

Abstract
Monogenic diabetes represents a heterogeneous group of disorders resulting from defects in single genes. Defects are categorized primarily into two groups: disruption of β‐cell function or a reduction in the number of β‐cells. A complex network of transcription factors control pancreas formation, and a dysfunction of regulators high in the hierarchy leads to pancreatic agenesis. Dysfunction among factors further downstream might cause organ hypoplasia, absence of islets of Langerhans or a reduction in the number of β‐cells. Many transcription factors have pleiotropic effects, explaining the association of diabetes with other congenital malformations, including cerebellar agenesis and pituitary agenesis. Monogenic diabetes variants are classified conventionally according to age of onset, with neonatal diabetes occurring before the age of 6 months and maturity onset diabetes of the young (MODY) manifesting before the age of 25 years. Recently, certain familial genetic defects were shown to manifest as neonatal diabetes, MODY or even adult onset diabetes. Patients with neonatal diabetes require a thorough genetic work‐up in any case, and because extensive phenotypic overlap exists between monogenic, type 2, and type 1 diabetes, genetic analysis will also help improve diagnosis in these cases. Next generation sequencing will facilitate rapid screening, leading to the discovery of digenic and oligogenic diabetes variants, and helping to improve our understanding of the genetics underlying other types of diabetes. An accurate diagnosis remains important, because it might lead to a change in the treatment of affected subjects and influence long‐term complications.
Keywords: Monogenic diabetes, Next generation sequencing
Monogenic diabetes represents a heterogeneous group of disorders resulting from defects in single genes. Defects are categorized primarily into two groups: disruption of beta cell function or a reduction in the number of beta cells. An accurate diagnosis is important because it may lead to a change in the treatment of affected subjects and influence long‐term complications.
Introduction
The prevalence of monogenic diabetes is estimated at 2–5% of all patients with diabetes1. The first description of a hereditary form dates back to 1928, when Cammidge identified families with autosomal dominant diabetes2. In 1975, Maturity Onset Diabetes of the Young (MODY) was defined as diabetes occurring before the age of 25 years with autosomal dominant inheritance as a result of an intrinsic β‐cell defect3. The first gene causally implicated was coded for the enzyme glucokinase (GCK)4. A few years later, two other monogenic forms of diabetes, MODY1 and MODY3, were attributed to mutations in transcription factor genes; the hepatocyte nuclear factor 4 and 1 alpha (HNF4A, HNF1A), respectively5.
Historically, the age at diabetes onset has been a criterion for classification. For example, neonatal diabetes is diagnosed within 6 months of birth, whereas MODY forms of diabetes occur before the age of 25 years. However, recent studies report that specific gene mutations occurring in the same family can present clinically as a neonatal form as well as ‘type 2‐like’ or ‘type 1‐like’ forms during adulthood.
Currently, many monogenic forms are missed or misclassified as type 2 or type 1 diabetes. Improved access to genetic testing will help determine the exact origin of diabetes. In the present review, I delineate the different gene defects using a functional approach, discussing developmental and cellular defects, glucose uptake at the cell surface, and then following the intracellular destiny of glucose molecules eliciting insulin secretion (Figure 1).
Figure 1.
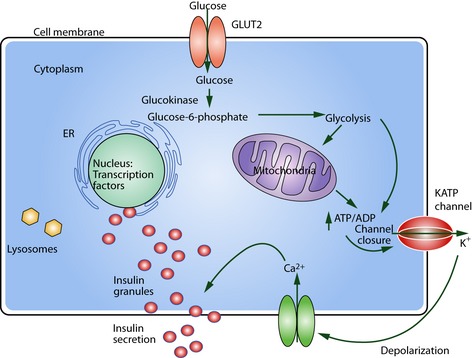
Schematic β‐cell. Subcellular localization of defects within the β‐cell leading to monogenic diabetes. Starting at glucose uptake at the GLUT2 transporter, during phosphorylation by the enzyme glucokinase or during glycolysis. Dysfunction of the adenosine triphosphate‐sensitive potassium (KATP) channel with the KIR6.2 subunits (brown) and SUR1 subunits (red) will interfere with insulin secretion. Malfunction of the transcription factors located in the nucleus will lead to the nucleopathies and finally endoplasmic reticulum (ER) stress and lysosomal defects can also cause diabetes. ADP, adenosine diphosphate; ATP, adenosine triphosphate; GLUT2, glucose transporter 2.
Path to Monogenic Diabetes
Nucleopathies Causing Developmental Pancreatic Defects
A network of nuclear transcription factors controls pancreatic development in humans and mice. Depending on their hierarchical position, defects lead to a severe phenotype, such as pancreatic agenesis with neonatal diabetes and exocrine insufficiency, or a milder phenotype, with diabetes onset during adolescence or adulthood. Pancreatic agenesis leads to severe intrauterine growth retardation as a result of the absence of insulin secretion, a major growth factor. Homozygous or compound heterozygous mutations usually cause more severe forms of diabetes, and many heterozygous mutations are associated with later‐onset diabetes (Table 1). Numerous transcription factors play a pleiotropic role, leading to syndromic forms of diabetes associated with malformations in other organ systems, such as congenital heart defects and gastrointestinal defects.
Table 1. Summary of mutations.
| Gene | Protein | Mutation | Phenotype | References |
|---|---|---|---|---|
| Nucleus | ||||
| PDX1/IPF1 | Pancreas/duodenum homeobox protein 1 |
Hom, CHet Het |
Pancreatic agenesis Adult onset |
7 |
| PTF1A | Pancreas transcription factor 1A | Hom | Pancreas and cerebellar agenesis | 13 |
| PTF1A Enhancer | Non‐coding region | Hom, CHet | Pancreatic agenesis | 15 |
| GLIS3 | Zinc finger protein GLIS3 | Hom | PNDM and hypothyroidism | 24 |
| NGN3 | Neurogenin 3 | Hom, CHet | PNDM or later onset diabetes, congenital diarrhea | 31 |
| RFX6 | DNA binding protein RFX6 | Hom | PNDM, variable pancreas hypoplasia, intestinal atresia, gall bladder hypoplasia | 39 |
| GATA6 | Transcription factor GATA6 | Het | PNDM and adult onset diabetes, variable exocrine pancreatic insufficiency | 16 |
| GATA4 | Transcription factor GATA4 | Het | Possible pancreatic agenesis and cardiac defects | 23 |
| NEUROD1 | Neurogenic differentiation factor 1 | Hom | PNDM, cerebellar hypoplasia, sensorineural deafness, retinal dystrophy | 44 |
| Het | Adult onset diabetes | 45 | ||
| PAX6 | Paired box protein Pax6 | CHet | PNDM with brain anomaly | 50 |
| Het | Diabetes and aniridia | 51 | ||
| PAX4 | Paired box protein Pax4 | Adult onset diabetes | 54 | |
| HNF1B | Hepatocyte nuclear factor 1beta | Het | PNDM with pancreas hypoplasia, RCAD syndrome | 55 |
| MNX1 | Motor neuron and pancreas homeobox protein 1 | Hom | PNDM | 66 |
| Het | Sacral dysgenesis without diabetes | 68 | ||
| KLF11 | Krueppel‐like factor 11 | Het | Adult onset diabetes | 84 |
| HNF1A | Hepatocyte nuclear factor 1alpha | Het | Macrosomia and hypoglycemia at birth, adolescent onset diabetes | 5 |
| HNF4A | Hepatocyte nuclear factor 4 alpha | Het | Macrosomia and hypoglycemia at birth, adolescent onset diabetes | 6 |
| Cell membrane and cytoplasm | ||||
| SLC2A2 | Glucose transporter 2 | Hom | Fanconi Bickel syndrome PNDM, TNDM | 87 |
| GCK | Glucokinase | Het | Mild non‐progressive hyperglycemia | 4 |
| Hom | PNDM | 73 | ||
| SLC19A2 | Thiamine transporter 1 | Hom | PNDM or early onset, megaloblastic anemia, sensorineural deafness | 97 |
| Lysosome | ||||
| SLC29A3 | Hom, CHet | Diabetes, pigmented hypertrichosis | 102 | |
| Endoplasmic reticulum | ||||
| WFS1 | Wolframin | CHet | Diabetes mellitus and insipidus, optic atrophy, deafness (Wolfram syndrome 1) | 105 |
| CISD2 | CDGSH iron‐sulfur domain‐containing protein 2 | Hom | Wolfram syndrome 2 without diabetes insipidus** | 145 |
| EIF2AK3 | Eukaryotic translation initiation factor 2‐alpha kinase 3 | Hom | PNDM, skeletal defect, growth retardation (Wollcot‐Rallison syndrome) | 113 |
| IER3IP1 | Immediate early response 3 interacting protein 1 | Hom | Microcephaly, epilepsy, PNDM (MEDS syndrome) | 115 |
| Insulin synthesis and secretion | ||||
| INS | Insulin | Hom, Het | PNDM, TNDM, adult onset | 120 |
| Het | Adult onset | 118 | ||
| BLK | Tyrosine‐protein kinase Blk | Het | Adult onset diabetes | 124 |
| KCNJ11 | KIR6.2 | Het | PNDM, TNDM, adult onset | 126 |
| ABCC8 | SUR1 | Het | PNDM, TNDM, adult onset | 127 |
| Exocrine pancreas | ||||
| CEL | Bile salt‐activated lipase | Het | Adult onset progressive diabetes, exocrine insufficiency | 128 |
| Autoimmune diabetes | ||||
| AIRE | Autoimmune regulator | Hom, Het | Systemic autoimmune disease | 146 |
| FOXP3 | FOXP3 protein | X‐linked | PNDM, diarrhea, eczema, thyroid autoimmunity | 74 |
| SIRT1 | NAD‐dependent protein deacetylase sirtuin‐1 | Het | Adult onset autoimmune diabetes, insulin resistance | 136 |
CHet, compound heterozygous; Het, heterozygous; Hom, homozygous; MEDS, microcephaly, epilepsy and permanent neonatal diabetes syndrome; NAD, nicotinamide adenine dinucleotide; PNDM, permanent neonatal diabetes mellitus; RACD, renal cysts associated with diabetes; TNDM, transient neonatal diabetes mellitus.
The first gene defect described in human pancreatic agenesis was pancreatic duodenal homeobox gene 1 (PDX1/IPF1)7. Homozygous and compound heterozygous mutations lead to a severe phenotype with neonatal diabetes and exocrine pancreatic insufficiency8. Heterozygous carriers present with late‐onset diabetes that can be misdiagnosed as type 2 diabetes9. PDX1 has dual functions. Early in embryogenesis, PDX1 is expressed in the forming pancreatic bud and controls the cell fate of pancreatic progenitors. During the postnatal period, PDX1 becomes restricted to β‐ and δ‐cells, where it is involved in β‐cell survival10 and regulates β‐cell susceptibility to endoplasmic reticulum (ER) stress11. This change in function could explain why diabetes worsens over time in heterozygous carriers.
Similarly, homozygous mutations in the pancreas‐specific transcription factor 1A gene (PTF1A) lead to pancreatic agenesis associated with cerebellar hypoplasia. PTF1A is important for pancreatic outgrowth in early embryogenesis and cerebellar formation12. Interestingly, low C‐peptide and insulin levels can be detected in the blood of patients with these homozygous mutations13. The source of insulin production has not been elucidated in humans; but in mice, insulin is thought to be secreted by scattered ectopic β‐cells in the spleen. Even if PTF1A mutations remain rare in human diabetes14, recessive mutations in a distal PTF1A enhancer are a frequent cause of pancreas agenesis in consanguineous families15.
Heterozygous mutations in the human GATA‐binding protein 6 gene (GATA6) can lead to pancreatic agenesis with neonatal diabetes and exocrine pancreatic insufficiency, or to later‐onset diabetes as well as type 2‐like diabetes with variable exocrine insufficiency16. GATA6 is expressed before PDX1 in the developing endoderm, the pleiotropic effects are explained by the expression in the developing heart, lung, allantois, muscle and gut18. Therefore, many of the human cases have heart malformations, and gastrointestinal, pituitary and cognitive deficits19. Homozygous mutations are probably lethal. The functions of GATA6 and GATA4 have been studied extensively in mouse models, but only the double GATA6/4 knockout replicates the human phenotype21. GATA factors bind the PDX1 promoter and are involved in the proliferation of pancreatic progenitor cells; the double knockout has a reduced number of PDX1‐positive cells during embryogenesis, resulting in pancreatic hypoplasia. During development, GATA4 is expressed in the pancreas, heart, liver and small intestine. Human GATA4 mutations mostly cause congenital heart malformations. A single report has associated an atrial septal defect with neonatal diabetes as a result of pancreatic agenesis and a heterozygous GATA4 mutation23.
Defective GLIS family zinc finger 3 (GLIS3), acting downstream of PDX1 and PTF1A, leads to neonatal diabetes combined with hypothyroidism, congenital glaucoma, hepatic fibrosis and polycystic kidneys24. GLIS3 directly transactivates the neurogenin 3 promoter, as well as the insulin promoter, and controls β‐cell expansion through transcriptional control of the cell cycle gene CCND225. This explains why targeted disruption of GLIS3 causes defective islet cell differentiation with a marked reduction in β‐cells. Intrauterine growth retardation points to insulin hyposecretion during pregnancy. Incomplete syndromes exist when residual transcripts are formed in a specific tissue, but neonatal diabetes and hypothyroidism persist throughout all families that have been described26. GLIS3 is vital for adult β‐cell function and mass, as conditional knockout of GLIS3 in adult β‐cells results in apoptosis and fulminant diabetes27. Interestingly, genome‐wide association studies have identified GLIS3 as a candidate gene in type 1 diabetes and type 2 diabetes28.
The transcription factor neurogenin 3 (NGN3), which acts downstream of PDX1, PTF1A and GLIS3 is the master gene controlling endocrine cell fate decisions in multipotent pancreatic endodermal progenitor cells. Targeted disruption leads to a failure of islet development, neonatal diabetes and early death. Therefore, NGN3 is required for the development of the four endocrine cell lineages30. In humans, loss‐of‐function mutations, such as compound heterozygosity for E28X and L135P, or homozygous mutations in the coding region (E123X), are associated with neonatal diabetes and congenital malabsorption diarrhea as a result of enteric anendocrinosis31. Thus, NGN3 is important in human islet and enteroendocrine cell development. Clinical characterization of these patients showed residual insulin secretion with a stimulated C‐peptide level of up to 546 pmol/L during a mixed meal test; however, the glucagon levels were not measurable. Interestingly, incomplete loss of NGN3 function still leads to severe diarrhea, but only to later‐onset diabetes at the age of 8 years33. Heterozygous NGN3 mutations rarely contribute to a type 2‐like diabetes in Japanese and Indian subjects35.
In 2004, several children from two different families were reported to have a syndrome comprising neonatal diabetes with a hypoplastic pancreas, intestinal atresia and gall bladder hypoplasia38. In 2010, the cause of this syndrome was attributed to mutations in regulatory factor X‐box binding 6 (RFX6) transcription factor39. All studied mutations except one were homozygous, and heterozygous parents had a normal oral glucose tolerance test40. The functional role of RFX6 was analyzed in mice harboring a targeted disruption of RFX6, these mice fail to generate islet cells, with the exception of pancreatic polypeptide (PP) cells41. During development, RFX6 acts downstream of NGN3, and directs the β‐cell fate. The size of the pancreas was reduced in most of the mice, as well as humans.
The transcription factor, NEUROD1, plays a multisystemic role in brain and pancreas development, and lies downstream of NGN3. Targeted disruption of NEUROD1 in mice results in a 74% reduction of insulin‐producing cells, as well as a 39% decrease in glucagon‐producing cells. The newborn mice develop diabetes and die after birth42. NEUROD1 also functions as an activator of both GCK and insulin (INS)43. In humans, a homozygous mutation leads to permanent neonatal diabetes associated with cerebellar hypoplasia, learning difficulties, profound sensorineural deafness, and visual impairment as a result of severe myopia and retinal dystrophy44. Malecki et al.45 were the first to describe a family with late onset diabetes associated with a heterozygous mutation in NEUROD1. More recently, a novel mutation was reported that led to autosomal dominant diabetes in a Chinese family with diabetes onset between 27 and 73 years‐of‐age46. In several families, NEUROD1 diabetes has been associated with obesity, increasing the difficulties in clinically differentiating between monogenic diabetes and type 2 diabetes47.
Paired box gene 6 (PAX6) is highly expressed in β‐cells, the developing brain, and eyes. In mice, targeted disruption of PAX6 leads to microphthalmia and congenital diabetes with a reduction in the number of insulin‐, glucagon‐, somatostatin‐, and PP‐producing cells48. PAX6 is also involved in the regulation of prohormone convertase 1/3 and contributes to proinsulin processing49. In humans, compound heterozygosity of PAX6 results in severe developmental defects in the brain with hypopituitarism and neonatal diabetes50. Heterozygous PAX6 mutations provoke aniridia associated with glucose intolerance51. Targeted disruption of the paired box gene 4 (PAX4) leads to an absence of β‐cells53. Surprisingly, only rare autosomal dominant diabetes cases have been associated with heterozygous PAX4 mutations, especially in some Asian populations54.
The first report implicating the transcription factor HNF1 homeobox B (HNF1B) was published in 1997 when Horikawa identified two Japanese families with diabetes associated with polycystic kidneys with heterozygous mutations55. The syndrome of renal cysts associated with diabetes (RCAD) sometimes includes genital tract abnormalities56. Defects in HNF1B can lead to neonatal diabetes with polycystic, dysplastic kidneys57. Histopathological analysis of an affected fetus with heterozygous frameshift mutations in HNF1B has shown pancreas hypoplasia, disorganized islets with decreased β‐cell density and a lack of GLUT2 expression58. This presentation can be explained by the loss of transcriptional activation of GLUT2 by HNF1B on its binding to the GLUT2 promoter. This work led to the conclusion that HNF1B is essential for human β‐cell maturation60. During embryogenesis, HNF1B is expressed widely in the visceral endoderm of all PDX1‐positive cells. After midgestation, HNF1B becomes a marker of ductal cells. In adults, HNF1B is expressed in the liver, stomach, ductal pancreatic cells, lungs and kidneys. HNF1B forms homodimers or heterodimers with the structurally similar HNF1A61. The β‐cell‐specific knockout of HNF1B confirmed the importance of HNF1B for glucose‐stimulated insulin secretion, but not arginine‐stimulated insulin secretion, which remains intact62.
Severe non‐diabetic renal disease can also be a phenotype of HNFB1 loss. Intriguingly, no phenotypic differences exist between large deletions, large genomic rearrangements and point mutations64. One contiguous gene deletion syndrome combining mental retardation, severe growth deficit, eye abnormalities and immune deficiency with RCAD was recognized by the identification of a chromosomal microdeletion involving 1.3–1.7 Mb on Chr17q1265.
Motor neuron and pancreas homeobox 1 transcription factor (MMNX1; also called HLXB9) is expressed in the pancreas during embryogenesis. The first described homozygous mutation in humans leads to permanent neonatal diabetes with normal pancreas morphology66. Earlier work showed dorsal pancreatic agenesis in knockout mice with disorganized islets and a marked reduction in the number of β‐cells67. Heterozygous deletions have been described in autosomal dominant sacral dysgenesis without diabetes68.
Nucleopathies Causing Functional Defects
Despite the expression of HNF1A and HNF4A during embryogenesis, their absence does not cause structural pancreatic defects, and diabetes manifests mostly during adolescence or young adulthood63.
In early embryogenesis, HNF1A is expressed in most epithelial cells and follows the pattern of HNF4A. After birth, HNF1A is localized predominantly in exocrine cells with lower expression in islet cells69. The functional HNF1A protein forms a dimer that is able to homodimerize or heterodimerize with HNF1B71. HNF1A is an essential transcription factor for the glucose‐stimulated insulin secretory response63. Progressive hyperglycemia is the hallmark of this diabetes phenotype. As the human phenotype can vary, even in the same family, especially in regards to diabetes onset, several factors that influence the phenotype have been identified. For example, the presence of maternal diabetes during pregnancy leads to an earlier manifestation of diabetes in offspring by more than 10 years.
Despite a favorable lipid profile, an increased risk of vascular complications is present in HNF1A‐diabetes75. The presence of HNF1A binding sites in the C‐reactive protein (CRP) promoter leads to a decrease in CRP levels when HNF1A is defective76. Therefore, CRP can be used as a biomarker, with a cut‐off for highly sensitive CRP levels of ≤0.2 mg/L, to distinguish HNF1A‐diabetes from type 2 diabetes with a sensitivity of 79% and specificity of 83%. Because of the decreased renal glucose absorption, an action controlled by HNF1A, renal glycosuria can assist in making the diagnosis77.
HNF4A is a nuclear transcription factor expressed in almost all PDX1‐positive cells in the pancreatic bud at very early stages of embryogenesis. At the end of pancreas development, HNF4A is expressed in all endocrine cell types as well as exocrine cells69, therefore mutations in HNF4A affect the function of the entire islet of Langerhans and is not restricted to the β‐cell. HNF4A functions primarily as a homodimer, and binds to the HNF1B promoter and HNF1A promoter78.
Subjects with HNF4A mutations can present with a dual phenotype, with hyperinsulinemic hypoglycemia at birth and diabetes many years later79. This paradoxical phenotype might be explained by functionally different HNF4A targets with sequential temporal expression leading to fetal and perinatal hyperinsulinemia and adolescent hypoinsulinemia80. Furthermore, progressive β‐cell exhaustion might contribute to the later onset of diabetes. Clinical studies have shown a concomitant decrease of insulin, glucagon, PP and amylin secretion in humans with HNF4 mutations81. Reduced HNF4A activity in humans is also associated with decreased lipoprotein (a) and apolipoprotein A‐II levels82, because HNF4A regulates the expression of a large number of genes involved in lipid metabolism83.
The transcription factor, Krüppel‐like factor 11 (KLF11), is responsible for an autosomal dominant form of diabetes84. Functional analysis has shown that KLF11 regulates PDX1 and INS transcription by binding to their respective promoters85. Interestingly, the diabetes‐causing mutation, c‐331, in the insulin gene promoter lies in the KLF11 binding site, showing the importance of KLF11 in humans86.
Cellular Defects in Non‐Nuclear Compartments
Defects in cellular structures, such as at the plasma membrane, the lysosome, the cytoplasm and the endoplasmic reticulum, are at the origin of many diabetes variants.
Glucose Uptake and Sensing
Glucose is taken up by the facilitative glucose transporter 2 (GLUT2) expressed at the surface of the human β‐cell, liver, kidney and intestine87. In 1997, Santer et al.89 reported the cause of Fanconi Bickel syndrome (FBS) as homozygous mutations in the solute carrier family 2 gene (SLC2A2) encoding the GLUT2 protein (Figure 1). FBS is an autosomal recessive disorder characterized by hyperglycemia, especially in the fed state, glycosuria and hepatorenal glycogen accumulation. Fasting hypoglycemia can also occur as a result of massive renal glucose loss. A defect in the glucose transporter leads to impaired monosaccharide uptake and accumulation in the blood. Furthermore, the rate limitation of glucose uptake by β‐cells leads to a decrease in insulin secretion, amplifying postprandial hyperglycemia. Diabetes onset varies greatly, but neonatal diabetes associated with galactosemia has been described90. Heterozygous mutations might lead to gestational diabetes91 or only renal glycosuria92.
After glucose enters the β‐cell, the enzyme GCK catalyzes the formation of glucose‐6‐phosphate and functions as a glucose sensor (Figure 1). GCK is also expressed in the liver and controls glycogen synthesis, gluconeogenesis, lipid synthesis and urea production. In the brain, GCK mediates glucose sensing93. Heterozygous loss‐of‐function mutations lead to mildly elevated fasting blood glucose levels, up to 6.7 mmol/L with a postprandial increase of 2 mmol/L up to 8.6 mmol/L4. Patients with one of the two specific mutations (GCK G261R and L184P) have exceptionally high postprandial glucose levels, sometimes exceeding 13 mmol/L94. No worsening occurs over time, and no long‐term complications have been described. However, homozygous inactivating mutations lead to severe neonatal diabetes, and insulin therapy is required73. Activating mutations of the same enzyme have the opposite effect, leading to neonatal hyperinsulinemic hypoglycemia95.
Cellular Metabolism
Solute carrier family 19 (thiamine transporter), member 2 (SLC19A2) encodes a high‐affinity thiamine transporter that is expressed in the pancreas, heart, skeletal muscle, placenta, brain, liver, retina, bone marrow and fibroblasts. Therefore, a loss‐of‐function of SLC19A2 results in manifestations such as megaloblastic anemia, diabetes, and sensorineural deafness, called thiamine‐responsive megaloblastic anemia (TRMA) or Rogers syndrome96. Diabetes can appear during the neonatal period, and has been found to be associated with visual system disturbances, neurological deficits, and cardiac abnormalities98. Adequate intracellular thiamine levels are important for mitochondrial adenosine triphosphate (ATP) synthesis and cellular function99.
Lysosome
Solute carrier family 29 (nucleoside transporter), member 3 (SLC29A3) encodes a nucleoside transporter localized to intracellular membrane compartments and expressed in the endocrine and exocrine pancreas101. Intracellular localization seems to be cell‐type dependent, and can involve lysosomes or mitochondria103. Mutation in SLC29A3 can lead to the autosomal recessive disorder with pigmented hypertrichosis and insulin‐dependent diabetes mellitus (PHID) manifesting in childhood103.
Endoplasmic Reticulum
Several forms of diabetes are due to dysfunction in the endoplasmic reticulum (ER); the first described form was Wolfram syndrome 1 (WFS1). WFS1 is an autosomal recessive, multisystem degenerative disorder also known as diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD). WFS1 was first reported in 1938, but the causative gene, WFS1, encoding the wolframin protein, was not identified until 1998104. Wolframin is expressed in the ER in many cell types, including the pancreas, heart, retina, brain, placenta, lung, liver, skeletal muscle and kidney. The predominant role of this protein is to protect the cell from ER stress and subsequent death; in β‐cells, wolframin associates with a cyclic adenosine monophosphate‐generating enzyme, increasing insulin production and release105. Generally, diabetes onset varies from age 3 weeks to 16 years, usually requiring insulin substitution. Optic atrophy starts around 11 years (range 6 weeks to 19 years), with most patients going blind109. Diabetes insipidus presents at an average age of 14 years (range 3 months to 40 years), and sensorineural deafness at an average of 16 years (range 5–39 years). Neurodegenerative symptoms, including cerebellar ataxia, peripheral neuropathy and psychiatric illnesses, manifest in the fourth decade. Most patients are compound heterozygous for two mutations. A rare autosomal dominant form of WFS1 also exists110.
Recently, CDGSH iron sulfur domain 2 (CISD2) was found to give rise to WFS2, a phenotype similar to WFS1, but without diabetes insipidus111. Some patients also show a significant bleeding tendency as a result of defective platelet aggregation with collagen. CISD2 encodes a protein that localizes in the ER and is involved in calcium homeostasis.
The eukaryotic translation initiation factor 2‐alpha kinase 3 (EIF2AK3) and immediate early response 3 interacting protein 1 (IER3IP1) are also located in the ER and play a role in the stress response. Gene defects in either of these genes leads to an overlapping autosomal recessive syndrome. Mutations in EIF2AK3 are associated with early onset diabetes, skeletal defects and growth retardation, also called Wollcot‐Rallison syndrome (WRS)112. Mutations in IER3IP1 lead to microcephaly, epilepsy and permanent neonatal diabetes (MEDS) in MEDS syndrome114. In the mouse, the targeted disruption of EIF2AK3 results in proinsulin accumulation in the ER of β‐cells and insulin deficiency. Therefore, EIF2AK3 seems to regulate ER‐to‐Golgi trafficking and proinsulin degradation in response to reduced insulin demand115. The pancreas‐specific knockout mouse model shows impaired β‐cell differentiation and lower insulin content at birth with a 50% reduction in β‐cell mass compared with wild type116. Postnatal proliferation of β‐cells is also reduced, leading to an 87% reduction in β‐cell mass at weaning. In addition, the β‐cells show a distended ER and squeezed mitochondria at birth. These results underline the importance of EIF2AK3 function in the prenatal and perinatal period.
Insulin Synthesis and Secretion
Mutations in INS were first described in a patient with mild diabetes and hyperinsulinemia, resembling type 2 diabetes117. In 2007, the first series of neonatal diabetes as a result of heterozygous INS mutations was reported119. The dominance of these heterozygous mutations is explained by the misfolding of preproinsulin, leading to intracellular accumulation and ER stress. In two diabetes mouse models harboring human mutations (C96S or C96Y in Ins2), the ultrastructure of the β‐cell is massively disrupted with dilatation of the ER, confirming increased ER stress and cell death120.
INS mutations, together with mutations in ABCC8, KCNJ11 and GATA6, are the most frequent cause of neonatal diabetes. The average age at diagnosis is 9 weeks, usually with ketoacidosis. However, some cases are diagnosed outside the neonatal period, between 6 months‐of‐age and 1 year. Over 80% of mutations are de novo. Interestingly, some family members carrying the same mutation have mild diabetes at the age of 30 years. Therefore, the phenotypic spectrum is quite broad. In 2010, recessive INS mutations were reported to have a slightly different phenotype: neonatal diabetes is diagnosed earlier, at 1 week‐of‐age, and growth retardation as a result of decreased insulin secretion in utero is more severe. Recessive mutations lead to decreased insulin biosynthesis through different mechanisms, such as a lack of translation initiation and decreased messenger ribonucleic acid stability. Recessive mutations might also cause transient neonatal diabetes, but these mutations are typically located in non‐coding regions, such as the insulin promoter122. Screening of over 1000 diabetic patients showed that INS mutations are rare after the neonatal period123. Later‐onset diabetes is mainly associated with mutations in the C‐peptide and signal peptide regions.
B lymphocyte kinase (BLK) expressed in pancreatic islets is an enhancer of insulin secretion, and the first mutation was described to cosegregate with diabetes in several families124. The human BLK mutant, Ala71Thr, leads to blunted insulin secretion in vitro, but BLK has not been confirmed in other cohorts with autosomally dominant diabetes125.
Channelopathies
Mutations in KCNJ11 and ABCC8, which encode the subunits of the ATP‐sensitive potassium (KATP) channel, lead to a similar phenotype as mutations in INS. Gain‐of‐function mutations that severely affect channel function result in permanent neonatal diabetes, and milder mutations result in transient neonatal diabetes126. All of the mutations impair KATP channel closure and, therefore, insulin secretion. As KCNJ11 is also expressed in the brain and skeletal muscle, diabetes might be associated with speech delay, epilepsy and muscular hypotonia. This syndrome is called DEND for developmental delay, epilepsy and neonatal diabetes, or intermediate DEND (iDEND) without epilepsy.
Exocrine Pancreas Defects Affecting Endocrine Function
The enzyme, carboxyl‐ester lipase (CEL), is involved in cholesterol ester hydrolysis in the duodenal lumen, and is expressed in the exocrine pancreas and lactating mammary glands, but not islet cells. A gene defect leads to pancreatic lipomatosis and exocrine pancreatic insufficiency in childhood, and progressive diabetes diagnosed at a mean age of 34 years128. Protein misfolding with intracellular and extracellular aggregation probably exerts a cytotoxic effect and lead to sustained disease progression involving the islets of Langerhans129.
Monogenic Autoimmune Diabetes
The first single gene defect associated with a systemic autoimmune disease, autoimmune polyendocrine syndrome type 1 (APS1) including diabetes, was found in the autoimmune regulator gene (AIRE)130. Mutations in AIRE lead to the highly variable APS1, affecting the pancreas, as well as the parathyroid, adrenal, thyroid, liver, ovary, stomach and skin. Dysfunctional fungal immunity gives rise to mucocutaneous candidiasis. The transcription factor, AIRE, is mainly expressed in lymphoid tissues, and is essential for generating central tolerance through negative selection of autoreactive T cells in the thymus. Mutant AIRE does not have the capability to maintain immunological tolerance, leading to the destruction of self, including β‐cells132.
Similarly, forkhead box P3 (FOXP3) defects lead to a systemic autoimmune disease, called immune dysregulation polyendocrinopathy enteropathy X‐linked (IPEX)74. This severe syndrome recognized in the neonatal period by diarrhea, diabetes, eczema, thyroid autoimmunity and an exaggerated response to viral infections often leads to death early in life. FOXP3 is critical in the development of regulatory T cells and the suppression of autoimmunity134. Female carriers have no established phenotype.
Sirtuin1 (SIRT1) is another gene responsible for a monogenic form of autoimmune diabetes associated with insulin resistance136. SIRT1 belongs to the family of histone deacetylases, regulating complex metabolic processes137. In β‐cells, SIRT1 likely regulates insulin secretion in response to glucose through downregulation of UCP2138. SIRT1 deacetylates p53, thereby inhibiting apoptosis; therefore, loss‐of‐function favors apoptosis, which occurs in autoimmune diabetes. Furthermore, SIRT1 has been proposed to act as an insulin sensitizer, which fits the human model in which SIRT1 mutation leads to insulin resistance associated with β‐cell destruction.
Implications for Treatment
Accurate diabetes diagnosis allows for improved treatment in at least five variants. Sulfonylurea drugs, such as glibenclamide, bind to the KATP channel and lead to channel closure thereby stimulating insulin secretion in β‐cells. These oral drugs overcome the impaired channel closure, the hallmark of gain‐of‐function mutations in the KCNJ11 and ABCC8 genes. This explains why a switch from insulin to sulfonylurea treatment improves metabolic control in most cases. As dysfunction of KIR6.2 protein, encoded by KCNJ11, is thought to be responsible for the neurological phenotype, several reports show an amelioration of neurological functions, at least in children141. In adults after decades of insulin treatment, a transfer to sulfonylurea can similarly restore endogenous insulin secretion141.
Patients carrying the HNF4A or HNF1A mutations are quite sensitive to sulfonylureas, these oral antidiabetics bypass the functional defect in β‐cells by acting downstream of the metabolic steps eliciting insulin secretion142.
Thiamine‐responsive diabetes as a result of decreased availability of cellular energy in the form of ATP responds to early substitution with thiamine, which enhances insulin secretion.
Finally, a new class of agents that activate GCK, enhancing glucose‐stimulated insulin release, is being developed for GCK‐deficient patients. These drugs could also be beneficial for type 2 diabetes143. However, the first clinical trials of the compound, GKA MK‐0941, were disappointing, reporting a loss of efficacy over time, and resulting in increased systolic blood pressure and serum triglycerides as a result of increased de novo lipogenesis144.
Conclusions
Over the past couple of years, discoveries about β‐cell genes in monogenic diabetes have led to a better understanding of the human β‐cell.
The availability of next‐generation sequencing will help unravel the full spectrum of genetic diabetes, ranging from truly monogenic to digenic, oligogenic and polygenic traits. This tool will certainly offer deeper comprehension of the different diabetes variations and lead to optimized treatment of the specific forms. The discovery of modifier genes will also be useful to better understand the different diabetes phenotypes. Functional analyses in vitro and in vivo will also help define specific gene‐related therapies. These results will prove to be relevant to the pathophysiology of β‐cell defects in type 2 diabetes. Broader knowledge of human diabetes will lead to improved treatment, outcomes, prevention and hopefully a cure.
Acknowledgments
The Swiss National Science Foundation, the Swiss Diabetes Foundation and the Federal Department of Foreign Affairs of Switzerland are acknowledged for supporting the author's research on monogenic diabetes. The author has no conflict of interest and nothing to disclose.
J Diabetes Invest 2014; 5: 121–133
References
- 1.Ledermann HM. Is maturity onset diabetes at young age (MODY) more common in Europe than previously assumed? Lancet 1995; 345: 648. [DOI] [PubMed] [Google Scholar]
- 2.Cammidge PJ. Diabetes mellitus and heredity. Br Med J 1928; 2: 738–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tattersall RB, Fajans SS. A difference between the inheritance of classical juvenile‐onset and maturity‐onset type diabetes of young people. Diabetes 1975; 24: 44–53 [DOI] [PubMed] [Google Scholar]
- 4.Vionnet N, Stoffel M, Takeda J, et al Nonsense mutation in the glucokinase gene causes early‐onset non‐insulin‐dependent diabetes mellitus. Nature 1992; 356: 721–722 [DOI] [PubMed] [Google Scholar]
- 5.Yamagata K, Oda N, Kaisaki PJ, et al Mutations in the hepatocyte nuclear factor‐1alpha gene in maturity‐onset diabetes of the young (MODY3). Nature 1996; 384: 455–458 [DOI] [PubMed] [Google Scholar]
- 6.Yamagata K, Furuta H, Oda N, et al Mutations in the hepatocyte nuclear factor‐4alpha gene in maturity‐onset diabetes of the young (MODY1). Nature 1996; 384: 458–460 [DOI] [PubMed] [Google Scholar]
- 7.Stoffers DA, Zinkin NT, Stanojevic V, et al Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet 1997; 15: 106–110 [DOI] [PubMed] [Google Scholar]
- 8.Schwitzgebel VM, Mamin A, Brun T, et al Agenesis of human pancreas due to decreased half‐life of insulin promoter factor 1. J Clin Endocrinol Metab 2003; 88: 4398–4406 [DOI] [PubMed] [Google Scholar]
- 9.Stoffers DA, Ferrer J, Clarke WL, et al Early‐onset type‐II diabetes mellitus (MODY4) linked to IPF1. Nat Genet 1997; 17: 138–139 [DOI] [PubMed] [Google Scholar]
- 10.Ahlgren U, Jonsson J, Jonsson L, et al beta‐cell‐specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta‐cell phenotype and maturity onset diabetes. Genes Dev 1998; 12: 1763–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sachdeva MM, Claiborn KC, Khoo C, et al Pdx1 (MODY4) regulates pancreatic beta cell susceptibility to ER stress. Proc Natl Acad Sci USA 2009; 106: 19090–19095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krapp A, Knöfler M, Ledermann B, et al The bHLH protein PTF1‐p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev 1998; 12: 3752–3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sellick GS, Barker KT, Stolte‐Dijkstra I, et al Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet 2004; 36: 1301–1305 [DOI] [PubMed] [Google Scholar]
- 14.Al‐Shammari M, Al‐Husain M, Al‐Kharfy T, et al A novel PTF1A mutation in a patient with severe pancreatic and cerebellar involvement. Clin Genet 2011; 80: 196–198 [DOI] [PubMed] [Google Scholar]
- 15.Weedon MN, Cebola I, Patch A‐M, et al Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet 2013; 46: 61–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen HL, Flanagan SE, Shaw‐Smith C, et al GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. Nature Publishing Group; 2011; 44: 20–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Franco E, Shaw‐Smith C, Flanagan SE, International NDM Consortium , Hattersley AT, et al GATA6 mutations cause a broad phenotypic spectrum of diabetes from pancreatic agenesis to adult‐onset diabetes without exocrine insufficiency. Diabetes 2013; 62: 993–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ketola I, Otonkoski T, Pulkkinen M‐A, et al Transcription factor GATA‐6 is expressed in the endocrine and GATA‐4 in the exocrine pancreas. Mol Cell Endocrinol 2004; 226: 51–57 [DOI] [PubMed] [Google Scholar]
- 19.Bonnefond A, Sand O, Guerin B, et al GATA6 inactivating mutations are associated with heart defects and, inconsistently, with pancreatic agenesis and diabetes. Diabetologia 2012; 55: 2845–2847 [DOI] [PubMed] [Google Scholar]
- 20.Catli G, Abaci A, Flanagan SE, et al A novel GATA6 mutation leading to congenital heart defects and permanent neonatal diabetes: a case report. Diabetes Metab [Internet]. Elsevier Masson SAS; 2013: 1–5 Available at: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=23639568&retmode=ref&cmd=prlinks [DOI] [PubMed] [Google Scholar]
- 21.Xuan S, Borok MJ, Decker KJ, et al Pancreas‐specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. J Clin Invest 2012; 122: 3516–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrasco M, Delgado I, Soria B, et al GATA4 and GATA6 control mouse pancreas organogenesis. J Clin Invest 2012; 122: 3504–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D'Amato E, Giacopelli F, Giannattasio A, et al Genetic investigation in an Italian child with an unusual association of atrial septal defect, attributable to a new familial GATA4 gene mutation, and neonatal diabetes due to pancreatic agenesis. Diabetic Med 2010; 27: 1195–1200 [DOI] [PubMed] [Google Scholar]
- 24.Senée V, Chelala C, Duchatelet S, et al Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet 2006; 38: 682–687 [DOI] [PubMed] [Google Scholar]
- 25.Kim Y‐S, Kang HS, Takeda Y, et al Glis3 regulates neurogenin 3 expression in pancreatic β‐cells and interacts with its activator, Hnf6. Mol Cells 2012; 34: 193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimitri P, Warner JT, Minton JAL, et al Novel GLIS3 mutations demonstrate an extended multisystem phenotype. Eur J Endocrinol 2011; 164: 437–443 [DOI] [PubMed] [Google Scholar]
- 27.Yang Y, Chang BH‐J, Chan L. Sustained expression of the transcription factor GLIS3 is required for normal beta cell function in adults. EMBO Mol Med 2012; 5: 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrett JC, Clayton DG, Concannon P, et al Genome‐wide association study and meta‐analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009; 41: 703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dupuis J, Langenberg C, Prokopenko I, et al New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010; 42: 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gradwohl G, Dierich A, LeMeur M, et al Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA 2000; 97: 1607–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pinney SE, Oliver‐Krasinski J, Ernst L, et al Neonatal diabetes and congenital malabsorptive diarrhea attributable to a novel mutation in the human neurogenin‐3 gene coding sequence. J Clin Endocrinol Metab 2011; 96: 1960–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rubio‐Cabezas O, Jensen JN, Hodgson MI, et al Permanent neonatal diabetes and enteric anendocrinosis associated with biallelic mutations in NEUROG3. Diabetes 2011; 60: 1349–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Cortina G, Wu SV, et al Mutant neurogenin‐3 in congenital malabsorptive diarrhea. N Engl J Med 2006; 355: 270–280 [DOI] [PubMed] [Google Scholar]
- 34.Jensen JN, Rosenberg LC, Hecksher‐Sørensen J, et al Mutant neurogenin‐3 in congenital malabsorptive diarrhea. N Engl J Med 2007; 356: 1781–1782 authorreply1782. [DOI] [PubMed] [Google Scholar]
- 35.del Bosque‐Plata L, Lin J, Horikawa Y, et al Mutations in the coding region of the neurogenin 3 gene (NEUROG3) are not a common cause of maturity‐onset diabetes of the young in Japanese subjects. Diabetes 2001; 50: 694–696 [DOI] [PubMed] [Google Scholar]
- 36.Milord E, Gragnoli C. NEUROG3 variants and type 2 diabetes in Italians. Minerva Med 2006; 97: 373–378 [PubMed] [Google Scholar]
- 37.Jackson AE, Cassell PG, North BV, et al Polymorphic variations in the neurogenic differentiation‐1, neurogenin‐3, and hepatocyte nuclear factor‐1alpha genes contribute to glucose intolerance in a South Indian population. Diabetes 2004; 53: 2122–2125 [DOI] [PubMed] [Google Scholar]
- 38.Mitchell J, Punthakee Z, Lo B, et al Neonatal diabetes, with hypoplastic pancreas, intestinal atresia and gall bladder hypoplasia: search for the aetiology of a new autosomal recessive syndrome. Diabetologia 2004; 47: 2160–2167 [DOI] [PubMed] [Google Scholar]
- 39.Smith SB, Qu H‐Q, Taleb N, et al Rfx6 directs islet formation and insulin production in mice and humans. Nature. Nature Publishing Group; 2010; 463: 775–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spiegel R, Dobbie A, Hartman C, et al Clinical characterization of a newly described neonatal diabetes syndrome caused by RFX6 mutations. Am J Med Genet 2011; 155A: 2821–2825 [DOI] [PubMed] [Google Scholar]
- 41.Soyer J, Flasse L, Raffelsberger W, et al Rfx6 is an Ngn3‐dependent winged helix transcription factor required for pancreatic islet cell development. Development 2010; 137: 203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naya FJ, Huang HP, Qiu Y, et al Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/NeuroD‐deficient mice. Genes Dev 1997; 11: 2323–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moates JM, Nanda S, Cissell MA, et al BETA2 activates transcription from the upstream glucokinase gene promoter in islet beta‐cells and gut endocrine cells. Diabetes 2003; 52: 403–408 [DOI] [PubMed] [Google Scholar]
- 44.Rubio‐Cabezas O, Minton JAL, Kantor I, et al Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes 2010; 59: 2326–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malecki MT, Jhala US, Antonellis A, et al Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet 1999; 23: 323–328 [DOI] [PubMed] [Google Scholar]
- 46.Liu L, Furuta H, Minami A, et al A novel mutation, Ser159Pro in the NeuroD1/BETA2 gene contributes to the development of diabetes in a Chinese potential MODY family. Mol Cell Biochem 2007; 303: 115–120 [DOI] [PubMed] [Google Scholar]
- 47.Gonsorcíková L, Průhová S, Cinek O, et al Autosomal inheritance of diabetes in two families characterized by obesity and a novel H241Q mutation in NEUROD1. Pediatr Diabetes 2008; 9(4 Pt 2): 367–372 [DOI] [PubMed] [Google Scholar]
- 48.Sander M, Neubuser A, Kalamaras J, et al Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev 1997; 11: 1662–1673 [DOI] [PubMed] [Google Scholar]
- 49.Liu T, Zhao Y, Tang N, et al Pax6 directly down‐regulates Pcsk1n expression thereby regulating PC1/3 dependent proinsulin processing. Kulkarni R, editor PLoS ONE 2012; 7: e46934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solomon BD, Pineda‐Alvarez DE, Balog JZ, et al Compound heterozygosity for mutations in PAX6in a patient with complex brain anomaly, neonatal diabetes mellitus, and microophthalmia. Am J Med Genet 2009; 149A: 2543–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yasuda T, Kajimoto Y, Fujitani Y, et al PAX6 mutation as a genetic factor common to aniridia and glucose intolerance. Diabetes 2001; 51: 224–230 [DOI] [PubMed] [Google Scholar]
- 52.Nishi M, Sasahara M, Shono T, et al A case of novel de novo paired box gene 6 (PAX6) mutation with early‐onset diabetes mellitus and aniridia. Diabet Med 2005; 22: 641–644 [DOI] [PubMed] [Google Scholar]
- 53.Sosa‐Pineda B, Chowdhury K, Torres M, et al The Pax4 gene is essential for differentiation of insulin‐producing beta cells in the mammalian pancreas. Nature 1997; 386: 399–402 [DOI] [PubMed] [Google Scholar]
- 54.Plengvidhya N, Kooptiwut S, Songtawee N, et al PAX4 mutations in Thais with maturity onset diabetes of the young. J Clin Endocrinol Metab 2007; 92: 2821–2826 [DOI] [PubMed] [Google Scholar]
- 55.Horikawa Y, Iwasaki N, Hara M, et al Mutation in hepatocyte nuclear factor‐1 beta gene (TCF2) associated with MODY. Nat Genet 1997; 17: 384–385 [DOI] [PubMed] [Google Scholar]
- 56.Lindner TH, Njolstad PR, Horikawa Y, et al A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo‐POU domain of hepatocyte nuclear factor‐1beta. Hum Mol Genet 1999; 8: 2001–2008 [DOI] [PubMed] [Google Scholar]
- 57.Yorifuji T, Kurokawa K, Mamada M, et al Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor‐1beta gene due to germline mosaicism. J Clin Endocrinol Metab 2004; 89: 2905–2908 [DOI] [PubMed] [Google Scholar]
- 58.Haumaitre C. Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1/MODY5 mutations. Hum Mol Genet 2006; 15: 2363–2375 [DOI] [PubMed] [Google Scholar]
- 59.Haldorsen IS, Vesterhus M, Raeder H, et al Lack of pancreatic body and tail in HNF1B mutation carriers. Diabet Med 2008; 25: 782–787 [DOI] [PubMed] [Google Scholar]
- 60.Cha JY, Kim H, Kim KS, et al Identification of transacting factors responsible for the tissue‐specific expression of human glucose transporter type 2 isoform gene. Cooperative role of hepatocyte nuclear factors 1alpha and 3beta. J Biol Chem 2000; 275: 18358–18365 [DOI] [PubMed] [Google Scholar]
- 61.Cereghini S. Liver‐enriched transcription factors and hepatocyte differentiation. FASEB J 1996; 10: 267–282 [PubMed] [Google Scholar]
- 62.Wang L, Coffinier C, Thomas MK, et al Selective deletion of the Hnf1beta (MODY5) gene in beta‐cells leads to altered gene expression and defective insulin release. Endocrinology 2004; 145: 3941–3949 [DOI] [PubMed] [Google Scholar]
- 63.Pontoglio M, Sreenan S, Roe M, et al Defective insulin secretion in hepatocyte nuclear factor 1alpha‐deficient mice. J Clin Invest 1998; 101: 2215–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bellanné‐Chantelot C, Clauin S, Chauveau D, et al Large genomic rearrangements in the hepatocyte nuclear factor‐1beta (TCF2) gene are the most frequent cause of maturity‐onset diabetes of the young type 5. Diabetes 2005; 54: 3126–3132 [DOI] [PubMed] [Google Scholar]
- 65.Raile K, Klopocki E, Holder M, et al Expanded clinical spectrum in hepatocyte nuclear factor 1B‐maturity‐onset diabetes of the young. J Clin Endocrinol Metab 2009; 94: 2658–2664 [DOI] [PubMed] [Google Scholar]
- 66.Bonnefond A, Vaillant E, Philippe J, et al Transcription factor gene MNX1 is a novel cause of permanent neonatal diabetes in a consanguineous family. Diabetes Metab [Internet]. Elsevier Masson SAS; 2013: 1–5 Available at: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=23562494&retmode=ref&cmd=prlinks [DOI] [PubMed] [Google Scholar]
- 67.Harrison KA, Thaler J, Pfaff SL, et al Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9‐deficient mice. Nat Genet 1999; 23: 71–75 [DOI] [PubMed] [Google Scholar]
- 68.Ross AJ, Ruiz‐Perez V, Wang Y, et al A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat Genet 1998; 20: 358–361 [DOI] [PubMed] [Google Scholar]
- 69.Nammo T, Yamagata K, Tanaka T, et al Expression of HNF‐4α (MODY1), HNF‐1β (MODY5), and HNF‐1α (MODY3) proteins in the developing mouse pancreas. Gene Expr Patterns 2008; 8: 96–106 [DOI] [PubMed] [Google Scholar]
- 70.Lee YH, Sauer B, Gonzalez FJ. Laron dwarfism and non‐insulin‐dependent diabetes mellitus in the Hnf‐1alpha knockout mouse. Mol Cell Biol 1998; 18: 3059–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Edghill EL, Bingham C, Ellard S, et al Mutations in hepatocyte nuclear factor‐1beta and their related phenotypes. J Med Genet 2005; 43: 84–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hagenfeldt‐Johansson KA, Herrera PL, Wang H, et al Beta‐cell‐targeted expression of a dominant‐negative hepatocyte nuclear factor‐1 alpha induces a maturity‐onset diabetes of the young (MODY)3‐like phenotype in transgenic mice. Endocrinology 2001; 142: 5311–5320 [DOI] [PubMed] [Google Scholar]
- 73.Njolstad PR, Sovik O, Cuesta‐Muñoz A, et al Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med 2001; 344: 1588–1592 [DOI] [PubMed] [Google Scholar]
- 74.Bennett CL, Christie J, Ramsdell F, et al The immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001; 27: 20–21 [DOI] [PubMed] [Google Scholar]
- 75.Steele AM, Shields BM, Shepherd M, et al Increased all‐cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabetic Med 2010; 27: 157–161 [DOI] [PubMed] [Google Scholar]
- 76.Owen KR, Thanabalasingham G, James TJ, et al Assessment of high‐sensitivity C‐reactive protein levels as diagnostic discriminator of maturity‐onset diabetes of the young due to HNF1A mutations. Diabetes Care 2010; 33: 1919–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pontoglio M, Prié D, Cheret C, et al HNF1alpha controls renal glucose reabsorption in mouse and man. EMBO Rep 2000; 1: 359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Odom DT. Control of pancreas and liver gene expression by HNF transcription factors. Science 2004; 303: 1378–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pearson ER, Boj SF, Steele AM, et al Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. Plos Med [Internet]. 2007; 4: e118 Available at: http://www.plosmedicine.org/article/fetchSingleRepresentation.action?uri=info:doi/10.1371/journal.pmed.0040118.sg001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Colclough K, Bellanné‐Chantelot C, Saint‐Martin C, et al Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity‐onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat 2013; 34: 669–685 [DOI] [PubMed] [Google Scholar]
- 81.McDonald TJ, McEneny J, Pearson ER, et al Lipoprotein composition in HNF1A‐MODY: differentiating between HNF1A‐MODY and Type 2 diabetes. Clin Chim Acta 2012; 413(9–10): 927–932 [DOI] [PubMed] [Google Scholar]
- 82.Shih DQ, Dansky HM, Fleisher M, et al Genotype/phenotype relationships in HNF‐4alpha/MODY1: haploinsufficiency is associated with reduced apolipoprotein (AII), apolipoprotein (CIII), lipoprotein(a), and triglyceride levels. Diabetes 2000; 49: 832–837 [DOI] [PubMed] [Google Scholar]
- 83.Stoffel M, Duncan SA. The maturity‐onset diabetes of the young (MODY1) transcription factor HNF4alpha regulates expression of genes required for glucose transport and metabolism. Proc Natl Acad Sci USA 1997; 94: 13209–13214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Neve B, Fernandez‐Zapico ME, Ashkenazi‐Katalan V, et al Role of transcription factor KLF11 and its diabetes‐associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci USA 2005; 102: 4807–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fernandez‐Zapico ME, van Velkinburgh JC, Gutiérrez‐Aguilar R, et al MODY7 gene, KLF11, is a novel p300‐dependent regulator of Pdx‐1 (MODY4) transcription in pancreatic islet beta cells. J Biol Chem 2009; 284: 36482–36490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bonnefond A, Lomberk G, Buttar N, et al Disruption of a novel Kruppel‐like transcription factor p300‐regulated pathway for insulin biosynthesis revealed by studies of the c.‐331 INS mutation found in neonatal diabetes mellitus. J Biol Chem 2011; 286: 28414–28424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fukumoto H, Seino S, Imura H, et al Sequence, tissue distribution, and chromosomal localization of mRNA encoding a human glucose transporter‐like protein. Proc Natl Acad Sci USA 1988; 85: 5434–5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Orci L, Thorens B, Ravazzola M, et al Localization of the pancreatic beta cell glucose transporter to specific plasma membrane domains. Science 1989; 245: 295–297 [DOI] [PubMed] [Google Scholar]
- 89.Santer R, Schneppenheim R, Dombrowski A, et al Mutations in GLUT2, the gene for the liver‐type glucose transporter, in patients with Fanconi‐Bickel syndrome. Nat Genet 1997; 17: 324–326 [DOI] [PubMed] [Google Scholar]
- 90.Yoo H‐W, Shin Y‐L, Seo E‐J, et al Identification of a novel mutation in the GLUT2 gene in a patient with Fanconi‐Bickel syndrome presenting with neonatal diabetes mellitus and galactosaemia. Eur J Pediatr 2002; 161: 351–353 [DOI] [PubMed] [Google Scholar]
- 91.Mueckler M, Kruse M, Strube M, et al A mutation in the Glut2 glucose transporter gene of a diabetic patient abolishes transport activity. J Biol Chem 1994; 269: 17765–17767 [PubMed] [Google Scholar]
- 92.Sakamoto O, Ogawa E, Ohura T, et al Mutation analysis of the GLUT2 gene in patients with Fanconi‐Bickel syndrome. Pediatr Res 2000; 48: 586–589 [DOI] [PubMed] [Google Scholar]
- 93.Dunn‐Meynell AA, Routh VH, Kang L, et al Glucokinase is the likely mediator of glucosensing in both glucose‐excited and glucose‐inhibited central neurons. Diabetes 2002; 51: 2056–2065 [DOI] [PubMed] [Google Scholar]
- 94.Cuesta‐Munoz AL, Tuomi T, Cobo‐Vuilleumier N, et al Clinical heterogeneity in monogenic diabetes caused by mutations in the glucokinase gene (GCK‐MODY). Diabetes Care 2010; 33: 290–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Osbak KK, Colclough K, Saint‐Martin C, et al Update on mutations in glucokinase (GCK), which cause maturity‐onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat 2009; 30: 1512–1526 [DOI] [PubMed] [Google Scholar]
- 96.Labay V, Raz T, Baron D, et al Mutations in SLC19A2 cause thiamine‐responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat Genet 1999; 22: 300–304 [DOI] [PubMed] [Google Scholar]
- 97.Fleming JC, Tartaglini E, Steinkamp MP, et al The gene mutated in thiamine‐responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat Genet 1999; 22: 305–308 [DOI] [PubMed] [Google Scholar]
- 98.Shaw‐Smith C, Flanagan SE, Patch A‐M, et al Recessive SLC19A2 mutations are a cause of neonatal diabetes mellitus in thiamine‐responsive megaloblastic anaemia. Pediatr Diabetes 2012; 13: 314–321 [DOI] [PubMed] [Google Scholar]
- 99.Depeint F, Bruce WR, Shangari N, et al Mitochondrial function and toxicity: role of the B vitamin family on mitochondrial energy metabolism. Chem Biol Interact 2006; 163: 94–112 [DOI] [PubMed] [Google Scholar]
- 100.Baldwin SA, Yao SYM, Hyde RJ, et al Functional characterization of novel human and mouse equilibrative nucleoside transporters (hENT3 and mENT3) located in intracellular membranes. J Biol Chem 2005; 280: 15880–15887 [DOI] [PubMed] [Google Scholar]
- 101.Edghill EL, Hameed S, Verge CF, et al Mutations in the SLC29A3 gene are not a common cause of isolated autoantibody negative type 1 diabetes. JOP. 2009; 10: 457–458 [PubMed] [Google Scholar]
- 102.Govindarajan R, Leung GPH, Zhou M, et al Facilitated mitochondrial import of antiviral and anticancer nucleoside drugs by human equilibrative nucleoside transporter‐3. Am J Physiol Gastrointest Liver Physiol 2009; 296: G910–G922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cliffe ST, Kramer JM, Hussain K, et al SLC29A3 gene is mutated in pigmented hypertrichosis with insulin‐dependent diabetes mellitus syndrome and interacts with the insulin signaling pathway. Hum Mol Genet 2009; 18: 2257–2265 [DOI] [PubMed] [Google Scholar]
- 104.Inoue H, Tanizawa Y, Wasson J, et al A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome) ‐ Nature Genetics. Nat Genet 1998; 20: 143–148 [DOI] [PubMed] [Google Scholar]
- 105.Ishihara H, Takeda S, Tamura A, et al Disruption of the WFS1 gene in mice causes progressive beta‐cell loss and impaired stimulus‐secretion coupling in insulin secretion. Hum Mol Genet 2004; 13: 1159–1170 [DOI] [PubMed] [Google Scholar]
- 106.Riggs AC, Bernal‐Mizrachi E, Ohsugi M, et al Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia 2005; 48: 2313–2321 [DOI] [PubMed] [Google Scholar]
- 107.Lemaire K, Schuit F. Integrating insulin secretion and ER stress in pancreatic β‐cells. Nat Cell Biol. Nature Publishing Group; 2012; 14: 979–981 [DOI] [PubMed] [Google Scholar]
- 108.Fonseca SG, Urano F, Weir GC, et al Wolfram syndrome 1 and adenylyl cyclase 8 interact at the plasma membrane to regulate insulin production and secretion. Nat Cell Biol. Nature Publishing Group; 2012; 14: 1105–1112 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 109.Rigoli L, Lombardo F, Di Bella C. Wolfram syndrome and WFS1 gene. Clin Genet 2010; 79: 103–117 [DOI] [PubMed] [Google Scholar]
- 110.Bonnycastle LL, Chines PS, Hara T, et al Autosomal dominant diabetes arising from a wolfram syndrome 1 mutation. Diabetes 2013; 62: 3943–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Amr S, Heisey C, Zhang M, et al A homozygous mutation in a novel zinc‐finger protein, ERIS, is responsible for Wolfram syndrome 2. Am J Hum Genet 2007; 81: 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Delepine M, Nicolino M, Barrett T, et al EIF2AK3, encoding translation initiation factor 2‐alpha kinase 3, is mutated in patients with Wolcott‐Rallison syndrome. Nat Genet 2000; 25: 406–409 [DOI] [PubMed] [Google Scholar]
- 113.Senée V, Vattem KM, Delépine M, et al Wolcott‐Rallison Syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes 2004; 53: 1876–1883 [DOI] [PubMed] [Google Scholar]
- 114.Abdel‐Salam GMH, Schaffer AE, Zaki MS, et al A homozygous IER3IP1mutation causes microcephaly with simplified gyral pattern, epilepsy, and permanent neonatal diabetes syndrome (MEDS). Am J Med Genet 2012; 158A: 2788–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gupta S, McGrath B, Cavener DR. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes 2010; 59: 1937–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang W, Feng D, Li Y, et al PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab 2006; 4: 491–497 [DOI] [PubMed] [Google Scholar]
- 117.Tager H, Given B, Baldwin D, et al A structurally abnormal insulin causing human diabetes. Nature 1979; 281: 122–125 [DOI] [PubMed] [Google Scholar]
- 118.Haneda M, Polonsky KS, Bergenstal RM, et al Familial hyperinsulinemia due to a structurally abnormal insulin. Definition of an emerging new clinical syndrome. N Engl J Med 1984; 310: 1288–1294 [DOI] [PubMed] [Google Scholar]
- 119.Støy J, Edghill EL, Flanagan SE, et al Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci USA 2007; 104: 15040–15044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang J, Takeuchi T, Tanaka S, et al A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta‐cell dysfunction in the Mody mouse. J Clin Invest 1998; 103: 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Herbach N, Rathkolb B, Kemter E, et al Dominant‐negative effects of a novel mutated Ins2 allele causes early‐onset diabetes and severe ‐cell loss in Munich Ins2C95S mutant mice. Diabetes 2007; 56: 1268–1276 [DOI] [PubMed] [Google Scholar]
- 122.Garin I, Edghill EL, Akerman I, et al Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci USA [Internet]. 2010; 107: 3105–3110 Available at: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=20133622&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Edghill EL, Flanagan SE, Patch AM, et al Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 2008; 57: 1034–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Borowiec M, Liew CW, Thompson R, et al Mutations at the BLK locus linked to maturity onset diabetes of the young and beta‐cell dysfunction. Proc Natl Acad Sci USA 2009; 106: 14460–14465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bonnefond A, Yengo L, Philippe J, et al Reassessment of the putative role of BLK‐p.A71T loss‐of‐function mutation in MODY and type 2 diabetes. Diabetologia 2012; 56: 492–496 [DOI] [PubMed] [Google Scholar]
- 126.Gloyn AL, Pearson ER, Antcliff JF, et al Activating mutations in the gene encoding the ATP‐sensitive potassium‐channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 2004; 350: 1838–1849 [DOI] [PubMed] [Google Scholar]
- 127.Babenko AP, Polak M, Cavé H, et al Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med 2006; 355: 456–466 [DOI] [PubMed] [Google Scholar]
- 128.Ræder H, Johansson S, Holm PI, et al Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet 2005; 38: 54–62 [DOI] [PubMed] [Google Scholar]
- 129.Johansson BB, Torsvik J, Bjørkhaug L, et al Diabetes and pancreatic exocrine dysfunction due to mutations in the carboxyl ester lipase gene‐maturity onset diabetes of the young (CEL‐MODY): a protein misfolding disease. J Biol Chem 2011; 286: 34593–34605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 2012; 13: 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Beier UH, Wang L, Bhatti TR, et al Sirtuin‐1 targeting promotes Foxp3+ T‐regulatory cell function and prolongs allograft survival. Mol Cell Biol 2011; 31: 1022–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cheng MH, Anderson MS. Monogenic autoimmunity. Annu Rev Immunol 2012; 30: 393–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Biason‐Lauber A, Lang‐Muritano M, Vaccaro T, et al Loss of kinase activity in a patient with Wolcott‐Rallison syndrome caused by a novel mutation in the EIF2AK3 gene. Diabetes 2002; 51: 2301–2305 [DOI] [PubMed] [Google Scholar]
- 134.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003; 4: 330–336 [DOI] [PubMed] [Google Scholar]
- 135.Khattri R, Cox T, Yasayko S‐A, et al An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol 2003; 4: 337–342 [DOI] [PubMed] [Google Scholar]
- 136.Biason‐Lauber A, Böni‐Schnetzler M, Hubbard BP, et al Identification of a SIRT1 mutation in a family with Type 1 diabetes. Cell Metab. Elsevier Inc; 2013; 17: 448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Houtkooper RH, Auwerx J. Exploring the therapeutic space around NAD+. J Cell Biol 2012; 199: 205–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Moynihan KA, Grimm AA, Plueger MM, et al Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose‐stimulated insulin secretion in mice. Cell Metab 2005; 2: 105–117 [DOI] [PubMed] [Google Scholar]
- 139.Pearson ER, Flechtner I, Njølstad PR, et al Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 2006; 355: 467–477 [DOI] [PubMed] [Google Scholar]
- 140.Mlynarski W, Tarasov AI, Gach A, et al Sulfonylurea improves CNS function in a case of intermediate DEND syndrome caused by a mutation in KCNJ11. Nat Clin Pract Neurol. 2007; 3: 640–645 [DOI] [PubMed] [Google Scholar]
- 141.Riveline J‐P, Rousseau E, Reznik Y, et al Clinical and metabolic features of adult‐onset diabetes caused by ABCC8 mutations. Diabetes Care 2012; 35: 248–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pearson ER, Starkey BJ, Powell RJ, et al Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 2003; 362: 1275–1281 [DOI] [PubMed] [Google Scholar]
- 143.Doliba NM, Fenner D, Zelent B, et al Repair of diverse diabetic defects of β‐cells in man and mouse by pharmacological glucokinase activation. Diabetes Obes Metab 2012; 14(Suppl 3): 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Meininger GE, Scott R, Alba M, et al Effects of MK‐0941, a novel glucokinase activator, on glycemic control in insulin‐treated patients with type 2 diabetes. Diabetes Care 2011; 34: 2560–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Amr S. Identification and Characterization of a Second Wolfram Syndrome Gene. Virginia Commonwealth University Richmond, Virginia, 2010 [Google Scholar]
- 146.Finnish‐German APECED Consortium . An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD‐type zinc‐finger domains. Nat Genet 1997; 17: 399–403 [DOI] [PubMed] [Google Scholar]