

Abstract
An emerging concept is the tight relationship between dysbiosis (microbiota imbalance) and disease. The increase in knowledge about alterations in microbial communities that reside within the host has made a strong impact not only on dental science, but also on immunology and microbiology as well as on our understanding of several diseases. Periodontitis is a well-characterized human disease associated with dysbiosis, characterized by the accumulation of multiple bacteria that play individual and critical roles in bone loss around the teeth. Dysbiosis is largely dependent on cooperative and competitive interactions among oral microbes during the formation of the pathogenic biofilm community at gingival sites. Oral pathobionts play different and synergistic roles in periodontitis development, depending on their host-damaging and immunostimulatory activities. Host immune responses to oral pathobionts act as a double-edged sword not only by protecting the host against pathobionts, but also by promoting alveolar bone loss. Recent studies have begun to elucidate the roles of individual oral bacteria, including a new type of pathobionts that possess strong immunostimulatory activity, which is critical for alveolar bone loss. Better understanding of the roles of oral pathobionts is expected to lead to a better understanding of periodontitis disease and to the development of novel preventive and therapeutic approaches for the disease.
Keywords: NOD1 ligands, revised keystone pathogen hypothesis, microbiota, alveolar bone resorption, commensal bacteria, Pasteurellaceae
Periodontitis, a Prototype of Dysbiosis-associated Disease
Direct attack is not the only way that microbes cause disease. Previous studies on infectious diseases have focused extensively on pathogenic microbes that directly damage tissues in the host. However, evidence is accumulating that another set of microbes can also induce disease or contribute critically to disease development. These microbes live as normal residents on the skin, and internal cavities, particularly the intestines of animals, including humans, are called commensals (Chow et al., 2011). Among them, a particular group of commensals can cause or promote disease, and these commensals are often called pathobionts. The concept “pathobiont” includes some opportunistic pathogens that live as commensals in healthy hosts but can cause disease in susceptible hosts (e.g., immunodeficient individuals). The overgrowth of pathobionts is often triggered by immunodeficiency, pathogen infection, and treatment with antibiotics and host-damaging drugs (Chow et al., 2011) (Fig. 1A). In addition, the overgrowth of some pathobionts can result in secondary infection or infection by opportunistic pathogens, but little is known about the roles of pathobionts in oral diseases including periodontitis, a common dental disease.
Figure 1.

Role of pathobionts and dysbiosis in periodontitis. (A) Pathobionts and associated disease. Pathobionts could be colonized as one of the resident bacteria (commensals) in human bodies without any obvious symptoms. Once dysbiosis is induced by environmental changes (e.g., antibiotic treatment, accumulation of other microbes or epithelial barrier disruption), pathobionts cause significant changes in host health. Alternatively, changes in hosts and bacteria, for instance, by genetic variations and immunological defects, affect the virulence of pathobionts, resulting in disease development. (B) Dysbiosis during periodontitis development. Even healthy individuals harbor about 700 bacterial species in the oral cavity, and oral bacteria constitute a community on enamel and epithelial surfaces, and in oral fluid. The first event at a periodontal site is colonization of the pioneer bacteria that form the biofilm. The pioneer bacteria include Streptococcus species, which strongly interact with host cells. Other indicated biofilm community members are also able to colonize and grow with co-aggregation. Putative pathogens such as F. nucleatum likely act as scaffolds to bridge multiple bacteria and to facilitate colonization by additional biofilm-forming community members. The poor availability of energy sources from foods enforces specific bacteria to obtain energy from host factors by damaging host tissues. These host-damaging bacteria include the red complex bacteria (P. gingivalis, T. forsythia, and T. denticola), which possess high protease activity, and toxin-secreting A. actinomycetemcomitans (Aa). Several keystone pathogens, including the red complex bacteria, possess the ability to avoid host detection to optimize their acquisition of energy sources from the host. In addition, the keystone pathogen hypothesis proposes that immunological interference by keystone pathogens further establishes dysbiosis. Finally, environmental changes induced by keystone pathogens facilitate colonization of additional pathobionts that stimulate host immune responses, which results in periodontitis.
Periodontitis is one of the most well-characterized human diseases associated with dysbiosis (Socransky et al., 1998; Jenkinson and Lamont, 2005; Darveau, 2010). Chronic periodontitis in adults is associated with poor dental hygiene, which induces dysbiosis with accumulation of a group of host-damaging bacteria called “red complex” bacteria that include Porphyromonas gingivalis (Pg), Tannerella forsythia, and Treponema denticola (Fig. 1B) (Socransky et al., 1998). Non-culture-based studies further identified Filifactor alocis, unnamed Treponema, Prevotella, Selenomonas, Peptostreptococcus, Anaeroglobus, and Desulfobulbus spp., unclassified Lachnospiraceae, Synergistetes, and TM7 species as the dominant bacterial species associated with periodontitis development (Paster et al., 2001; Griffen et al., 2012). Red complex bacteria possess high levels of protein-degrading activity that is largely mediated by proteases including gingipains (Pg), PrtH (T. forsythia), and dentilisin (T. denticola), and these bacterial proteases appear to be important for virulence (Saito et al., 1997; O’Brien-Simpson et al., 2001; Bamford et al., 2007). Pg is an assaccharolytic bacterium that grows poorly on glucose as an energy source, but grows well in the presence of amino acids derived from cleaved products of host proteins (Shah and Williams, 1987). Pg also damages the barrier function of gingival epithelium via the production of gingipains (Katz et al., 2000; Groeger et al., 2010), which is important for the induction of inflammation. Accumulation of red complex bacteria is supported by other oral commensals that physically and metabolically interact with red complex bacteria. These include streptococci, the pioneer colonizers on the surfaces of host epithelium and tooth, and Fusobacteria, which interact with red complex and other bacteria to facilitate the formation of a more complex microbial community at anaerobic periodontal pockets (Socransky et al., 1998; Kolenbrander et al., 2002) (Fig. 1B). Colonization of Pg also contributes to dysbiosis by interfering with the complement-mediated immune system (Hajishengallis et al., 2011), which led to the hypothesis that keystone bacteria such as Pg trigger dysbiosis and the alteration of host immune responses, and that other bacteria orchestrate inflammatory disease (Hajishengallis et al., 2012). Aggregatibacter actinomycetemcomitans (Aa) is another bacterium which is tightly, but not completely, associated with a particular form of periodontitis, called aggressive or juvenile periodontitis, rather than with chronic periodontitis (Henderson et al., 2010; Fine et al., 2013). Aa JP2 strains secrete high levels of leukotoxin, an RTX-type toxin that damages host cells (Henderson et al., 2010). Therefore, both chronic and aggressive periodontitis are associated with bacteria that damage host soft tissues that is critical for the development of alveolar bone loss.
Metabolic Interactions of Pathobionts at Gingival Sites
Dysbiosis in periodontitis development is dependent on metabolic and physical interactions and competitive toxicity among oral bacteria (Fig. 2). For example, red complex bacteria are obligate anaerobes, and many in the periodontitis-associated non-red complex are obligate anaerobes or microaerobes. Therefore, anaerobic growth conditions are required for the accumulation of red complex bacteria. The genomes of Pg (W83, ATCC 33277, and TDC60; GenBank accession NC_002950, NC_015571, and NC_010729, respectively) and T. forsythia ATCC 43037 (GenBank accession NC_016610) lack several orthologues of E. coli heme biosynthesis genes (Schobert and Jahn, 2002). Therefore, growth of these bacteria requires exogenous heme. Potential sources of heme are other bacteria that synthesize heme, and this might be one of the reasons why Pg and T. forsythia are dependent on other oral bacteria for growth. The dependency of T. forsythia on N-acetyl muramic acid also suggests metabolic interaction of T. forsythia with other bacteria that release small peptidoglycan-related molecules (Wyss, 1989). However, Pg also possesses the ability to sense and recover heme from host tissues, suggesting that another source of heme in vivo is potentially the host (Scott et al., 2013). At the bottom pocket of the cemento-enamel junction, host factors are the only available major energy source for red complex bacteria. Although the genetic basis of the assaccharolytic feature of Pg remains poorly understood, it could be explained at least in part by the fact that the major energy source for Pg are amino acids, which are derived from the degradation of host proteins by bacterial proteases (Schobert and Jahn, 2002; Henderson et al., 2010; Hajishengallis et al., 2011, 2012; Fine et al., 2013). Metabolomic analysis showed that there are increased amounts of amino acids and other digested macromolecules in oral fluid from periodontitis patients (Barnes et al., 2011), suggesting that bacteria can commonly share energy sources produced by red complex and other bacteria. Conversely, these common nutritional sources are competitively used by several bacteria for their growth, and the latter is likely to affect the composition of the microbiota in addition to other competitive mechanisms, described below. Several studies with probiotic bacteria in vitro suggest that understanding of the inhibitory interactions between oral pathobionts and other oral bacteria may provide new therapeutic approaches against periodontitis (Bizzini et al., 2012).
Figure 2.
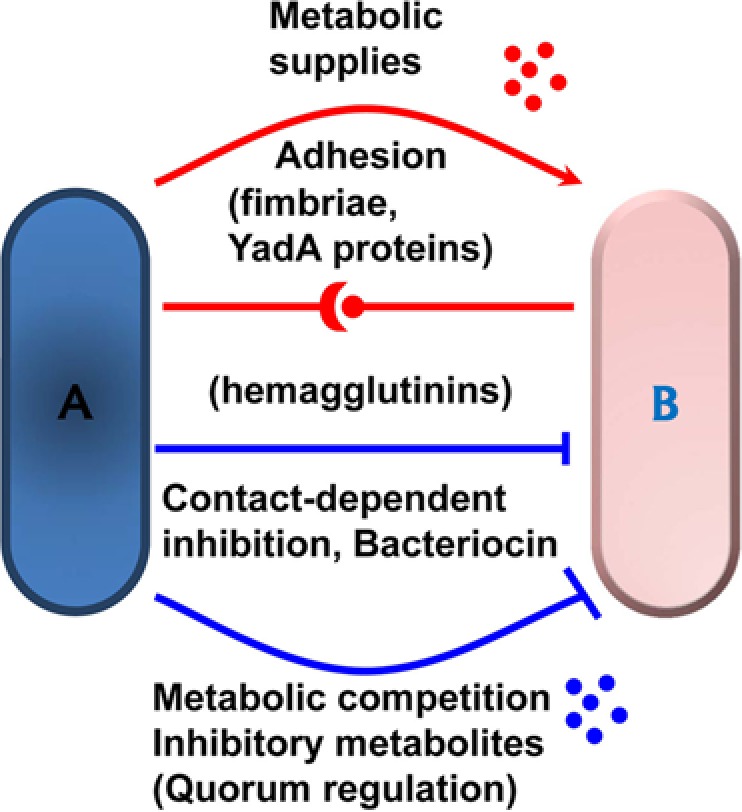
Bacterial-bacterial interactions that regulate dysbiosis. Dysbiosis is largely dependent on cooperative and competitive metabolic and physiological interactions among bacteria. Individual bacteria depicted as (A) and (B). affect cooperative and inhibitory colonization of neighboring bacteria by regulating the production of metabolic supplies including nutrients, pH modifiers, and chemical sensor ligands at bacterial habitats. Bacteria also compete with each other for common resources required for growth at shared niches. Physical interactions among bacteria can also affect growth of neighboring bacteria positively and negatively. For example, whereas fimbriae, YadA- and some hemagglutinin-domain-containing proteins are important for adhesion and cooperative co-colonization, other hemagglutinin-domain-containing proteins (such as high-molecular-weight bacteriocins) are known to regulate neighboring bacteria negatively by contact-dependent inhibition. Bacteria also inhibit growth of neighboring bacteria and, in particular, related bacteria by the production of small-molecular-weight bacteriocins. Some metabolites also negatively regulate the adhesiveness of neighboring bacteria through quorum regulation to establish a functional biofilm.
Regulation of Bacteria-Bacteria and Bacteria-Host Cell Interactions by Adhesion Factors Produced by Periodontitis-associated Pathobionts
Other mechanisms utilized by specific bacteria at gingival sites located near the area of bone loss are dependent on physiological interactions among bacteria. Red complex bacteria possess several proteins which interact with other bacteria. For example, biofilm-forming Streptococcus gordonii produce SspB to bind the minor fimbrial Mfa1 protein of Pg in addition to the interaction between the major fimbrial FimA protein of Pg and S. gordonii GAPDH, which facilitates Pg colonization and Pg-induced alveolar bone loss (Maeda et al., 2004; Daep et al., 2011). Furthermore, hemagglutinin A (HagA) and gingipains mediate the interaction of Pg with T. denticola (Ito et al., 2010). Similarly, structural analysis of biofilms in vivo and co-aggregation analysis in vitro with periodontitis-associated bacteria provided evidence that physiological interactions of red complex bacteria with non–red complex bacteria facilitate their colonization (Jenkinson and Lamont, 2005). Periodontitis-associated bacteria express several putative adhesins, and Pg produces hemagglutinins that are important for the interaction of bacteria with host cells or bacterial invasion into host cells (Song et al., 2005). Similarly, Flp fimbriae, YadA-containing ApiA/Omp100, and two non-fimbriae autotransporter proteins, EmaA and Aae, are important for the interaction of Aa with host factors and/or host cells (Henderson et al., 2010). Importantly, intra-bacterial interactions can modify bacteria/host interactions. For example, in vitro studies showed that S. gordonii provides H2O2 to enhance the expression of ApiA in Aa (Ramsey and Whiteley, 2009). F. nucleatum possesses FadA, which can mediate its interaction with host epithelial cells (Han et al., 2005) and facilitates the penetration of non-invasive bacteria into endothelial cell layers in vitro (Fardini et al., 2011), suggesting that intra-bacterial interactions are potentially important for immunostimulation by non-invasive oral bacteria.
Negative Interactions of Oral Bacteria with Periodontitis-associated Pathobionts
Accumulation of specific bacteria is not only dependent on cooperative interaction between and among bacteria, but also is regulated by competitive interactions. In an effort to identify probiotics, bacteriocins produced by oral resident and non-resident bacteria, including Lactobacillus paracasei HL32 and Bacillus amyloliquefaciens, were found to inhibit Pg growth (Hammami et al., 2013). Although the relevance of these findings to how competitive interactions work in dysbiosis during periodontitis development is unclear, these studies suggest a potential mechanism of dysbiosis through the production of bacteriocins. Close inspection of PFAM databases indicates that many oral bacteria produce small-size (lantibiotic-type) bacteriocins, suggesting that competition between and among oral bacteria might involve lantibiotic-type bacteriocins. Notably, bacteriocins from S. salivarius are effective in the treatment of the oral bacterial species involved in halitosis (Burton et al., 2006). Bacteriocins from different bacteria possess different specificity against oral pathobionts. For example, bacteriocin from Prevotella nigrescens is bactericidal against P. gingivalis, but bacteriocin from P. intermedia is not (Takada et al., 1991; Kaewsrichan et al., 2004). Further analysis is required for the role of bacteriocins in the regulation of intra-species interactions and dysbiosis in the oral cavity to be understood.
As described earlier, particular autotransporter proteins (type Va secretion system) of Aa have been shown to mediate invasiveness into host cells (Henderson et al., 2010). Aa and other proteobacteria possess multiple putative types V and VI secretion system (T5SS and T6SS) effector proteins with unknown functions in PFAM databases, although types III and IV secretion systems are restricted to particular species. Importantly, T5SS- and T6SS-mediated pathways have been shown to be important for competition between and among proteobacteria species (Hayes et al., 2010). Hemagglutination and related domains that mediate cooperative colonization exist not only in adhesins, but also in high-molecular-weight bacteriocins that are important for contact-dependent inhibition of neighboring bacteria in airborne and enteric proteobacteria (Simionato et al., 2006). Because of the similarity among proteobacteria, it is possible that oral proteobacteria might also control neighboring bacteria through T5SS- and T6SS-dependent mechanisms. Co-incubation of Pg with S. gordonii induces expression of Ltp1, a native regulator of Mfa fimbriae and LuxS in Pg, perhaps for optimization of bacterial ratio during biofilm formation (Simionato et al., 2006). Therefore, inhibitory interactions among bacteria may not only regulate competition among neighboring bacteria, but also stabilize the bacterial community or change the composition of the microbiota during periodontitis development.
A Double-edged Sword: the Role of Bacteria-induced Immune Responses in Alveolar Bone Loss
Accumulating evidence is mounting suggesting that host immune responses to oral bacteria mediate alveolar bone loss in periodontitis. Genetic analyses have implicated polymorphisms of IL1B, IL1RN, IL6, IL10, FcγRIIIb, VDR, CD14, and TLR4 genes in susceptibility to periodontitis, although conclusive association is still lacking (Laine et al., 2012). More definitive evidence for a role of immune responses to oral bacteria in periodontitis was provided by experiments with mice deficient in several genes that are critical for innate and acquired immunity. Enforced infection of mice with Pg, Aa, or a combination of multiple bacteria including T. denticola, T. forsythia, and F. nucleatum induces alveolar bone loss in mice (Graves et al., 2008). Mice lacking iNOS, P-selectin, or ICAM1 are susceptible to alveolar bone loss after Pg infection (Baker et al., 2000; Fukada et al., 2008). Moreover, pre-immunization of mice with Pg reduces Pg-induced alveolar bone loss (Gibson et al., 2004). These observations suggest that some immune responses to oral bacteria are protective against periodontitis development. Further evidence for a protective role of host immunity against periodontitis is the finding that strains of periodontitis-associated bacteria that are resistant to host immunity-mediated elimination are more virulent in periodontitis. For example, Pg strains that possess capsules or the ability to invade and hide inside host cells are more resistant against complement and more virulent in an experimental model (Bostanci and Belibasakis, 2012). This also suggests that some host immune responses are protective against periodontitis development by eliminating pathobionts involved in periodontitis.
However, mice lacking several innate immune receptors, including TLR2 and NOD1, show decreased alveolar bone loss in mouse experimental periodontitis models (Burns et al., 2006; Papadopoulos et al., 2013; Jiao et al., 2013). TLR2 and NOD2 are pattern recognition receptors (PRRs) that recognize bacteria-specific molecules, lipoproteins, and small peptidoglycan-related molecules, respectively (Takeuchi and Akira, 2010). Like TLR2 and NOD1 KO mice, mice lacking C3aR and C5aR, which are receptors for activated and cleaved complement factors, also showed decreased alveolar bone loss in Pg-infected periodontitis models (Liang et al., 2011; Abe et al., 2012). C3 and C5 processing are produced during complement-mediated bacterial elimination (Ricklin et al., 2010). Furthermore, Pg gingipains process C3 (Popadiak et al., 2007). This suggests that bacteria-triggered immune responses are critical for induction of alveolar bone loss, although some bacteria-induced immune responses are beneficial to the host (Fig. 3). The loss of alveolar bone in periodontitis is primarily mediated by a series of immune responses that result in increased osteoclast differentiation and activation. Osteoclast differentiation is controlled by RANK activation via the balanced effects of its ligand, RANKL. and the inhibitor osteoprotegerin (OPG) (Darveau, 2010). Signal blockage of inflammatory cytokines, IL-1 and TNF, inhibits RANK activation and alveolar bone loss in experimental periodontitis models, suggesting that IL-1 and TNF mediate RANK activation (Assuma et al., 1998; Cochran, 2008). Stimulation of activated CD4+ T- and B-cells and osteoblasts by IL-1β and TNFα induces RANKL expression and inhibits OPG expression (Stolina et al., 2009). In response to oral bacteria, IL-6, TNFα, and IL-1β are secreted from neutrophils and macrophages that are recruited to damaged gingival tissue (Assuma et al., 1998; Cochran, 2008). Bacteria possess different and multiple types of immunostimulatory molecules, some of which induce recruitment of immune cells and others induce secretion of TNFα and IL-1β from immune cells (Takeuchi and Akira, 2010). For example, NOD1 ligands produced by certain bacteria, strongly induce chemokine secretion from non-hematopoietic cells, but do not induce secretion of TNFα and IL-1β from hematopoietic cells (Hasegawa et al., 2006; Masumoto et al., 2006). Thus, the induction of differential immune responses by gingival and recruited immune cells to particular PRR ligands affects specific and distinguishable processes in the sequence of events that result in alveolar bone loss. For example, NOD1 stimulation of epithelial cells mediates the recruitment of neutrophils to inflammatory sites, whereas recruited neutrophils require other PRR ligands (e.g., LPS) to secrete IL-1β (Hasegawa et al., 2011).
Figure 3.
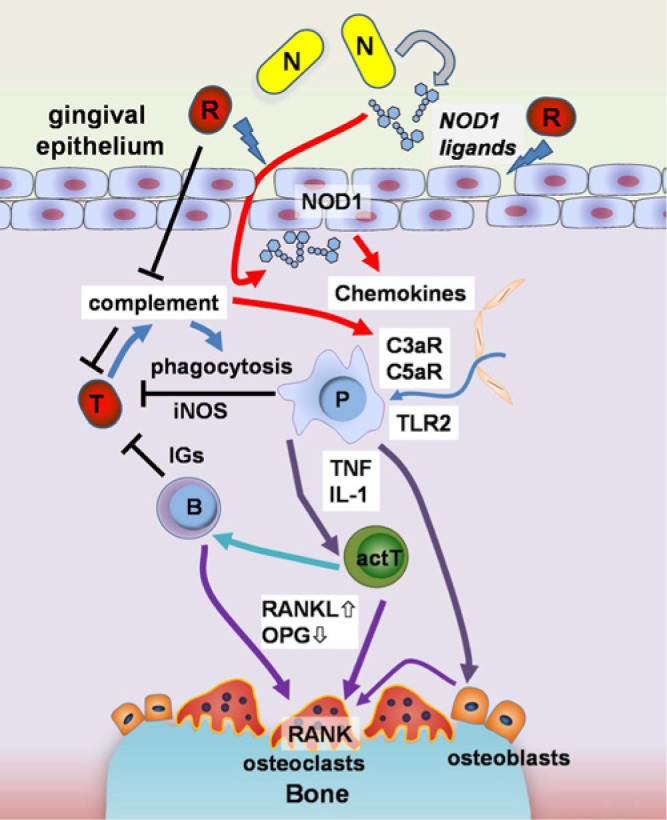
A proposed model for the role of immunostimulatory pathobionts in periodontitis. Development of periodontitis is associated with immune responses to oral bacteria. Complement, phagocytosis, iNOS-mediated immune responses and production of antigen-specific immunoglobulin (IGs) protect hosts from translocated harmful bacteria (T). Meanwhile, alveolar bone resorption by increased osteoclast differentiation and activation is triggered by two types of pathobionts. Many pathobionts, including red complex bacteria (R), subvert the host immune system and/or are immunosuppressive. Red complex pathobionts damage the epithelial tissue through the production of high protease activity which allows for the translocation of immunostimulatory bacterial molecules into tissues. P. gingivalis gingipain proteases also inactivate the complement system by cleaving C3 and C5. NOD1 ligands produced by specific pathobionts (N) are released from bacteria and function as immunostimulants away from bacteria, to cause alveolar bone loss at damaged gingival sites. NOD1 ligands possess the ability to recruit neutrophils which secrete inflammatory cytokines such as TNF and IL-1 to alter the RANKL/osteoprotegerin (OPG) expression balance in activated T- (actT), B-cells, and osteoblasts. Neutrophils and other phagocytic cells (P) also express innate immune receptors such as Toll-like receptor 2 (TLR2) and complement C3a and C5a receptors (C3aR and C5aR) at high levels. C3aR and C5aR also mediate recruitment of the phagocytic cells. The increased level of RANKL and the decreased level of OPG increase osteoclast differentiation, which results in alveolar bone loss.
Individual Bacteria Possess Different Immunostimulatory Activity to Modulate Different Immune Responses
Different bacteria produce different types of immunostimulatory molecules to stimulate different PRRs. Individual bacteria that express specific ligands for PRRs contribute to particular immune responses involved in the sequential induction of alveolar bone loss. For example, proteobacteria and specific Firmicutes release high levels of NOD1 ligands (Hasegawa et al., 2006). NOD1 and NOD2 ligands can induce chemokines from host cells that are insensitive to TLR ligands or tolerized to TLR signaling by prolonged exposure to TLR ligands (Hasegawa et al., 2011). Cells that are part of gingival tissue such as epithelial and stromal cells have a greater ability to respond to NOD1 ligands than to TLR4 ligands, whereas macrophages respond more robustly to TLR ligands than to NOD1/2 ligands and secrete TNFα, IL-6, and other cytokines (Kim et al., 2008). TLR4 is preferentially stimulated by a particular type of LPS that exists in many proteobacteria but is absent in Bacteroidetes, including Pg (Bryant et al., 2010). IL-1β secretion from macrophages requires activation of the inflammasome, which is controlled by two signals: (1) TLR ligands that are present in many bacteria and prime macrophages to induce pro-IL-1β and (2) activation of the inflammasome, which is induced by danger signals such as toxins specific to particular types of bacteria (Franchi et al., 2009). The activation of the complement system, which yields C3a and C5a production, is also dependent on particular bacterial strains (Franchi et al., 2009). Even within identical bacterial species, only certain strains of bacteria are resistant against the complement system by covering their cell surfaces with a capsule, complement-resistant LPS, and other molecules (Rautemaa and Meri, 1999; Bostanci and Belibasakis, 2012). Thus, individual bacteria are involved in the activation of specific immune responses in the sequence of events that results in alveolar bone loss.
Novel Oral Pathobionts with Strong Immunostimulatory Activity
The importance of a novel type of pathobionts in periodontitis was suggested by experiments with mouse models of periodontitis. Pg-infected mice under conventional SPF conditions, but not Pg-monocolonized mice, show significant alveolar bone loss (Hajishengallis et al., 2011). This indicates that, in addition to host-damaging bacteria, some pathobionts are needed to induce alveolar bone loss. In SPF mice, ligature placement around or between the molars also results in alveolar bone loss (Graves et al., 2008). Although gingival damage is bacteria independent in the ligature model, and SPF mice are free of red complex bacteria, bone loss induced by ligature placement is still dependent on oral bacteria (Jiao et al., 2013). Thus, non-red-complex bacteria have the ability to induce alveolar bone directly in the presence of gingival damage in addition to playing an indirect role in disease by facilitating the colonization of host-damaging red complex bacteria. Analysis of microbiota showed that ligature placement induced dysbiosis at damaged gingival sites (Jiao et al., 2013). An important event in the ligature model is the marked accumulation (more than 40% of total oral bacteria) of one bacterium identified as a novel Pasteurellaceae species, named NI1060 (Jiao et al., 2013). NI1060 is related to Aa, and genome sequencing of NI1060 showed that it contains several orthologues of Aa virulence genes (Jiao et al., 2013). NI1060 possesses the ability to induce alveolar bone loss in the ligature-induced model in a NOD1-dependent manner. Importantly, both NI1060 and Aa release high levels of NOD1 ligands when compared with Pg and other oral bacteria. Monocolonization of GF with NI1060 is sufficient to increase CXCL1 secretion, leading to neutrophil recruitment, and to induce several inflammatory downstream events, including production of IL-1β, TNFα, and RANKL, which are important for alveolar bone loss (Jiao et al., 2013). Because NI1060 is not an invasive pathogen, and major NOD1 ligands are soluble molecules, NOD1 ligands cannot stimulate host tissues to induce inflammatory molecules without the loss of the epithelial barrier (Hasegawa et al., 2011; Jiao et al., 2013). In another words, alveolar bone loss requires both host damage and immunostimulation, which is presumably induced by host-damaging bacteria and NOD1-stimulatory bacteria, respectively (Fig. 3). Red complex bacteria possess specific virulence factors to allow the bacteria to avoid recognition by host immune receptors and to subvert innate and acquired immune responses (Rautemaa and Meri, 1999; Bostanci and Belibasakis, 2012). Although we still do not know which immunostimulatory oral bacteria are important for the development of periodontitis in humans, the mouse studies suggest that host-damaging and immunostimulatory bacteria belong to different species and play distinct but cooperative roles in the induction of alveolar bone loss. Aa JP2, whose infection is associated with aggressive periodontitis in humans, possesses dual function in that it can damage host tissue via the production of cytotoxic leukotoxin, but also releases high levels of NOD1-stimulatory activity. Thus, the association of Aa with aggressive periodontitis may be explained by the ability of Aa to induce several activities involved in alveolar bone loss. Analysis of Aa-monocolonized mice is needed to test the unique role of Aa in periodontitis. Pasteurellaceae is not the only genus that stimulates NOD1, since several Proteobacteria, Peptostreptococcaceae, and Bacillaleceae species also possess high NOD1-stimulatory activity (Hasegawa et al., 2006). Therefore, it will be important to determine which pathobionts possess NOD1-stimulatory activity and their role in human periodontitis.
Perspectives
Accumulating evidence supports the “keystone-pathogen hypothesis” in which colonization of keystone bacteria such as Pg triggers dysbiosis and alteration of host immune responses, and other bacteria orchestrate inflammatory disease leading to bone loss (Hajishengallis et al., 2012). The finding that NOD1-stimulatory pathobionts can induce alveolar bone loss further refines the “keystone-pathogen hypothesis” by suggesting that individual oral pathobionts that accumulate during dysbiosis play a critical and specific role in periodontitis development. One of the major differences between known pathobionts and NOD1-stimulatory pathobionts is the ability of the latter to stimulate host cells without direct bacteria-host cell contact, because the majority of NOD1 ligands are released from bacteria (Hasegawa et al., 2011; Jiao et al., 2013). Therefore, tissue translocation or invasiveness of bacteria is not the only way to stimulate the host NOD1 receptor. Clostridium difficile, an enteric NOD1-stimulatory pathobiont, induces NOD1 stimulation but does not translocate into tissues (Hasegawa et al., 2011). These facts suggest that host-protective responses inside tissues might be ineffective to prevent alveolar bone loss, although NOD1-mediated signaling plays a role in the elimination of invasive pathogens including Listeria monocytogenes (Hasegawa et al., 2011). Thus, this novel type of immunostimulatory pathobiont is beneficial to the host in the absence of host-damaging bacteria, but can also promote pathology under certain conditions such as in periodontitis. Further investigation is needed to understand the role of immunostimulatory pathobionts in heath and disease.
Accumulation of immunostimulatory pathobionts appears to be dependent on interactions with other oral bacteria and host cells. In the case of NI1060, the accumulation at gingival sites above the bone loss might be dependent on nutrients from damaged tissue. NI1060 accumulates at damaged gingival sites over 400-fold more than at the healthy gingiva, although NI1060 represents only ~2 % of the total bacterial population in the oral cavities of healthy adult mice (Jiao et al., 2013). Therefore, it is likely that accumulation of at least some immunostimulatory pathobionts depends on nutrients derived from the host. However, NI1060 and related Aa, unlike red complex bacteria, are facultative anaerobes, and therefore anaerobic conditions are not essential for their growth (Jiao et al., 2013). Moreover, NI1060 and Aa do not require heme and NAD for growth as is the case for other Pasteurellaceae species such as H. influenzae (Garrity et al., 1984; Jiao et al., 2013). Importantly, the healthy mouse gingiva also harbor abundant numbers of Actinobacillus muris, another Pasteurellaceae species that possesses metabolic profiles similar to that of NI1060 (Garrity et al., 1984; Jiao et al., 2013). Therefore, the remarkable dominance of Aa in aggressive human periodontitis and NI1060 at the damaged gingiva in the ligature model of mouse periodontitis cannot be simply explained by the presence of nutrients released from damaged host cells. Colonization of GF, but not conventional SPF, mice with Staphylococcus xylosus, a dominant species in healthy adult mice, induces significant accumulations of bacteria in the oral cavity (Jiao et al., 2013), suggesting that NI1060 possesses a mechanism to acquire dominancy by outcompeting other commensals. Like other pathobionts, NI1060 produces several putative adhesins, including Flp orthologues, YadA, and Hemagglutinin proteins (Jiao et al., 2013), which are critical for virulence in other bacteria, suggesting that physical interactions of immunostimulatory pathobionts with other oral bacteria and host cells might play an important role in bone-loss-inducing virulence activity. Investigation of the interactions of immunostimulatory pathobionts with other members of the oral bacterial community should improve our understanding of the pathogenesis of periodontitis and of bacteria-specific immune responses.
Acknowledgments
The authors thank Drs. Gabriel Núñez and Jessica Werner for critical review and stimulating discussions.
Footnotes
This work was supported by grant R01DE018503 from the National Institutes of Health (NIH).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Abe T, Hosur KB, Hajishengallis E, Reis ES, Ricklin D, Lambris JD, et al. (2012). Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. J Immunol 189:5442-5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assuma R, Oates T, Cochran D, Amar S, Graves DT. (1998). IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immunol 160:403-409. [PubMed] [Google Scholar]
- Baker PJ, DuFour L, Dixon M, Roopenian DC. (2000). Adhesion molecule deficiencies increase Porphyromonas gingivalis-induced alveolar bone loss in mice. Infect Immun 68:3103-3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford CV, Fenno JC, Jenkinson HF, Dymock D. (2007). The chymotrypsin-like protease complex of Treponema denticola ATCC 35405 mediates fibrinogen adherence and degradation. Infect Immun 75:4364-4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes VM, Ciancio SG, Shibly O, Xu T, Devizio W, Trivedi HM, et al. (2011). Metabolomics reveals elevated macromolecular degradation in periodontal disease. J Dent Res 90:1293-1297. [DOI] [PubMed] [Google Scholar]
- Bizzini B, Pizzo G, Scapagnini G, Nuzzo D, Vasto S. (2012). Probiotics and oral health. Curr Pharm Des 18:5522-5531. [DOI] [PubMed] [Google Scholar]
- Bostanci N, Belibasakis GN. (2012). Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol Lett 333:1-9. [DOI] [PubMed] [Google Scholar]
- Bryant CE, Spring DR, Gangloff M, Gay NJ. (2010). The molecular basis of the host response to lipopolysaccharide. Nat Rev Microbiol 8:8-14. [DOI] [PubMed] [Google Scholar]
- Burns E, Bachrach G, Shapira L, Nussbaum G. (2006). Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol 177:8296-8300. [DOI] [PubMed] [Google Scholar]
- Burton JP, Chilcott CN, Moore CJ, Speiser G, Tagg JR. (2006). A preliminary study of the effect of probiotic Streptococcus salivarius K12 on oral malodour parameters. J Appl Microbiol 100:754-764. [DOI] [PubMed] [Google Scholar]
- Chow J, Tang H, Mazmanian SK. (2011). Pathobionts of the gastrointestinal microbiota and inflammatory disease. Curr Opin Immunol 23:473-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran DL. (2008). Inflammation and bone loss in periodontal disease. J Periodontol 79:1569-1576. [DOI] [PubMed] [Google Scholar]
- Daep CA, Novak EA, Lamont RJ, Demuth DR. (2011). Structural dissection and in vivo effectiveness of a peptide inhibitor of Porphyromonas gingivalis adherence to Streptococcus gordonii. Infect Immun 79:67-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveau RP. (2010). Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol 8:481-490. [DOI] [PubMed] [Google Scholar]
- Fardini Y, Wang X, Témoin S, Nithianantham S, Lee D, Shoham M, et al. (2011). Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol 82:1468-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine DH, Markowitz K, Fairlie K, Tischio-Bereski D, Ferrendiz J, Furgang D, et al. (2013). A consortium of Aggregatibacter actinomycetemcomitans, Streptococcus parasanguinis, and Filifactor alocis is present in sites prior to bone loss in a longitudinal study of localized aggressive periodontitis. J Clin Microbiol 51:2850-2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. (2009). The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10:241-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada SY, Silva TA, Saconato IF, Garlet GP, Avila-Campos MJ, Silva JS, et al. (2008). iNOS-derived nitric oxide modulates infection-stimulated bone loss. J Dent Res 87:1155-1159. [DOI] [PubMed] [Google Scholar]
- Garrity GM, Bell JA, Lilburn T. (1984). Genus I Family I Pasteurellaceae. In: Bergey’s Manual of Systematic Bacteriology. Vol. I Boone DR, Castenholz RW, Garrity GM, editors. New York, NY: Springer, pp. 851-912. [Google Scholar]
- Gibson FC, 3rd, Gonzalez DA, Wong J, Genco CA. (2004). Porphyromonas gingivalis-specific immunoglobulin G prevents P. gingivalis-elicited oral bone loss in a murine model. Infect Immun 72:2408-2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves DT, Fine D, Teng YT, Van Dyke TE, Hajishengallis G. (2008). The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol 35:89-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, et al. (2012). Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J 6:1176-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeger S, Doman E, Chakraborty T, Meyle J. (2010). Effects of Porphyromonas gingivalis infection on human gingival epithelial barrier function in vitro. Eur J Oral Sci 118:582-589. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. (2011). Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10:497-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Darveau RP, Curtis MA. (2012). The keystone-pathogen hypothesis. Nat Rev Microbiol 10:717-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammami R, Fernandez B, Lacroix C, Fliss I. (2013). Anti-infective properties of bacteriocins: an update. Cell Mol Life Sci 70:2947-2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YW, Ikegami A, Rajanna C, Kawsar HI, Zhou Y, Li M, et al. (2005). Identification and characterization of a novel adhesin unique to oral fusobacteria. J Bacteriol 187:5330-5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes CS, Aoki SK, Low DA. (2010). Bacterial contact-dependent delivery systems. Annu Rev Genet 44:71-90. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Yang K, Hashimoto M, Park JH, Kim YG, Fujimoto Y, et al. (2006). Differential release and distribution of NOD1 and NOD2 immunostimulatory molecules among bacterial species and environments. J Biol Chem 281:29054-29063. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim YG, Núñez G, et al. (2011). Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol 186:4872-4880. [DOI] [PubMed] [Google Scholar]
- Henderson B, Ward JM, Ready D. (2010). Aggregatibacter (Actinobacillus) actinomycetemcomitans: a triple A* periodontopathogen? Periodontol 2000 54:78-105. [DOI] [PubMed] [Google Scholar]
- Ito R, Ishihara K, Shoji M, Nakayama K, Okuda K. (2010). Hemagglutinin/adhesin domains of Porphyromonas gingivalis play key roles in coaggregation with Treponema denticola. FEMS Immunol Med Microbiol 60:251-260. [DOI] [PubMed] [Google Scholar]
- Jenkinson HF, Lamont RJ. (2005). Oral microbial communities in sickness and in health. Trends Microbiol 13:589-595. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Darzi Y, Tawaratsumida K, Marchesan JT, Hasegawa M, Moon H, et al. (2013). Induction of bone loss by pathobiont-mediated NOD1 signaling in the oral cavity. Cell Host Microbe 13:595-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaewsrichan J, Douglas CW, Nissen-Meyer J, Fimland G, Teanpaisan R. (2004). Characterization of a bacteriocin produced by Prevotella nigrescens ATCC 25261. Lett Appl Microbiol 39:451-458. [DOI] [PubMed] [Google Scholar]
- Katz J, Sambandam V, Wu JH, Michalek SM, Balkovetz DF. (2000). Characterization of Porphyromonas gingivalis-induced degradation of epithelial cell junctional complexes. Infect Immun 68:1441-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YG, Park JH, Shaw MH, Franchi L, Inohara N, Núñez G. (2008). The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity 28:246-257. [DOI] [PubMed] [Google Scholar]
- Kolenbrander PE, Andersen RN, Blehert DS, Egland PG, Foster JS, Palmer RJ., Jr (2002). Communication among oral bacteria. Microbiol Mol Biol Rev 66:486-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine ML, Crielaard W, Loos BG. (2012). Genetic susceptibility to periodontitis. Periodontol 2000 58:37-68. [DOI] [PubMed] [Google Scholar]
- Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, et al. (2011). The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol 186:869-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Nagata H, Yamamoto Y, Tanaka M, Tanaka J, Minamino N, et al. (2004). Glyceraldehyde-3-phosphate dehydrogenase of Streptococcus oralis functions as a coadhesin for Porphyromonas gingivalis major fimbriae. Infect Immun 72:1341-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto J, Yang K, Varambally S, Hasegawa M, Tomlins SA, Qiu S, et al. (2006). NOD1 acts as an intracellular receptor to stimulate chemokine production and neutrophil recruitment in vivo. J Exp Med 203:203-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien-Simpson NM, Paolini RA, Hoffmann B, Slakeski N, Dashper SG, Reynolds EC. (2001). Role of RgpA, RgpB, and Kgp proteinases in virulence of Porphyromonas gingivalis W50 in a murine lesion model. Infect Immun 69:7527-7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos G, Weinberg EO, Massari P, Gibson FC, 3rd, Wetzler LM, Morgan EF, et al. (2013). Macrophage-specific TLR2 signaling mediates pathogen-induced TNF-dependent inflammatory oral bone loss. J Immunol 190:1148-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, et al. (2001). Bacterial diversity in human subgingival plaque. J Bacteriol 183:3770-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popadiak K, Potempa J, Riesbeck K, Blom AM. (2007). Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J Immunol 178:7242-7250. [DOI] [PubMed] [Google Scholar]
- Ramsey MM, Whiteley M. (2009). Polymicrobial interactions stimulate resistance to host innate immunity through metabolite perception. Proc Natl Acad Sci USA 106:1578-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautemaa R, Meri S. (1999). Complement-resistance mechanisms of bacteria. Microbes Infect 1:785-794. [DOI] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. (2010). Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11:785-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Ishihara K, Kato T, Okuda K. (1997). Cloning, expression, and sequencing of a protease gene from Bacteroides forsythus ATCC 43037 in Escherichia coli. Infect Immun 65:4888-4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobert M, Jahn D. (2002). Regulation of heme biosynthesis in nonphototrophic bacteria. J Mol Microbiol Biotechnol 4:287-294. [PubMed] [Google Scholar]
- Scott JC, Klein BA, Duran-Pinedo A, Hu L, Duncan MJ. (2013). A two-component system regulates hemin acquisition in Porphyromonas gingivalis. PLoS One 8:e73351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah HN, Williams RA. (1987). Utilization of glucose and amino acids by Bacteroides intermedius and Bacteroides gingivalis. Curr Microbiol 15:241-246. [Google Scholar]
- Simionato MR, Tucker CM, Kuboniwa M, Lamont G, Demuth DR, Tribble GD, et al. (2006). Porphyromonas gingivalis genes involved in community development with Streptococcus gordonii. Infect Immun 74:6419-6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr (1998). Microbial complexes in subgingival plaque. J Clin Periodontol 25:134-144. [DOI] [PubMed] [Google Scholar]
- Song H, Belanger M, Whitlock J, Kozarov E, Progulske-Fox A. (2005). Hemagglutinin B is involved in the adherence of Porphyromonas gingivalis to human coronary artery endothelial cells. Infect Immun 73:7267-7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolina M, Schett G, Dwyer D, Vonderfecht S, Middleton S, Duryea D, et al. (2009). RANKL inhibition by osteoprotegerin prevents bone loss without affecting local or systemic inflammation parameters in two rat arthritis models: comparison with anti-TNFα or anti-IL-1 therapies. Arthritis Res Ther 11:R187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K, Hirasawa M, Ikeda T. (1991). Isolation and purification of bacteriocin from Prevotella intermedia (Bacteroides intermedius). J Periodontol 62:439-444. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. (2010). Pattern recognition receptors and inflammation. Cell 140:805-820. [DOI] [PubMed] [Google Scholar]
- Wyss C. (1989). Dependence of proliferation of Bacteroides forsythus on exogenous N-acetylmuramic acid. Infect Immun 57:1757-1759. [DOI] [PMC free article] [PubMed] [Google Scholar]