

Abstract
Recent phase I studies have reported single-agent activities of poly (ADP-ribose) polymerase (PARP) inhibitor in sporadic and in BRCA-mutant prostate cancers. Two of the most common genetic alterations in prostate cancer, ETS gene rearrangement and loss of PTEN, have been linked to increased sensitivity to PARP inhibitor in preclinical models. Emerging evidence also suggests that PARP1 plays an important role in mediating the transcriptional activities of androgen receptor (AR) and ETS gene rearrangement. In this article, the preclinical work and early-phase clinical trials in developing PARP inhibitor-based therapy as a new treatment paradigm for metastatic prostate cancer are reviewed.
Keywords: biomarker, PARP inhibitor, prostate cancer, therapy
INTRODUCTION
During evolution, mammalian cells have developed robust repairing mechanisms to correct DNA damages from sources like ultraviolet light, ionizing radiation and reactive oxygen species.1 The odds of an uncorrected error during DNA replication is about 1:106 in normal cells. The early onset of cancers in Lynch syndrome (mismatch repair gene deficiency) and familial breast-ovarian cancer syndrome (defective homologous recombination repair (HRR) due to BRCA mutations) are well-known examples of how diminished DNA damage repair (DDR) pathways lead to genomic instability, which is considered the driving force of tumorgenesis.2,3,4 On the other hand, certain DDR pathways are reserved to avoid excessive damage to cancer genome. As our knowledge on DNA base excision repair (BER), mismatch repair, HRR, nonhomologous end joining and Fanconi anemia repair pathways accumulates, several small molecules have been developed, with many of them being tested in early-phase clinical trials.5,6 Among these, the poly (ADP-ribose) polymerase (PARP) inhibitor is the most extensively studied in the clinical setting.7 Targeting the diminished DDR pathways that cancer cells rely on would provide the maximum therapeutic index, with minimum collateral damage to normal tissues.
Despite its genetic and phenotypic heterogeneity, prostate cancer, like most other cancers, acquires the hallmarks of cancer through gene mutations and chromosomal alterations.8,9,10,11,12 Through targeted exon sequencing, global copy number and transcriptome profiling; Taylor et al. reported that DNA copy number variations are enriched in metastatic prostate cancers and in cell/xenograft lines compared to primary tumors.9 These increased gene copy number gains and losses in end-stage prostate cancer have also been confirmed in a recent study by Grasso et al.11 Such enrichment of genomic instability could be attributed to diminished DNA repair in late-stage prostate cancer. Indeed, diminished DNA repair has been reported in cell lines developed from metastatic lesions of prostate cancer.13,14 In addition, p53, a key DNA damage check point control gene, is among the frequently mutated genes in treatment-naïve as well as treatment-refractory lethal prostate cancers.11,15 Furthermore, two of the most common genetic alterations in prostate cancer, ETS gene rearrangement and loss of PTEN, have been linked to enhanced DNA damage and defective DNA repair.16,17
Between 2010 and 2013, six new agents have been approved for treating metastatic castration-resistant prostate cancer (CRPC).18 Although the overall survival of patients has been improved with these newly developed androgen deprivation therapies,19,20,21 immunotherapies,22 chemotherapies,23 and bone targeting radiopharmaceuticals,24 metastatic CRPC remains an incurable disease. Cancer cells with high genomic instability tend to develop early treatment resistance and run a more aggressive clinical course.25 This is evidenced by the poor prognosis in patients with BRCA2-mutant prostate cancer.26 DNA copy number alterations have also been shown to be better than Gleason grade in predicting the risk of biochemical recurrence of primary prostate cancers.9 By blocking the diminished DDR, PAPR inhibitor-based therapy could potentially provide a high therapeutic index to lethal prostate cancer with genomic instability. In addition to its well-documented role in BER, emerging evidence has indicated that PARP1 plays an important role in mediating the transcriptional activities of androgen receptor (AR) and ETS gene rearrangement.16,27 In this article, the preclinical work and early-phase clinical trials in developing the PARP inhibitor as a new treatment for metastatic CRPC will be reviewed.
The search for ‘brca-ness’ for parp inhibition in prostate cancer
In the landmark phase I study with the PARP inhibitor olaparib, of the three prostate cancer patients, one patient was a BRCA2 mutation carrier. This patient showed more than 50% reduction in prostate-specific antigen (PSA) levels and resolution of bone metastases, as shown by MRI.28 In a follow-up phase II study presented at the American Society for Clinical Oncology 2013 Annual Meeting, among eight prostate cancer patients with BRCA1 or BRCA2 mutations, four (50%) had either complete or partial response to olaparib and one patient (25%) demonstrated stable disease for at least 8 weeks. These impressive results were consistent with the preclinical data of ‘synthetic lethality’, where cancer cells with defective HRR are sensitive to additional blockage of DNA repair with PARP inhibitor. Given that familiar BRCA mutations are rare in prostate cancer, identifying sporadic genetic alterations that could diminish the HRR and sensitize prostate cancer cells to PARP inhibitor based therapy has been an area of active research.
Loss of PTEN and Rad51
The tumor suppressor PTEN negatively regulates the PI3K/AKT/mTOR (phosphoinositide kinase-3/protein kinase B/mammalian target of rapamycin) tumor survival pathway. Data from array comparative genome hybridization and targeted exome sequencing have reported enrichment of loss of PTEN in metastatic prostate cancer.9,11 Preclinical studies with colon, breast and prostate cancer cell lines have shown that loss of PTEN was associated with reduced Rad51 levels and subsequent sensitivity to PARP inhibition.17 Rad51 acts downstream of the BRCA1 and BRCA2 and is a key protein in the HRR pathway.29 Such correlations between loss of PTEN and Rad51 level; however, were not observed in a recent study with radical prostatectomy samples.30 Although one could argue that radical prostatectomy samples may not recapitulate the downstream events of PTEN loss in late-stage prostate cancer, a recent phase I study with PARP inhibitor niraparib/MK4827 did not observe associations between loss of PTEN and antitumor activities in sporadic metastatic prostate cancer patients.31
With 23 CRPC patients (only one had documented BRCA mutation); this recently reported phase I study with niraparib/MK4827 had by far the largest prostate cancer cohort among the single-agent PARP inhibitor trials. Although no radiological responses were noted, nine of the 21 patients (43%) treated at either 290 or 300 mg day−1 had stable disease for a median duration of 254 days (range 124–375 days).31 This is the first study to document clinical activity of a PARP inhibitor in sporadic CRPC. More importantly, this study took great effort to explore the association between clinical benefit and loss of PTEN expression or ETS rearrangement in circulating tumor cells as well as in tumor tissue. Although the marker studies are exploratory in this phase I trial, loss of PTEN by itself is probably not sufficient to predict defective HRR or sensitivity to PARP inhibition.
Among other potential biomarkers, reduced Rad51 levels have been linked to loss of PTEN and subsequent sensitivity to PARP inhibition in preclinical models.17 As a key protein in HRR, mutation or copy number changes in Rad51 are rare; whereas, overexpression of Rad51 is common in cancer, particularly in cancers that harbor mutant p53.32 Wild-type p53 can suppress Rad51 transcription either directly or indirectly.33,34 Other than p53 mutations, RB mutation is another frequent genetic event in metastatic CRPC.11 Loss of functional RB could lead to enhanced E2F1 activity, and E2F1 has been shown to bind to Rad51 promoter and increases its transcription.35 Overexpression of Rad51 in cancer cells with p53 or RB mutations could potentially rescue the HRR defects caused by BRCA mutation or loss of PTEN.36,37,38,39 The p53 and Rad51 status can be studied in formalin-fixed, paraffin-embedded samples with established immunohistochemistry protocols. Efforts are underway to detect Rad51 nuclear foci formation, a widely accepted marker for activation of the HRR pathway, in circulating tumor cells. Incorporating biomarkers like p53 and Rad51 in PARP inhibitor-based clinical trials would help to delineate the mechanism of prostate cancer cells’ sensitivity or resistance to PARP inhibition.
Hypoxia and p53
Intratumoral hypoxia has been well-documented in prostate cancer,40 and downregulation of DNA repair proteins like Rad51 and BRCA1 has been reported in prostate cancer cells grown under chronic hypoxia culture.41,42 Such downregulation of DNA repair proteins under chronic hypoxia could sensitize prostate cancer cells to PARP inhibitor. Although preclinical studies have reported increased cell death and DNA damage in non-prostate cancer cell lines grown under hypoxia,43 our study showed that prostate cancer cells with mutant p53 were resistant to PARP inhibitor veliparib and the DNA damaging topoisomerase I inhibitor irrinotecan/CPT-11 under hypoxia (Figure 1). Our data support that hypoxia contributes to drug resistance, but such resistance was not due to increased mutation rate secondary to diminished DNA repair as previously suggested.25,44,45 Upregulation of Rad51 was observed soon after challenging these p53-mutant prostate cancer cells with DNA-damaging agents (Figure 1). We also assessed genomic stability by looking at chromosomal copy number alterations with nCounter molecular karyotyping. When compared to the untreated control under chronic hypoxia culture, we did not detect increased chromosomal copy number alterations in PC3 cells with up to 3 weeks of hypoxia culture and veliparib treatment (Figure 2). Furthermore, recent high-throughput sequencing studies have reported that mutation rates in metastatic CRPC (2.0 mutations per mega base) were not significantly increased compared to primary tumors (0.9–1.5 mutations per mega base). The enrichment of genomic instability in late-stage prostate cancer could result from increasing tumor heterogeneity after a prolonged selection process by the tumor microenvironment and treatment.
Figure 1.
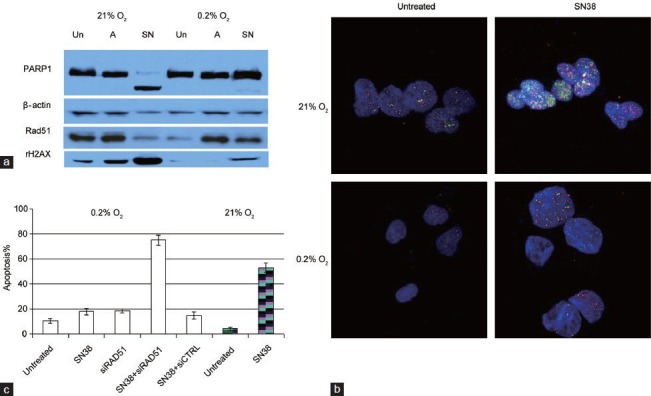
(a) Western blots comparing PARP1 cleavage (apoptosis marker) and levels of Rad51 and rH2AX (marker for double strand DNA breaks) in untreated (Un), ABT888-treated (A) or SN38-treated (SN) PC3 cells grown under 21% or 0.2% oxygen for 4 days. SN38 is the active metabolite of irinotecan. Compared to drug-treated PC3 cells under normoxia, less DNA damage and apoptosis were observed in treated PC3 cells under hypoxia. (b) Immunofluorescence of Rad51 (red) and γ-H2AX (green) foci in untreated or SN38-treated (0.1 μmol l–1 for 4 h) PC3 cells grown under normoxia (21% O2) or chronic hypoxia (0.2% O2 for 72 h). Increased Rad51 nuclear foci formation was observed soon after PC3 cells were challenged with SN38. Compared to SN38-treated cells under normoxia, there was less DNA damage under hypoxia. (c) Blocking Rad51 upregulation with siRNA-resensitized PC3 cells to SN38. Percentage of apoptosis was detected by flow cytometry with propidium iodide and annexin V. PC3 cells grown in 0.2% O2 were transfected with Rad51 siRNA or scrambled siRNA control (siCTRL); inhibiting Rad51 with siRad51 reversed the resistance to SN38 under hypoxic culture. Untreated and SN38-treated PC3 cells grown in 21% O2(hatched column) and 0.2% O2 were shown for comparison.
Figure 2.
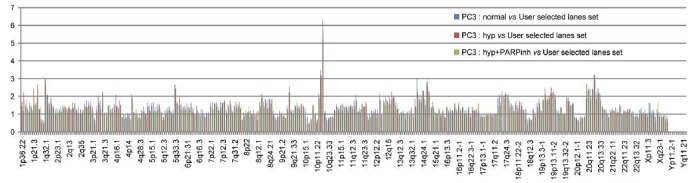
Distribution of the copy numbers of 338 probes covering 24 chromosomes in PC3 cells as detected by nCounter human karotyping. PC3 cells are known to have Y-chromosome deletion. These copy numbers were normalized to human foreskin fibroblasts (user selected lane). Compared to PC3 cells grown under normoxia, no significant alterations in chromosomal copy numbers were observed in PARP inhibitor-treated or untreated PC3 cells grown under 0.2% O2(hyp) for 3 weeks.
As a DNA damage checkpoint gene, p53 is among the most frequently mutated genes in cancer. Based on the International Agency for Research on Cancer TP53 database, the frequency of p53 mutations is 17.5% among 1106 sporadic prostate cancers. In the study by Barbieri et al., six missense mutations and one frameshift mutation in p53 were detected by exome sequencing of 111 treatment-naïve primary prostate cancers.15 Although this 6% mutation frequency is lower than the historical data, mutation in p53 was the second most common mutation in this dataset. The exome sequencing study by Grasso et al.11 reported two point mutations and two frame shift mutations in 11 treatment-naïve high-grade primary prostate cancers (36% mutation frequency) and 14 point mutations and five frame shift mutations in 50 heavily treated lethal CRPC patients (38% mutation frequency). Copy number loss of p53 was also observed in nine of these 50 lethal cases.15 Given that wild-type p53 can suppress the transcription of Rad51,33,34 prostate cancer cells with mutant p53 can evade not only apoptosis but will likely have more effective HRR due to lack of suppression of Rad51 transcription by wild-type p53.
Disruption of transcriptional regulation with parp inhibitor
As an abundant and ubiquitous nuclear protein, PARP1 has been shown to bind to promoters and enhancers and to interact with a wide variety of proteins that regulate gene transcription.46 Most of these regulations require its enzymatic activity, i.e. poly (ADP-ribosyl) ation. Other than diminishing DNA repair, emerging preclinical data have indicated that PARP inhibitor can disrupt transcriptional regulation by AR and the ETS fusion protein.16,27
The recurrent fusions between the androgen-regulated TMPRSS2 and ETS transcription factor genes (primarily ERG) occur in about 50% of primary prostate cancers.10,12 ERG was recently shown to interact with PARP1, with PARP1 activity required for ERG-mediated transcription, cell invasion and metastasis.16 Inhibition of PARP1 enhanced DNA double-strand breaks induced by ERG overexpression and slowed the growth of ERG-positive prostate cancer cells.16 Such enhanced PARP inhibitor activity can be contributed to downregulation of ERG-mediated transcription, the genomic instability in ERG fusion-positive tumors, or a combination of both. Another study with prostate cancer cell line models showed that PARP inhibition reduced AR occupancy of chromatin and suppressed the expression of AR target genes, including UBE2C, a key regulator of cell cycle progression.27 Unlike its interaction with ERG, PARP1 occupied the AR target gene promoter without forming a complex with AR. The authors also reported that treatment with PARP inhibitor slowed down the proliferation (Ki67 index) of prostate cancer cells grown in an ex vivo culture system.27
Despite this strong preclinical rationale for targeting ETS fusion-positive prostate cancer with PARP inhibitor, no correlations were observed between ETS gene rearrangement and antitumor activities (time to disease progression, PSA response rate or decline in circulating tumor cells) in the phase I study with niraparib in 23 CRPC patients.31 As an early genetic alteration in prostate cancer, ETS gene rearrangement by itself may not be sufficient to predict response of late-stage CRPC to PARP inhibition.
RATIONALE FOR PARP INHIBITOR-BASED COMBINATION THERAPIES
Grade 3 or 4 toxicities are rare in early-phase clinical trials with single-agent PARP inhibitor.28,31 This has led to preclinical and clinical studies with various PARP inhibitor-based regimens in prostate cancer with the goal to maximize the DNA damage or to disrupt the transcriptional regulation through AR and ETS fusion proteins.
Combining the PARP inhibitor with cytotoxic chemotherapy
PARP inhibitor veliparib/ABT-888 enhanced the activity of multiple DNA-damaging agents, including cisplatin, carboplatin, cyclophosphamide, irinotecan and temozolomide, in various solid tumor preclinical models.47 Among these agents, temozolomide was shown to have the most synergistic antitumor activity when combined with veliparib/ABT-888.48 As an alkylating agent, temozolomide induces DNA methylation at guanine O6 (O6-MeG), guanine N7 (N7-MeG) and adenine N3 (N3-MeA). BER is involved in repairing the N3-MeA and N7-MeG DNA adducts.49 Blocking BER with PARP inhibitor would therefore enhance the DNA damage caused by temozolomide. Combining veliparib with temozolomide, not temozolomide alone, inhibited the growth of orthotopic and intratibial mouse prostate cancer xenografts made of luciferase-labeled PC3 cells.48 The veliparib and temozolomide combination was subsequently tested in patients with metastatic CRPC who have failed up to two non-hormonal systemic therapies in a multi-institutional pilot study (NCT01085422). The primary objective of this study is to assess the efficacy of this combination based on the rate of PSA decline of 30% or greater. Despite the promising preclinical activity, only two of the 25 evaluable patients had a confirmed PSA response: 1 had a 37% decrease in PSA, while the other had a 96% decrease in PSA and a 40% reduction in tumor size. Four of the 25 patients had stable disease for a minimum of 4 months. Median progression-free survival was 2.1 months. Of note, temozolomide showed no activity as a single agent in prostate cancer. The updated results and biomarker studies of this trial are not published yet. If feasible, a comprehensive genetic analysis with exome sequencing on the patient with both PSA and radiographic responses may help uncover genetic alterations that sensitize his CRPC to the temozolomide-veliparib combination.
Although none of the DNA-damaging chemotherapy agents has been approved for prostate cancer treatment, such agents have been used to treat small cell prostate cancer and ‘anaplastic’ prostate cancer. Most recently, the carboplatin-docetaxel combination followed by cisplatin and etoposide at disease progression have shown meaningful clinical activity in a phase II study of 120 patients who met the predefined criteria of ‘anaplastic’ prostate cancers.50 For the majority of these patients, their cancer became castration resistant within 6 months of androgen deprivation therapy (45.6%), with a bulky (≥5 cm) lymphadenopathy or bulky (≥5 cm) high-grade (Gleason ≥ 8) tumor mass in the prostate or pelvis (43%). This subset of prostate cancer shares several clinical features of treatment-related neuroendocrine prostate cancer (NEPC) and has a very poor prognosis. Using next-generation RNA-sequencing and oligonucleotide arrays, Beltran et al. profiled seven NEPC, 30 prostate adenocarcinoma and five benign prostate tissue samples and found significant overexpression and gene amplification of AURKA and MYCN in NEPC compared to primary prostate cancer. This finding was validated from a large cohort of prostate tumors with immunohistochemistry and fluorescent in situ hybridization.51 A follow-up study also indicated that prostate adenocarcinomas with AURKA and MYCN coamplification are at risk to develop NEPC after androgen deprivation therapy.52 It would be important to check AURKA and MYCN status in the ‘anaplastic’ prostate cancers and correlate these amplifications with their response to chemotherapy in the clinical study by Aparicio et al. If feasible, assessment of DNA damage and DNA repair markers in pre- and posttreatment tumor samples would also be informative. Based on its fast proliferation and the rapid development of treatment resistance, studies of anaplastic prostate cancer at the DNA level will likely reveal genetic alterations in cell cycle checkpoint and DNA repair genes. PARP inhibitor may enhance the chemotherapy-induced DNA damage in anaplastic prostate cancer.
Combining the PARP inhibitor with androgen deprivation therapy
The success of abiraterone and enzalutamide in treating metastatic CRPC indicates persistent AR signaling in this late stage of prostate cancer. Based on the emerging role of PARP1 in mediating transcriptional regulation by AR and the ETS fusion protein,16,27 a multicenter randomized phase II trial is evaluating whether adding the PARP inhibitor veliparib to abiraterone acetate and prednisone would improve the PSA response rate (primary objective) of the standard abiraterone acetate and prednisone regimen in patients with metastatic CRPC (NCT01576172). The availability of metastatic tumor tissue that is evaluable for ETS fusion status is required for this study, and a logistic model will be used to determine the association of ETS fusion status with the PSA response in the veliparib-abiraterone-prednisone arm. PSA decline rate, objective response rate, progression-free survival and toxicity are among the secondary objectives. This study will provide the much needed clinical validation on the value of ETS gene rearrangement as a predicative biomarker for PARP inhibitor-based therapy for metastatic CRPC.
In addition to disruption of AR transcription, combining veliparib with abiraterone for ETS fusion-positive prostate cancer could lead to enhanced DNA damage and apoptosis, as indicated in the preclinical study by Brenner et al.16 This potential benefit, however, will not be well captured by PSA response rate or by progression-free survival (secondary endpoint) of the NCT01576172 study. Even if this combination improves the PSA response rate, it is still unclear whether this combination is more effective or safer than combining abiraterone with enzalutamide, which is being tested in a phase II study for patients with progressive metastatic CRPC in the bone (NCT01650194). Assessment of DNA damage in posttreatment tumor biopsies or circulating tumor cells, if built into the NCT01576172 study, would help to distinguish the valiparib-abiraterone combination from the enzalutamide-abiraterone combination.
Combining the PARP inhibitor with radiotherapy
A preclinical study with prostate cancer cell lines reported that combining the PARP inhibitor rucaparib with radiation enhanced the DNA damage and antitumor effects compared to radiation alone.53 The strongest synergistic activities were observed in LNCaP and VCaP cells, which express ETS fusion proteins. Although combining a PARP inhibitor with radiation would be an attractive strategy to increase the cure rate of newly diagnosed ETS fusion-positive nonmetastatic prostate cancer, overtreatment and long-term safety are the main concerns of testing this strategy in the clinic. The biochemical recurrence rate after initial definitive surgery or radiation is between 20% and 30%. Only a portion of patients with biochemical recurrence will die from prostate cancer. There is also a lack of long-term safety data on the PAPR inhibitors tested in early-phase clinical trials. Enhanced DNA damage with a PARP inhibitor and radiation could lead to genomic instability and more aggressive prostate cancer when it recurs. Secondary malignancy would be another concern.
Combining PARP inhibitors with radiopharmaceuticals like radium 223 could be a reasonable combination for patients with metastatic CRPC to the bone, as concerns for overtreatment or for long-term safety are insignificant at this lethal stage of prostate cancer. If markers like p53 mutation, ETS gene rearrangement and/or loss of PTEN are able to identify the late-stage prostate cancers enriched with genomic instability, combining radium 223 with a PARP inhibitor could be more effective than combing radium 223 with docetaxel, enzalutamide or abiraterone in this group of lethal prostate cancer.
CONCLUSIONS
The activity of the PARP inhibitor niraparib in sporadic prostate cancer provides a strong clinical rationale for developing PARP inhibitor-based therapies for metastatic CRPC. As presently shown, loss of PTEN or presence of ETS gene rearrangement by itself is unlikely to be sufficient to predict response to PARP inhibitor-based therapies. Other than blocking DDR, PARP inhibitors have been shown to disrupt transcriptional regulation through AR and ETS fusion proteins in CRPC preclinical models. This preclinical finding is being tested in an ongoing phase II study of abiraterone with or without veliparib in patients with metastatic CRPC (NCT01576172). Whether this combination can lead to greater PSA response than abiraterone alone in ETS fusion-positive prostate cancer remains to be seen. Moving forward, we will continue to learn from PARP inhibitor-based trials and evaluate the biomarkers being embedded in these studies. Based on the recent success of platinum-based regimens in treating anaplastic prostate cancer, I would propose focusing future PARP inhibitor-based trials and biomarker studies on this subtype of lethal prostate cancer where there is truly an unmet need.
COMPETING INTERESTS
The author declares no competing interests.
ACKNOWLEDGMENTS
JZ is supported by the National Institutes of Health Moffitt Cancer Center Support Grant for new faculty recruitment (P30 CA076292-11S5), and part of his work referred by this review was supported by the American Society of Clinical Oncology, Conquer Cancer Foundation 2010 Young Investigator Award. The author thanks Abbott Laboratories for providing veliparib (ABT888) and Rasa Hamilton at Moffitt Cancer Center for editorial assistance.
REFERENCES
- 1.Ohnishi T, Mori E, Takahashi A. DNA double-strand breaks: their production, recognition, and repair in eukaryotes. Mutat Res. 2009;669:8–12. doi: 10.1016/j.mrfmmm.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 2.Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, et al. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76:1–18. doi: 10.1111/j.1399-0004.2009.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lynch HT, Silva E, Snyder C, Lynch JF. Hereditary breast cancer: part I. Diagnosing hereditary breast cancer syndromes. Breast J. 2008;14:3–13. doi: 10.1111/j.1524-4741.2007.00515.x. [DOI] [PubMed] [Google Scholar]
- 4.Lynch HT, Casey MJ, Snyder CL, Bewtra C, Lynch JF, et al. Hereditary ovarian carcinoma: heterogeneity, molecular genetics, pathology, and management. Mol Oncol. 2009;3:97–137. doi: 10.1016/j.molonc.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 6.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–94. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 7.Yap TA, Sandhu SK, Carden CP, de Bono JS. Poly (ADP-ribose) polymerase (PARP) inhibitors: exploiting a synthetic lethal strategy in the clinic. CA Cancer J Clin. 2011;61:31–49. doi: 10.3322/caac.20095. [DOI] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 9.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–9. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 11.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–77. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, Wang J, Fraig MM, Metcalf J, Turner WR, et al. Defects of DNA mismatch repair in human prostate cancer. Cancer Res. 2001;61:4112–21. [PubMed] [Google Scholar]
- 14.Fan R, Kumaravel TS, Jalali F, Marrano P, Squire JA, et al. Defective DNA strand break repair after DNA damage in prostate cancer cells: implications for genetic instability and prostate cancer progression. Cancer Res. 2004;64:8526–33. doi: 10.1158/0008-5472.CAN-04-1601. [DOI] [PubMed] [Google Scholar]
- 15.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, et al. Mechanistic rationale for inhibition of poly (ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–78. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–22. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu JJ, Zhang J. Sequencing systemic therapies in metastatic castration-resistant prostate cancer. Cancer Control. 2013;20:181–7. doi: 10.1177/107327481302000306. [DOI] [PubMed] [Google Scholar]
- 19.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 21.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 23.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lance. 2010;376:1147–54. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 24.Parker C, Nilsson S, Heinrich D, Helle SI, O’Sullivan JM, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369:213–23. doi: 10.1056/NEJMoa1213755. [DOI] [PubMed] [Google Scholar]
- 25.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–92. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 26.Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31:1748–57. doi: 10.1200/JCO.2012.43.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134–49. doi: 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, et al. Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 29.Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraser M, Zhao H, Luoto KR, Lundin C, Coackley C, et al. PTEN Deletion in Prostate Cancer Cells Does Not Associate With Loss of RAD51 Function: implications for Radiotherapy and Chemotherapy. Clin Cancer Res. 2011;18:1015–27. doi: 10.1158/1078-0432.CCR-11-2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, et al. The poly (ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14:882–92. doi: 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 32.Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst) 2008;7:686–93. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arias-Lopez C, Lazaro-Trueba I, Kerr P, Lord CJ, Dexter T, et al. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006;7:219–24. doi: 10.1038/sj.embor.7400587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fong V, Osterbur M, Capella C, Kim YE, Hine C, et al. Adenoviral vector driven by a minimal Rad51 promoter is selective for p53-deficient tumor cells. PloS ONE. 2011;6:e28714. doi: 10.1371/journal.pone.0028714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kachhap SK, Rosmus N, Collis SJ, Kortenhorst MS, Wissing MD, et al. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PloS ONE. 2010;5:e11208. doi: 10.1371/journal.pone.0011208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown ET, Holt JT. Rad51 overexpression rescues radiation resistance in BRCA2-defective cancer cells. Mol Carcinog. 2009;48:105–9. doi: 10.1002/mc.20463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Z, Waldman AS, Wyatt MD. Expression and regulation of RAD51 mediate cellular responses to chemotherapeutics. Biochem Pharmacol. 2012;83:741–6. doi: 10.1016/j.bcp.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee SA, Roques C, Magwood AC, Masson JY, Baker MD. Recovery of deficient homologous recombination in Brca2-depleted mouse cells by wild-type Rad51 expression. DNA Repair (Amst) 2009;8:170–81. doi: 10.1016/j.dnarep.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 39.Martin RW, Orelli BJ, Yamazoe M, Minn AJ, Takeda S, et al. RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors. Cancer Res. 2007;67:9658–65. doi: 10.1158/0008-5472.CAN-07-0290. [DOI] [PubMed] [Google Scholar]
- 40.Marignol L, Coffey M, Lawler M, Hollywood D. Hypoxia in prostate cancer: a powerful shield against tumour destruction? Cancer Treat Rev. 2008;34:313–27. doi: 10.1016/j.ctrv.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 41.Bindra RS, Schaffer PJ, Meng A, Woo J, Maseide K, et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol. 2004;24:8504–18. doi: 10.1128/MCB.24.19.8504-8518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meng AX, Jalali F, Cuddihy A, Chan N, Bindra RS, et al. Hypoxia down-regulates DNA double strand break repair gene expression in prostate cancer cells. Radiother Oncol. 2005;76:168–76. doi: 10.1016/j.radonc.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 43.Chan N, Pires IM, Bencokova Z, Coackley C, Luoto KR, et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010;70:8045–54. doi: 10.1158/0008-5472.CAN-10-2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Papp-Szabó E, Josephy PD, Coomber BL. Microenvironmental influences on mutagenesis in mammary epithelial cells. Int J Cancer. 2005;116:679–85. doi: 10.1002/ijc.21088. [DOI] [PubMed] [Google Scholar]
- 45.Reynolds TY, Rockwell S, Glazer PM. Genetic instability induced by the tumor microenvironment. Cancer Res. 1996;56:5754–7. [PubMed] [Google Scholar]
- 46.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, et al. ABT-888, an orally active poly (ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–37. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 48.Palma JP, Wang YC, Rodriguez LE, Montgomery D, Ellis PA, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009;15:7277–90. doi: 10.1158/1078-0432.CCR-09-1245. [DOI] [PubMed] [Google Scholar]
- 49.Trivedi RN, Almeida KH, Fornsaglio JL, Schamus S, Sobol RW. The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res. 2005;65:6394–400. doi: 10.1158/0008-5472.CAN-05-0715. [DOI] [PubMed] [Google Scholar]
- 50.Aparicio AM, Harzstark AL, Corn PG, Wen S, Araujo JC, et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin Cancer Res. 2013;19:3621–30. doi: 10.1158/1078-0432.CCR-12-3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;6:487–95. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia. 2013;15:1–10. doi: 10.1593/neo.121550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chatterjee P, Choudhary GS, Sharma A, Singh K, Heston WD, et al. PARP inhibition sensitizes to low dose-rate radiation TMPRSS2-ERG fusion gene-expressing and PTEN-deficient prostate cancer cells. PLoS ONE. 2013;8:e60408. doi: 10.1371/journal.pone.0060408. [DOI] [PMC free article] [PubMed] [Google Scholar]