ortl) without prednisone in men with nonmetastatic castration-resistant prostate cancer (nmCRPC) and rising prostate-specific antigen (PSA): updated results of a phase II study. ASCO Meeting Abstr. 2012;30:4549. [Google Scholar]
- 69.Eisner JR, Abbott DH, Bird IM, Rafferty SW, Moore WR, et al. VT-464: a novel, selective inhibitor of P450c17(CYP17)-17,20 lyase for castration-refractory prostate cancer (CRPC) ASCO Meeting Abstr. 2012;30:e15167. [Google Scholar]
- 70.Abbott DH, Eisner JR, Bird IM, Rafferty SW, Moore WR, et al. Plasma steroid concentrations in male Rhesus monkeys following treatment with the P450c17 (CYP17) inhibitors VT-464 and abiraterone acetate: a comparison to human 17,20-Lyase (lyase) and combined Lyase/17α-hydroxylase (hydroxylase) deficiencies. Endocr Rev. 2012;33 SAT-256 (03_MeetingAbstracts) [Google Scholar]
- 71.Eisner JR, Abbott DH, Bird IM, Rafferty SW, Moore WR, et al. Assessment of steroid hormones upstream of p450c17 (CYP17) in chemically castrate male rhesus monkeys following treatment with the CYP17 inhibitors VT-464 and abiraterone acetate (AA) Endocr Rev. 2012;33 SAT-266 (03_MeetingAbstracts) [Google Scholar]
- 72.Figg WD, Spencer SD, Pisle ST, Pressler HM, Troutman SM, et al. Activity of oral VT-464, a selective CYP17-lyase inhibitor, in the LNCaP prostate cancer xenograft. ASCO Meeting Abstr. 2012;30:4671. [Google Scholar]
- 73.Handratta VD, Vasaitis TS, Njar VC, Gediya LK, Kataria R, et al. Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model. J Med Chem. 2005;48:2972–84. doi: 10.1021/jm040202w. [DOI] [PubMed] [Google Scholar]
- 74.Vasaitis T, Belosay A, Schayowitz A, Khandelwal A, Chopra P, et al. Androgen receptor inactivation contributes to antitumor efficacy of 17{alpha}-hydroxylase/17,20-lyase inhibitor 3beta-hydroxy-17-(1H-benzimidazole-1-yl) androsta-5,16-diene in prostate cancer. Mol Cancer Ther. 2008;7:2348–57. doi: 10.1158/1535-7163.MCT-08-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jacoby DB, Williams M. Differential effects of galeterone, abiraterone, orteronel, and ketoconazole on CYP17 and steroidogenesis. ASCO Meeting Abstr. 2013;31:184. [Google Scholar]
- 76.Soifer HS, Souleimanian N, Wu S, Voskresenskiy AM, Collak FK, et al. Direct regulation of androgen receptor activity by potent CYP17 inhibitors in prostate cancer cells. J Biol Chem. 2012;287:3777–87. doi: 10.1074/jbc.M111.261933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Montgomery RB, Eisenberger MA, Rettig M, Chu F, Pili R, et al. Phase I clinical trial of galeterone (TOK-001), a multifunctional antiandrogen and CYP17 inhibitor in castration resistant prostate cancer (CRPC) ASCO Meeting Abstr. 2012;30:4665. [Google Scholar]
- 78.Bruno RD, Vasaitis TS, Gediya LK, Purushottamachar P, Godbole AM, et al. Synthesis and biological evaluations of putative metabolically stable analogs of VN/124-1 (TOK-001): head to head anti-tumor efficacy evaluation of VN/124-1 (TOK-001) and abiraterone in LAPC-4 human prostate cancer xenograft model. Steroids. 2011;76:1268–79. doi: 10.1016/j.steroids.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Taplin ME, Chu F, Morrison J, Pili R, Rettig M, et al. Cancer Res. Vol. 72. Chicago, IL. Philadelphia (PA): proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; 2012, AACR; 2012. ARMOR1: safety of galeterone (TOK-001) in a phase 1 clinical trial in chemotherapy naive patients with castration resistant prostate cancer (CRPC) nr CT.07. [Google Scholar]
- 80.Kramer WG, Vince B, McGarry C. Comparison of the pharmacokinetics (PK) of galeterone novel oral formulations. ASCO Meeting Abstr. 2013;31:e16075. [Google Scholar]
- 81.Yuan X, Cai C, Chen S, Chen S, Yu Z, et al. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2013 Jun 10; doi: 10.1038/onc.2013.235. doi: 10.1038/onc.2013.235. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–25. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li R, Evaul K, Sharma KK, Chang KH, Yoshimoto J, et al. Abiraterone inhibits 3b-hydroxysteroid dehydrogenase: a rationale for increasing drug exposure in castration-resistant prostate cancer. Clin Cancer Res. 2012;18:3571–9. doi: 10.1158/1078-0432.CCR-12-0908. [DOI] [PubMed] [Google Scholar]
- 84.Wright JL, Kwon EM, Ostrander EA, Montgomery RB, Lin DW, et al. Expression of SLCO transport genes in castration-resistant prostate cancer and impact of genetic variation in SLCO1B3 and SLCO2B1 on prostate cancer outcomes. Cancer Epidemiol Biomarkers Prev. 2011;20:619–27. doi: 10.1158/1055-9965.EPI-10-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang M, Xie W, Mostaghel E, Nakabayashi M, Werner L, et al. SLCO2B1 and SLCO1B3 may determine time to progression for patients receiving androgen deprivation therapy for prostate cancer. J Clin Oncol. 2011;29:2565–73. doi: 10.1200/JCO.2010.31.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Egan A, Dong Y, Zhang H, Qi Y, Balk SP, et al. Castration-resistant prostate cancer: adaptive responses in the androgen axis. Oncogene. 2014;40:426–33. doi: 10.1016/j.ctrv.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 87.Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074–84. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ferraldeschi R, Sharifi N, Auchus RJ, Attard G. Molecular pathways: inhibiting steroid biosynthesis in prostate cancer. Clin Cancer Res. 2013;19:3353–9. doi: 10.1158/1078-0432.CCR-12-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2011;108:13728–33. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Todd M, Meyers ML, Charnas R, Acharya M, Molina A. Fast and flawed or scientifically sound: the argument for administering oral oncology drugs during fasting. J Clin Oncol. 2012;30:888–9. doi: 10.1200/JCO.2011.39.7851. [DOI] [PubMed] [Google Scholar]
- 91.Richards J, Lim AC, Hay CW, Taylor AE, Wingate A, et al. Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: a rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res. 2012;72:2176–82. doi: 10.1158/0008-5472.CAN-11-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Veldscholte J, Berrevoets CA, Ris-Stalpers C, Kuiper GG, Jenster G, et al. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J Steroid Biochem Mol Biol. 1992;41:665–9. doi: 10.1016/0960-0760(92)90401-4. [DOI] [PubMed] [Google Scholar]
- 93.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–8. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 94.Mao HL, Zhu ZQ, Chen CD. The androgen receptor in hormone-refractory prostate cancer. Asian J Androl. 2009;11:69–73. doi: 10.1038/aja.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cai H, Babic I, Wei X, Huang J, Witte ON. Invasive prostate carcinoma driven by c-Src and androgen receptor synergy. Cancer Res. 2011;71:862–72. doi: 10.1158/0008-5472.CAN-10-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Logothetis CJ, Gallick GE, Maity SN, Kim J, Aparicio A, et al. Molecular classification of prostate cancer progression: foundation for marker-driven treatment of prostate cancer. Cancer Discov. 2013;3:849–61. doi: 10.1158/2159-8290.CD-12-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134–49. doi: 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, et al. Mechanistic rationale for inhibition of poly (ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–78. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–86. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hodgson MC, Shao LJ, Frolov A, Li R, Peterson LE, et al. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res. 2011;71:572–82. doi: 10.1158/0008-5472.CAN-10-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 102.Efstathiou E, Titus MA, Tsavachidou D, Wen S, Hoang A, et al. Cancer Res. Vol. 72. Chicago, IL. Philadelphia, (PA): AACR: proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; 2012 Mar 31-Apr 4; 2012. Modulation of intracrine androgen signaling in bone metastatic castrate resistant prostate cancer (bmCRPC) by MDV3100. nr 2696. [Google Scholar]
- 103.Efstathiou E, Titus MA, Wen AS, San Miguel A, Hoang A, et al. The effects of enzalutamide (ENZA) in combination with abiraterone acetate (AA) in patients with bone metastatic castration resistant prostate cancer (mCRPC): presented at: European Cancer Congress 2013; September 27-October 1, 2013, Amsterdam. Eur J Cancer. 2013;49 Abstract 2854. [Google Scholar]
- 104.Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011;18:R183–96. doi: 10.1530/ERC-11-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–46. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 106.Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–22. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Press Release: medivation and astellas announce the phase 3 PREVAIL trial of enzalutamide meets both co-primary endpoints of overall survival and radiographic progression-free survival in chemotherapy-naive patients with advanced prostate cancer [Google Scholar]
- 108.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 109.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 110.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–54. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 111.Tannock IF, Osoba D, Stockler MR, Ernst DS, Neville AJ, et al. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J Clin Oncol. 1996;14:1756–64. doi: 10.1200/JCO.1996.14.6.1756. [DOI] [PubMed] [Google Scholar]
- 112.Parker C, Nilsson S, Heinrich D, Helle SI, O’Sullivan JM, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369:213–23. doi: 10.1056/NEJMoa1213755. [DOI] [PubMed] [Google Scholar]
- 113.Small EJ, Lance RS, Redfern CH, Millard FE, Gardner TA, et al. A randomized phase II trial of sipuleucel-T with concurrent or sequential abiraterone acetate (AA) plus prednisone (P) in metastatic castrate-resistant prostate cancer (mCRPC) ASCO Meeting Abstr. 2013;31:5047. doi: 10.1158/1078-0432.CCR-15-0079. [DOI] [PubMed] [Google Scholar]
- 114.Sheikh NA, Jones LA. CD54 is a surrogate marker of antigen presenting cell activation. Cancer Immunol Immunother. 2008;57:1381–90. doi: 10.1007/s00262-008-0474-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hanania NA, Sockrider M, Castro M, Holbrook JT, Tonascia J, et al. Immune response to influenza vaccination in children and adults with asthma: effect of corticosteroid therapy. J Allergy Clin Immunol. 2004;113:717–24. doi: 10.1016/j.jaci.2003.12.584. [DOI] [PubMed] [Google Scholar]
- 116.Antonarakis ES, Kibel AS, Adams G, Karsh LI, Elfiky A, et al. A randomized phase II study evaluating the optimal sequencing of sipuleucel-T and androgen deprivation therapy (ADT) in biochemically recurrent prostate cancer (BRPC): immune results. ASCO Meeting Abstr. 2013;31:5016. [Google Scholar]
- 117.Madan RA, Gulley JL, Schlom J, Steinberg SM, Liewehr DJ, et al. Analysis of overall survival in patients with nonmetastatic castration-resistant prostate cancer treated with vaccine, nilutamide, and combination therapy. Clin Cancer Res. 2008;14:4526–31. doi: 10.1158/1078-0432.CCR-07-5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mistry SJ, Oh WK. New paradigms in microtubule-mediated endocrine signaling in prostate cancer. Mol Cancer Ther. 2013;12:555–66. doi: 10.1158/1535-7163.MCT-12-0871. [DOI] [PubMed] [Google Scholar]
- 119.Fitzpatrick JM, de Wit R. Taxane mechanisms of action: potential implications for treatment sequencing in metastatic castration-resistant prostate cancer. Eur Urol. 2013 Jul 25; doi: 10.1016/j.eururo.2013.07.022. doi: 10.1016/jeururo.2013.07.022. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 120.Darshan MS, Loftus MS, Thadani-Mulero M, Levy BP, Escuin D, et al. Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res. 2011;71:6019–29. doi: 10.1158/0008-5472.CAN-11-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhu ML, Horbinski CM, Garzotto M, Qian DZ, Beer TM, et al. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. 2010;70:7992–8002. doi: 10.1158/0008-5472.CAN-10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gan L, Chen S, Wang Y, Watahiki A, Bohrer L, et al. Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer Res. 2009;69:8386–94. doi: 10.1158/0008-5472.CAN-09-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.van Soest RJ, van Royen ME, de Morrée ES, Moll JM, Teubel W, et al. Cross-resistance between taxanes and new hormonal agents abiraterone and enzalutamide may affect drug sequence choices in metastatic castration-resistant prostate cancer. Eur J Cancer. 2013;49:3821–30. doi: 10.1016/j.ejca.2013.09.026. [DOI] [PubMed] [Google Scholar]
- 124.Mezynski J, Pezaro C, Bianchini D, Zivi A, Sandhu S, et al. Antitumour activity of docetaxel following treatment with the CYP17A1 inhibitor abiraterone: clinical evidence for cross-resistance? Ann Oncol. 2012;23:2943–7. doi: 10.1093/annonc/mds119. [DOI] [PubMed] [Google Scholar]
- 125.Loriot Y, Bianchini D, Ileana E, Sandhu S, Patrikidou A, et al. Antitumour activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100) Ann Oncol. 2013;24:1807–12. doi: 10.1093/annonc/mdt136. [DOI] [PubMed] [Google Scholar]
- 126.Noonan KL, North S, Bitting RL, Armstrong AJ, Ellard SL, et al. Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann Oncol. 2013;24:1802–7. doi: 10.1093/annonc/mdt138. [DOI] [PubMed] [Google Scholar]
- 127.Bianchini D, Lorente D, Rodriguez-Vida A, Omlin A, Pezaro C, et al. Antitumour activity of enzalutamide (MDV3100) in patients with metastatic castration-resistant prostate cancer (CRPC) pre-treated with docetaxel and abiraterone. Eur J Cancer. 2014;50:78–84. doi: 10.1016/j.ejca.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 128.Schrader AJ, Boegemann M, Ohlmann CH, Schnoeller TJ, Krabbe LM, et al. Enzalutamide in castration-resistant prostate cancer patients progressing after docetaxel and abiraterone. Eur Urol. 2014;65:30–6. doi: 10.1016/j.eururo.2013.06.042. [DOI] [PubMed] [Google Scholar]
- 129.Miyamoto DT, Lee RJ, Stott SL, Ting DT, Wittner BS, et al. Androgen receptor signaling in circulating tumor cells as a marker of hormonally responsive prostate cancer. Cancer Discov. 2012;2:995–1003. doi: 10.1158/2159-8290.CD-12-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]