

Abstract
The emergence of extensively drug-resistant tuberculosis (XDR-TB) necessitates the need to identify new anti-tuberculosis drug targets as well as to better understand essential biosynthetic pathways. GlgE is a Mycobacterium tuberculosis (Mtb) encoded maltosyltransferase involved in α-glucan biosynthesis. Deletion of GlgE in Mtb results in the accumulation of M1P within cells leading to rapid death of the organism. To inhibit GlgE a maltose-C-phosphonate (MCP) 13 was designed to act as an isosteric non-hydrolysable mimic of M1P. MCP 13, the only known inhibitor of Mtb GlgE, was successfully synthesized using a Wittig olefination as a key step in transforming maltose to the desired product. MCP 13 inhibited Mtb GlgE with an IC50 = 230 ± 24 μM determined using a coupled enzyme assay which measures orthophosphate release. The requirement of M1P for the assay necessitated the development of an expedited synthetic route to M1P from an intermediate used in the MCP 13 synthesis. In conclusion, we designed a substrate analogue of M1P that is the first to exhibit Mtb GlgE inhibition.
Keywords: Mycobacterium tuberculosis, GlgE, C-phosphonates, Enzyme inhibition, Maltose-1-phosphate
1. Introduction
Tuberculosis (TB) is an infectious disease caused by various strains of mycobacteria, most commonly, Mycobacterium tuberculosis (Mtb)1 which is the leading cause of death in the world due to bacterial infection. TB attacks the lungs and also affects other parts of the body. It is easily diffused through the air when people who have an active TB infection cough, sneeze, or otherwise transmit their saliva through the air2. In 2011, 5.8 million newly diagnosed TB cases were reported to national TB control programs (NTPs) and to the World Health Organization (WHO). In addition, there were 990,000 deaths reported1. At one time TB was believed to be readily treatable and easily eradicated, but in recent years due to poverty and co-infection with HIV, extensively drug-resistant strains (XDR) of TB have emerged which make treatment increasingly difficult. Therefore, there is an urgent need to discover targets for treating this disease that are different from antibiotics currently in use.
Trehalose, the common name of α-D-glucopyranosyl-(1→1)-α-D-glucopyranoside, is a pivotal metabolite present in mycobacteria. For example, it is used as mycolic acid carrier molecule for biosynthesis of mycolylarabinogalactan and trehalose-6,6′-dimycolate (TDM)3. Following the mycolyltransfer reaction, trehalose is transported from the periplasmic space back into the cytoplasm and recycled for use in the production of various trehalolipids and sulfolipids4. Most important for this paper, trehalose can be incorporated into a branched α-glucan5. This pathway requires three enzymes trehalose synthase (TreS), maltokinase (Pep2), and GlgE a maltosyltransferase (Scheme 1)5. In this pathway, trehalose is first isomerized to maltose (4-O-α-D-glucopyranosyl-D-glucose) by TreS and then maltose is phosphorylated by Pep2 to produce maltose-1-phosphate (M1P)6. The maltosyltransfer catalyzed by GlgE using M1P generates linear α-glucans that function as substrates for GlgB, which introduces α-1,6 branches into the α-glucan.
Scheme 1.
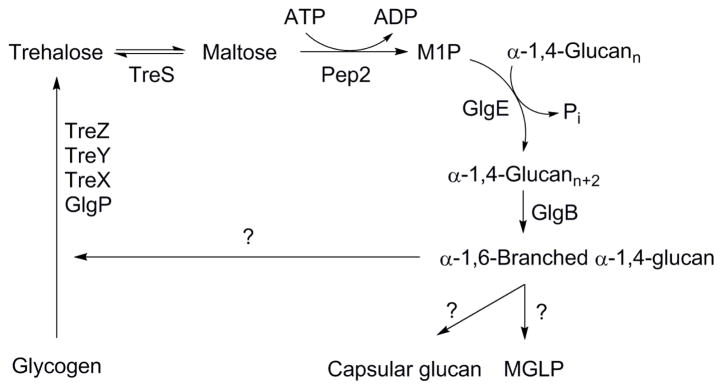
The GlgE pathway from trehalose to α-glucan
Kalscheuer and co-workers reported that inactivation of GlgE led to self-poisoning by the accumulation of phosphosugar M1P, facilitated by a self-amplifying feedback response6. GlgE has a unique combination of gene essentiality within a synthetic lethal pathway which substantiates GlgE as a new potential drug target candidate for the treatment of TB. Since GlgE is the first enzyme involved in α-glucan synthesis suggested to be a potential target for developing antimicrobials, to date, no inhibitors have been described
Syson and co-workers determined the structure of GlgE isoform I from Streptomyces coelicolor (Sco) which placed GlgE in the glycosylhydrolase family GH13_3, which use a double displacement enzymatic mechanism consistent with that of the related retaining glycosyl hydrolases7. The Syson study demonstrated that Sco GlgE and Mtb GlgE have similar catalytic properties and suggest that the Sco GlgE can be used as a surrogate for understanding the Mtb GlgE8. GlgE was predicted to bind M1P within the so-called sucrose phosphorylase +1 subsite and catalysis is proposed to use phosphate as a leaving group. GlgE exhibits an α-retaining catalytic mechanism9 forming linear α-glucans as shown in Scheme 28. Based on this information, the authors hypothesized that aspartic acid 394 and glutamic acid 423 represent key catalytic residues that function in the enzymatic reaction as the nucleophile and general acid, respectively (Sco numbering).
Scheme 2.
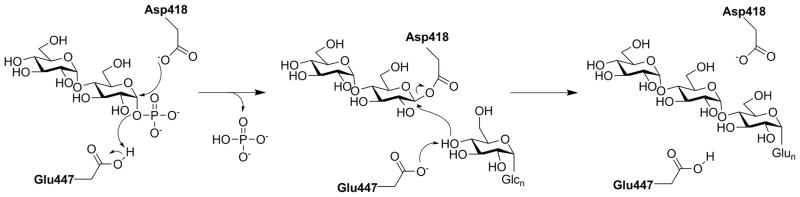
Proposed mechanism of GlgE. The numbering Asp418 and Glu447 are based on the numbering of Mycobacterium tuberculosis GlgE.
Based on the above mechanism, we expected a non-hydrolyzable and isosteric analog of M1P, maltose C-phosphonate (MCP) 13 would interact sufficiently with the substrate binding pocket to produce an inhibitory effect. Such a compound, since it would be non-hydrolyzable, is expected to have future value in structural biology studies that could aid inhibitor development. Synthesis of glycosyl C-phosphonates has been accomplished before for glucose and ribose monosaccharides. The most common routes for synthesis of such compounds comprise (i) a Horner-Wadsworth-Emmons approach that involves treating glycosyl donor 5 with tetramethyl methylene diphosphate10 or (ii) a Michaelis–Arbuzov reaction between an anomeric halomethylene moiety and a trialkyl phosphite. We investigated both synthetic strategies in order to elaborate maltose into the desired target MCP 13.
2. Results
2.1. Attempted synthesis of maltose-C-phosphonate 13 (MCP) using a Horner-Wadsworth-Emmons (HWE) approach
In order to synthesize the target molecule, maltose (1) was first peracetylated with acetic anhydride followed by glycosylation with allyl alcohol and boron trifluoride diethyl etherate to generate a peracetylated allyl maltoside11, Scheme 3.
Scheme 3.

Reagents and conditions: (i) Ac2O, pyridine, cat. DMAP, rt (95%); (ii) BF3-Et2O, allyl alcohol, CH2Cl2, rt; (iii) NaOMe, MeOH, 1 h, rt (90%); (iv) NaH, BnBr, DMF, rt (85%); (v) PdCl2, MeOH, rt (97%); (vi) CH2[P(O)(OMe)2]2, 50% NaOH, DCM, rt.
Subsequently, deacetylation under Zemplén conditions afforded glycoside 3 followed by benzylation to afford a mixture of perbenzylated allyl maltoside 412 in 85% yield. To deprotect the anomeric allyl group, we treated 4 with N-bromosuccinimide13. Instead of obtaining the reducing sugar 5 we obtained a bromo-substituted adduct (not shown) which was confirmed by ESI-MS. As an alternative, we treated 4 with palladium chloride in methanol to obtain 5 in 97% yield 14. The target C-phosphonate 13 would have been accessed by a Horner-Wadsworth-Emmons9 reaction mediated by tetramethyl methylenediphosphonate followed by global deprotection; however, the major product was the elimination product 7. The initial base used in the HWE was 50% NaOH (aq). As alternatives, we explored LiHMDS, LDA and sodium methoxide however these alternatives did not mitigate the problem. We also explored the Lewis acid catalyzed reaction15 of a maltose β-acetate and propargyltrimethylsilane to afford an α-C-allenyl maltoside (not shown). C-allenes can be converted to aldehydes by ozonolysis15 followed by reduction to give alcohols. The alcohols can be transformed to iodides16 and phosphorylated by using trimethyl phosphite to afford phosphonates; however, the reaction yields were modest.
2.2. Synthesis of maltose-C-phosphonate 13 using Wittig olefination followed by Michealis–Arbuzov reaction
Walker and Vederas have reported the generation of α-C-glycoside phosphonates17 via a Wittig olefination followed by an intra-molecular oxymercuration with introduction of the phosphonate moiety by a Michaelis–Arbuzov reaction in order to synthesize mono- and disaccharide analogs of moenomycin and lipid II for inhibition of the transglycosylase activity of penicillin-binding protein 1B. Thus, we saw an opportunity to preserve some of Scheme 3 by reacting reducing sugar 5 with the ylide formed from deprotonation of methyltriphenylphosphonium bromide with n-butyllithium to produce alkene18 8, Scheme 4. Cyclization of the alkene with mercury trifluoroacetate followed by anion exchange with KCl to afforded alkylmercuric chloride 9. Iodo-demercuration mediated by iodine afforded the alkyl iodide 10. The primary alkyl iodide was displaced with trimethyl phosphite using a Michaelis–Arbuzov reaction17 to afford the phosphonate 11. By global deprotection of the dimethyl phosphonates with trimethylsilyl bromide followed by hydrogenolysis using 20% Pd(OH)2 on carbon under 1 atm hydrogen gas yield the target molecule 13.
Scheme 4.

Reagents and conditions: (i) nBuLi, Ph3PCH3Br PPh3=CH2, dry THF, rt (51%); (ii) (a) Hg(CF3CO2)2; (b) KCl, dry THF, rt (97%); (iii) I2, CH2Cl2, rt (90%); (iv) P(OCH3)3, reflux, 24 h, rt (86%); (v) TMSBr, CH2Cl2, rt (75%); (vi) 20% Pd(OH)2 on carbon/H2, THF/EtOH, rt, quantitative.
2.3. Synthesis of maltose-1-phosphate disodium salt 16
In order to perform inhibition studies, Syson et al. synthesized the substrate molecule maltose-1-phosphate with acetyl protection in which they obtained the β-anomer as the major product and used HPLC to purify out the desired α-anomer. HPLC is a powerful yet tedious process. Thus, to access M1P more readily for future assay development we synthesized substrate 16 by treating compound 5 with tetrabenzyl pyrophosphate to provide phosphate ester 14 in 56% yield (Scheme 5) with the α-anomer as the principle product. Purification was performed easily using flash column chromatography on silica gel. Further, intermediate 14 could be globally deprotected in a single step with 20% Pd(OH)2 on carbon under H2 gas. The phosphonic acid was converted to the stable sodium salt 16 by treatment with triethylamine followed by cation exchange through Dowex® 50WX8 H+ that had been converted to the Na+ form8.
Scheme 5.
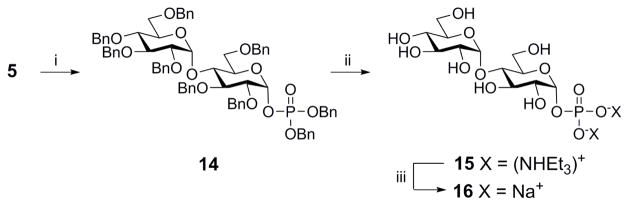
Reagents and conditions: (i) Tetrabenzyl pyrophosphate, LDA, −72–0 °C, (56%); (ii) (a) 20% Pd(OH)2 on carbon/H2, THF/EtOH, rt; (b) Et3N, rt; (iii) Dowex Na+ resin, quantitative.
2.4. Mtb GlgE inhibition studies
An EnzChek® Phosphate Assay Kit (Molecular Probes) was used to couple the phosphate produced from Mtb GlgE activity into a colorimetric readout. The phosphate assay can be performed continuously as a coupled assay or an endpoint assay and is based on work originally described by Webb19. Briefly, purine nucleoside phosphorylase (PNP) converts the substrate 2-amino-6-mercapto-7-methylpurine riboside (MESG) to ribose-1-phosphate and 2-amino-6-mercapto-7-methyl-purine (Figure 1)18. Conversion of MESG to 2-amino-6-mercapto-7-methyl-purine shifts absorbance from 330 nm to 360 nm, respectively. The assay is described in Figure 1.
Figure 1.

Mtb GlgE coupled enzyme assay utilizing PNP. (a) M1P and glycogen (n = a number of maltose units) serve as substrates for GlgE. (b) PNP consumes Pi and MESG to form 2-amino-6-mercapto-7-methyl-purine.
Using this assay we determined an IC50 for compound 13. Assays were performed under atmospheric pressure at 22 °C in a 96 well format on a Spectra max 340PC (Molecular Devices). All 1 mM MESG stock solutions were prepared and stored in dH2O at −20 °C. Phosphate release, catalyzed by Mtb GlgE, was monitored by the production of 2-amino-6-mercapto-7-methyl-purine in a coupled assay catalyzed by purine nucleoside phosphorylase. The reaction was monitored at 5 s intervals for 30 min. Each reaction consisted of 1 mM MESG, 0.2 U of PNP, 20X reaction buffer (1.0 M Tris-HCl, 20 mM MgCl2, pH 7.5, containing 2 mM sodium azide), 50 nm GlgE, 250 μM M1P (16) and varied concentration of inhibitor MCP 13. Inhibition was determined by comparing the relative rate of the reaction performed with inhibitor against a reaction that contained no inhibitor (Vo′/Vo), where Vo′ and Vo are steady-state rates with and without inhibitor, respectively. The inhibitor concentration that reduced enzyme activity by 50% (IC50) was found from the graph (Figure 2) to be 230 μM ± 24. This value is near the reported Mtb GlgE Km of 250 μM ± 58.
Figure 2.

IC50 determination of MCP 13. Vo′ and Vo are steady-state rates with and without inhibitor. Error bars are calculated from the result of reactions performed in triplicate.
3. Discussion
Mtb GlgE is considered a validated target for killing Mtb. We initiated efforts to develop a carbohydrate-based substrate analog of M1P, the substrate of the enzyme. We anticipated that maltose-C-phosphonate 13 would be isosteric and isoelectronic with M1P; however, it would also be non-hydrolyzable and would therefore inhibit GlgE. We were able to access the phosphonate inhibitor by subjecting the C1 halomethylene to a Michealis-Arbuzov reaction using a trialkylphosphite. Coupling constants for the C1* methylene protons maltose-C-phosphonate 13 were at δ 2.18 ppm (ddd, J1*a, 1*b = 15.87 Hz, J1*a, 1 = 11.72 Hz, J1*a, P = 27.34 Hz) for H1*a, 2.00 ppm (ddd, J1*a, 1*b = 15.87 Hz, J1*b, 1 = 5.08 Hz, J1*b, P = 19.78 Hz) for H1*b protons. In comparison to literature20 which is reported for a galactose C-phosphonate the α–anomer possessed resonances at 2.06 ppm (ABXX′, J1*a, 1*b = 15.6 Hz, J1*a, 1 = 10.7 Hz, J1*a, P = 26.5 Hz) for H1*a, and 1.93 ppm (ABXX′, J1*b, 1 = 4.0 Hz, J1*b, P = 19.6 Hz, J1*a, 1*b = 15.6 Hz) for H1*b. The β –anomer possessed resonances at 2.17 ppm (ABXX′, J1*a, 1 = 2.8 Hz, J1*a, P = 19.0 Hz, J1*a, 1*b = 15.4 Hz) for H1*a, and 1.82 ppm (ABXX′. J1*b, 1 = 9.4 Hz, J1*b, P = 21.9 Hz, J1*a, 1*b = 15.4 Hz) for H1*b. Comparing the coupling constant of J1*a, 1 for the three compounds strongly supports the assignment of the α–anomer. We attempted to evaluate the inhibitory activity of maltose-C-phosphonate against Mtb GlgE using a malachite green-base assay6 but encountered elevated background levels that hindered data analysis. Therefore, we moved on to a PNP-based coupled enzyme assay. The PNP coupled assay was simple to operate and exhibited a clear inhibitor concentration dependent decrease in enzymatic activity; furthermore, it has the potential to be performed in a continuous mode. We could synthesize M1P 16 easily from intermediate 5 without the need of HPLC purification. The α-anomer was the principle product and had having spectroscopic properties identical to those reported in literature8. We are currently in the process of addressing the detailed mode of interaction of MCP 13 with Mtb GlgE and Sco GlgE via co-crystallization studies. The data from those ongoing studies are expected to provide valuable insight concerning the development of more potent Mtb GlgE inhibitors.
4. Conclusion
We have synthesized a substrate analog of maltose-1-phosphate the substrate for Mtb GlgE. Key steps in the synthesis of the target C-phosphonate were the use of a Wittig olefination, an intramolecular oxymercuration, and a Michealis-Arbuzov reaction between alkylhalide and a trialkylphosphite. MCP 13 inhibited Mtb GlgE at IC50 = 230 ± 24 μM. We were also able to synthesize M1P as the desired α-anomer as the exclusive product. The purification of the protected M1P was performed easily. Rapid access to M1P is critical to our efforts on development high throughput screening capabilities for identifying inhibitors of Mtb GlgE. The ongoing crystallization studies of MCP 13 Mtb and Sco GlgE are expected to provide information that with aid us in the development of new anti-tuberculosis drugs.
5. Experimentals
5.1. Materials
All fine chemicals such as D-(+)-maltose monohydrate, benzyl bromide, methyltriphenylphosphonium bromide, 20% palladium hydroxide on carbon powder, palladium chloride, trimethyl silylbromide and anhydrous solvents such as anhydrous methanol and N,N-dimethylformamide were purchased from Acros and used without further purification. Trimethylphosphite and n-butyl lithium were purchased from Sigma-Aldrich. Tetramethyl methylenediphosphate, tetrabenzyl pyrophosphate and mercury (II) trifluoroacetate were purchased from Alfa Aesar. Potassium chloride and all other the solvents were obtained from Fisher. Silica (230–400 mesh) for column chromatography was obtained from Sorbent Technologies; Thin-layer chromatography (TLC) precoated plates were from EMD. TLCs (silica gel 60, f254) were visualized under UV light or by charring (5% H2SO4 – MeOH). Flash column chromatography was performed on silica gel (230–400 mesh) using solvents as received. 1H NMR were recorded either on INOVA 600 MHz spectrometer in CDCl3 using residual CHCl3 and TMS as internal references, respectively. 13C NMR were recorded on the VXRS 400, INOVA 600 and AVANCE 600 in CDCl3 using the triplet centered at δ 77.27 as internal reference. High resolution mass spectrometry (HRMS) was performed on a micro mass Q-TOF2 instrument. EnzChek® Phosphate Assay Kit was purchased from Molecular Probes for use in the Mtb GlgE inhibition assay.
5.2. Synthesis
5.2.1. 1,2,3,6,2′,3′,4′,6′-octa-O-acetyl-D-maltose (2)21
D(+)-Maltose monohydrate (1.00 g, 2.78 mmol) was suspended in dry pyridine (3.60 mL, 44.5 mmol) and acetic anhydride (3.14 mL, 33.4 mmol) and a catalytic amount of 4-dimethylaminopyridine was added. The solution was stirred at ambient temperature for 16 h. The reaction mixture was diluted with ethyl acetate and washed successively with 1N HCl (10 mL) and saturated. aq. NaHCO3 (20 mL). The resulting organic phase was dried (anhydrous Na2SO4), filtered and the filtrate concentrated under reduced pressure; yield: 95% (1.79 g); silica gel TLC Rf = 0.5 (1:1 acetone-hexanes).
5.2.2. Allyl 4-O-α-D-glucopyranosyl-α/β-D-glucopyranose11 (3)
Peracetylated maltose 2 (500 mg, 0.74 mmol) was dissolved in dichloromethane. To the solution was added by allyl alcohol (0.16 mL, 2.22 mmol) and boron trifluoride diethyl etherate (0.28 mL, 2.22 mmol) at 0 °C. The resulting solution was stirred at ambient temperature under nitrogen atmosphere. The reaction was monitored by TLC and appeared to be complete after 4 h. The reaction mixture was diluted with dichloromethane (7 mL) and successively washed with saturated aq. NaHCO3 (8 mL), 10% aq. NaCl (4 mL), and water (8 mL). The resulting organic phase was dried (anhydrous Na2SO4), filtered and the filtrate was concentrated under reduced pressure. The product was dried overnight and deacetylated by dissolving in dry methanol followed by addition of a catalytic amount of sodium metal until the solution reached pH 9. The reaction was monitored for completion using TLC. The reaction was neutralized by adding Amberlite IRA-118H H+ resin until the pH reached 7. Then resin was filtered away and the filtrate concentrated under reduced pressure to afford the allyl maltoside 3; yield: 90 % (0.45 g).
5.2.3. Allyl 2,3,6-Tri-O-benzyl-4-O-(2′,3′,4′,6′-tetra-O-benzyl-α-D-glucopyranosyl)-α/β-D-glucopyranoside (4)12
A solution of allyl 4-O-α-D-glucopyranosyl-α/β-D-glucopyranose (3) (840 mg, 2.19 mmol) in dry N,N-dimethylformamide (40 mL) was cooled to 0 °C. The solution was treated drop-wise with a suspension of sodium hydride (60% dispersion in mineral oil) (1.073 g, 26.82 mmol) in dry N,N-dimethylformamide. Benzyl bromide (2.73 mL, 23.0 mmol) was added drop-wise over 15 min and the solution stirred at room temperature for 16 h. The reaction was poured over ice and extracted with diethyl ether (20 mL). The combined organic layers were washed with brine. The resulting organic phase was dried (anhydrous Na2SO4), filtered and the filtrate concentrated under reduced pressure to obtain a product. The product was purified by silica gel flash column chromatography by eluting with 9:1 hexanes-ethyl acetate. The product fractions were combined, concentrated, and dried in vacuum to afford a yellow oily product; yield: 85% (1.87g); silica gel TLC Rf = 0.61 (8:2 hexanes-ethyl acetate).
5.2.4. 2,3,6-Tri-O-benzyl-4-O-(2′,3′,4′,6′-tetra-O-benzyl-α-D-glucopyranosyl)-α/β-D-glucopyranoside (5)12
Allyl 2,3,6-tri-O-benzyl-4-O-(2′,3′,4′,6′-tetra-O-benzyl-α-D-glucopyranosyl)-α/β-D-glucopyranoside (4) (0.4 g, 0.4 mmol) and palladium (II) chloride (0.2 mg, 0.6 mmol) in dry methanol (16 mL) were stirred vigorously for overnight at ambient temperature until consumption of starting material was observed by TLC. The reaction was diluted with diethyl ether, filtered through Celite™ 545 filter aid and concentrated under reduced pressure to obtain the crude product. The crude product was purified by silica gel flash column chromatography using 9:1 hexanes-ethyl acetate. The product fractions were combined, concentrated, and dried under reduced pressure to afford a colorless glassy solid; yield: 97% (0.384 g); silica gel TLC Rf = 0.19 (3:7 ethyl acetate-hexanes).
5.2.5. 3,4,7-Tri-O-benyl-5-O-(2′,3′,4′,6′-tetra-O-benzyl-α-D-glucopyranosyl)-D-glucphept-1-enitol (8)
2.22 M n-Butyl lithium in hexanes (1.5 mL, 3.3 mmol) was added drop-wise to a suspension of methyltriphenylphosphonium bromide (0.95 g, 2.67 mmol) in tetrahydrofuran (14 mL) at −20 °C. The solution was stirred at −20 °C for 15 min and raised to ambient temperature for 1 h. The solution was cooled to −20 °C and 2,3,6-tri-O-benzyl-4-O-(2′,3′,4′,6′-tetra-O-benzyl-α-D-glucopyranosyl)-α/β-D-glucopyranoside (5) (0.65 g, 0.67 mmol) in 13 mL of tetrahydrofuran was added drop-wise. The solution was stirred at −20 °C for 15 min and allowed to warm to ambient temperature and stirred for 6 h. The solution was diluted with acetone (6 mL) and stirred for 30 min. Diethyl ether (22 mL) was added to precipitate of triphenylphosphine oxide. The latter was removed by filtration through Celite™ 545 filter aid. The filtrate was washed successively with saturated aq. NaHCO3 and brine. The solution was dried (anhydrous Na2SO4), filtered and filtrate concentrated under reduced pressure to obtain the crude product. The product was purified by silica gel flash column chromatography by eluting with 9:1 hexanes-ethyl acetate. The product fractions were combined, concentrated, and dried in vacuum to afford a yellow oil; yield: 51.3% (0.33 g); silica gel TLC Rf = 0.63 (3:7 ethyl acetate-hexanes); [α]D23 = 58.4° (c = 0.5, CHCl3); 1H NMR (CDCl3, 600 MHz): δ 3.52 (m, 3H, H-6b, H-6b′, H-2′), 3.64(m, 2H, H-6a, H-4′), 3.76 (dd, 1H, J = 4.2, 5.4 Hz, H-3), 3.92(t, 1H, J = 4.8 Hz, H-4), 3.95 (t, 1H, J = 9 Hz, H-3′), 4.02 (m, 1H, H-5′), 4.12 (m, 1H, H-5), 4.28 (dd, 1H, J = 6, 12 Hz, H-2), 4.92-4.38 (m, 14H, -CH2Ph), 4.96 (d, 1H, J = 3.6 Hz, H-1), 5.22 (dd, 2H, J = 17.4, 29.4 Hz, =CH2), 5.91 (m, 1H, H-1′), 7.32-7.09 (m, 35H, Aromatic H’s); 13C NMR (150 MHz, CDCl3): δ 68.54, 70.77, 71.18, 71.55, 71.60, 73.39, 73.51, 73.66, 74.31, 75.21, 75.70, 78.03, 79.32, 80.60, 81.63, 82.13, 99.12, 118.86, 127.69, 127.73 (2 C’s), 127.76 (2 C’s), 127.82 (2 C’s), 127.83 (2 C’s), 127.85 (2 C’s), 127.86 (2 C’s), 127.87 (2 C’s), 128.04 (2 C’s), 128.05 (2 C’s), 128.08 (2 C’s), 128.19 (2 C’s), 128.36 (2 C’s), 128.51 (2 C’s), 128.52 (2 C’s), 128.53 (2 C’s), 128.57 (2 C’s), 135.74 (2 C’s), 138.12 (2 C’s), 138.35 (2 C’s), 138.41 (2 C’s), 138.48, 138.52, 138.93 ppm; mass spectrum (HRMS), m/z = 993.4581 (M+Na)+, C61H64O11 requires 993.4554.
5.2.6. 2,3,6,2′,3′,4′,6′-Hepta-O-benzyl-α-D-maltosyl methyl mercuric chloride (9)
A solution of 3,4,7-tri-O-benyl-5-O-(2′,3′,4′,6′-tetra-O-benzyl-α-D-glucopyranosyl)-D-glucphept-1-enitol (8) (0.3 g, 0.3 mmol) and mercuric trifluoroacetate (0.1 g, 0.3 mmol) in dry tetrahydrofuran (21 mL) was stirred at room temperature for 3 h. A solution of potassium chloride (0.179 g) in 3 mL of water was added and the mixture was stirred for another 3 h. The tetrahydrofuran was removed under reduced pressure and the remaining aqueous solution was extracted with dichloromethane. The combined extracts were washed with brine, dried (anhydrous MgSO4), filtered and the filtrate was concentrated under reduced pressure to obtain the crude material. The crude material was purified by silica gel flash column chromatography eluting with 9:1 hexanes-ethyl acetate. The product fractions were combined, concentrated, and dried in vacuum to afford yellow oily liquid; yield: 97% (0.35 g); silica gel TLC Rf = 0.45 (3:7 ethyl acetate-hexanes); [α]D23 = 27.6° (c = 0.5, CHCl3); 1H NMR (CDCl3, 600 MHz): δ 1.83 (dd, 1H, J = 6, 12Hz, H1*a), 2.06 (dd, 1H, J = 9, 11.4 Hz, H1*b), 3.46 (dd, 1H, J = 1.2, 10.2 Hz, H-4′), 3.51 (dd, 1H, J = 3.6, 9.6 Hz, H-2′), 3.61 (dd, 1H, J = 3, 8.4 Hz, H-2), 3.64 (m, 2H, H-6b, H-6b′), 3.71 (dd, 1H, J = 5.4, 10.8 Hz, H-3), 3.76 (m, 1H, H-5′), 3.98 (m, 1H, H-6a), 3.94 (m, 2H, H-6a, H-6a′), 3.94 (m, 1H, H-5), 4.18 (m, 1H, H-1), 4.85-4.37 (m, 14H, -CH2Ph), 5.40 (d, J = 3.6 Hz, H-1′), 7.32–7.39 (m, 35H, Aromatic H’s); 13C NMR (150 MHz, CDCl3): δ 68.35, 69.33, 71.31, 71.47, 71.92, 73.38, 73.65, 73.71, 73.78, 73.97, 75.15, 75.73, 76.91, 77.94 (2 C’s), 79.63, 80.08, 82.12, 97.62, 127.03 (2 C’s), 127.66 (2 C’s), 127.71 (2 C’s), 127.73 (2 C’s), 127.79 (2 C’s), 127.84 (2 C’s), 127.88 (2 C’s), 127.91 (2 C’s), 127.99 (2 C’s), 128.02 (2 C’s), 128.23 (2 C’s), 128.47 (2 C’s), 128.51 (2 C’s), 128.54 (2 C’s), 128.59 (2 C’s), 128.86 (2 C’s), 128.92 (2 C’s), 137.26, 138.03, 138.15, 138.19, 138.42, 138.57, 138.85 ppm; mass spectrum (HRMS), m/z = 1229.3959 (M+H)+, C62H65ClHgO10 requires 1229.3870.
5.2.7. 2,3,6,2′,3′,4′,6′-Hepta-O-benzyl-α-D-maltosyl Methyl iodide (10)
A solution of 2,3,6,2′,3′,4′,6′-hepta-O-benzyl-α-D-maltosyl methyl mercuric chloride (9) (0.32 g, 0.26 mmol) in dry dichloromethane (20 mL) was treated with iodine (0.21 g, 0.81 mmol). After stirring for 1 h the reaction was quenched by addition of sodium thiosulfate (500 mg in 6 mL water) and the mixture was stirred until the solution become colorless. The organic phase was separated and washed with water (5 mL), brine (5 mL), dried (anhydrous MgSO4) and filtered. The filtrate was evaporated under reduced pressure and purified by silica gel flash column chromatography eluting with 9:1 hexanes-ethyl acetate. The product fractions were combined, concentrated, and dried in vacuum to afford yellow oily liquid; yield: 89.5% (0.255 g); silica gel TLC Rf = 0.7 (3:7 ethyl acetate-hexanes); [α]D23 = 37.1° (c = 0.5, CHCl3); 1H NMR (CDCl3, 600 MHz): δ 3.38 (m, 2H, H-6a, H1*a), 3.52 (m, 4H, H-6b, H-6b′, H-2′, H1*b), 3.65 (t, 1H, J = 9.6 Hz, H-4), 3.71 (dd, 1H, J = 2.4, 10.8 Hz, H-3), 3.76 (m, 1H, H-5′), 3.81 (dd, 1H, J = 4.2, 10.8 Hz, H-2), 3.84 (m, 1H, H-5), 3.89 (dd, 1H, J = 3, 9.6 Hz, H-3′), 4.01 (dd, 1H, J = 6, 9 Hz, H-4′), 4.16 (m, 1H, H-1), 4.84-4.28 (m, 14H, -CH2Ph), 5.41 (d, J = 3.6 Hz, 1H), 7.32-7.09 (m, 35H, Aromatic H’s); 13C NMR (150 MHz, CDCl3): δ 68.22, 69.69, 71.18, 71.90, 72.97, 73.40, 73.44, 73.50, 73.62, 73.71, 73.73, 75.14, 75.70, 77.81, 77.86, 79.63, 79.91, 82.14, 97.25, 127.14 (2 C’s), 127.55 (2 C’s), 127.67 (2 C’s), 127.73 (2 C’s), 127.80 (2 C’s), 127.87 (2 C’s), 127.91 (2 C’s), 127.96 (2 C’s), 128.00 (2 C’s), 128.07 (2 C’s), 128.09 (2 C’s), 128.10 (2 C’s), 128.23 (2 C’s), 128.42 (2 C’s), 128.48 (2 C’s), 128.52 (2 C’s), 128.56 (2 C’s), 128.59 (2 C’s), 128.63 (2 C’s), 137.86, 138.04, 138.14, 138.47, 138.53, 138.55, 138.88 ppm; mass spectrum (HRMS), m/z = 1119.3505 (M+H)+, C62H65IO10 requires 1119.3520.
5.2.8. Dimethyl (2,3,6,2′,3′,4′,6′-Hepta-O-benzyl-α-D-maltosyl)methylphosphonate (11)
Trimethyl phosphite (7 mL) was added to 2,3,6,2′,3′,4′,6′-hepta-O-benzyl-α-D-maltosyl methyl iodide (0.24 g, 0.22 mmol) and the solution was refluxed for 24 h. The resulting solution was concentrated at 35 °C under reduced pressure to get the crude material. The crude material was purified by silica gel flash column chromatography eluting with 1:1 hexanes-ethyl acetate. The product fractions were combined, concentrated, and dried in vacuum to afford yellow oily liquid; yield: 86% (0.20 g); silica gel TLC Rf = 0.2 (1:1 ethyl acetate-hexanes); [α]D23 = 30.8° (c = 1, CHCl3); 1H NMR (CDCl3, 600 MHz): δ 2.18 (m, 1H, H1*a), 2.28 (m, 1H, H1*b), 3.42 (dd, 1H, J = 1.2, 10.2 Hz, H-4), 3.51 (dd, 1H, J = 3.6, 10.2, H-2′), 3.55 (dd, 1H, J = 3, 10.8 Hz, H-3), 3.65 (dd, 1H, J = 3, 8.4 Hz, H-4′), 3.69 (t, J = 10.8 Hz, -OCH3), 3.79 (m, 3H, H-5, H-5′, H-2), 3.91 (t, 1H, J = 9.6Hz, H-3′), 4.86-4.28 (m, 14H, -CH2Ph), 5.54 (d, 1H, J = 3.6 Hz, H-1′), 7.32-7.09 (m, 35H, Aromatic H’s); 13C NMR (150 MHz, CDCl3): δ 52.61, 52.66, 68.27, 68.93, 68.96, 69.35, 71.16, 72.14, 72.86, 73.08, 73.42, 73.59, 73.66, 73.72, 75.15, 75.72, 77.02, 77.23, 77.44, 77.84, 78.71, 79.64, 80.67, 82.18, 97.27, 127.03(2 C’s), 127.53 (2 C’s), 127.61 (2 C’s), 127.67 (2 C’s), 127.72 (2 C’s), 127.80 (2 C’s), 127.85 (2 C’s), 127.88 (2 C’s), 127.99 (2 C’s), 128.02 (2 C’s), 128.13 (2 C’s), 128.26 (2 C’s), 128.42 (2 C’s), 128.49 (2 C’s), 128.53 (2 C’s), 128.61 (2 C’s), 137.87, 138.08, 138.17, 138.46, 138.62, 138.66, 138.93 ppm; 31P NMR (162 MHz, CDCl3): δ 33.19 ppm; mass spectrum (HRMS), m/z = 1101.4530 (M+H)+, C64H71O13P requires 1101.4507.
5.2.9. (2,3,6,2′,3′,4′,6′-Hepta-O-benzyl-α-D-maltosyl)methylphosphonic acid (12)
Bromotrimethylsilane (0.4 mL, 2.8 mmol) was added to a solution of dimethyl 2,3,6,2′,3′,4′,6′-hepta-O-benzyl-α-D-maltosyl phosphonate (11) in dichloromethane (3 mL) and stirred at ambient temperature for 1.5 h. A solution of methanol-water (1:1, 1.8 mL) was added to the reaction and the resulting solution was concentrated under reduced pressure to dryness to afford phosphonic acid 12; yield: 75% (0.147 g); silica gel TLC Rf = 0.16 (1:3 ethyl acetate-hexanes); [α]D23 = 26.8° (c = 1, CHCl3); 1H NMR (CDCl3, 600 MHz): δ 2.12 (m, 1H, H1*a), 2.44 (m, H1*b), 3.31 (d, 1H, J = 10.2 Hz, H-4), 3.51 (m, 4H, H-4′, H-6b, H-6b′, H-2′), 3.71 (m, 1H, H-5′), 3.77 (t, 1H, J = 8.4 Hz, H-2), 3.82 (t, J = 7.2 Hz, H-3′), 3.92 (t, J = 7.8 Hz, H-3), 4.78-4.32 (m, 15H, -CH2Ph, H-1), 5.36 (d, 1H, J = 3Hz, H-1′), 7.32-7.09 (m, 35H, Aromatic H’s); 13C NMR (150 MHz, CDCl3): δ 68.22, 71.02, 71.54, 72.57, 73.46, 73.62, 73.69, 73.74, 75.19, 75.71, 77.75, 79.37, 81.98, 97.68, 125.51 (2 C’s), 126.96 (2 C’s), 127.63 (2 C’s), 127.77 (2 C’s), 127.91 (2 C’s), 127.98 (2 C’s), 128.04 (2 C’s), 128.12 (2 C’s), 128.20 (2 C’s), 128.24 (2 C’s), 128.33 (2 C’s), 128.44 (2 C’s), 128.51 (2 C’s), 128.57 (2 C’s), 128.61 (2 C’s), 128.75 (2 C’s), 129.25(2 C’s), 137.62, 137.97, 138.46, 138.78; 31P NMR (162 MHz, CDCl3): δ 30.89 ppm; mass spectrum (HRMS), m/z = 1073.4192 (M+H)+, C62H67O13P requires 1073.4217.
5.2.10. α-D-Maltose C-Phosphonate (13)
Palladium hydroxide (10%) on carbon (catalytic amount) was added to a solution of 2,3,6,2′,3′,4′,6′-Hepta-O-benzyl-α-D-maltosyl)methylphosphonic acid (12) (90.0 mg, 0.08 mmol) in tetrahydrofuran (0.2 mL) and ethanol (1.8 mL), The reaction mixture was stirred at ambient temperature under hydrogen atmosphere overnight. The catalyst was filtered away and washed with 20% methanol/dichloromethane and methanol. The filtrate was concentrated to dryness to afford maltose-C-phosphonate (13); yield: 100% (35 g);[α]D23 = 81.5° (c = 1, H2O); 1H NMR (CDCl3, 600 MHz): δ 2.00 (ddd, 1H, J1*a, 1*b = 15.87 Hz, J1*b, 1 = 5.08 Hz, J1*b, P = 19.78 Hz, H1*b), 2.18 (ddd, 1H, J1*a, 1*b = 15.87 Hz, J1*a, 1 = 11.72 Hz, J1*a, P = 27.34 Hz, H1*a), 3.35 (t, 1H, J = 9.6 Hz, H-4′), 3.52 (dd, 1H, J = 7.2 Hz, 10.8 Hz, H-2′), 3.80 (m, 5H, H-6a, b, H-5, H-2, H-3′, H-4), 4.32 (m, 1H, H-1), 5.15 (d, 1H, J = 6 Hz); 13C NMR (150 MHz, CDCl3): δ 24.62, 60.23, 60.32, 69.22, 70.39, 70.48, 70.85, 71.58, 72.02, 72.47, 72.75, 76.54, 99.34. 31P NMR (162 MHz, CDCl3): δ 26.34 ppm; mass spectrum (HRMS), m/z = 443.0930 (M+H)+, C13H25O13P requires 443.0942.
5.2.11. Dimethyl 3,7-di-O-benyl-5-O-(2′,3′,4′,6′-tetra-O-benzyl-α-D-glucopyranosyl)-D-glucphept-1,3-dienitol phosphonate (7)
A 50% aq. solution NaOH (2 mL) was slowly added to a vigorously stirring mixture of 2,3,6-tri-O-benzyl-4-O-(2′,3′,4′,6′-tetra-O-benzyl-α-D-glucopyranosyl)-α/β-D-glucopyranoside (5) (40.0 mg, 0.04 mmol) and tetramethyl methylenediphosphonate (10.4 mg, 0.04 mmol) in dichloromethane (2 mL). After 18 h at ambient temperature the reaction was complete by TLC. The reaction was diluted with dichloromethane and the aqueous phase was extracted with dichloromethane. The combined organic phases were dried (anhydrous MgSO4), filtered and the filtrate concentrated under reduced pressure to get the crude material. The crude material was purified by silica gel flash column chromatography eluting with 9:1 hexanes-ethyl acetate. The product fractions were combined, concentrated, and dried in vacuum to afford yellow oily liquid; yield: 45% (20 mg); silica gel TLC Rf = 0.7 (1:9 ethyl acetate-hexanes); 1H NMR (CDCl3, 600 MHz): δ 3.63-3.52 (m, 4H, H′-5, H′-4, H′-6a, b), 3.72 (m, 6H, -OCH3), 3.88 (t, 1H, J = 9.6 Hz, H′-3), 4.231 (d, 1H, J = 12 Hz, H-3), 4.46 (dd, 1H, J = 2.4 Hz, 9.6 Hz, H-4), 4.69–4.74 (m, 4H, H-2, H-5, H-6a, b), 4.94-4.76 (m, 12H, -CH2Ph), 5.948 (dd, 1H, J = 3.6 Hz, 13.8 Hz, H-1), 6.949 (dd, 1H, J = 4.8 Hz, 16.8 Hz, H-2), 7.34-6.98 (m, 30H, aromatic H’s); 13C NMR (150 MHz, CDCl3): δ 20.93, 21.92, 55.65, 68.98, 71.45, 75.96, 78.85, 80.77, 81.16, 81.12, 107.04, 128.19, 128.25, 128.32, 130.17, 130.16, 133.01, 145.26, 170.17, 170.53, 170.74 ppm; 31P NMR (162 MHz, CDCl3): δ 23.69 ppm; mass spectrum (ESIMS), m/z = 993.7 (M+Na)+, C57H63O12P requires 970.41.
5.2.12. Dibenzyl 2,3,6,2′,3′,4′,6′ Hepta-O-benzyl-α-D-maltosyl Phosphate (14)
2.22 M n-Butyl lithium in hexanes (0.63 mL, 1.39 mmol) was added drop-wise to a suspension of diisopropyl amine (0.19 mL, 1.39 mmol) in tetrahydrofuran (6 mL) at −78 °C. The solution was stirred and warmed to 0 °C in 15 min. 2,3,6-tri-O-benzyl-4-O-(2′,3′,4′,6′-tetra-O-benzyl-α-D-glucopyranosyl)-α/β-D-glucopyranoside (0.8 g, 0.8 mmol) in tetrahydrofuran (12 mL) was added slowly and cooled to −80 °C under nitrogen atmosphere. After 10 min stirring a solution of tetrabenzyl pyrophosphate (0.66 g, 1.23 mmol) in tetrahydrofuran (6 mL) was added and stirred continuously before being warm to +4 °C followed by stirring for 16 h. An off-white precipitate of LiOP(O)(OBn)2 was removed by filtration. The resulting solution was evaporated to dryness under reduced pressure and the product was re-dissolved in ethyl acetate. The organic layer was washed successively with saturated aq. NaHCO3 and brine. The solution was dried (anhydrous Na2SO4), filtered and filtrate concentrated under reduced pressure to obtain the crude product. The crude product was purified by silica gel flash chromatography eluting with 1:9 ethyl acetate-toluene. The product fraction were combined, concentrated, and dried in vacuum to afford yellow oily liquid; yield: 30% (0.3 g); silica gel TLC Rf = 0.6 (3:7 ethyl acetate-toluene); [α]D23 = 42.7° (c = 1, CHCl3); 1H NMR (CDCl3, 600 MHz): δ 3.37 (d, 1H, J = 10.2 Hz, H-3), 3.49 (m,3H, H′-2, H′-5, H′-4), 3.67 (m, 3H, H′-4, H′-6a, b, H-2), 3.77 (dd, 1H, J1, 2 = 3 Hz, J = 11.4 Hz, H-5), 3.89 (t, 1H, J = 9.0 Hz, H-3), 4.02 (m, 2H, H-6a, b), 5.07-4.24 (m, 18H, -CH2Ph), 5.69 (d, 1H, J = 3 Hz, H′-1), 5.98 (dd, 1H, J = 3 Hz, 6 Hz, H-1), 7.27-7.07 (m, 45H, Aromatic H’s); 13C NMR (150 MHz, CDCl3): δ 68.24, 68.44, 69.22, 69.26, 69.41, 71.16, 72.28, 73.06, 73.37, 73.57, 74.06, 75.07, 75.62, 77.74, 79.47, 81.24, 82.03, 95.32, 97.14, 125.39(2 C’s), 126.67(2 C’s), 126.78(2 C’s), 127.45(2 C’s), 127.86(2 C’s), 127.87(2 C’s), 127.88(2 C’s), 128.61(2 C’s), 128.69(2 C’s), 129.12(2 C’s), 135.84, 135.89, 137.43, 138.19, 138.82 ppm; 31P NMR (162 MHz, CDCl3): δ 0.90 ppm; mass spectrum (HRMS), m/z = 1255.484 (M+Na)+, C75H77O14P requires 1232.484.
5.2.13. α-D-Maltose-1-Phosphate, Disodium salt8 (16)
Palladium hydroxide (10%) on carbon (catalytic amount) was added to a solution of dibenzyl 2,3,6,2′,3′,4′,6′-hepta-O-benzyl-α-D-maltosyl phosphate (0.27 g, 0.21 mmol) in tetrahydrofuran (0.6 mL) and ethanol (5.4 mL). The reaction mixture was stirred at ambient temperature under hydrogen atmosphere overnight. The catalyst was filtered away and washed with 20% methanol/dichloromethane and methanol. Ten drops of triethylamine were added to the filtrate followed by concentration under reduced pressure to afford a colorless triethylamine salt. The triethylamine salt was dissolved in water and eluted through Dowex® 50WX8 H+ that had been converted to the Na+ form. The filtrate containing the product was concentrated under reduced pressure to afford colorless α-D-maltose-1-phosphate, disodium salt; yield: 100% (92 mg); mass spectrum (ESI-MS), m/z = 421.1(M-H)−, C12H21Na2O14P requires 466.2.
Protein expression and purification
The glgE (Rv1327c) gene from M. tuberculosis strain H37Rv was PCR amplified using the following primers:
5′-CAC CAT ATG AGT GGC CGG GCA AT-3′
5′-AAA GGA TCC TCA CCT GCG CAG CA-3′
The product was placed between the NdeI and BamHI cut sites of a modified pET-28 plasmid. The resulting pDR28-glgE encodes a recombinant GlgE enzyme possessing an N-terminal polyhistidine tag. The sequence of the glgE expression plasmid was confirmed by DNA sequencing.
The pDR28-glgE plasmid was used to transform T7 Rosetta cells. The bacterial cells were cultured at 37 °C in Luria Broth to an O.D. of 1.2–1.6 at 600 nm. Protein expression was induced by the addition of IPTG to a final concentration of 1 mM. Cells were harvested by centrifugation after incubating for 24 hours at 16 °C. Pelleted cells were re-suspended in buffer A containing 5 mM imidazole, 500 mM NaCl, 20 mM Tris pH 7.5, 10% glycerol, and 5 mM β-mercaptoethanol. Lysozyme (10 μM) and DNaseI (100 μM) were added to the cell re-suspension and incubated for one hour on ice prior to lysis by sonication. The resulting suspension was centrifuged at 15,000 x g for 30 minutes. The supernatant was applied to a 5 mL HiTrap Talon Crude column (GE Healthcare) previously equilibrated with buffer A. Proteins were eluted from the column by applying a linear gradient of imidizole from 5–500 mM over 20 column volumes. Fractions containing GlgE were pooled and applied to a Hi-Load Superdex200 size exclusion column for further purification. GlgE was eluted from the column isocratically with a buffer containing 150 mM NaCl, 20 mM Tris pH 7.5, and 0.3 mM TCEP. Fractions containing only GlgE were subsequently pooled. The purity of the protein was confirmed using SDS-PAGE and concentrated to 5.28 mg/mL.
Inhibition Studies
Assays were performed under atmospheric pressure at 22 °C in a 96 well format on a Spectra max 340PC (Molecular Devices). All 1 mM MESG stock solutions were prepared and stored in dH2O at −20 °C. Phosphate release catalyzed by Mtb GlgE was monitored using a coupled assay utilizing purine nucleoside phosphorylase at 5 s intervals for 30 min. For each reaction, a master mix was made consisting of 1 mM MESG (20 μL), 0.2 U of PNP (1 μL), 20X reaction buffer (1.0 M Tris-HCl, 20 mM MgCl2, pH 7.5, containing 2 mM sodium azide) (5 μL), 50 nm GlgE (5 μL), 250 μM M1P (16)(3.125 μL), 40 mM stock solution of inhibitor MCP (13) was made in dH2O and was added for each corresponding stock solution to reach the desired inhibitor concentration (31.25–500 μM) and varied volumes of dH2O for an overall reaction volume of 100 μL. Inhibitor MCP 13 was not added to the positive control and the negative control lacked GlgE. Inhibition was determined by comparing the relative rate of the reaction performed with inhibitor against a reaction that contained no inhibitor (Vo′/Vo), where Vo′ and Vo are steady-state rates with and without inhibitor, respectively. The inhibitory concentration (IC50) was obtained by fitting the data into the equation below using Microsoft Excel:
A1 is the Vo′/Vo value of the bottom plateau of the curve; A2 is the Vo′/Vo value of the top plateau of the curve; [I] and P are inhibitor concentration and hill slope, respectively.
Supplementary Material
Acknowledgments
This work was supported in part by a DeArce Memorial Fund grant to S.J.S. from the University of Toledo and a grant from the National Institutes of Health (grant number AI105084) to S.J.S and D.R.R. We thank Max O. Funk and Ronald E. Viola for their helpful advice on enzyme kinetics.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.World Health Organization. The Global Tuberculosis Report: 2012, Geneva. World Health Organization; 2012. [Google Scholar]
- 2.Konstantinos A. Testing for tuberculosis. Australian Prescriber. 2010;33:12. [Google Scholar]
- 3.Grzegorzewicz AE, Pham H, Gundi VA, Scherman MS, North EJ, Hess T, Jones V, Gruppo V, Born SE, Korduláková J. Nat Chem Bio. 2012;8:334. doi: 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hatzios SK, Bertozzi CR. PLoS Pathog. 2011;7:e1002036. doi: 10.1371/journal.ppat.1002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalscheuer R, Syson K, Veeraraghavan U, Weinrick B, Biermann KE, Liu Z, Sacchettini JC, Besra G, Bornemann S, Jacobs WR., Jr Nat Chem Bio. 2010;6:376. doi: 10.1038/nchembio.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalscheuer R, Weinrick B, Veeraraghavan U, Besra GS, Jacobs WR., Jr Proc Natl Acad Sci USA. 2010;107:21761. doi: 10.1073/pnas.1014642108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mc Carter JD, Withers SG. Curr Opin Struct Biol. 1994;4:885. doi: 10.1016/0959-440x(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 8.Syson K, Stevenson CE, Rejzek M, Fairhurst SA, Nair A, Bruton CJ, Field RA, Chater KF, Lawson DM, Bornemann S. J Bio Chem. 2011;286:38298. doi: 10.1074/jbc.M111.279315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee SS, Hong SY, Errey JC, Izumi A, Davies GJ, Davis BG. Nat Chem Bio. 2011;7:631. doi: 10.1038/nchembio.628. [DOI] [PubMed] [Google Scholar]
- 10.Nasomjai P, O’Hagan D, Slawin AM. Beilstein J Org Chem. 2009;5:37. doi: 10.3762/bjoc.5.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morales JC, Zurita D, Penadés S. J Org Chem. 1998;63:9212. [Google Scholar]
- 12.Wessel HP, Mayer B, Englert G. Carbohydr Res. 1993;242:141. doi: 10.1016/0008-6215(93)80028-d. [DOI] [PubMed] [Google Scholar]
- 13.Panchadhayee R, Misra AKJ. Carbohydr Chem. 2010;29:76–83. [Google Scholar]
- 14.Zeng J, Vedachalam S, Xiang S, Liu XW. Org Lett. 2010;13:42. doi: 10.1021/ol102473k. [DOI] [PubMed] [Google Scholar]
- 15.Kolympadi M, Fontanella M, Venturi C, Andre S, Gabius HJ, Jiménez-Barbero J, Vogel P. Chem Eur J. 2009;15:2861. doi: 10.1002/chem.200801394. [DOI] [PubMed] [Google Scholar]
- 16.Classon B, Liu Z, Samuelsson B. J Org Chem. 1988;53:6126. [Google Scholar]
- 17.Garneau S, Qiao L, Chen L, Walker S, Vederas JC. Bioorg Med Chem. 2004;12:6473. doi: 10.1016/j.bmc.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 18.Chiara JL, Bobo S, Sesmilo E. Synthesis. 2008;19:3160. [Google Scholar]
- 19.Webb MR. Proc Natl Acad Sci USA. 1992;89:4884. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caravano A, Vincent SP. Eur J Org Chem. 2009;11:1771. [Google Scholar]
- 21.Braitsch M, Kählig H, Kontaxis G, Fischer M, Kawada T, Konrat R, Schmid W. Beilstein J Org Chem. 2012;8:448. doi: 10.3762/bjoc.8.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.