

Key Points
Total body irradiation causes long-term bone marrow suppression by selectively inducing HSC senescence.
The induction of HSC senescence is independent of telomere shortening and p16Ink4a and Arf.
Abstract
Exposure to total body irradiation (TBI) induces not only acute hematopoietic radiation syndrome but also long-term or residual bone marrow (BM) injury. This residual BM injury is mainly attributed to permanent damage to hematopoietic stem cells (HSCs), including impaired self-renewal, decreased long-term repopulating capacity, and myeloid skewing. These HSC defects were associated with significant increases in production of reactive oxygen species (ROS), expression of p16Ink4a (p16) and Arf mRNA, and senescence-associated β-galacotosidase (SA-β-gal) activity, but not with telomere shortening or increased apoptosis, suggesting that TBI induces residual BM injury via induction of HSC premature senescence. This suggestion is supported by the finding that SA-β-gal+ HSC-enriched LSK cells showed more pronounced defects in clonogenic activity in vitro and long-term engraftment after transplantation than SA-β-gal– LSK cells isolated from irradiated mice. However, genetic deletion of p16 and/or Arf had no effect on TBI-induced residual BM suppression and HSC senescence, because HSCs from irradiated p16 and/or Arf knockout (KO) mice exhibited changes similar to those seen in HSCs from wild-type mice after exposure to TBI. These findings provide important new insights into the mechanism by which TBI causes long-term BM suppression (eg, via induction of premature senescence of HSCs in a p16-Arf–independent manner).
Introduction
Bone marrow (BM) suppression is one of the common side effects of radiotherapy and the primary cause of death after exposure to a moderate or high dose of total body irradiation (TBI).1,2 Acute BM suppression occurs within days after exposure to ionizing radiation (IR), primarily as a result of induction of apoptosis in the rapidly proliferating hematopoietic progenitor cells (HPCs).3 Its clinical manifestations recently have been more successfully managed by the use of hematopoietic growth factors (HGFs).4 However, some irradiated patients also develop long-term or residual BM injuries manifested by decreases in HSC reserves and impairments in HSC self-renewal after recovering from IR-induced acute myelosuppression. Unlike acute myelosuppression, residual BM damage is latent and the patients with residual BM injuries usually have an extended period of normal blood cell counts under homeostatic conditions, despite decreases in HSC reserves.4,5 Because of this latency, the clinical implications of residual BM injury have been largely overlooked. Moreover, the importance of residual BM damage is further obscured by the seemingly complete recovery of peripheral blood cell counts and BM cellularity, especially after treatment with HGFs. In fact, the use of HGFs may worsen IR-induced residual BM damage by promoting proliferation and differentiation of HSCs and HPCs at the expense of HSC self-renewal.6 This could lead to accelerated exhaustion of HSCs and further compromise the long-term recovery of BM hematopoietic function. Although residual BM damage is latent, it is long lasting, shows little tendency for recovery, and can lead to the development of hypoplastic anemia or a myelodysplastic syndrome at a later time or after additional hematopoietic stress.4,5 In addition, residual BM injury can predispose irradiated individuals to develop leukemia and lymphoma by reducing the fitness of HSCs.7 However, the mechanisms by which IR induces residual BM suppression have not been clearly defined, which hampers development of effective treatments to ameliorate the injury.
IR-induced residual BM injury has been attributed to induction of HSC senescence. This assumption is supported by our recent findings that LSK cells (ie, Lin–Sca1+c-kit+) isolated from the BM of sublethally irradiated mice expressed increased levels of biomarkers for senescent cells, such as SA-β-gal and p16.8,9 However, LSK cells are heterogeneous and only a small proportion of the LSK population is made up of HSCs, with the rest being composed of multipotent progenitor cells (MPPs). Therefore, it remains to be determined whether IR can actually induce HSCs to undergo senescence and whether IR induces HSC senescence prematurely or via telomere shortening resulting from increased HSC proliferation after IR.
The p16-Arf locus encodes 2 tumor suppressors, p16 and Arf.10-12 p16 functions as a cyclin-dependent kinase (CDK) 4/6 inhibitor.10 By inhibiting CDK4/6 activity, p16 causes retinoblastoma protein (Rb) hypophosphorylation and suppresses expression of E2F-dependent genes,13 resulting in restriction of G1/S cell cycle progression and induction of senescence. It has been suggested that diverse stimuli can induce cellular senescence via various upstream signal transduction cascades (including the p53-p21 and p38 pathways) that eventually converge on p16, whose induction provides an inescapable barrier to prevent senescent cells from re-entering the cell cycle. In contrast, the biological action of Arf relies on the p53 pathway. This is because Arf can directly bind to mouse double minute 2 homolog (MDM2) and cause the accumulation of p53 by segregating MDM2 from p53 and by inhibiting MDM2’s E3 ubiquitin protein ligase activity for p53.10,11,14 Therefore, activation of p53 by Arf can induce not only senescence but also apoptosis, depending on which gene downstream of p53 is induced after p53 activation.
Upregulation of p16 and Arf has been implicated in mediating the induction of cellular senescence in a variety of cells including HSCs. For example, increased expression of p16 and Arf was found in HSCs from Bmi1−/− mice.15 However, it appears that p16, but not Arf, plays an important role in mediating induction of Bmi1−/− HSC senescence, whereas Arf mainly promotes HSC apoptosis.15 In addition, it has been found that knockout of both the p16 and Arf genes in mice significantly increases the clonal expansion of HSCs in vitro but modestly promotes HSC self-renewal in vivo.16 Knockout of the Arf gene alone provides no significant advantage for HSC/HPC expansion and self-renewal,16 but knockout of p16 increases the life-span of HSCs by promoting HSC self-renewal.16-19 Furthermore, mutation of the ataxia telangiectasia mutated (ATM) gene also results in upregulation of p16 and Arf in HSCs. Inactivation of the p16-Rb pathway restores the reproductive function of ATM−/− HSCs, whereas inhibition of the Arf-p53 pathway has no such effect.20,21 These findings suggest that p16 plays a more significant role than Arf in regulating HSC self-renewal and inducing HSC senescence, even though both proteins are overexpressed in senescent HSCs. Although increased expression of p16 and Arf has been found in IR-induced senescent LSK cells, the roles of these proteins in mediating IR-induced HSC senescence and residual BM suppression are unknown and thus were also investigated in the present study.
Materials and methods
Reagents
All the antibodies used to stain various types of hematopoietic cells are presented in supplemental Table 1.
Animals
The following strains of mice were used in our studies with the protocols approved by the Institutional Animal Care and Use Committees (IACUC) of Medical University of South Carolina (MUSC) and University of Arkansas for Medical Sciences (UAMS): C57BL/6J (or CD45.2), B6.SJL-PtprcaPep3b/BoyJ (or CD45.1), p16−/− (Ink4a Δexon 1α), Arf −/− (Ink4a Δexon 1β), and p16Arf −/− (Ink4a Δexon 2 and 3) mice. More details about the mice can be found in supplemental Methods.
TBI
Mice were exposed to a sublethal (6.0 Gy) dose of TBI in a Mark I 137Ce γ-irradiator (JL Shepherd, Glendale, CA) at a rate of 2.4 Gy/min. Mice were irradiated on a rotating platform.
Isolation of BM mononuclear cells (BM-MNCs), lineage-negative hematopoietic cells (Lin– cells), and HSCs
BM-MNCs, Lin– cells, and HSCs (CD150+CD48–LSK+ cells) were isolated as described in supplemental Methods.
Analysis of the frequencies and numbers of different hematopoietic cell populations by flow cytometry
The frequencies of HPCs (Lin–Sca1–c-kit+ cells), LSK cells (Lin–Sca1+c-kit+ cells), MPPs (CD150–CD48–LSK cells), HSCs (CD150+CD48–LSK cells), short-term HSCs (ST-HSCs, CD34+CD150+CD48–LSK cells), and long-term HSCs (LT-HSCs, CD34–CD150+CD48–LSK cells) were analyzed with an Aria II cell sorter after immunostaining with appropriate antibodies as described in supplemental Methods.
Cobblestone area-forming cell (CAFC) assay and competitive repopulation assay (CRA)
The details of these assays can be found in supplemental Methods.
SA-β-gal activity analysis and isolation of SA-β-gal+ and SA-β-gal– LSK cells for characterization, single cell culture, and transplantation
The details of SA-β-gal staining and isolation of SA-β-gal+ and SA-β-gal– LSK cells by cell sorting can be found in supplemental Methods.
Quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR)
Expression of various genes was measured by qRT-PCR and calculated by the comparative CT method, as described previously.8 The sequences of all primers used in the qRT-PCR assays are listed in supplemental Table 2.
Analysis of apoptosis, the levels of intracellular ROS production, cell cycle distribution, and BrdU incorporation
These assays were done using the methods described in supplemental Methods.
Telomere length measurements by fluorescence in situ hybridization (FISH)
Relative telomere lengths in T cells and myeloid cells were measured by flow-FISH using the Telomere DNA Kit/FITC for Flow Cytometry (Dako Denmark A/S, Glostrup, Denmark), according to the manufacturer’s protocol, and were expressed as mean fluorescent intensity (MFI).
Telomere length measurements by quantitative PCR (qPCR)
Average telomere lengths in T cells, myeloid cells, LSK cells, and HSCs were measured by qPCR as previously described.22 The details of this assay can be found in supplemental Methods.
Statistical analysis
The details of data statistical analysis can be found in supplemental Methods.
Results
TBI causes residual BM suppression by selectively inducing HSC senescence
Our previous studies showed that exposure of mice to a sublethal dose of TBI caused a sustained reduction in BM LSK cells and long-term suppression of BM function.8,9,23 These changes were associated with significant increases in p16 and Arf expression and SA-β-gal staining in LSK cells.8 Therefore, we hypothesized that TBI causes residual BM suppression via induction of HSC senescence. However, LSK cells are heterogeneous and contain MPPs and HSCs. It is not known whether TBI actually induces HSC senescence. To clarify this issue, we exposed C57BL/6 mice to 6 Gy TBI. Two months after TBI when various peripheral blood cell counts and BM cell numbers in the irradiated mice had returned to normal levels,8 we harvested BM from these irradiated mice and unirradiated controls and analyzed the frequencies and numbers of different hematopoietic cell populations in BM cells by flow cytometry, as shown in Figure 1A. The results from this assay revealed that the frequencies and numbers of HPCs (Lin–Sca1–c-kit+ cells) in the irradiated mice had nearly reached normal levels, but the frequencies and numbers of LSK cells in these mice remained significantly lower than those in controls (Figure 1B). These changes in LSK cells were mainly attributed to the significant reduction in MPPs because the level of HSCs was relatively normal in the irradiated mice (Figure 1B). Further analysis revealed that exposure to TBI did not significantly change the frequencies and numbers of ST-HSCs and LT-HSCs in BM cells (Figure 1B).
Figure 1.
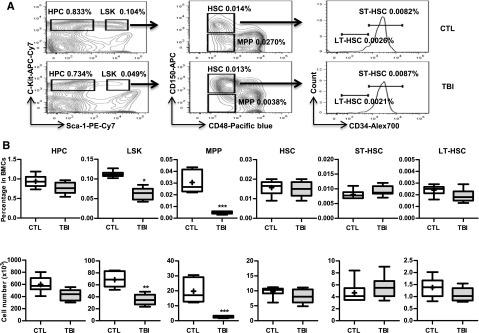
TBI causes sustained quantitative reduction of MPPs but not of HPCs and HSCs. C57BL/6 mice were exposed to a sublethal dose (6 Gy) of TBI or were sham irradiated as a control (CTL). Two months after TBI, BM cells (BMCs) were harvested from the 2 hind legs of individual mice for analysis. (A) Representative gating strategy of flow cytometric analysis for HPCs (Lin–Sca1–c-kit+ cells), LSK cells (Lin–Sca1+c-kit+ cells), MPPs (CD150–CD48–LSK cells), HSCs (CD150+CD48–LSK cells), ST-HSCs (CD34+CD150+CD48–LSK cells), and LT-HSCs (CD34–CD150+CD48–LSK cells) in lineage-negative (Lin–) BM-MNCs is shown. (B) Frequencies (upper panel) and total numbers (lower panel) of HPCs, LSK cells, MPPs, HSCs, ST-HSCs, and LT-HSCs in BMCs from each mouse are presented as mean ± SD (n = 6-8 mice/group). *P < .05, **P < .01, and ***P < .001, TBI vs CTL.
Although TBI does not cause sustained quantitative reduction in HSCs, it induces long-term alterations in HSC functions. When measured by 4- and 6-week CAFCs, the frequencies of functional HSCs were significantly lower in BM cells from the irradiated mice than in those from normal controls (Figure 2A). However, the frequencies of functional HPCs represented by 1- and 2-week CAFCs were relatively normal in BM cells from the irradiated mice compared with those from unirradiated controls (Figure 2A). Furthermore, after transplantation into lethally irradiated recipients (Figure 2B), HSCs from irradiated mice exhibited significant functional defects compared with normal HSCs, including reduced ability to generate long-term donor cell engraftment and myeloid lineage skewing (Figure 2C-D).
Figure 2.
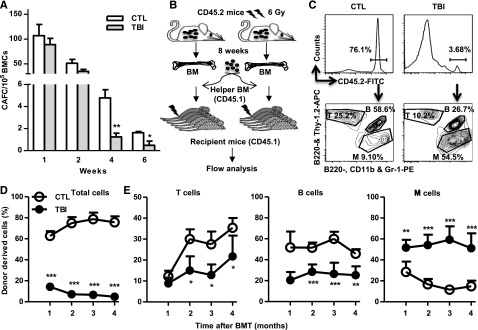
TBI causes sustained reduction of HSC clonogenic activity and long-term repopulating ability. (A) Two months after 6 Gy TBI, total BM cells (BMCs) were harvested from control (CTL) and irradiated (TBI) mice and were analyzed by CAFC assays. The numbers of 1-, 2-, 4-, and 6-week CAFCs were counted and expressed as mean ± SD (n = 3-4 mice per group) of CAFCs per 100 000 BMCs. *P < .05 and **P < .01, TBI vs CTL. (B) Diagram illustrating the competitive repopulating assay. (C) Representative gating strategy for flow cytometric analysis of CD45.2 donor cell engraftment with anti-CD45.2-FITC antibody. B cells were labeled with anti-B220-APC and B220-PE, T cells with anti-Thy1.2-APC, and myeloid (M) cells with anti-CD11b-PE and Gr-1-PE. (D) Percentages of total donor-derived (CD45.2) hematopoietic cells in peripheral blood of the recipients are presented as mean ± SD (CTL: n = 6; TBI: n = 8). ***P < .001, TBI vs CTL. (E) Percentages of T, B, and M cells in donor-derived hematopoietic cells in peripheral blood of the recipients are presented as mean ± SD (CTL: n = 6; TBI: n = 8). *P < .05, **P < .01, and ***P < .001, TBI vs CTL.
To determine whether the dysfunction of HSCs from irradiated animals is attributable to induction of HSC senescence by IR, the activity of SA-β-gal in HSCs and their progeny was directly measured by a newly developed SA-β-gal flow cytometric assay. As we showed previously,8 increased SA-β-gal staining was found after TBI mainly in LSK cells but not in HPCs (Figure 3A and supplemental Figure 1). Further analysis revealed that more HSCs (particularly LT-HSCs) than MPPs were stained positive for SA-β-gal after TBI, indicating that increased SA-β-gal staining in LSK cells is primarily attributable to the induction of HSC senescence by IR. In contrast, no significant changes in apoptosis were found in HSCs from mice 2 months after TBI based on the results from the Annexin V staining assay (Figure 3B). These findings suggest that TBI induced residual BM suppression, at least in part, via selective induction of HSC senescence. This suggestion is supported by the finding that irradiated HSCs expressed about 18-fold higher levels of p16 and 14-fold higher levels of Arf mRNA than did unirradiated HSCs (Figure 3C). Increased expression of p16 and Arf has been implicated in mediating induction of senescence in a variety of cells including HSCs and is the most robust biomarker of senescence cells besides SA-β-gal staining.8,24,25 In addition, expression of p21 mRNA, but not that of p27 and p57 mRNA, was also moderately higher in HSCs from irradiated mice than in those from control mice. Increased expression of p21 also plays a role in induction of senescence, particularly during its initiation.26
Figure 3.
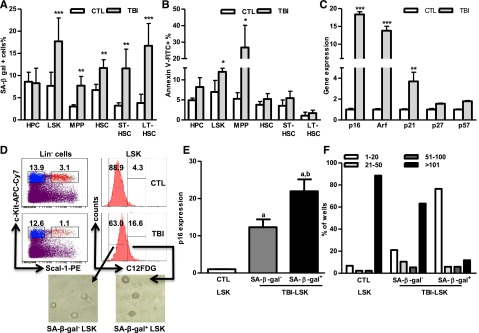
TBI induces senescence but not apoptosis in HSCs. BMCs were harvested from control (CTL) and irradiated (TBI) mice 2 months after 6 Gy TBI as described. (A-B) Percentages of SA-β-gal+ cells (A) and Annexin V–positive apoptotic cells (B) in HPCs, LSK cells, MPPs, HSCs, ST-HSCs, and LT-HSCs are presented as mean ± SD (CTL: n = 4; TBI: n = 6). *P < .05, **P < .01, and ***P < .001, TBI vs CTL. (C) Fold increases in relative gene expression for various CDK inhibitors in sorted HSCs after TBI. Data from 3 independent experiments using sorted HSCs pooled from 3 to 4 mice per group are presented as mean ± SD (n = 3). **P < .01 and ***P < .001, TBI vs CTL. (D) SA-β-gal+ and SA-β-gal– LSK cells were isolated from irradiated mice (TBI) 2 months after 6 Gy TBI by cell sorting according to the intensity of C12FDG staining. Their differential SA-β-gal activities were confirmed by SA-β-gal enzymatic activity assay after isolation as shown in the inserted microscopic images. (E) The expression of p16 in SA-β-gal+ and SA-β-gal– LSK cells and in control unirradiated LSK cells (CTL) was measured with qRT-PCR and expressed as fold increases compared with CTL. Data are presented as mean ± SD (n = 3). a, P < .05 vs CTL-LSK cells; b, P < .05 vs TBI-LSK β-gal– cells. (F) Cumulative production of number of cells from wells with single TBI SA-β-gal+ and SA-β-gal– LSK cells and control (CTL) LSK cells is shown as a representative of 3 separated assays.
However, not all HSCs became senescent 2 months after TBI because only about 12% and 17% of HSCs and LSK cells from irradiated mice exhibited significant increases in SA-β-gal staining, respectively (Figure 3A and supplemental Figure 1), which was also confirmed by analysis of p16 and p21 mRNA expression at the single-cell level in HSCs, as shown in supplemental Figure 2. Therefore, we sorted SA-β-gal– and SA-β-gal+ LSK cells from irradiated mice and compared their reproductive functions in vitro in a single cell culture and in vivo after transplantation to determine whether SA-β-gal+ LSK cells represent true senescent cells. As is shown in Figure 3D-F and Figure 4A-C, SA-β-gal+ LSK cells expressed significantly higher levels of p16, exhibited diminished ability to proliferate in vitro, and produced substantially lower levels of long-term engraftment after transplantation compared with SA-β-gal– LSK cells and LSK cells from control unirradiated mice. In addition, SA-β-gal– LSK cells from irradiated mice also expressed slightly higher levels of p16 and exhibited some deficiencies in their proliferative and repopulating functions compared with those of control unirradiated LSK cells.
Figure 4.
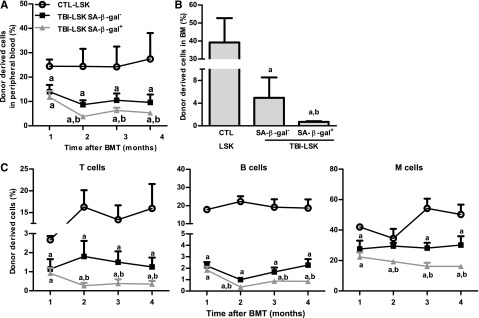
SA-β-gal+ LSK cells from total body–irradiated mice are highly deficient in long-term engraftment after transplantation. Five hundred CD45.2 SA-β-gal+ or SA-β-gal– LSK cells isolated from irradiated mice (TBI) or 500 control unirradiated LSK cells (CTL) along with 2 × 105 competitive CD45.1 BM cells were transplanted into lethally irradiated CD45.1 recipient. (A) Percentages of total donor-derived cells in peripheral blood at various times after transplantation. (B) Percentage of donor cell engraftment in BM 4 months after transplantation. (C) Percentages of peripheral blood T-cell, B-cell, and myeloid (M) cell engraftment at various times after transplantation. The data are presented as mean ± SD (n = 6 recipients/group). a, P < .05 vs CTL-LSK cells; b, P < .05 vs TBI-LSK β-gal– cells.
TBI induces HSC senescence independent of telomere shortening
Induction of cellular senescence occurs as a result of extensive replication, which results in telomere shortening or which occurs prematurely after cells are exposed to oxidative, genotoxic, or oncogenic stress.27-29 Our previous studies suggest that IR induces HSC senescence primarily via induction of oxidative stress,9,23,30 which may induce HSC senescence prematurely, via induction of oxidative damage, or by increasing HSC cycling that results in telomere shortening. To investigate these possibilities, intracellular production of ROS in different populations of hematopoietic cell from irradiated mouse BM was compared with that from normal controls. As shown in Figure 5A and supplemental Figure 3A, ROS production in HPCs and MPPs from irradiated mice was slightly higher (∼1.2- to 1.3-fold) than in cells from unirradiated mice. However, after TBI, ROS production in irradiated HSCs including ST-HSCs and LT-HSCs was more than 1.7-fold higher than in respective unirradiated control cell populations, confirming that TBI can selectively induce chronic oxidative stress in HSCs. The increased production of ROS in irradiated LT-HSCs was associated with a significant reduction in cell quiescence (eg, fewer G0 cells) and an increase in DNA synthesis (eg, more BrdU-labeled cells) (Figure 5B-C and supplemental Figure 3B-C), indicating that LT-HSCs are highly responsive to ROS-stimulated cell proliferation. Alternatively, increased cycling of irradiated LT-HSCs may represent a mechanism for nonsenescent LT-HSCs to compensate for the loss of senescent LT-HSCs after TBI; however, this increased cycling did not result in telomere shortening in HSCs and their progeny (Figure 5D-F), providing key evidence that TBI induces HSC senescence in a telomere shortening–independent manner. These findings suggest that HSCs mainly undergo premature senescence after exposure to IR.
Figure 5.
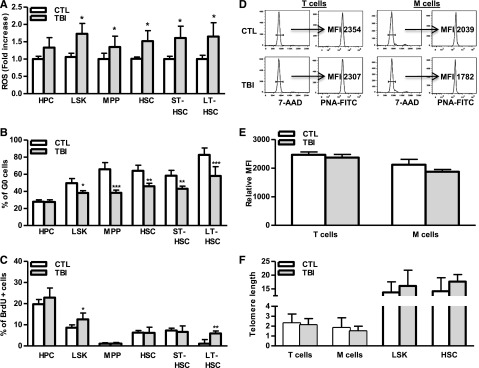
TBI causes persistent increases in ROS production and cell cycling in HSCs without induction of telomere shortening. BMCs were harvested from control (CTL) and irradiated (TBI) mice 2 months after 6 Gy TBI as described. (A) Fold increases in ROS production in BM HPCs, LSK cells, MPPs, HSCs, ST-HSCs, and LT-HSCs after TBI are presented as mean ± SD (CTL: n = 4 ; TBI: n = 6). *P < .05, TBI vs CTL. (B-C) Percentages of G0 (B) and BrdU-positive cells (C) in BM HPCs, LSK cells, MPPs, HSCs, ST-HSCs, and LT-HSCs from control (CTL) and irradiated (TBI) mice are presented as mean ± SD (CTL: n = 4 ; TBI: n = 6). *P < .05, **P < .01, and ***P < .001, TBI vs CTL. (D) Representative flow plots for telomere length measurement in T cells and myeloid cells (M cells) by flow FISH. (E) The relative telomere lengths of these cells from 3 independent experiments using sorted T and M cells pooled from 3 to 4 TBI or control (CTL) mice per group are presented as mean ± SD of relative MFI of telomere fluorescent staining. (F) Relative telomere lengths of HSCs, LSK cells, T cells, and myeloid cells (M cells) from control (CTL) and irradiated (TBI) mice analyzed by qPCR-based telomere length assays. Data from 3 independent experiments using sorted HSC, LSK cells, T cells, and M cells pooled from 3 to 4 mice per group are presented as mean ± SD of the ratios of telomere vs the 36B4 single-copy gene.
Knockout of p16 and/or Arf fails to protect HSCs from TBI-induced senescence
The p16-Arf locus encodes 2 tumor suppressors, p16 and Arf,10,11 which have been implicated in mediating induction of cellular senescence in a variety of cells including HSCs. Because increased expression of p16 and Arf was found in irradiated HSCs in association with elevated SA-β-gal activity and significantly reduced HSC function, we next examined whether p16 and/or Arf play important roles in IR-induced HSC senescence using the well-characterized p16 and/or Arf KO mouse models. In basal conditions, all of these KO mice had similar hematologic profiles as those of wild-type (WT) controls, including comparable frequencies and numbers of HPCs, LSK cells, MPPs, and HSCs in BM (Figure 6 and supplemental Figures 4 and 5). Two months after TBI, they also exhibited the same degrees of reductions in BM LSK cells and MPPs, with no significant changes in HPCs and HSCs, except that p16 KO mice showed a significant reduction in HSCs after TBI, compared with unirradiated controls. Before exposure to TBI, BM cells from the KO mice also generated similar numbers of various CAFCs; after TBI, they exhibited comparable reductions in HSC clonogenicity, manifested by decreases in 4- and 6-week CAFCs, which were similar to those seen in cells from irradiated WT mice (Figure 7A-C). These findings suggest that p16 and Arf do not mediate induction of HSC senescence by TBI, even though TBI can upregulate their expression in HSCs.
Figure 6.
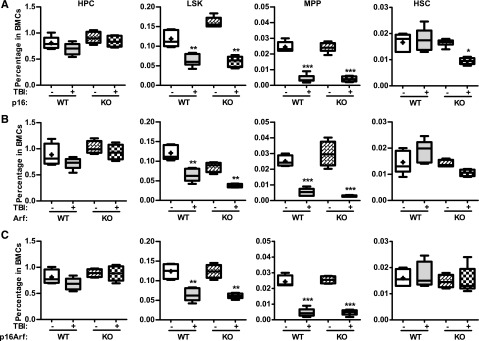
Knockout of p16 and/or Arf has no effect on TBI-induced reduction of MPPs. p16−/− (KO), Arf−/− (KO), p16Arf−/− (KO), and WT mice were exposed to a sublethal dose (6 Gy) of TBI or were sham irradiated as a control. Two months after TBI, BM cells (BMCs) harvested from the 2 hind legs of individual mice were analyzed by flow cytometry for HPCs, LSK cells, MPPs, and HSCs. The frequencies of HPCs, LSK cells, MPPs, and HSCs in BMCs from each mouse strain are presented as mean ± SD (CTL: n = 4; TBI: n = 6). *P < .05, **P < .01, and ***P < .001, TBI vs control unirradiated mice.
Figure 7.

Knockout of p16 and/or Arf has no effect on TBI-induced HSC senescence and long-term injury. (A-C) Two months after 6 Gy TBI, total BM cells (BMCs) were harvested from control and irradiated (TBI) p16−/−, Arf−/−, and p16Arf−/− mice and p16+/+, Arf+/+, and p16Arf+/+ mice for clonogenic function analysis by CAFC assays. The numbers of 1-, 2-, 4-, and 6-week CAFCs were counted and expressed as mean ± SD (n = 3 mice/group) of CAFCs per 100 000 BMCs. *P < .05 and **P < .01, TBI vs CTL. (D) Two months after 6 Gy TBI, total BM cells (BMCs) were harvested from control and irradiated (TBI) p16Arf−/− and p16Arf+/+ mice for analyses of SA-β-gal activity in various populations of BM hematopoietic cells using C12FDG as a substrate. Percentages of SA-β-gal–positive cells in HPCs, LSK cells, MPPs, HSCs, ST-HSCs, and LT-HSCs are presented as mean ± SD (CTL: n = 3 and TBI: n = 4). **P < .01 and ***P < .001, TBI vs CTL. (E) Two months after 6 Gy TBI, total BM cells (BMCs) were harvested from control and irradiated (TBI) p16−/− and p16+/+ mice for analysis of HSC long-term repopulating ability by CRA as described in Figure 2B. Percentages of total donor-derived (CD45.2) hematopoietic cells in peripheral blood of the recipients and percentages of T cells, B cells, and myeloid (M) cells in donor-derived hematopoietic cells in the peripheral blood of recipients are presented as mean ± SD (n = 9-10 recipients from 2 independent experiments). ***P < .001, TBI vs CTL. (F) Repopulating units (RU) calculated according to the engraftment data presented in (E) are presented as mean ± SD. ***P < .001, TBI vs CTL.
This suggestion is further supported by the finding that similar numbers of BM ST-HSCs and LT-HSCs from both WT and p16-Arf KO mice became SA-β-gal+ after TBI (Figure 7D), despite the fact that HSCs from p16-Arf KO mice express no detectable p16 and Arf mRNA (supplemental Figure 6). In addition, when BM cells from irradiated p16 KO mice were transplanted into lethally irradiated recipients, they demonstrated defects in HSC function that were similar to those of cells from irradiated WT mice, including reduced long-term donor cell engraftment and myeloid lineage skewing (Figure 7). Because the BM cells from Arf and p16-Arf KO mice have a higher propensity for malignant transformation, particularly after TBI, we did not perform the competitive repopulation assay on irradiated BM cells from these mice. Therefore, these data demonstrate that IR induces residual BM suppression and HSC senescence in a p16/Arf-independent manner.
Discussion
Because of improvements in early detection and treatment of cancer, the number of cancer survivors is increasing. Unfortunately, long-term cancer survivors are at increased risk for developing late effects related to cancer treatment, including IR-induced residual BM injury. IR-induced residual BM injury can deteriorate to become hypoplastic anemia or myelodysplastic syndrome over time or after additional treatment with IR and/or chemotherapy or undergoing autologous BM transplantation.1,2 To address this emerging problem and develop effective interventions, we must have a better understanding of the pathogenesis of this late effect. Therefore, in the present study, we investigated how residual BM suppression was induced by IR in a mouse model.
The results from our study showed that exposure of mice to a sublethal dose of TBI did not induce a reduction in the numbers of BM HSCs but resulted in long-term alterations in HSC functions, including reduced clonogenicity, diminished ability to generate long-term donor cell engraftment after transplantation, and skewed differentiation toward myeloid lineages. Some of these changes are likely attributable to induction of HSC senescence. This is supported by the finding that HSCs isolated from the sublethally irradiated mice expressed increased levels of SA-β-gal and p16, two of the most widely used biomarkers for senescent cells, but the cells showed minimal changes in apoptosis compared with HSCs from unirradiated control animals. However, induction of HSC differentiation in response to IR-induced DNA damage via activation of the granulocyte colony-stimulating factor/Stat3/BATF–dependent differentiation checkpoint also contribute to IR-induced myeloid-skewed HSCs by selectively depleting lymphoid-biased HSCs, as was recently reported.31 In addition, damage to the HSC niche, such as induction of BM stromal cell senescence, and a negative bystander effect of TBI on HSCs, also play a role in IR-induced residual BM injury.32,33
Our studies also revealed that only a small fraction of HSCs became senescent at a time after TBI. This may be in part attributable to the immunosurveillance of senescent cells by immune cells that can rapidly remove senescent cells to prevent the accumulation of senescent cells in vivo.34 In addition, it is possible that induction of HSC senescence by IR is a dynamic process in which some HSCs undergo senescence more readily than others. This suggestion is supported by the finding that SA-β-gal– LSK cells from irradiated mice, compared with control nonirradiated LSK cells, also expressed slightly higher levels of p16 and exhibited some deficiencies in reproductive functions, indicating that they too are abnormal. Even though they have not become senescent but may be on their way to becoming senescent cells with time or after exposure to additional stress, as SA-β-gal– LSK cells from irradiated mice also exhibited a significant reduction in long-term engraftment after transplantation compared with LSK cells from control mice.
A growing body of evidence suggests that ROS plays an important role in mediating induction of HSC senescence under various pathological conditions, such as mutation of ATM and various Fanconi anemia genes; deletion of Bmi1, FoxOs, Mdm2, and Tsc1; and aging.15,19,20,35-38 Our recent studies showed that exposure of mice to TBI induced persistent increases in ROS production in HSC-enriched LSK cells, in part via upregulation of nicotinamide adenine dinucleotide phosphate-oxidase 4.23 Treating the irradiated mice with various antioxidants after IR significantly attenuated TBI-induced LSK cell senescence and residual BM suppression.9,30 In addition, ectopic expression of manganese superoxide dismutase and catalase in HSCs reduces IR-induced HSC injury.39
However, the molecular mechanisms by which ROS mediates the induction of HSC senescence by IR were unknown. The results from our current study suggest that ROS may mediate IR-induced HSC senescence in a telomere-independent manner, because increased production of ROS by irradiated HSCs was not associated with telomere shortening in HSCs and their progeny, even though increased production of ROS was associated with a significant reduction in HSC quiescence and an increase in HSC proliferation after TBI. Similar results were also observed recently in senescent HSCs from Atm-mutated mice.21 Alternatively, ROS may mediate IR-induced premature senescence of HSCs via activation of the p38-p16 pathway. It has been shown that p38 plays an important role in regulating HSC self-renewal, and activation of p38 by oxidative stress can mediate induction of HSC senescence in Atm mutants and FoxO3 KO mice via upregulation of p16.21,36 Our previous studies showed that activation of p38 was involved in IR-induced BM hematopoietic cell senescence and dysfunction.30,40
However, the present study showed that induction of HSC senescence by IR was independent of p16, because knockout of p16 in mice had no significant effects on TBI-induced HSC senescence and residual BM suppression. Furthermore, the lack of effect of p16 deletion cannot be attributed to compensation by Arf, because HSCs from irradiated Arf and p16/Arf KO mice exhibited changes similar to those seen in the cells from WT mice after TBI. The finding that IR induced HSC senescence and residual BM suppression in a p16 and Arf-independent manner is intriguing, because upregulation of p16 and Arf has been implicated in mediating induction of HSC senescence in a variety of pathological conditions and during aging,8,41,42 and it has been shown that IR induced senescence in BM stromal cells in a p16- and Arf-dependent manner.32 Furthermore, early T-lymphoid progenitors are exquisitely dependent on CDK4/6 activity and, therefore, are very sensitive to p16 upregulation for induction of senescence during aging.42,43 Collectively, these findings suggest that p16/Arf can mediate induction of cellular senescence in a manner dependent on cell type and cell context.
It remains of great interest to determine whether other CDK inhibitors may be able to functionally substitute for p16 to mediate IR-induced HSC senescence. One of the inhibitors that may substitute p16 is p21, because TBI also caused a sustained increase in p21 expression in HSCs, and increased expression of p21 has been implicated in induction of cellular senescence independent of p16.44,45 However, it was recently reported that p21 upregulation actually protected HSCs against IR by limiting IR-induced DNA damage and p53 activation and by promoting HSC cell cycle entry and self-renewing expansion, suggesting that p21 and p16 may have opposite effects on IR-induced HSC senescence.46-48 In addition, we did not detect significant increases in p21 and p18Ink4c (p18) mRNA expression in HSCs from irradiated p16/Arf KO mice (supplemental Figure 6), although p18 can also negatively regulate HSC self-renewal.49 Nor was the expression of p27 and p57 significantly elevated by TBI in HSCs from the KO mice. Instead, a >80-fold increase in the expression of p15Ink4b (p15) mRNA was observed in HSCs from irradiated p16/Arf KO mice, whereas a <tenfold increase in p15 mRNA expression was detected in HSCs from irradiated WT mice, compared with the cells from control animals (supplemental Figure 6). It has been shown that p15, encoded by the p15 gene adjacent to the p16-Arf locus, assumes a significant role in tumor suppression by mediating oncogenic stress-induced senescence in the absence of p16.50 Therefore, it will be of great interest to determine whether p15 can function as a substitute for p16 to mediate TBI-induced HSC senescence in p16/Arf KO mice when Cdkn2ab−/− mice that are deficient for p15, p16, and Arf expression become available for our study in the future.
In summary, the results from this study provide important new insights into the mechanism by which IR causes long-term BM suppression via induction of premature senescence of HSCs in a p16-Arf–independent manner. A better understanding of the mechanism will allow us to develop new interventions to circumvent IR-induced long-term toxicity to BM via molecularly targeted inhibition of IR-induced HSC senescence, providing the interventions do not cause increased risk of tumorigenesis. Because some chemotherapeutic agents can also induce HSC senescence and residual BM injury, these interventions may also benefit cancer patients undergoing chemotherapy.
Supplementary Material
Acknowledgments
This study was supported in part by grants from the National Institutes of Health (R01-CA122023 and AI080421), a grant from the Edward P. Evans Foundation, a grant from the National Natural Science Foundation of China (81129020), the National Program on Key Basic Research Project (2011CB964800-G), and the Arkansas Research Alliance Scholarship from the Arkansas Science & Technology Authority.
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: L.S. designed, performed, and analyzed experiments and wrote the manuscript; W.F., H.L., D.G., Y.L., Y.W., and L.L. performed experiments; A.M. analyzed experiments; N.E.S. analyzed experiments and wrote the manuscript; and D.Z. designed and analyzed experiments and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for L.L. is Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Correspondence: Daohong Zhou, MD, Winthrop P. Rockefeller Cancer Institute, University of Arkansas for Medical Sciences, 4301 W Markham, #607, Little Rock, AR 72205; e-mail: dzhou@uams.edu.
References
- 1.Mauch P, Constine L, Greenberger J, et al. Hematopoietic stem cell compartment: acute and late effects of radiation therapy and chemotherapy. Int J Radiat Oncol Biol Phys. 1995;31(5):1319–1339. doi: 10.1016/0360-3016(94)00430-S. [DOI] [PubMed] [Google Scholar]
- 2.Testa NG, Hendry JH, Molineux G. Long-term bone marrow damage in experimental systems and in patients after radiation or chemotherapy. Anticancer Res. 1985;5(1):101–110. [PubMed] [Google Scholar]
- 3.Mohrin M, Bourke E, Alexander D, et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7(2):174–185. doi: 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner RV, Begue R, McKinnon E. The effect of granulocyte-macrophage colony-stimulating factor (GM-CSF) on primitive hematopoietic stem cell (PHSC) function and numbers, after chemotherapy. Exp Hematol. 2001;29(9):1053–1059. doi: 10.1016/s0301-472x(01)00685-3. [DOI] [PubMed] [Google Scholar]
- 5.van Os R, Robinson S, Sheridan T, Mislow JM, Dawes D, Mauch PM. Granulocyte colony-stimulating factor enhances bone marrow stem cell damage caused by repeated administration of cytotoxic agents. Blood. 1998;92(6):1950–1956. [PubMed] [Google Scholar]
- 6.van Os R, Robinson S, Sheridan T, Mauch PM. Granulocyte-colony stimulating factor impedes recovery from damage caused by cytotoxic agents through increased differentiation at the expense of self-renewal. Stem Cells. 2000;18(2):120–127. doi: 10.1634/stemcells.18-2-120. [DOI] [PubMed] [Google Scholar]
- 7.Marusyk A, Porter CC, Zaberezhnyy V, DeGregori J. Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol. 2010;8(3):e1000324. doi: 10.1371/journal.pbio.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Schulte BA, LaRue AC, Ogawa M, Zhou D. Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood. 2006;107(1):358–366. doi: 10.1182/blood-2005-04-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Wang Y, Pazhanisamy SK, et al. Mn(III) meso-tetrakis-(N-ethylpyridinium-2-yl) porphyrin mitigates total body irradiation-induced long-term bone marrow suppression. Free Radic Biol Med. 2011;51(1):30–37. doi: 10.1016/j.freeradbiomed.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13(1):77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 11.Sharpless NE, DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev. 1999;9(1):22–30. doi: 10.1016/s0959-437x(99)80004-5. [DOI] [PubMed] [Google Scholar]
- 12.Cheng T. Cell cycle inhibitors in normal and tumor stem cells. Oncogene. 2004;23(43):7256–7266. doi: 10.1038/sj.onc.1207945. [DOI] [PubMed] [Google Scholar]
- 13.Narita M, Nũnez S, Heard E, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113(6):703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 14.Sherr CJ, Weber JD. The ARF/p53 pathway. Curr Opin Genet Dev. 2000;10(1):94–99. doi: 10.1016/s0959-437x(99)00038-6. [DOI] [PubMed] [Google Scholar]
- 15.Park IK, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 16.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423(6937):255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 17.Lewis JL, Chinswangwatanakul W, Zheng B, et al. The influence of INK4 proteins on growth and self-renewal kinetics of hematopoietic progenitor cells. Blood. 2001;97(9):2604–2610. doi: 10.1182/blood.v97.9.2604. [DOI] [PubMed] [Google Scholar]
- 18.Stepanova L, Sorrentino BP. A limited role for p16Ink4a and p19Arf in the loss of hematopoietic stem cells during proliferative stress. Blood. 2005;106(3):827–832. doi: 10.1182/blood-2004-06-2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janzen V, Forkert R, Fleming HE, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443(7110):421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 20.Ito K, Hirao A, Arai F, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431(7011):997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 21.Ito K, Hirao A, Arai F, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12(4):446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 22.Callicott RJ, Womack JE. Real-time PCR assay for measurement of mouse telomeres. Comp Med. 2006;56(1):17–22. [PubMed] [Google Scholar]
- 23.Wang Y, Liu L, Pazhanisamy SK, Li H, Meng A, Zhou D. Total body irradiation causes residual bone marrow injury by induction of persistent oxidative stress in murine hematopoietic stem cells. Free Radic Biol Med. 2010;48(2):348–356. doi: 10.1016/j.freeradbiomed.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92(20):9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krishnamurthy J, Torrice C, Ramsey MR, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114(9):1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pantoja C, Serrano M. Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras. Oncogene. 1999;18(35):4974–4982. doi: 10.1038/sj.onc.1202880. [DOI] [PubMed] [Google Scholar]
- 27.Serrano M, Blasco MA. Putting the stress on senescence. Curr Opin Cell Biol. 2001;13(6):748–753. doi: 10.1016/s0955-0674(00)00278-7. [DOI] [PubMed] [Google Scholar]
- 28.Marcotte R, Wang E. Replicative senescence revisited. J Gerontol A Biol Sci Med Sci. 2002;57(7):B257–B269. doi: 10.1093/gerona/57.7.b257. [DOI] [PubMed] [Google Scholar]
- 29.Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Liu L, Zhou D. Inhibition of p38 MAPK attenuates ionizing radiation-induced hematopoietic cell senescence and residual bone marrow injury. Radiat Res. 2011;176(6):743–752. doi: 10.1667/rr2727.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Sun Q, Morita Y, et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell. 2012;148(5):1001–1014. doi: 10.1016/j.cell.2012.01.040. [DOI] [PubMed] [Google Scholar]
- 32.Carbonneau CL, Despars G, Rojas-Sutterlin S, et al. Ionizing radiation-induced expression of INK4a/ARF in murine bone marrow-derived stromal cell populations interferes with bone marrow homeostasis. Blood. 2012;119(3):717–726. doi: 10.1182/blood-2011-06-361626. [DOI] [PubMed] [Google Scholar]
- 33.Shen H, Yu H, Liang PH, et al. An acute negative bystander effect of γ-irradiated recipients on transplanted hematopoietic stem cells. Blood. 2012;119(15):3629–3637. doi: 10.1182/blood-2011-08-373621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ceccaldi R, Parmar K, Mouly E, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012;11(1):36–49. doi: 10.1016/j.stem.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyamoto K, Araki KY, Naka K, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1(1):101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Abbas HA, Maccio DR, Coskun S, et al. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell. 2010;7(5):606–617. doi: 10.1016/j.stem.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen C, Liu Y, Liu R, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205(10):2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miao W, Xufeng R, Park MR, et al. Hematopoietic stem cell regeneration enhanced by ectopic expression of ROS-detoxifying enzymes in transplant mice. Mol Ther. 2013;21(2):423–432. doi: 10.1038/mt.2012.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li D, Wang Y, Wu H, et al. Mitigation of ionizing radiation-induced bone marrow suppression by p38 inhibition and G-CSF administration. J Radiat Res (Tokyo) 2011;52(6):712–716. doi: 10.1269/jrr.11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Johnson SM, Fedoriw Y, et al. Expression of p16(INK4a) prevents cancer and promotes aging in lymphocytes. Blood. 2011;117(12):3257–3267. doi: 10.1182/blood-2010-09-304402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Sanoff HK, Cho H, et al. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell. 2009;8(4):439–448. doi: 10.1111/j.1474-9726.2009.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Signer RA, Montecino-Rodriguez E, Witte ON, Dorshkind K. Aging and cancer resistance in lymphoid progenitors are linked processes conferred by p16Ink4a and Arf. Genes Dev. 2008;22(22):3115–3120. doi: 10.1101/gad.1715808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Keizer PL, Packer LM, Szypowska AA, et al. Activation of forkhead box O transcription factors by oncogenic BRAF promotes p21cip1-dependent senescence. Cancer Res. 2010;70(21):8526–8536. doi: 10.1158/0008-5472.CAN-10-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takeuchi S, Takahashi A, Motoi N, et al. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression in vivo. Cancer Res. 2010;70(22):9381–9390. doi: 10.1158/0008-5472.CAN-10-0801. [DOI] [PubMed] [Google Scholar]
- 46.Insinga A, Cicalese A, Faretta M, et al. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proc Natl Acad Sci USA. 2013;110(10):3931–3936. doi: 10.1073/pnas.1213394110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baker DJ, Weaver RL, van Deursen JM. p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell Rep. 2013;3(4):1164–1174. doi: 10.1016/j.celrep.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson SM, Torrice CD, Bell JF, et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest. 2010;120(7):2528–2536. doi: 10.1172/JCI41402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu H, Yuan Y, Shen H, Cheng T. Hematopoietic stem cell exhaustion impacted by p18 INK4C and p21 Cip1/Waf1 in opposite manners. Blood. 2006;107(3):1200–1206. doi: 10.1182/blood-2005-02-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krimpenfort P, Ijpenberg A, Song JY, et al. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature. 2007;448(7156):943–946. doi: 10.1038/nature06084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.