

Abstract
Alzheimer’s disease (AD) is a complex neurodegenerative disorder with a multi-faceted pathogenesis. So far, the therapeutic paradigm “one-compound-one-target” has failed and despite enormous efforts to elucidate the pathophysiology of AD, the disease is still incurable.
The multiple factors involved in AD include amyloid aggregation to form insoluble neurotoxic plaques of Aβ, hyperphosphorylation of tau protein, oxidative stress, calcium imbalance, mitochondrial dysfunction and deterioration of synaptic transmission. These factors together, accentuate changes in the CNS homeostasis, starting a complex process of interconnected physiological damage, leading to cognitive and memory impairment and neuronal death.
A recent approach for the rational design of new drug candidates, also called multitarget-directed ligand (MTDL) approach, has gained increasing attention by many research groups, which have developed a variety of hybrid compounds acting simultaneously on diverse biological targets. This review aims to show some recent advances and examples of the exploitation of MTDL approach in the rational design of novel drug candidate prototypes for the treatment of AD.
Keywords: Alzheimer’s disease, multi-target directed drugs, neurodegenerative disorders, rational drug design.
1. INTRODUCTION
Alzheimer's disease (AD) is one of the biggest challenges of current research for new drugs [1-3]. In addition to cancer, diabetes, rheumatoid arthritis and other chronic inflammatory and neurodegenerative diseases, AD is characterized by multiple factors involving physiological, biochemical, chemical mediators operating concurrently with, caused by the same or different pathways [4, 5].
In recent years, advances in biochemistry, neuro-pharmacology and other biological fields have been responsible for new insights in the high complexity multifactorial pathophysiological hallmarks of AD. Thus, the development of innovative and effective multi-target directed drugs for AD represents a new paradigm in the discovery and drug design. Until then, the question that guided the planning of a new drug was “What is the best target for the treatment or prevention of a disease?” which supported, since the decade of 90, the origin of the reductionist strategy. By this concept, a drug should have its action limited to individual genes, a single protein or enzyme and a specific ligand to a target, following the current paradigm “one gene, one target, one drug” [1, 4, 6].
The limitations imposed by the low therapeutic efficacy against most of these diseases, coupled with the apparent reduction in the approval of new bioactive chemical entities, despite significant investments in the Pharmaceutical Industry, have reinforced the need to look up new strategies for planning and discovery of more effective and safer drugs.
In this context, three therapeutic strategies are possible: the first would be the use in combination of multiple active ingredients, like the associations of natural ingredients used for centuries in Traditional Chinese Medicine; or cocktails of drugs, such as used in the treatment of the acquired immunodeficiency syndrome (AIDS) and cancer. Another alternative would be a combination of more than one active ingredient in the same formulation. In a third and more recent approach, it would be a single molecule assembling structural requirements to allow molecular recognition of more than one target simultaneously, featuring a multi-target drug [4, 5]. When multiple targets related to the same pathology are associated with distinct biochemical cascades, the active ligand can be also called as a symbiotic compound [4].
2. SOCIO-ECONOMIC IMPACT AND PREVALENCE OF ALZHEIMER'S DISEASE
With the increase in average life expectancy worldwide, neurodegenerative diseases such as AD have attracted great attention, especially in more developed countries. As a progressive, disabling and incurable disease, patients die within average of 10 years after its installation [7]. The age is its main risk factor, with a prevalence of 0.7% among individuals 60-65 years of age and about 40% in the age groups above 90 years [8, 9]. In 2012, about 5.4 million of American people were carriers of AD. Despite the very imprecise estimates available, it is believed that by 2050, over 16 million Americans will have AD, with an annual cost that could exceeds U$ 1.1 trillion [11, 12]. Other estimates point to a global epidemic of AD, reaching 26 million people by 2050, if a cure is not yet discovered and available [10, 14].
The increase in life quality, as a natural consequence of recent advances in medicine and technological development, has been reflected directly in the increased longevity worldwide. The United Nations (UN) believes that the “age of aging” began in 1975 and this trend is expected to extend until 2025 [14, 15]. According to the Brazilian Institute of Geography and Statistics, Europe occupies the 1st position in the proportion of the population above 60 years (19.8%), followed by North America (16.3%) [16].
3. MULTIFACTORIAL ASPECTS OF ALZHEIMER'S DISEASE
AD is a degenerative disorder of the central nervous system (CNS) and is one of the most common dementia among the population above age 65. AD is characterized by a progressive loss of memory, cognition, motor and functional capacity, gradually undermining social behavior and ability to perform routine tasks such as feeding, personal hygiene and interpersonal relationships. Although the etiology of AD is not completely understood, it is well established that operation of multiple interconnected factors are related to the installation, development and evolution of the disease [13].
Apparently, physiological deregulation in some brain regions, which the origin remains not entirely understood, is responsible for an overproduction of amyloid peptide (Aβ). Then, β- e γ-secretase enzymes abnormally cleave Aβ, producing insoluble fragments of 39-43 amino acid residues. Fragments Aβ1-42, even at low concentrations, seem to be more prone to oligomerization and formation of insoluble neurotoxic aggregates, the so-called amyloid plaques, and are the main therapeutic target advocated by the “Amyloid Hypothesis” [5, 6, 17].
A secondary event due to neurotoxicity of amyloid plaques is the hyperphosphorylation of the neuronal microtubule constitutive tau protein. This abnormal phosphorylation of tau leads to structural collapse of microtubules and the consequent release of tau protein fragments, which take the form of insoluble fibrillar coiled, depositing intracellular as neurofibrillary tangles [6, 12, 18-20].
These two biochemical factors acting together lead to accentuate changes in the CNS homeostasis and function, causing numerous cellular processes and organelles activation, acting as starting points of a complex process of interconnected physiological damage, also leading to neuronal death [19]. Although most studies have emphasized the neurotoxicity of amyloid plaques, recent evidences indicate that increasing the concentration of soluble amyloid oligomers generated by overproduction of Aβ can be also responsible for increasing neuronal injury [6, 21].
Mitochondria are the major intracellular targets of soluble Aβ oligomers (sAβ). In excess, sAβ eventually interfere with normal function of mitochondria, causing overproduction of reactive oxygen species (ROS), inhibition of cellular respiration, ATP production and damage in mitochondrial structure [21, 22]. It seems that, the deleterious effects of sAβ the mitochondria are the result of changes in homeostasis and intracellular Ca2+ signaling, as evidenced by the induction of massive influx of Ca+2 in cultured neurons, causing its concentration increasing in mitochondria and neuronal apoptosis [21]. Accumulation of calcium in mitochondria causes opening of the mitochondrial permeability transition pore (mPTP), a wide mitochondrial membrane channel that allows the passage of large molecules bidirectional uncontrolled, resulting in disintegration of organelles and functional structure [21].
Recent studies have shown that the dephosphorylation of neurofibrillary tangles of tau protein was able to restore its ability to bind to microtubules neurons, indicating that the kinetic mechanisms that regulate the phosphorylation/dephosphorylation process are altered in AD [23]. The nature of the protein kinases, phosphatases and tau sites involved in these lesions was recently unveiled, suggesting that activation of protein phosphatase fosfoseril or fosfotreonil (PP-2a) or inhibition of glycogen synthase-kinase 3β (GSK-3β) protein as cyclin-dependent kinase 5 (CDK5) may be required for inhibiting the degeneration caused by neurofibrils in AD [23].
In recent years, many other hypotheses have been proposed to explain the complexity and multifactorial pathogenesis of AD, including oxidative stress, disruption of homeostasis by metal ions and neuroinflammation [19, 24, 25]. Currently, oxidative stress is considered one of the major causative factors of AD, unifying a number of other sequential or individual pathophysiologic events. Oxidative damage in the brain of AD patients is a result of excessive production of free radicals induced by Aβ, functional alteration in mitochondria, inadequacy in energy supplement, production of inflammatory mediators and alteration of antioxidant defenses [26, 28]. Modulation of cellular oxidative processes is closely related to the redox properties of some metals. Change in concentration of such ions can lead to oxidative stress and increased ROS production. Cupper (Cu2+) and zinc (Zn2+) and other metal ions influence the processes of protein aggregation, a critical step in many neurodegenerative diseases. In the case of AD, amyloid precursor protein (APP) and Aβ are able to form complexes with and reduce Cu2+, which forms a high affinity complex with Aβ, promoting their aggregation.
Furthermore, in vitro studies showed that Aβ neuro-toxicity is dependent on the catalytic generation of H2O2, which is enhanced by the presence of Aβ-Cu+2 complexes. In addition, Cu2+, Zn2+ and Fe3+ are present in amyloid plaques in brains with AD, which can be dissolved by the action of metal chelating substances [6, 20, 29].
The production of Aβ also depends on the bioavailability of cholesterol in nerve cells, since the activity balance of the α- and β-secretases is related to the lipid composition of cells. High concentrations of cholesterol into the cells lead to an increase in amyloidogenic APP process by β-secretase, whereas at lower levels, cholesterol metabolism stimulates the increasing the physiological α-secretase action on APP. The hypothesis that the control of plasmatic cholesterol levels would be beneficial for treating AD has been demonstrated by using anticholesterolemic drugs, such as statins, which act as HMG-CoA reductase inhibitors [20].
Finally, deposition of Aβ fragments and neurofibrils, coupled with the uncontrolled production of ROS, are crucial for the installation of a neuroinflammatory process, with the same complexity observed in peripheral tissues. The scope and relevance of this process in the establishment and development of chronic DA have been demonstrated in several recent studies in the literature [30-33]. Among all the brain cells, microglia appears to have fundamental importance in CNS inflammation associated with AD. These cells could be activated by Aβ and modulate the production of cytokines, chemokines and neurotoxins that are highly neurotoxic, contributing to neuronal degeneration [19, 30-33].
Given the variety of factors associated with the onset, progress and severity of AD, increasing their degree of pathophysiological complexity, and associated to the inefficiency of the current therapeutic arsenal available, it becomes unavoidable to adopt a new concept for the rational design of new drugs against DA. In this context, drug candidate prototypes with dual mode of action were the first attempts to look up ligands recognized by more than one molecular target or more than one site on the same macromolecular target. Currently, a new strategy of multi-target directed ligands is gaining special attention in the scientific community, which has been seeking in molecular hybridization, a tool for designing new molecular patterns. These molecular hybrids could lead to the identification of new bioactive chemical entities with selectively affinity for multiple targets, preferably in different biochemical cascades. Therefore, these innovative ligands could play a singular role in the advance of a broadly and more efficient therapy, and perhaps, in the cure of AD.
4. RECENT MULTI-TARGET DIRECTED DRUG CANDIDATES DESIGNED FOR THE TREATMENT OF AD
This new approach, considering multifunctional drugs or ligands directed at multiple targets associated with the same disease (symbiotic drugs), has gained special importance and introducing a new thinking approach in the design of new drug candidates for AD. Molecular hybridization of pharmacophoric subunits of different bioactive molecules, is the main tool for structural planning, and have provided the discovery of numerous ligands with multiple properties, including antioxidant, neuroprotective, metal chelation, anti-inflammatory, anti-Aβ aggregation, and cholinesterase and secretase inhibitory activities. Therefore, a set of other potential therapeutic targets have been studied for simultaneous intervention, seeking for a more efficacy in relieve the symptoms and slowing the progression of AD, and why not, its definitive control and cure [34-38].
Since 2005, the literature has shown several results from applying this innovative approach of drug design. Drugs such as donepezil, tacrine and rivastigmine [39, 40] have been used as structural models for molecular hybridization with bioactive synthetic and natural products such as curcumine [41, 42], berberine [43, 44], 8-hydroxyquinolines [45], among others in the search for new chemical entities with multiple bioactive properties useful for the treatment of AD.
As one example of this strategy, berberine (1) was used as a basic skeleton for the construction of a series of hybrid compounds with molecular subunits that include melatonin (2) and ferulic acid (3, Fig. 1). The aiming was to obtain new derivatives with antioxidant properties and anti-Aβ aggregation, but with reduced inhibitory potency of acetylcholinesterase (AChE). The hybrid compound hidroquine 4 demonstrated the best ability for suppression of Aβ aggregation, plus excellent antioxidant effect and reasonable ability to inhibit acetylcholinesterase (AChE) and butirylcholinesterase (BuChE). In addition, derivative 5 (Fig. 1) was identified as a potent inhibitor of AChE, with strong antioxidant properties, confirming the multi-target potential of these substances in comparison to berberine, the original prototype [44].
Fig. (1).

Series of hybrid derivatives (4-7) structurally designed from berberine (1).
Memoquin (8, Fig. 2), a 1,4-benzoquinone-poliamine hybrid compound, was reported by Cavalli and co-workers as a promising drug prototype for AD. The structural design of compound 8 was based on a polyamine core, derived from coproctamine (9), a dimeric agent with anticholinesterasic and antimuscarinic. Aiming to add to these properties, the ability to neutralize ROS (reactive oxygen species) and neuroprotection, authors proposed the insertion of a 1,4-benzoquinone, from coenzyme Q10 (CoQ10), a fragment with a recognized potent mitochondrial antioxidant property and a protector of the hippocampus neurons against Aβ(1–40)–induced neurotoxicity. The inner polymethylene chain was replaced by the benzoquinone nucleus. The insertion of the benzoquinone nucleus contributes to increase the neuroprotective activity, because a hydrophobic and planar π system is generated, which is able in principle to bind Aβ [42, 47, 48, 50].
Fig. (2).

Design strategy for memoquin (8) and their derivatives (12-14).
Memoquin (8) exhibited ability to inhibit AChE (IC50= 1.55 nM), almost 15-fold more potent than the reference compound (coproctamine, 9). Memoquin (8) also inhibited AChE-induced Aβ1-40 aggregation with an IC50 of 28.3 µM and the self-induced Aβ 1–42 aggregation with an IC50 of 5.93 µM. In addition, compound 8 is also capable to inhibit BACE-1 (β secretase) activity in range nanomolar concentrations (IC50= 108 nM) and showed antioxidant properties, reducing the formation of free radicals by 44.1%. It was demonstrated that the 1,4-benzoquinone moiety can be reduced in vivo to the 1,4-dihydroquinone form, increasing its antioxidant potential and scavenging properties. Finally, in vivo assays showed the potential of memoquin to restore the cholinergic deficit and successfully reduce Aβ expression and accumulation, also reducing hyperphosphorylation of tau protein [48, 49].
Considering all these multi-target profile of memoquin (8), Bolognesi and co-workers reported in 2009 the design of new hybrid lipoic acid-memoquin (11) derivatives inspired on the memoquin structure and lipoic acid structure which is a potent antioxidant. In addition to the observed biological properties of memoquin, compound 12 also exhibited a good neuroprotective effect against oxidative stress, greater than the reference control lipoic acid (10) [49].
Curcumine (15, Fig. 3) is the major constituent of Curcuma longa, a plant species used in Indian Traditional Medicine, with Aβ-aggregation inhibitory properties have been used as a structural model for molecular to generate new analogs of memoquin with enhanced neuroprotective AChE and BuChE inhibitory properties. Among the novel memoquin hybrid derivatives, compounds 16 and 17 (Fig. 3) showed the best multiple-target action profile with selective inhibitory activities of AChE and BuChE, with modulatory effects on the amyloid aggregation and inhibition of neurofibrils formation [46].
Fig. (3).

Compounds 16 and 17 designed as new analogues of memoquin (8) with neuroprotective properties, anti-amyloid, and selective AChE and BuChE inhibitory activities.
Tacrine (19, Fig. 4) is the first approved ChEs inhibitor by the FDA for the treatment of AD. However, due to some undesirable side effects this drug have its therapeutic use restricted and, therefore, the search for more effective and secure tacrine derivatives is still of interest. In recent years, many studies have focused on the combining the potent ChEs inhibitory properties of tacrine with additional biological properties to search for new MTDLs.
Fig. (4).

Chemical structure of tacrine (19) and tacrine-hybrid derivatives (21-24).
Based on this strategy, Contelles and co-workers synthesized a new series of 4H-pyrano [2,3-b] quinolone derivatives (20), planned by molecular hybridization of tacrine fused to a benzylamino-3-pyridyl ring moiety, aiming to obtain new AChEIs with an innovative molecular scaffold and to explore its pharmacophoric contributions to additional neuroprotective properties and modulatory effects on voltage-dependent Ca+2 channels. In general, the new tacrine-hybrid compounds showed to be better selective for AChE inhibition than tacrine. It seems that the substitution at C-20 position of the 3-pyridyl ring system (compounds 22, 23, 24) was deleterious for AChE inhibition as observed for derivative 21 (X= H). In contrast, compounds 22-24 showed a good neuroprotective profile, as the derivative 23 exhibiting the best neuroprotective effect, with only 46% of cell-death suppression [51].
The dimer bis-(7)-tacrine (25, Fig. 5), was one the first homodimers reported in the literature with increased AChE affinity, exhibiting a 1000-fold higher inhibitory potency than tacrine. This best inhibitory profile is due to a dual simultaneously interaction with active and peripheral sites of AChE. Further studies, demonstrated that this compound was also capable to inhibit the AChE-induced Aβ aggregation with an IC50 of 41.7 µM. Because of this symbiotic profile, compound 25 has been described as an important structural model for rational design of novel MTDLs [52].
Fig. (5).

Design of the new series of derivatives of bis-(7)-tacrine (25) with improved activities for inhibition of AChE inhibition and AChEinduced amyloid-β aggregation, and ion metal chelation.
In 2007, Bolognesi and co-workers proposed modifications on the structure of bis-(7)-tacrine (25) with an insertion of a spacer subunit with functional groups capable to of chelate metals, which are involved in the degenerative process. For this purpose, compounds 26 (BW284c51) and ambenonium (27) were selected as model prototypes due to their cholinesterase inhibitory activity and their singular structural feature, carrying carbonyl and oxalamide functionalities with could lead to a the desired metal chelation property (Fig. 5). The multifunctional compounds maintained a potent AChEIs in the nanomolar range, showed additional inhibitory effect of AChE-induced amyloid-β aggregation. The results showed the ability for chelation of Cu2+ and Fe3+ ions and suggest that these compounds might act against AD by a chelation mechanism [40].
Minarini and co-workers also explored the structure of bis-(7)-tacrine (25), in an attempt to obtain the hybrid derivative cystamine bis-(7)-tacrine (31), with an insertion of the cystamine moiety 32 as a linker between two acrydine subunits of the bis-tacrine framework of 25 (Fig. 6). Cystamine (31) have shown important biological properties such as antioxidant and neuroprotective. This study revealed the hybrid dimer cystamine-tacrine 31 was able to inhibit AChE (IC50= 5.04 nM), BuChE (IC50= 4.23 nM), self-Aβ aggregation (IC50= 24.2 µM) and AChE-induced Aβ aggregation (52,6%) in the same range of the reference compound, with additional neuroprotective effect on SH-SY5Y cell line against H2O2-induced oxidative injury, with low toxicity [52].
Fig. (6).

Design of the multipotent cystamine-tacrine dimer (31) by molecular hybridization of bis-(7)-tacrine (25) and cystamine (32).
Fang and co-workers reported in 2008 the synthesis and pharmacologic evaluation of a new series of tacrine-ferulic acid hybrid derivatives (33) (Fig. 7). They planned a new structural pattern by the connection of ferulic acid (34) to the structure of tacrine (18) via an alkylenediamine as a linker side chain. This approach was used aiming to obtain new multi-target hybrid compounds which could conjugate the AChE inhibitory activity originating from the tacrine template and the antioxidant activity from the ferulic acid moiety. The compounds showed lower antioxidant activity than ferulic acid, but all hybrids tested showed moderate to good antioxidant activity. All compounds effectively inhibited AChE in in vitro assays. Particularly, compounds 35 showed a 10-fold higher AChE inhibitory activity (IC50= 4.4 nM) than tacrine. Enzymatic kinetics studies suggest that this compound possess high affinity binding to the peripheral anionic site (PAS) of AChE and, therefore, could inhibit Aβ-peptide fibril formation [53].
Fig. (7).

Design of the new series of hybrid tacrine-ferulic acid (34) derivatives with dual antioxidant and IAChE properties.
In another approach, Rosini and co-workers also used tacrine as a model for the planning of a new series of hybrid compounds (36-39) based on the structural feature of carvedilol (35). This compound had been identified by endows a significant neuroprotective efficacy, probably related to its modulatory action as a low-affinity antagonist of N-methyl-D-aspartate (NMDAR). Thus, carbazole moiety of 35 was elected as a probable structural pharmacophoric subunit responsible by its antioxidant properties and efficient inhibitory activity of Aβ-fibril formation (Fig. 8). Biological evaluation revealed that all compounds showed effective AChE-inhibiting activity in a nanomolar range, being more potent than tacrine. Particularly, compounds 36-39 showed an adequate biological profile for multi-target drugs, inhibiting AChE activity and also capable to block in vitro Aβ self-aggregation, AChE-mediated Aβ-aggregation, NMDARs antagonistic effect and to reduce cerebral oxidative stress [54].
Fig. (8).

Chemical structure of carvelodiol (35) and their active derivatives 36-39.
In another approach, tacripyrines (40) were designed by combining tacrine with a calcium antagonist such as nimodipine (41) (Fig. 9). In this study, the AChE inhibitory activity of the nimodipine–tacrine hybrids 40 is potentiated, with IC50 in the nanomolar range for AChE. Besides acting as an inhibitor of AChE, these compounds also showed significant neuroprotective effects. In particular, compound 42 (IC50= 105 ±15 nM), proved to be moderately active in models AChE-induced and Aβ-self aggregation models (30 and 35% inhibition, respectively). Further kinetic studies and molecular modeling data suggest that compound 42 could interact with the PAS of the AChE, exerting its inhibitory properties by noncompetitive mode. Additionally, the most of the compounds showed moderate Ca2+ channel blocking effect and therefore the compounds are neuroprotective agents [55].
Fig. (9).

Structure of tacripyrines (40) and the most active compound 42 designed by molecular hybridization of Tacrine (19) and Nimodipine (41).
A novel series of tacrine–caffeic acid hybrids (43) were designed by combining the structure of caffeic acid (44) and tacrine (19) (Fig. 10) as multitarget-directed ligands against Alzheimer’s disease. In vitro studies showed that most of the target molecules exhibited significant antioxidant activities on oxygen radical absorbance capacity method (ORAC). The hybrid molecules showed to be good metal chelators and also exhibited higher antioxidant capacity than the caffeic acid with values of 2.75−9.37 µM. In particular, compound 45 exhibited the greatest ability in inhibition of self- or AChE-induced β-amyloid1–40 aggregation, as well as showed potent neuroprotective effects against H2O2- and glutamate- induced cell death, with low toxicity in HT22 cells. Further molecular modeling and enzymatic kinetics studies clarified that these inhibitors may act in two binding sites of AChE, which could explain the inhibitory effect exerted over Aβ-aggregation [56].
Fig. (10).

Chemical structure of caffeic acid (43) and their hybrid derivatives 44 and 45.
In another work, Wang and collaborators reported a series of tacrine-based hybrid compounds, containing an additional pharmacophoric subunit 4-phenyl-2-aminothiazole with different spacer subunits. The pharmacophoric groupment 4-phenyl-2-aminothiazole was elected based on data form literature that suggest this functionality as responsible for inhibitory effects on tau protein aggregation and on Aβ self-aggregation, neuroprotective and anti-inflammatory.
All 10 compounds of the series of phenylthiazole–tacrine hybrids (47, Fig. 11) were potent inhibitors of cholinesterases with IC50 values ranging from 5.78 ± 0.05 to 7.14 ± 0.01 nM for AChE, and from 5.75 ± 0.03 to 10.35 ± 0.15 nM for BuChE. Moreover, a structure-activity relationship study was conducted to study the influence on the type of middle linker and substitutions at 40-position of 4-phenyl-2-aminothiazole. The results were indicative that the length of middle linker affected the AChE inhibitory potency. Compound 48, with the largest linker subunit exhibited showed the greater potency in AChE inhibition (IC50= 7.14 ± 0.01 nM). Data from kinetic studies have shown that this compound is a mixed-type inhibitor and could bind simultaneously at the catalytic and the peripheral sites of AChE. Most of phenylthiazole–tacrine hybrids showed a good inhibitory potency on Aβ1–42 self-aggregation, although the activity was lower when compared with subunit 4-phenyl-2-aminothiazole. Additionally, compound 48 displayed a blockade effect on Ca2+ overload in the primary cultured cortical neurons [57].
Fig. (11).

Design of the phenylthiazole–tacrine hybrid compounds 47 and structure of the most active derivative 48.
According to the studies of Simoni and co-workers, a series of hybrid derivatives of galanthamine (49), an AChE inhibitors and memantine (50), a noncompetitive NMDA receptor antagonist, showed potent AChE inhibitory activities with multiple neuroprotective effects in vitro (Fig. 12). Some of these hybrid compounds were also capable to prevent glutamate-induced neurotoxicity by moderately blocking glutamate receptor NMDA subtype and neuronal cell death via NMDA receptor-dependent effect at nanomolar concentrations (e.g., memagal (52), IC50 = 0.28 nM). These molecules were the first examples reported of dual AChE/NR2B drug prototype candidates [58].
Fig. (12).

Chemical structure of galanthamine-memantine hybrid compounds 51 with dual AChE/NR2B effect.
A new series of 30 bivalent β-carboline derivatives (53) was synthesized by Rook and co-workers based on previous studies that disclosed monovalent β-carbolines as potent dual AChE/BuChE inhibitors (Fig. 13). During an investigative study for the ability of these β-carboline derivatives to interact with multiple targets, the researchers also found that several compounds were also potent NMDA receptor blockers, in addition to the potent inhibitory properties of both AChE/BuChE enzymes. A structure-activity study revealed that the inhibitory activity of quaternary N9-bivalent β-carbolines was increased compared to their monovalent analogues. Compound 55 showed the higher AChE inhibitory activity (IC50= 0.5 nM) and proved to be a potent inhibitor of the transient induced-glutamate Ca+2 with an IC50= 1.4 µM, which is 4-fold more potent than memantine (IC50= 5 5.6 µM) in the same assay conditions [59].
Fig. (13).
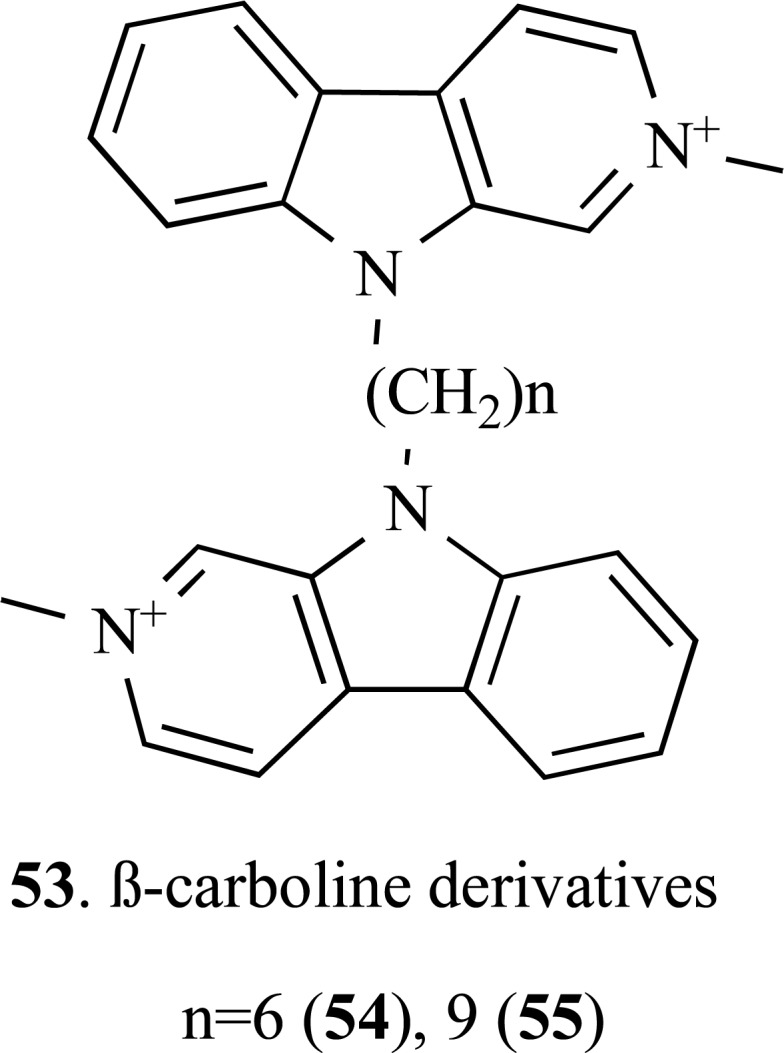
Chemical structure of new bivalent β-carboline derivatives 54 and 55.
In another approach for drug design of MDTLs, Rizzo and co-workers reported a new series of hybrid molecules based on the structure of the AChE inhibitor N-Methyl-N-benzylamine (56), and compound SKF-64346 (57), a benzofuran derivative with good inhibitory properties of Aβ-fibril formation, using a heptyloxy moiety as a linker subunit. All compounds evaluated have shown to be good AChE and Aβ-fibril formation inhibitors, reducing the Aβ neurotoxicity. Compound 58 showed an IC50 of 1.82 µM in BuChE inhibition, being 5.7-fold more selective toward BuChE than AChE (IC50 =10.5 µM). Furthermore, this compound was also able to inhibit Aβ self-aggregation with an IC50 of 13 µM and showed a neuroprotective effect of 63% at 30 mM [60, 61]. Some years later, aiming to improve its pharmacological profile the researchers modified the original structure of compound 58 by variation of the spacer subunit between the 2-arylbenzofuran and the N-methyl-N-benzylamine moieties. They proposed a variation in the length of the spacer subunit, modification in the substituents in 3 position of the benzofuran scaffold and change of the N-methyl-N-benzylamine heptyloxy side chain from the para to meta position (Fig. 14) [61]. These structural modifications led to a new series 59 that exhibited an increased in activity for most of the modified compounds.
Fig. (14).

Design of compound 58 by molecular hybridization of SKF-64346 (57) and N-Methyl-N-benzylamines (56) and its molecular derivative series 59.
Among the most active compounds of the series 59, compounds 60 and 61 exhibited the best BuChE inhibitory properties, with carbamate 62 also showing higher dual inhibitory activity of AChE and BuChE in comparison to rivastigmine. In addition, compounds 63 and 64 also showed significant in vitro inhibitory activities on Aβ-fibril formation up to 81% (Fig. 15) [61, 62].
Fig. (15).

Chemical structures of the improved derivatives 60-64 designed by molecular optimization of the hybrid prototype 59.
In another work, Piazza and co-workers also searching for multifunctional drug candidates for AD planned a series dual of inhibitors of human AChE (hAChE) and β-secretase (BACE-1). The molecular design was based in a dual binding site AChE inhibitor, AP2238 (65), which is able to simultaneously interact with both the central and the peripheral anionic sites of AChE. It was synthesized a series of compound 65 derivatives by replacing the methoxy substituents of the coumarin moiety by an amidic chain subunit aiming to extend the activity to BACE-1 (Fig. 16). The positions 6 or 7 of the coumarin moiety were also replaced by a dihalophenyl acid subunit, considering that this moiety emerged as a leitmotif in different BACE-1 inhibitors reported in the literature. All compounds were potent inhibitors, particularly compounds 67 (IC50 hAChE = 0.551 µM, (IC50 BACE-1= 0.149 µM), and 65 (IC50 hAChE= 0.181 µM, IC50 BACE-1= 0.150 µM) that exhibited the best inhibitory profile in the target series. Biological data clearly indicated that the introduction of a halophenyl alkylamidic subunit on the scaffold of 65 allowed obtaining more potent BACE-1 inhibitors, and the presence of substituents in the positions 6 or 7 on the coumarin nucleus seems to equally contribute to the relative binding affinity of this molecular series [62].
Fig. (16).

Chemical structure of compounds 68 and 69, designed as dual hybrid inhibitors of hAChE and BACE-1 based on the AChE inhibitor AP2238 (65).
A new family of multitarget molecules able to interact with both AChE and BuChE, as well as with monoamine oxidase (MAO) A and B, has been synthesized by Samadi and collaborators. Rational structural pattern of these compounds considered conjunctive approach by combination of benzylpiperidine and N-propargylamine moieties present in the AChE inhibitor donepezil (70) and the MAO inhibitor PF9601N (71), respectively, connected by a central pyridine or naphthyridine ring system (Fig. 17). The most promising derivative 72 showed a potent inhibitory activity of AChE (IC50= 37 nM), but less potent than donepezil. This compound also showed a moderate inhibitory potency of MAO-A (IC50= 41 μM). Moreover, molecular modeling showed that these inhibitors probably act in two binding sites of AChE, and that the length of the spacer subunit is of particular relevance in the modulation of AChE inhibition, being crucial for the dual interaction of these molecules with both CAS and PAS sites the enzyme [63].
Fig. (17).

Chemical structure of the most active dual AChE and MAO-A inhibitor designed by molecular hybridization of donepezil and PF9601N (72).
In 2011, another recent publication by Bolea and collaborators also reported the synthesis and pharmacological evaluation of a new family of multifunctional molecules capable to interact simultaneously with the AD-related enzymes AChE, BuChE, MAO-A and MAO-B. The structural architecture of the series 74 (Fig. 18) was based on the molecular hybridization of the benzylpiperidine moiety of donepezil (70) and the indolyl-propapylamino subunit present in the structure of the MAO inhibitor N-(5benzyloxy-1-methyl-1H-indol-2-yl)methyl-N-methylprop-2-yn-1-amine (73), connected a central oxy-methylene chain [64]. Compound 75 was identified from series 74 as the most promising ligand, showing a good selectivity and high potency in the inhibition of MAO-A with an IC50 = 5.2 nM (IC50 MAO-B = 43.0 nM), but less potent and with poor selectivity for the AChE (IC50 = 0.35 µM) and BuChE (IC50 = 0.46 µM). Further molecular modeling and enzymatic kinetics studies clarified that these inhibitors act in two binding sites of AChE, which could explain the inhibitory effect over Aβ-self-induced (47.8 ± 2.1 %) and AChE-dependent aggregation (32.4 ± 7 %) exerted compound 75 [64].
Fig. (18).

Chemical structure of the lead-compound 75 designed as a MDTL ligand for ChEs and MAO-A and B.
Based on the remarkable capacity of tacrine to interact in the active site of AChE, Martins and collaborators proposed a new series of tacrine-derived analogues by the insertion of a furo [2,3-b] quinolin-4-amine (76) and pyrrolo [2,3-b] quinolin-4-amine (77) subunits (Fig. 19). Pharmacological results revealed that the furanotacrines and pyrrolotacrines showed selective inhibition of BuChE, in a micromolar range, but these compounds have lower potency than tacrine. The lead-compound 79, exhibited the best inhibitory profile with IC50= 0.61 nM and IC50 BuChE= 0.074 nM, for AChE from Electrophorus electricus and BuChE from equine serum, with significant neuroprotective effects against Aβ-induced toxicity in concentrations below 300 nM. Molecular modelling studies have corroborate that the hybrid derivatives containing pyrrole or furan ring subunits interacts with AChE and BuChE and the presence of a phenyl ring at the position 1 of the pyrrole ring is beneficial for both AChE and BuChE inhibition [65].
Fig. (19).

Chemical structure of the compound 79, the lead-molecule form the pyrrolo[2,3-b] quinolin-4-amine series.
Taking the structure of donepezil (70) as a model, Huang and co-workers reported in 2012 a new family of derivatives based on the structure of benzylidene-indanone (80) pharma-cophoric subunit of 70 with the insertion of set of substituted benzene ring systems (81) (Fig. 20). In vitro evaluation showed that most of the compounds were potent inhibitors of MAO-B (IC50 of 7.5−40.5 μM), besides antioxidant (ORAC-FL value of 2.75−9.37 µM) and metal chelator properties. In addition, they showed a great ability for inhibition self-induced β-amyloid aggregation (10.5−80.1%, 20 μM). Compound 82, was identified as the most potent inhibitor agent of Aβ1−42 aggregation (80.1%) and MAO-B (IC50 = 7.5 μM). Moreover, compound 82 proved to be an excellent antioxidant and metal chelator agent, being able to inhibit Cu2+-induced Aβ1−42 aggregation and disassembling the Aβ fibrils [66]. Thus compound 82 is one of the most current example from literature of a genuine MDTL drug prototype candidate for AD treatment.
Fig. (20).

Chemical structure of compound 72, a substitutedbenzylidene indanone derivative with a wide range of multi-activity profile.
Resveratrol (83), a potent phenolic natural plant meta-bolite, with remarkable antioxidant and anti-inflammatory properties, have been recently described as an inhibitor of Aβ aggregation. Thus, its molecular scaffold has been also explored for assembly of innovative MTDLs with antioxidant, anti-inflammatory and inhibitory of Aβ aggregation properties. Compounds 86 and 87 were the lead-compounds identified among a series of resveratrol-derived molecular hybrids (85), planned by the combination of the catechol pharmacophore of 83 with the well-known metal chelator clioquinol (84) (Fig. 21). These two hybrid-compounds showed high potency in the inhibition of self-induced Aβ aggregation (86, IC50 = 7.56 μM and 87, IC50 = 6.51 μM) in comparison to resveratrol (IC50 = 15.11 μM). Furthermore, antioxidant assays performed by the ORAC method, showed the ability of compounds 86 and 87 in decreasing the generation of ROS species in 4.72 and 4.70 trolox equivalent, respectively. These compounds were also capable to disassembling the highly structured Aβ fibrils generated by self- and Cu (II)-induced Aβ aggregation and exhibited significant inhibitory effect on MAO-A and MAO-B with a moderate AChE inhibition and low neurotoxicity [67].
5. CONCLUSION
The development of drugs for a more effective treatment, and hopefully curing, AD is still a challenge for Medicinal Chemistry. The more recent insights about the pathophysiology of AD have proven a complex inter-connected variety of deleterious events, with multiple pathways involving in its etiology and a variety of factors that are associated with the installation, progress and severity of AD. This complex multifactorial aspect of AD and other neuronal disorders have supported the new paradigm of MTDLs (or one-compound-multi-targets strategy) as a more rational strategy approach to the development of new drug candidates, than the one-target-one drug concept. The low efficacy of the therapeutic approach currently employed could be, in part attributed to this reductionist concept, and it becomes inevitable to adopt a new concept for the rational design of new drugs against AD. Therefore, a drug for the treatment of AD will only be effective if modulate multiple targets simultaneously involved in the disease. In the last decade efforts have being employed in the design and search for new biological chemical entities capable to direct simultaneously with different molecular targets involved in the pathogenesis of AD and many new significantly active and promise molecules have been discovered. Consequently, new drug prototype candidates have being identified and they are the starting point of a revolutionary new way of thinking drug design and some drug candidates are under pre-clinical evaluation. Despite none of these MTDL drug candidates have reached clinical phase of development, we hope that in the next few years the system biology approach could effectively contribute to the Medicinal Chemists in the discovery of innovative chemical entities able to understand and effectively modify the course of this devastating neurodegenerative disorder.
Fig. (21).

Chemical structures of the very active MTDL resveratrolderived compounds 73 and 74.
ACKNOWLEDGEMENTS
This work was supported by grants from INCT-INOFAR Program (CNPq, Brazil, #573.564/2008-6) and Universal Project (FAPEMIG, Brazil, #APQ-CEX-00807-12). The authors are also grateful for the fellowships granted by CNPq to CVJ.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ABBREVIATIONS
- AChE
= acetylcholinesterase
- Aβ
= amyloid-beta
- BACE 1
= beta secretase 1
- BuChE
= butirylcholinesterase
- CNS
= central nervous system
- DA
= doença de Alzheimer
- IAChEs
= acetilcolinesterase inhibitor
- IC50
= minimum inhibitory concentration of 50%
- MAO A
= monoamine oxidase A
- MAO B
= monoamine oxidase B
- MTDLs
= multitarget-directed ligand
- NMDARs
= N-methyl-D-aspartate
- PAS
= peripheral anionic site
- ROS
= reactive oxygen species
- UN
= United Nations
REFERENCES
- 1.Bolognesi M L, Matera R, Minarini A, Rosini M, Melchiorre C. Alzheimer's disease: new approaches to drug discovery. Curr. Opin. Chem. Biol. 2009;13:303–308. doi: 10.1016/j.cbpa.2009.04.619. [DOI] [PubMed] [Google Scholar]
- 2.Youdim M B H, Buccafusco J J. Multi- functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol. Sci. 2005;26:27–35. doi: 10.1016/j.tips.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Alzheimer’s Dementia. 2011;7:2–68. doi: 10.1016/j.jalz.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Barreiro E J, Fraga C A M. New Insights for Multifactorial Disease Therapy: The Challenge of the Symbiotic Drugs. Curr. Drug Ther. 2008;3:1–13. [Google Scholar]
- 5.Zhang H-Y. One-compound-multiple-targets strategy to combat Alzheimer's disease. FEBS Lett. 2005;579:5260–5264. doi: 10.1016/j.febslet.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Cavalli A, Bolognesi M L, Minarini A, Rosini M, Tumiatti V, Recanatini M, Melchiorre C. Inhibition of Acetylcholinesterase, ß-Amyloid Aggregation, and NMDA Receptors in Alzheimer’s Disease: A Promising Direction for the Multi-target-Directed Ligands Gold Rush. J. Med. Chem. 2008;51:4381–4384. doi: 10.1021/jm800577j. [DOI] [PubMed] [Google Scholar]
- 7.Moller H J, Graeber M B. The case described by Alois Alzheimer in 1911. Eur. Arch. Psychiatry Clin. Neurosci. 1998;248:111–122. doi: 10.1007/s004060050027. [DOI] [PubMed] [Google Scholar]
- 8.Site of the Brazilian Institute of Geography and Statistics. Available in: http://biblioteca.ibge.gov.br/visualizacao/periodicos/93/cd_2010_ caracteristicas_populacao_domicilios.pdf. [Accessed on: Agust 2013]. Available in http://www.ibge.gov.br/home/presidencia/noticias/ noticia_visualiza.php?id_noticia=1722&id_pagina=1. Accessed: 08/ 2012,
- 9.Site of Alzheimer Med. Available in: http://www.alzheimermed. com.br/conceitos/aspectos-socioeconomicos. Accessed on: August. 2013.
- 10.Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Alzheimer’s Dementia. 2011;7:2. doi: 10.1016/j.jalz.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi H M. Forecasting the global burden of Alzheimer's disease. Alzheimers Dement. 2007;3:186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 12.Alzheimers Association . Available in: http://www.lz.org/downloads/Facts_Figures_2012.pdf. Accessed on: Agust. 2012.
- 13.Piau A, Nourhashémi F, Hein C, Caillaud C, Vellas B. Progress in the development of new drugs in Alzheimer’s disease. J. Nutr. Health Aging. 2011;15:45–57. doi: 10.1007/s12603-011-0012-x. [DOI] [PubMed] [Google Scholar]
- 14.Site of World Health Organization. Population ageing: a public health challenge. Available in: https://apps.who.int/inf-fs/en/fact135. html. Accessed on: August. 2013.
- 15.Kalache A, Keller I. In: Tllis R. Increasing longevity medical, social and political implications. London: Royal College Of Physicians of London; 1998. Ageing in developing countries. pp. 69–80. [PMC free article] [PubMed] [Google Scholar]
- 16.Site of the Brazilian Institute of Geography and Statistics. Available in: http://www.ibge.gov.br/home/estatistica/populacao/ censo2000/populacao/censo2000_populacao.pdf. Accessed on: August. 2013.
- 17.Chen S Y, Chen Y, Li Y P, Chen S H, Tan J H, Ou T M, Gu L Q, Huang Z S. Design, synthesis, and biological evaluation of curcumin analogues as multifunctional agents for the treatment of Alzheimer's disease. Bioorg. Med. Chem. 2011;19:5596–5604. doi: 10.1016/j.bmc.2011.07.033. [DOI] [PubMed] [Google Scholar]
- 18.Ray B, Lahiri D K. Neuroinflammation in Alzheimer's disease: different molecular targets and potential therapeutic agents including curcumin. Curr. Opin. Pharmacol. 2009;9:434–444. doi: 10.1016/j.coph.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 19.Viegas F P D, Simões M C R, Rocha M D, Castelli M R, Moreira M S, Viegas Junior C. Doença de Alzheimer: Caracterização, Evolução e Implicações do Processo Neuro- inflamatório. Rev. Virtual Quim. 2011;3:286–306. [Google Scholar]
- 20.Schmitt B, Bernhardt T, Moeller H-J, Heuser I, Frölich L. Combination Therapy in Alzheimer's disease. CNS Drugs. 2004;18:827–844. doi: 10.2165/00023210-200418130-00001. [DOI] [PubMed] [Google Scholar]
- 21.Starkov A A, Beal F M. Portal to Alzheimer's disease. Nat. Med. 2008;14:1020–1021. doi: 10.1038/nm1008-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reddy P H, Beal M F. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol. Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J Z, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blennow K, de Leon M J, Zetterberg H. Plasma Aß in Alzheimer's disease—up or down?. Lancet. 2006;368:387–388. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 25.da Rocha M D, Viegas F P D, Campos H C, Nicastro P C, Fossaluzza P C, Fraga C A M, Barreiro E J, Viegas Jr C. The Role of Natural Products in the Discovery of New Drug Candidates for the Treatment Neurodegenerative Disorders II: Alzheimers Disease. CNS Neurol Disorders - Drug Targets. 2011;10:251–270. doi: 10.2174/187152711794480429. [DOI] [PubMed] [Google Scholar]
- 26.Liu Q, Xie F, Rolston R, Moreira P I, Nunomura A, Zhu X, Smith MA, Perry G. Prevention and treatment of Alzheimer disease and aging antioxidants. Mini Rev. Med. Chem. 2007;7:171–180. doi: 10.2174/138955707779802552. [DOI] [PubMed] [Google Scholar]
- 27.Azzi A. Oxidative stress A dead end or a laboratory hypothesis?. Biochem Biophys Res. Comm. 2007;362:230–232. doi: 10.1016/j.bbrc.2007.07.124. [DOI] [PubMed] [Google Scholar]
- 28.Legg K. Neurodegenerative diseases An alternative path to reduce neuroinflammation. Nat. Rev. Drug Disc. 2011;10:901–910. doi: 10.1038/nrd3607. [DOI] [PubMed] [Google Scholar]
- 29.Gaggelli E, Kozlowski H, Valensin G. Copper homeostasis and neurodegenerative disorders (Alzheimer's prion and Parkinson's diseases and amyotrophic lateral sclerosis) Chem. Rev. 2006;106:1995–2044. doi: 10.1021/cr040410w. [DOI] [PubMed] [Google Scholar]
- 30.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole G M, Cooper N R, Eikelenboom P, Emmerling M, Fiebich B L, Finch C E, Frautschy S, Griffin W S T, Hampel H, Hull M, Landreth G, Lue LF, Mrak R, Mackenzie IR, McGeer PL, O’Banion M K, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel F L, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol. Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heneka MT, O’Banion MK. Inflammatory processes in Alzheimer's disease. J. Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 32.Giunta B, Fernandez F, Nikolic WV, Obregon D, Rrapo E, Town T, Tan JJ. Inflammaging as a prodrome to Alzheimer's disease. Neuroinflammation. 2008;5:51–66. doi: 10.1186/1742-2094-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamal MA, Greig NH, Reale M. Anti-Inflammatory Properties of Acetylcholinesterase Inhibitors Administred in Alzheimer's Disease Anti-inflamm. Anti-Allergy Med. Chem. 2009;8:85–100. [Google Scholar]
- 34.Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9:702–716. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- 35.Youdim MBH, Buccafusco JJ. Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol. Sci. 2005;26:27–35. doi: 10.1016/j.tips.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 36.Bolognesi ML, Matera R, Minarini A, Rosini M, Melchiorre C. Alzheimer's disease new approaches to drug discovery. Curr. Op. Chem. Biol. 2009;13:303–308. doi: 10.1016/j.cbpa.2009.04.619. [DOI] [PubMed] [Google Scholar]
- 37.Piau A, Nourhashemmi F, Hein C, Caillaud C, Vellas B. Progress in the development of new drugs in Alzheimer’s disease, J. Nut Health Aging. 2010;15:45–57. doi: 10.1007/s12603-011-0012-x. [DOI] [PubMed] [Google Scholar]
- 38.Cavalli A, Bolognesi M L, Minarini A, Rosini M, Tumiatti V, Recanatini M, Melchiorre C. Multi-target-Directed Ligands To Combat Neurodegenerative Diseases. J Med Chem. 2008;51:347. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 39.Samadi A, Valderas C, de los Ríos C, Bastida A, Chioua M, González-Lafuente L, Colmena I, Gandía L, Romero A, del Barrio L, Martín-de-Saavedra M D, López MG, Villarroya M, Contelles JM. Cholinergic and neuroprotective drugs for the treatment of Alzheimer and neuronal vascular diseases II.Synthesis biological assessment and molecular modelling of new tacrine analogues from highly substituted 2-aminopyridine-3-carbonitriles. . Bioorg. Med. Chem. 2011;19:122–133. doi: 10.1016/j.bmc.2010.11.040. [DOI] [PubMed] [Google Scholar]
- 40.Bolognesi M L, Cavalli A, Luca Valgimigli L, Bartolini M, Rosini M, Andrisano V, Recanatini M, Melchiorre C. Multi-Target-Directed Drug Design Strategy: From a Dual Binding Site Acetylcholinesterase Inhibitor to a Trifunctional Compound against Alzheimer’s Disease. J. Med. Chem. 2007;50:6446–6449. doi: 10.1021/jm701225u. [DOI] [PubMed] [Google Scholar]
- 41.Chen S-Y, Chen Y, Li Y-P, Chen S-H, Tan J-H, Ou T-M, Gu L-Q, Huang Z-S. Design, synthesis, and biological evaluation of curcumin analogues as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2011;19:5596–5604. doi: 10.1016/j.bmc.2011.07.033. [DOI] [PubMed] [Google Scholar]
- 42.Cavalli A, Bolognesi ML, Capsoni S, Andrisano V, Bartolini M, Margotti E, Cattaneo A, Recanatini M, Melchiorre C. A small molecule targeting the multifactorial nature of Alzheimer’s disease. Angew Chem. Int. 2007;46:3689–3692. doi: 10.1002/anie.200700256. [DOI] [PubMed] [Google Scholar]
- 43.Shan W-J, Huang L, Zhou Q, Meng F-C, Li X-S. Synthesis, biological evaluation of 9-N-substituted berberine derivatives as multi-functional agents of antioxidant, inhibitors of acetylcholinesterase, butyrylcholinesterase and amyloid-ß aggregation. Eur. J. Med. Chem. 2011;46:5885–5893. doi: 10.1016/j.ejmech.2011.09.051. [DOI] [PubMed] [Google Scholar]
- 44.Jiang H, Wang X, Huang L, Luo Z, Su T, Ding K, Li X. Benzenediol-berberine hybrids: Multifunctional agents for Alzheimer’s disease. Bioorg. Med. Chem. 2011;19:7228–7235. doi: 10.1016/j.bmc.2011.09.040. [DOI] [PubMed] [Google Scholar]
- 45.Fernandez-Bachiller M I, Pérez C, González-Muñoz G C, Conde S, López M G, Villarroya M, Garcia A G, Rodriguez-Franco M I. Novel Tacrine-8-hydroxyquinoline hybrids as multifunctional agents for the treatment of alzheimer’s disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J. Med. Chem. 2010;53:4927–4937. doi: 10.1021/jm100329q. [DOI] [PubMed] [Google Scholar]
- 46.Bolognesi M L, Bartolini M, Tarozzi A, Morroni F, Lizzi F, Milelli A, Minarini A, Rosini M, Hrelia P, Andrisano V, Melchiorre C. Multitargeted drugs discovery: Balancing anti-amyloid and anticholinesterase capacity in a single chemical entity. Bioorg. Med. Chem. Lett. 2011;21:2655–2658. doi: 10.1016/j.bmcl.2010.12.093. [DOI] [PubMed] [Google Scholar]
- 47.Bolognesi M L, Bartolini M, Rosini M, Andrisano V. Melchiorre, C.Structure-activity relationships of memoquin: Influence of the chain chirality in the multi-target mechanism of action. Bioorg. Med. Chem. Lett. 2009;19:4312–4315. doi: 10.1016/j.bmcl.2009.05.087. [DOI] [PubMed] [Google Scholar]
- 48.Bolognesi M L, Cavalli A, Melchiorre C. Memoquin: a multi-target-directed ligand as an innovative therapeutic opportunity for Alzheimer’s disease. Neurotherapeutics. 2009;6:152–162. doi: 10.1016/j.nurt.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolognesi M L, Cavalli A, Bergamini C, Fato R, Lenaz G, Rosini M, Bartolini M, Andrisano V, Melchiorre C. Toward a rational design of multitarget-directed antioxidants: merging memoquin and lipoic acid molecular frameworks. J. Med. Chem. 2009;52:7883–7886. doi: 10.1021/jm901123n. [DOI] [PubMed] [Google Scholar]
- 50.Bolognesi M L, Andrisano V, Bartolini M, Banzi R, Melchiorre C. Propidium-based polyamine ligands as potent inhibitors of acetylcholinesterase and acetylcholinesterase-induced amyloid-beta aggregation. J. Med. Chem. 2005;48:24–27. doi: 10.1021/jm049156q. [DOI] [PubMed] [Google Scholar]
- 51.Contelles J M, Leo R, Lopez M G, Garci´a A G, Villarroya M. Synthesis and biological evaluation of new 4H-pyrano[2,3-b]quinolone derivatives that block acetylcholinesterase and cell calcium signals, and cause neuroprotection against calcium overload and free radicals. Eur. J. Med. Chem. 2006;41:1464–1469. doi: 10.1016/j.ejmech.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 52.Minarini A, Milelli A, Tumiatti V, Rosini M, Simoni E, Bolognesi a M L, Andrisano V, Bartolini M, Motori E, Angeloni C, Hrelia S. Cystamine-tacrine dimer: A new multi-target-directed ligand as potential therapeutic agent for Alzheimer’s disease treatment. Neuropharmacology . 2012;62:997–1003. doi: 10.1016/j.neuropharm.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 53.Fang L, Kraus B, Lehmann J, Heilmann J, Zhang Y, Decker M. Design and synthesis of tacrine-ferulic acid hybrids as multi-potent anti-Alzheimer drug candidates. Bioorg. Med. Chem. Lett. 2008;18:2905–2909. doi: 10.1016/j.bmcl.2008.03.073. [DOI] [PubMed] [Google Scholar]
- 54.Rosini M, Simoni , Bartolini M, Cavalli A, Ceccarini L, Pascu N, McClymont D W, Tarozzi A, Bolognesi ML, Minarini A, Tumiatti V, Andrisano V, Mellor I R, Melchiorre C. Inhibition of Acetylcholinesterase, ß-Amyloid Aggregation, and NMDA Receptors in Alzheimer’s Disease: A Promising Direction for the Multi-target-Directed Ligands Gold Rush. J. Med. Chem. 2008;51:4381–4384. doi: 10.1021/jm800577j. [DOI] [PubMed] [Google Scholar]
- 55.Contelles J M, Leon L, de los Rios C, Samadi A, Bartolini M, Andrisano V, Huertas O, Barril X, Luque FJ, Rodriguez-Franco M I, Lopez B, Lopez MB, Garcia AG, Carreiras MC, Villarroya M. Tacripyrines the First Tacrine-Dihydropyridine Hybrids as Multitarget-Directed Ligands for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2009;52:2724–2732. doi: 10.1021/jm801292b. [DOI] [PubMed] [Google Scholar]
- 56.Chao X, He X, Yang Y, Zhou X, Jin M, Liu S, Cheng Z, Liu P, Wang Y, Yu J, Tan Y, Huang Y, Qin J, Rapposelli S, Pi R. Design synthesis and pharmacological evaluation of novel tacrine-caffeic acid hybrids as multi-targeted compounds against Alzheimer’s disease. Bioog. Med. Chem.ett. . 2012;22:6498–6502. doi: 10.1016/j.bmcl.2012.08.036. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y, Wang F, Yu J P, Jiang F C, Guan X L, Wang C M, Li L, Cao H, Li MX, Chen JG. Novel multipotent phenylthiazole-tacrine hybrids for the inhibition of cholinesterase activity, b-amyloid aggregation and Ca2+ overload. Bioorg. Med. Chem. 2012;20:6513–6522. doi: 10.1016/j.bmc.2012.08.040. [DOI] [PubMed] [Google Scholar]
- 58.Simoni E, Daniele S, Bottegoni G, Pizzirani D, Trincavelli ML, Goldoni L, Tarozzo G, Reggiani A, Martini C, Piomelli D, Melchiorre C, Rosini M, Cavalli A. Combining Galantamine and Memantine in Multitargeted New Chemical Entities Potentially Useful in Alzheimer’s Disease. J. Med Chem. 2012;55:9708–9721. doi: 10.1021/jm3009458. [DOI] [PubMed] [Google Scholar]
- 59.Rook Y, Schmidtke K U, Gaube F, Schepmann D, Wunsch B, Heilmann J, Lehmann J, Winckler T. Bivalent ß-Carbolines as Potential Multitarget Anti-Alzheimer Agents. J. Med. Chem. 2010;53:3611–3617. doi: 10.1021/jm1000024. [DOI] [PubMed] [Google Scholar]
- 60.Rizzo S, Riviere C, Piazzi L, Bisi A, Gobbi S, Bartolini M, Andrisano V, Morroni F, Tarozzi A, Monti J P, Rampa A. Benzofuran-Based Hybrid Compounds for the Inhibition of Cholinesterase Activity ß Amyloid Aggregation and Aß Neurotoxicity. J. Med Chem. 2008;51:2883–2886. doi: 10.1021/jm8002747. [DOI] [PubMed] [Google Scholar]
- 61.Rizzo S, Tarozzi A, Bartolini M, Costa G, Bisi A, Gobbi S, Belluti F, Ligresti A, Allarà M, Monti J P, Andrisano V, Di Marzo V, Hrelia P, Rampa A. 2-Arylbenzofuran-based molecules as multipotent Alzheimer’s disease modifying agentes. Eur. J. Med. Chem. 2012;58:519–532. doi: 10.1016/j.ejmech.2012.10.045. [DOI] [PubMed] [Google Scholar]
- 62.Piazzi L, Cavalli A, Colizzi F, Belluti F, Bartolini M, Mancini F, Recanatini M, Andrisano V, Rampa A. Multi-target-directed coumarin derivatives hAChE and BACE1 inhibitors as potential anti-Alzheimer compounds. Bioorg. Med Chem Lett. 2008;18:423–426. doi: 10.1016/j.bmcl.2007.09.100. [DOI] [PubMed] [Google Scholar]
- 63.Samadi A, Chioua M, Bolea I, de los Ríos C, Iriepa I, Moraleda I, Bastida A, Esteban G, Unzeta M, Gálvez E, Contelles J M. Synthesis biological assessment and molecular modeling of new multipotente MAO and cholinesterase inhibitors as potential drugs for the treatment of Alzheimer’s disease. Eur J Med Chem. 2011;46:4665–4668. doi: 10.1016/j.ejmech.2011.05.048. [DOI] [PubMed] [Google Scholar]
- 64.Bolea I, Jimenez JJ, de los Rios C, Chioua M, Pouplana R, Luque J, Unzeta M, Contelles J M, Samadi A. Synthesis Biological Evaluation, and Molecular Modeling of Donepezil and N-[(5-(Benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine Hybrids as New Multipotent Cholinesterase/ Monoamine Oxidase Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2011;54:8251–8270. doi: 10.1021/jm200853t. [DOI] [PubMed] [Google Scholar]
- 65.Martins C, Carreiras MC, León R, de los Ríos C, Bartolini M, Andrisano V, Iriepa I, Moraleda I, Gálvez E, García M, Egea J, Samadi A, Chioua M, Contelles J M. Synthesis and biological assessment of diversely substituted furo[2,3-b]quinolin-4-amine and pyrrolo[2,3-b]quinolin-4-amine derivatives as novel tacrine analogues. Eur. J. Med. Chem. 2011;46:6119–6130. doi: 10.1016/j.ejmech.2011.09.038. [DOI] [PubMed] [Google Scholar]
- 66.Minarini A, Milelli A, Tumiatti V, Rosini M, Simoni E, Bolognesi ME, Andrisano V, Bartolini M, Motori E, Angeloni C, Hrelia S. Multitarget-Directed Benzylideneindanone Derivatives Anti-ß-Amyloid (Aß) Aggregation Antioxidant Metal Chelation and Monoamine Oxidase B (MAO-B) Inhibition Properties against Alzheimer’s Disease. Neuropharmacology. 2012;62:997–1003. [Google Scholar]
- 67.Lu C, Guo Y, Yan J, Luo Z, Luo H B, Yan M, Huang L, Li X. Design Synthesis and Evaluation of Multitarget-Directed Resveratrol Derivatives for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2013;56:5843–5859. doi: 10.1021/jm400567s. [DOI] [PubMed] [Google Scholar]