

Abstract
The mitochondrial DNA (mtDNA) encompasses two classes of functionally important sequence variants: recent pathogenic mutations and ancient adaptive polymorphisms. To rapidly and cheaply evaluate both classes of single nucleotide variants (SNVs), we have developed an integrated system in which mtDNA SNVs are analyzed by multiplex primer extension using the SNaPshot system. A multiplex PCR amplification strategy was used to amplify the entire mtDNA, a computer program identifies optimal extension primers, and a complete global haplotyping system is also proposed. This system genotypes SNVs on multiplexed mtDNA PCR products or directly from enriched mtDNA samples and can quantify heteroplasmic variants down to 0.8% using a standard curve. With this system, we have developed assays for testing the common pathogenic mutations in four multiplex panels: two genotype the 13 most common pathogenic mtDNA mutations and two genotype the 10 most common Leber Hereditary Optic Neuropathy mutations along with haplogroups J and T. We use a hierarchal system of 140 SNVs to delineate the major global mtDNA haplogroups based on a global phylogenetic tree of coding region polymorphisms. This system should permit rapid and inexpensive genotyping of pathogenic and lineage-specific mtDNA SNVs by clinical and research laboratories.
Keywords: mtDNA, multiplex assay, pathogenic mutations, polymorphisms, SNaPshot
Introduction
Both recent deleterious mutations and ancient functional polymorphisms in the mtDNA need to be considered when evaluating patients for mitochondrial disease or in various clinical and research applications. The mtDNA has a very high mutation rate and as mtDNA proteins are highly evolutionarily conserved, most de novo mtDNA mutations are deleterious. Occasionally, an mtDNA mutation alters OXPHOS in a manner that is not deleterious and therefore is not purified from the population. These variants can be neutral or beneficial in certain environments and are therefore enriched by natural selection. This results in mtDNA lineages being associated with indigenous populations of specific geographic regions and these sequence polymorphisms in mtDNA are then accumulated along maternally inherited lineages. The resulting groups of related haplotypes are known as haplogroups (Ruiz-Pesini et al., 2004; Ruiz-Pesini and Wallace, 2006).
The maternally inherited mtDNA is present in thousands of copies per cell and has a very high sequence evolution rate (Wallace, 2005a, 2007). As a result, new mtDNA mutations arise frequently in the maternal germline, initially present as a mixture of the normal and mutant mtDNAs known as heteroplasmy. Heteroplasmic mutations then segregate during mitotic and meiotic replication to yield pure mutant or normal mtDNAs, homoplasmy.
Because deleterious mutations are continuously being removed by selection, existing pathogenic mutations must have arisen recently. Initially, they are heteroplasmic and as the percentage of mutant mtDNAs increases mitochondrial dysfunction occurs, resulting in disease. As the more severe mutations are lethal at higher percentages of heteroplasmy, they are generally heteroplasmic in clinical specimens. This class of mutations includes the tRNALys m.8344A>G mutation associated with myoclonic epilepsy and ragged red fiber (MERRF) disease (Wallace et al., 1988b; Shoffner et al., 1990); the tRNALeu(UUR) m.3243A>G mutation associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms (MELAS) (Goto et al., 1990); the ND6 m.14459G>A mutation associated with Leber Hereditary Optic Neuropathy (LHON) and dystonia (Jun et al., 1994); and the ATP6 m.8993T>G and T>C mutations associated with neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP) and Leigh Syndrome (LS) (Holt et al., 1990). Milder deleterious mutations can segregate to homoplasmy, without blocking reproduction. Examples of these mutations include the 12S rRNA m.1555A>G deafness mutation (Prezant et al., 1993; Fischel-Ghodsian et al., 1997), the ND4 m.11778G>A LHON mutation (Wallace et al., 1988a), the ND1 m.3460G>A LHON mutation (Huoponen et al., 1991), the ND6 m.14484 T>C LHON mutation (Johns et al., 1992), and the ND3 m.10663T>C LHON mutation (Brown et al., 2002). Currently, there are dozens of confirmed pathogenic mtDNA mutations. In total, over 200 have been reported but still require further work to confirm their pathogenicity (http://www.mitomap.org) (Wallace et al., 2007).
To become polymorphic, mutations must become enriched in a particular population over many generations. Hence, they are generally ancient and most commonly associated with a particular haplogroup. In sub-Saharan Africans most mtDNAs belong to haplogroups L0, L1, L2, and L3, all of which are encompassed within macro-haplogroup L. In Euroasians, most mtDNAs radiated from two macro-haplogroups, M and N, both derived from L3. Macro-haplogroup N radiated into Europe giving rise to haplogroups H, I, J, Uk, T, U, V, W and X, and both macro-haplogroups M and N radiated into Asia, M giving rise to haplogroups C, D, G and many others and N to haplogroups A, B, F and others. Of the Asian haplogroups, only A, C, and D crossed the Bering land bridge to be the first human inhabitants of the Americas. A, C, and D were subsequently joined in the Americas by haplogroups B and X (Wallace et al., 1999).
The functional importance of mtDNA haplogroups has been supported by their association with a broad spectrum of clinical phenotypes. LHON mutations m.10663T>C, m.11778G>A, and m.14484T>C are frequently associated with European mtDNA haplogroup J (Brown et al., 1995, 1997, 2002; Torroni et al., 1997). Haplogroup J is also associated with longevity in Europeans (Ivanova et al., 1998; De Benedictis et al., 1999; Rose et al., 2001; Niemi et al., 2003) and D with longevity in Asians (Tanaka et al., 1998, 2000). The haplogroup H correlates with increased risk, and haplogroups J and Uk with decreased risk, for developing Parkinson's disease (van der Walt et al., 2003; Ghezzi et al., 2005; Khusnutdinova et al., 2008). The haplogroup H m.4336T>C variant is associated with increased risk of Alzheimer's disease and haplogroups U and T with decreased risk (Shoffner et al., 1993; Chagnon et al., 1999; Carrieri et al., 2001; van der Walt et al., 2004). Haplogroup H is also associated with reduced risk of age-related macular degeneration, whereas haplogroups J and U are associated with increased risk of macular degeneration, as well as increased drusen levels and retinal pigment abnormalities (Jones et al., 2007; Udar et al., 2009). Haplogroup J has been associated with increased risk of diabetes in certain European populations (Mohlke et al., 2005; Crispim et al., 2006; Saxena et al., 2006), and N9a with decreased risk of diabetes, metabolic syndrome, and myocardial infarction in Asians (Fuku et al., 2007; Nishigaki et al., 2007). Haplogroup H has been associated with protection against sepsis (Baudouin et al., 2005), U with increased serum IgE levels (Raby et al., 2007), and certain subtypes of H and Uk with protection against the diverse complications of AIDs and antiretroviral induced lipodystrophy (Hendrickson et al., 2008, 2009). Finally, various haplogroups and single nucleotide variants (SNVs) have been correlated with altered risk for cancer (Gottlieb and Tomlinson, 2005; Wallace, 2005b; Booker et al., 2006; Brandon et al., 2006; Bai et al., 2007; Darvishi et al., 2007).
A variety of methods have been developed to detect pathogenic and polymorphic SNVs. These include high pressure liquid chromatography (dHPLC) of heteroduplexes (van den Bosch et al., 2000), restriction analysis resolved by dHPLC (Procaccio et al., 2006), luminex genotyping (Fuku et al., 2007), Surveyor nuclease-mediated heteroduplex digestion (Bannwarth et al., 2005), etc. Unfortunately, these methods can be limited by low throughput, high cost, inability to quantify heteroplasmy, or complexity in interpreting the results. To permit rapid and inexpensive screening for pathogenic and haplogroup-specific SNVs, we report the application of the SNaPshot primer extension system to mtDNA SNVs. This system permits the interrogation of any mtDNA SNV as well as the quantification of heteroplasmy which can be useful for clinical investigation. There are several previous examples of using the SNaPshot system to test for population genetic, clinical, or forensic uses (Brandstatter et al., 2003; Quintans et al., 2004; Crespillo et al., 2005; Salas et al., 2005; Filippini et al., 2007). In the past, haplotyping has been restricted to specialists in the field of mitochondrial biology. The current paper extends previous efforts by providing key simplifications that generalize the technique allowing anyone to test for mtDNA based SNV for use in haplotyping or mutation detection in a multiplex manner. First, we have introduced a rho-zero tested set of amplicons which eliminate the potential for problems with nuclear DNA pseudogene amplification. Second, we provide an amplification system that encompasses the full coding region of the mtDNA so that amplicons no longer need to be redesigned or added to permit interrogation of additional SNVs. Third, we developed a web accessible primer design tool that quickly allows the selection of compatible primer extension sequences for a multiplex assay. Finally, we propose a haplotyping strategy that provides a straightforward method for classifying mtDNA haplogroups, thus permitting distinguishing between ancient polymorphisms and recent pathogenic mutations.
In contrast to limitations in previous systems utilizing SNaPshot to haplotype samples, our system is universal and able to utilize any designed extension primer found in mtDNA. Similar to previous designs, our design can determine the limits of detection for heteroplasmic variants (Brandstatter et al., 2003; Vallone et al., 2004; Nelson et al., 2007). All of the previously designed systems have utilized many small amplicons to enrich amplification of low copy number samples. Although this is crucial for forensic work, it is needlessly complex for research or diagnostic use. In addition, each primer added to a multiplex mix complicates the mixture and the possibility of mispriming, pseudogene amplification, primer dimers, and interference during extension. For these reasons, we took an opposing track requiring a higher initial sample input (less than 200 ng total DNA per genome), but in turn amplifying the entire mtDNA coding region and therefore allowing for the interrogation of any coding region SNV of interest without switching amplification primer sets. This strategy also affords greater sensitivity in heteroplasmic detection. When combined with the newly developed primer design program and the phylogenetic tree included as part of MITOMAP and MITOMASTER (Figure 1), this system permits an integrated analysis of mtDNA variation appropriate for the individual clinical or research laboratory (Brandon et al., 2009).
Figure 1.
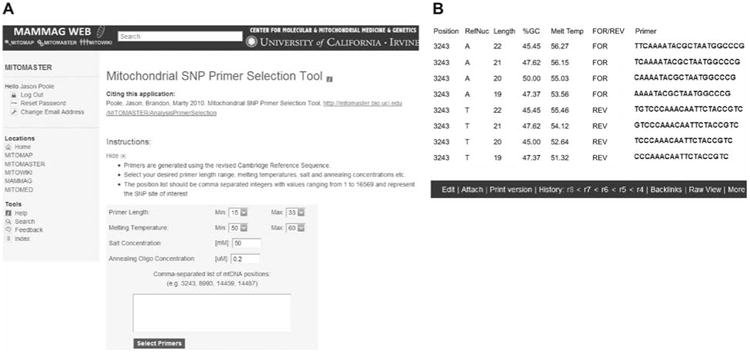
Primer selection tool for mtDNA SNVs.
This tool is accessible through the web via MITOMASTER. (A) The user inputs SNV positions based on the rCRS as well as selecting other basic parameters such as desired length, melting temperature, salt concentration, and oligo concentration. (B) Program output displays the results for the inputted positions (in this case the 3243 position). Output fields include position, rCRS reference nucleotide, percent GC content of the resultant primer, melting temperature, forward or reverse strand, and the nucleotide sequence.
Results
Genotyping procedure
A flow sheet for SNaPshot analysis of mtDNA pathogenic and haplogroup-associated SNVs is provided in Figure 2. The sample mtDNA is either PCR amplified in two multiplex reactions containing seven coding region amplicons plus one control region amplicon, or the mtDNA is chemically purified and tested directly without amplification. Different length primers are used for each nucleotide locus to be interrogated, such that multiple different SNV tests can be resolved on a single ABI 3130xl capillary (Life Technologies, Inc., Carlsbad, CA, USA). The various primers for the SNVs to be interrogated together are mixed with the amplicons containing the appropriate template regions, and the primers extended with Taq polymerase in the presence of the four fluorescent-labeled dideoxynucleotides. The extended primers are then resolved according to their various lengths on an ABI 3130×l automated sequencer capillary. The fluorescent color incorporated into the 3′ end of each primer and thus present in the primer bin indicates the SNV allele.
Figure 2.
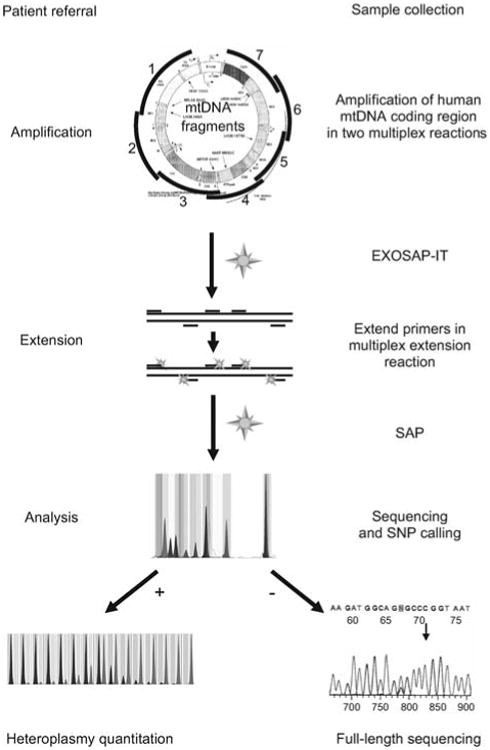
Flow sheet for SNaPshot identification of mtDNA SNVs.
The entire coding region is PCR amplified using multiplex amplification. Amplicons are EXOSAP-treated, mixed with non-overlapping primers designed to end at the base 5′ to the SNV site of interest, and the primers cyclically extended using the four differentially fluorescently labeled dideoxyribonucleotides. The reactions are then treated with shrimp alkaline phosphatase (SAP) and resolved on an ABI 3130×1 capillary. The fluorescent base or bases incorporated into the 3′ end of each primer then reveal the allele present for that SNV. By using multiple non-overlapping primers, multiple SNVs can be interrogated in one reaction. Individual SNV alleles can then be confirmed by quantitative SNaPshot analysis or by direct dideoxyribonucleotide sequencing. Heteroplasmy can be quantified by the relative fluorescence of the mutant and normal bases, compared against a standard curve containing SNV mixtures of known percentages. MITOMAP (Ruiz-Pesini et al., 2007) and MITOMASTER (Brandon et al., 2009) are then used for data interpretation.
This system permits the direct identification of homoplas-mic pathogenic mutations and haplogroup-specific polymorphisms which incorporate one dideoxynucleotide and fluoresce at a single wavelength. It also detects heteroplasmic pathogenic mutations which incorporate two dideoxynucleotides, one mutant and the other normal, and thus fluoresce at two wavelengths. The proportion of the two fluorescent colors can be used to determine the percentage of heteroplasmy, when compared with a standard curve.
SNV identity and heteroplasmy can be confirmed by various procedures including PCR amplification of the mtDNA surrounding the SNV using a FAM-labeled primer, digestion with a restriction enzyme that distinguishes the presence of the normal versus mutant base, and resolution of the fragments on an ABI 3130×1 sequencer (Sarzi et al., 2007) or by direct dideoxy sequencing.
Multiplex amplification of the two halves of the mtDNA
Clinically important SNVs are dispersed around the mtDNA, yet full-length mtDNA can be difficult to amplify from clinical samples. We therefore developed a system for amplifying the mtDNA in two parallel PCR reactions that when combined contained the entire coding region (Table 1). Overlapping primer sets were chosen which function at the same primer concentrations and do not result in loss in signal strength relative to singleplex amplifications. Thus, two multiplex mixtures permit interrogation of all clinically relevant alleles.
Table 1.
Primer sequences used for multiplex amplification of the whole mtDNA.
| Amplicon | Position | Sequence (5′-3′) | Size (bp) | Strand |
|---|---|---|---|---|
| 1 | 550–569 | AACCAAACCCCAAAGACACC | 2452 | F |
| 1 | 3001–2982 | CTGATCCAACATCGAGGTCG | R | |
| 2 | 2777–2797 | GTCCTAAACTACCAAACCTGC | 2417 | F |
| 2 | 5193–5174 | GTGTTAGTCATGTTAGCTTG | R | |
| 3 | 5042–5061 | AGCAGTTCTACCGTACAACC | 2476 | F |
| 3 | 7517–7497 | TTTGAAAAAGTCATGGAGGCC | R | |
| 4 | 7316–7336 | GATTTGAGAAGCCTTCGCTTC | 2524 | F |
| 4 | 9839–9819 | GCCAATAATGACGTGAAGTCC | R | |
| 5 | 9592–9611 | TCCCACTCCTAAACACATCC | 2539 | F |
| 5 | 12 130–12 111 | AAACCCGGTAATGATGTCGG | R | |
| 6 | 11 711–11 727 | GCCCACGGGCTTACATC | 2868 | F |
| 6 | 14 578–14 559 | GATTGTTAGCGGTGTGGTCG | R | |
| 7 | 14 111–14 130 | TCTTCCCACTCATCCTAACC | 2590 | F |
| 7 | 131–112 | ACAGATACTGCGACATAGGG | R | |
| 8 | 15 572–15 591 | TTCGCCTACACAATTCTCCG | 1643 | F |
| 8 | 645–626 | TTTATGGGGTGATGTGAGCC | R |
For, forward strand; Rev, reverse strand; nt, nucleotide; bp, base pair.
Testing for pseudogene amplification using rho-zero cells
To avoid errors owing to nuclear-mtDNA pseudogene amplification, the regional PCR amplification primers were chosen such that no primer pairs were encompassed within a contiguous pseudogene (Wallace et al., 1997). Similar primer sets have now been published. The nucleotide positions for 247 nDNA-mtDNA pseudogenes (Mishmar et al., 2004) are available in MITOMASTER (Brandon et al., 2009). Each primer pair was also tested against DNA extracted from cells which had been cured of their resident mtDNAs (ρ° cells) (King and Attardi, 1989) to confirm that the primer pair did not amplify any nDNA-encoded sequences. The primers listed in Table 1 fulfill these criteria.
Multiplex tests of common pathogenic alleles
Four pathogenic mutation multiplex reactions (MD1, MD2, LD1, and LD2) have been developed to test for the common pathogenic mtDNA mutations (Table 2). The peak distributions of the normal alleles for all four reactions are shown in Figure 3A–D.
Table 2.
SNV panels used for interrogating the 23 most common pathogenic mtDNA SNV alleles using multiplex extension primers.
| Peak number | Mutation | As tested | Disease | Sequence (5′-3′) | Strand | Length w/tail |
|---|---|---|---|---|---|---|
| Panel MD1 | ||||||
| 1 | m.3460G>A | LHON | CTACAACCCTTCGCTGAC | F | 18 | |
| 2 | m.3271T>C | A>G | MELAS | TAAGAAGAGGAATTGAACCTCTGACTGTAA | R | 30 |
| 3 | m.8356T>C | MERRF | (T)10AAAGATTAAGAGAACCAACACCTCT | F | 35 | |
| 4 | m.l555A>G | DEAF | (T)4CTAAAACCCCTACGCATTTATATAGAGGAG | F | 34 | |
| 5 | m.8344A>G | MERRF | GTAAAGCTAACTTAGCATTAACCTTTTAAGTrAAAGATTAAGAGA | F | 45 | |
| 6 | m.H778G>A | LHON | (T)35CTACGAACGCACTCACAGTC | F | 55 | |
| 7 | m.7445A>G, C Panel MD2 | T>C, G | DEAF | (T)47GGGTTCGATTCCTTCCTTTrTrG | R | 70 |
| 1 | m.l4459G>A | LHON | CTCAGGATACTCCTCAATAGCCATC | F | 25 | |
| 2 | m.14484T>C | LHON | CGCTGTAGTATATCCAAAGACAACCA | F | 26 | |
| 3 | m.3243A>G, T | T>C | MELAS/MM/CPEO | GTTTTATGCGATTACCGGGC | R | 20 |
| 4 | m.8993T>G, C | NARP | (T)9AGCCTACTCATTCAACCAATAGCCC | F | 34 | |
| 5 | m.l4487T>C | A>G | LS | AGGTTATATGGGTTTAATAGTTrTrTrAATrTATTrAGGGGGA | R | 43 |
| 6 | m.l0191T>C Panel LD1 | LS | (T)43GCGGCTTCGACCCTATA | F | 60 | |
| 1 | m.3460G>A | LHON | CTACAACCCTTCGCTGAC | F | 18 | |
| 2 | m.3733G>A | C>T | LHON | GAATGATGGCTAGGGTGACTT | R | 21 |
| 3 | m.l4568C>T | G>A | LHON | GGGGGTTTAGTATTGATTGTTAGC | R | 24 |
| 4 | m.l0663T>C | LHON | CAATATTGTGCCTATTGCCATACTAG | F | 26 | |
| 5 | m.l4482C>G, A | LHON | (T)10CGCTGTAGTATATCCAAAGACAAC | F | 34 | |
| 6 | m.l4495A>G | T>C | LHON | GGGGAGGTTATATGGGTTTAATAGTTTTTTTAATTTATT | R | 39 |
| 7 | m.H778G>A Panel LD2 | LHON | (T)35CTACGAACGCACTCACAGTC | F | 55 | |
| 1 | m.l3708G>A | Haplotype | CTAAACCCCATTAAACGCCTG | F | 21 | |
| 2 | m.4917A>G | Haplotype | ATCATATACCAAATCTCTCCCTCACTA | F | 27 | |
| 3 | m.14484T>C | LHON | CGCTGTAGTATATCCAAAGACAACCA | F | 26 | |
| 4 | m.4216T>C | Haplotype | CCACTCACCCTAGCATTACTTATATGA | F | 27 | |
| 5 | m.4171C>A | LHON | (T)14ACGACCAACTCATACACCTC | F | 34 | |
| m.l4459G>A | LHON | TTTTCTCAGGATACTCCTCAATAGCCATC | F | 29 |
All polymorphisms are listed using the forward strand SNV. The ‘As tested’ column indicates which nucleotides are detected during the assay. MD, mitochondrial disease; LD, LHON disease; ‘Length with tail’ includes annealing length plus poly(T) tail.
MD1: la-3460 (WT); lb-3460 (LHON); 2-3271 (MELAS); 3-8356 (MERRF); 4-1555 (DEAF) 5-8344 (MERFF); 6-11778 (LHON); 7-7445 (DEAF/SNHL). MD2: 1-14459 (LHON); 2-14484 (LHON); 3-3243 (MELAS/MM/CPEO); 4a-8993 (WT); 4b-8993 (LS-NARP 4c-8993 (NARP); 5-14487 (LS); 6-10191 (LS). LD1: (all LHON) la-3460 (WT); lb-3460 (LHON) 2-3733 (LHON) 3-14568 4-10663; 5-14482; 6-14459 7-14495; 8-11778. LD2: 1-13708 (J) 2-4917 (T); 3a-14484 (WT); 3b-14484 (LHON); 4-4216 (JT); 5-4171 (LHON).
Figure 3.
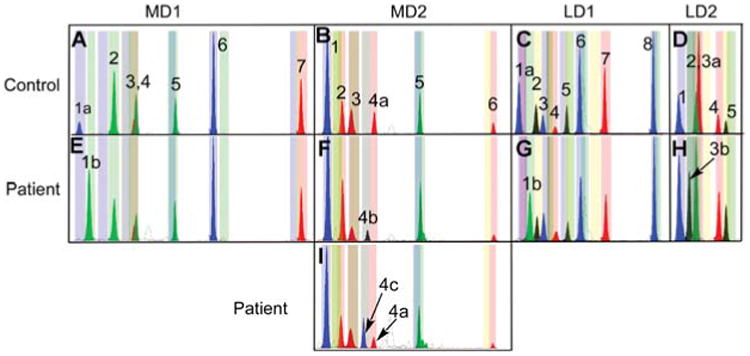
Four SNaPshot multiplex panels for interrogating pathogenic mtDNA mutations.
(A, B) MD1 and MD2 interrogate an array of common pathogenic mutations. (C, D) LD1 and LD2 interrogate the common LHON alleles. Peaks are numbered for each multiplex starting from the peak furthest to the left in each panel. Corresponding peak positions are listed in Table 2. Panels (A–D) show the peak distributions in a healthy individual. Panels (E–I) show the position of representative mutant alleles, identified by the labeled peaks. MD1: 1a-3460G (WT); 1b-3460A (LHON). MD2: 4a-8993T (WT); 4b-8993G (LS-NARP) 4c-8993C (NARP); LD1: 1a-3460G (WT); 1b-3460A (LHON). LD2: 3a-14484T (WT); 3b-14484C (LHON).
Multiplex assays MD1 and MD2 detect the 13 most common pathogenic mtDNA mutations (Wallace et al., 2007) (http://www.mitomap.org). The LD1 and LD2 multiplex assays detect the 10 most prevalent LHON mutations plus the three coding region SNVs which are commonly used to discriminate between haplogroups J and T (Brown et al., 1995, 1997, 2002; Torroni et al., 1997). Most of the interrogated SNVs are biallelic. However, four pathogenic nucleotides have more than one allele: m.7445A>C or G for deafness, m.8993T>C or G for NARP/LS, m.3243A>G or T for MELAS, and m.14482C>A or G for LHON (Besch et al., 1999; Wallace et al., 2007).
Primers for interrogating mutant alleles, which can be generated ranging from 18 to 70 nts, are displayed migrating from left to right in Figure 3. The range of migrations for each primer following incorporation of the wild type (WT) or mutant bases has been determined to assure non-overlapping bins for the various primer alleles. The GeneMapper software (Life Technologies Inc., Carlsbad, CA, USA) has been customized to automatically identify the presence of a control or mutant peak for each SNV. The allele-specific bin settings are shown in Figure 3 as the faint pastel colored bands matching the color of the extended primer allele. To validate the accuracy of individual (not run on multiplex panels necessarily) SNV assays for pathogenic mutations and haplogroup SNVs, DNAs harboring the mutant and control base were analyzed in a small randomized blind study designed to contain both WT and mutant bases. In this case, samples of known pathogenic origin were provided to a researcher in coded format so that patient and control samples were masked. The SNaPshot assay was run and samples analyzed for the presence of any mutations. The results of the coded samples were compared to a key of the samples revealing the known mutations which could then be matched to the SNaPshot results. Two of the 19 samples were unable to amplify and therefore excluded from the study. The other 17 samples were successfully typed including 10 mutations: 8993T>C, 8993T>G, 3243A>G, 8344A>G, 14459G>A, 11778G>A, 1555A>G, 3460G>A, 14484T>C, and 8356T>C (data not shown).
The peak distribution of representative mutant alleles is presented in Figure 3E–I, indicated by a number and letter. MD1 1a shows the LHON nt 3460G normal allele and 1b the mutant nt 3460A allele. MD2 4a shows the normal nt 8993 allele (8993T), 4b the NARP-LS mutant allele 8993G, and 4c the alternative NARP-LS mutant allele 8993C found in a heteroplasmic state. LD1 1a shows the LHON nt 3460G normal allele and 1b is the 3460A mutant allele, LD2 3a the LHON nt 14484T normal allele and 3b is the nt 14484C mutant allele.
Quantification of heteroplasmy by primer extension
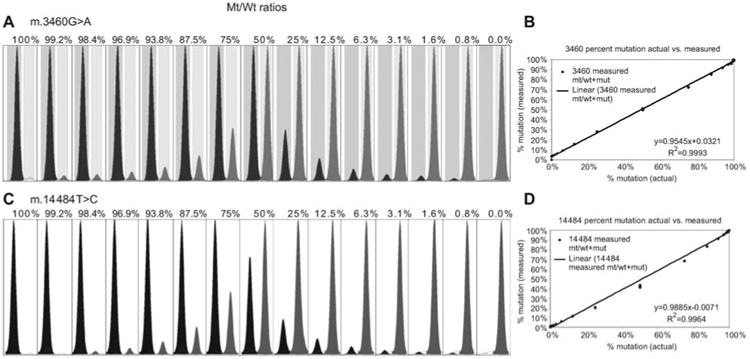
For the more severe pathogenic mutations it is generally important to quantify the percentage heteroplasmy, an influential factor in the severity of an individual's clinical symptoms (Genasetti et al., 2007). However, the efficiency of incorporation of the different fluorescent-labeled nucleotides varies, so the patient results must be compared to a standard curve generated using known mixtures of cloned mutant and normal mtDNA fragments. The standard curves for varying mixtures of the common LHON m.3460G>A and m.14484T>C mutations, total of 1 ng DNA, have been generated by two-fold serial dilution from 50% heteroplasmy down to 0.8% for each of the two alleles, Figure 4A and C. In this experiment, a total of 1 ng of DNA was used. For example, for a 50% heteroplasmy, 0.5 ng of each plasmid was used. Therefore, to achieve a 25% heteroplasmy, 0.75 ng of one form was used and only 0.25 ng of the other would be added. Extension peaks were normalized using the area under the curve by comparison of the total area under the curve for both peaks to the area under the curve for each peak resulting in a percentage scale that corresponds directly to the percent heteroplasmy found in the sample. Correlation coefficients (R2) for both SNV loci were greater than 0.99 (Figure 4B and D), confirming the accuracy and efficiency of the SNaPshot system for detecting heteroplasmy (Cassandrini et al., 2006) over a range of at least two orders of magnitude. This compares favorably with other methods for quantifying heteroplasmy including FAM labeling, DHPLC, real-time PCR, and pyrosequencing (Procaccio et al., 2006; Sarzi et al., 2007).
Figure 4.
SNaPshot quantification of mtDNA heteroplasmy.
(A) Relative peak sizes obtained using known mixtures of cloned 3460 mutant and normal allele mtDNA fragments. Actual percent heteroplasmy is listed above each peak mixture. Heteroplasmic quantity was determined using the area under the curve for each peak divided by the sum of the WT and mutant peak areas. (B) XY scatter plot of data shows the linear regression of measured vs. actual heteroplasmy from panel (A), m.3460G>A, determined by the goodness of fit for our method. (C, D) Heteroplasmic quantification of the m.14484T>C LHON mutation.
Analysis of mtDNA SNVs without PCR amplification
PCR amplification of multiple independent mtDNA products (for multiplex detection) adds several steps to an optimized allelic analysis strategy, and in a clinical setting requires confirmation that nuclear pseudogenes are not amplified. Pseudogene amplification can inadvertently bias heteroplasmic alleles and introduce spurious bases (Wallace et al., 1997; Parfait et al., 1998). Although this issue can be subverted through the use of excess template and high fidelity polymerase, these concerns could also be eliminated by direct allelic primer extension from enriched mtDNA using plasmid purification procedures. For this type of analysis, closed circular mtDNA was prepared from isolated mitochondria of both lymphoblast and muscle samples using plasmid enrichment procedures or mtDNA isolation kits (Biovision, Mountain View, CA, USA). This resulted in a dominant DNA component migrating at 16 kb and detected by agarose gel electrophoresis (Figure 5C).
Figure 5.
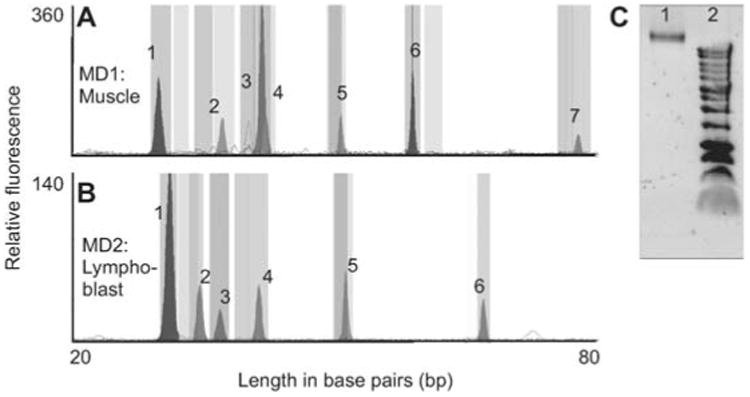
Analysis of SNVs using mtDNA enriched template.
Multiplex primer extensions were performed on DNA extracted from isolated mitochondria using standard plasmid purification procedures or a commercial mtDNA isolation kit (Biovision). (A) MD1 analysis of muscle mtDNA. (B) MD2 analysis of lymphoblast mtDNA. Patterns are identical to amplified DNA, peak heights are lower overall but non-specific peaks are eliminated. (C) Agarose gel showing purification of mtDNA. Lane 1 shows a 16 kb band of mtDNA isolated from human muscle and lane 2 is a DNA ladder showing the highest molecular weight band at 12 kb.
The enriched mtDNA samples were then used as a template for extension of SNaPshot primers. A total of 50 extension cycles were used, versus the 25 cycles used for PCR amplified products. We also increased the ABI 3130×1 sequencer injection voltage to 2.2 volts from the standard 1.2 volts. Both modifications increased the peak signal strength. The MD1 extension results for mtDNA isolated from a muscle biopsy are shown in Figure 5A, and for MD2 mtDNA isolated from lymphoblast cells in Figure 5B.
SNaPshot analysis of mtDNA haplogroups
Because mtDNA haplogroups can modify the penetrance of the milder mtDNA mutations, predispose individuals to a broad range of common diseases, and be important for individual identification and for population studies, we used MITOMASTER (Brandon et al., 2009) to develop a SNaPshot SNV haplogrouping strategy. We took advantage of our sequential mtDNA mutational tree (Ruiz-Pesini et al., 2007) to identify key coding region variants that founded and thus defined the branching pattern of the mtDNA tree. We then selected a hierarchical set of SNVs that would sequentially subdivide the tree into successively smaller branches (Figure 6).
Figure 6.
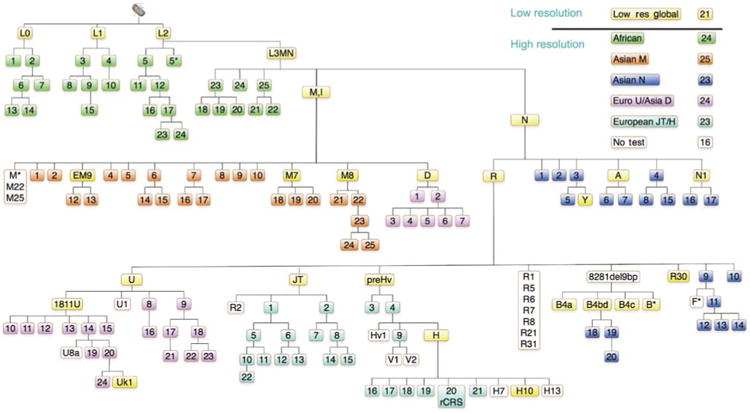
Hierarchical SNV classification of human mtDNA haplogroups.
Aggregates of SNVs are used to define the branches of different portions of the mtDNA tree (Ruiz-Pesini et al., 2007) and are color coded. To classify unknown mtDNAs, they are first divided into major macro-haplogroups and haplogroups using the 21 ‘low resolution’ SNVs, shown in yellow. The mtDNAs for each macro-haplogroup or major haplogroup branch of the tree are then assembled and further subclassified together using the appropriate ‘high resolution’ haplogroup screens. The full version of this tree containing the nucleotide positions and variant nucleotides can be found at the following website: http://www.mitomap.org/pub/MITOMAP/MITOMAPFigures/SNaPshot-Sets.pdf. The SNVs used to define each region of the mtDNA tree are as follows: low resolution global SNVs (yellow): L0(m.13276G), L1(m.13789C), L2(m.7146G), L3MN(m.3594C), M(m.10400T), EM9(m.4491A), M7(m.9824C), M8(m.8584A), D(m.5178A), N(m.9540T), A(m.1736G), R(m.12705C), R30(m.8584A), U(m.12308G), U1811(m.1811G), JT(m.4216C), preHv(m.11719G), H(m.7028C), H10(m.4216C), B4a(m.5465C), B4bd(m.15535T), B4c(m.15346C), B*(m.8584A), N1 (m.10238C). African SNVs (green): 1(m.4232T) 2(m.9818G), 3(m.8027G), 4(m.5046C), 5(m.8206G), 5(m.13590G), 6(m.9755G+m.15431G), 7(m.7257A), 8(m.6917G), 9(m.10321T), 10(m.3693G), 11(m.5069(T)), 12(m.11944T), 13(m.5096A), 14(m.5147G),15(m.6150G), 16(m.7771A), 17(m.7624(A)), 18(m.14905C), 19(m.9554G), 20(m.6221A), 21(m.5147A), 22 (m.6221C) 23(m.4767A), 24(m.15849C). Asian M SNVs (orange): 1(m.12403G), 2(m.15431C), 4(m.15218A), 5(m.8108), 6(m.4833A), 7(m.12940G), 8(m.10986), 9(m.5319T), 10(m.11482), 12(m.7598G), 13(m.3394T), 14(m.15497G), 15(m.13563T), 16(m.14025A), 17(m.11061C), 18(m.4958A), 19(m.7853G), 20(m.5442T), 21(m.9090A), 22(m.14318T), 23(m.15204A), 24(m.12672A), 25(m.3816T). Asian N SNVs (dark blue): 1(m.8404A), 2(m.9140C), 3(m.5417G), 4(m.5460G+8994G), 5(m.12358T), 6(m.8563T), 7(m.8027G), 8(m.3505A), 9(m.3970G+13928C), 10(m.10118A), 11(m.10310C), 12(m.10609T), 13(m.12338T+13708G), 14(m.5263G), 15(m.7864C), 16(m.4529A), 17(m.6221T+14470T), 18(m.11914C), 19(m.4820G), 20(m.9950T). Asian D/European U SNVs (purple): 1(m.5301T), 2(m.3010G+m.8414G), 3(m.2092C), 4(m.3316G), 5(m.14979T), 6(m.9824A), 7(m.10181C), 8(m.3348T), 9(m.9477G), 10(m.13020A), 11(m.15454A), 12(m.5360C), 13(m.15693T), 14(m.9698T), 15(m.6386C), 16(m.7805C), 17(m.14793A), 18(m.14182A), 19(m.14167C), 20(m.14798A), 21(m.15218G), 22(m.13637A), 23(m.5656T), 24(m.9716T). European JT/H SNVs (turquoise): 1(m.13708G), 2(m.4917A), 3(m.13188C), 4(m.14766C), 5(m.3010C), 6(m.15257G), 7(m.12633C), 8(m.14233A), 9(m.4580G), 10(m.14798T), 11(m.5460G+13879A), 12(m.15812G), 13(m.10499T), 14(m.13965A), 15(m.5147G), 16(m.3010C), 17(m.4024A+5004A), 18(m.4336T), 19(m.3915G), 20(m.1438A+4769T), 21(m.6776T), 22(m.3394).
First, the mtDNAs are subdivided into the most ancient macro-haplogroup categories L (African), M and N (Asian), N (European), and the most common haplogroups using 21 (Table 3) SNVs interrogated in two multiplexed panels, HT1 and HT2 (Figure 6, yellow). Each macro-haplogroup was further subdivided into haplogroups by six sets of 23–25 SNVs (Figure 6). By multiplex interrogation of successive groups of SNVs from the top of the tree to the bottom, this hierarchical system permits the maximum resolution of the mtDNA lineages with the minimum number of SNV tests. Therefore, the multiplex nature of SNaPshot provides a perfect platform for a hierarchal analysis of mtDNA haplogroups.
Table 3.
Extension primers for the multiplex detection of 21 haplogroups defining SNVs in two panels.
| Peak # | Hg | Polymorphism | For/Rev | Tested nt | bp | Sequence (59-39) |
|---|---|---|---|---|---|---|
| Panel | HT1 | |||||
| 1 | Hv | m.11719A>G | 19 | TTCTCATAATCGCCCACGG | ||
| 2 | U1811 | m.1811A>G | 27 | ACCGCAAGGGAAAGATGAAAAATTATA | ||
| 3 | N | m.9540C>T | 20 | TTCTAGCCCCTACCCCCCAA | ||
| 4 | B4bd | m.15535C>T | 27 | CAATTATACCCTAGCCAACCCCTTAAA | ||
| 5 | M | m.10400C>T | 33 | CTATGAGTGACTACAAAAAGGATTAGACTGAAC | ||
| 6 | L0 | m.13276A>G | 34 | (T)6CTCCACTTCAAGTCAACTAGGACTCATA | ||
| 7 | L2 | m.7146A>G | R | T>C | 33 | AGAAAGTTAGATTTACGCCGATGAATATGATAG |
| 8 | M7/D4 | m.9824T>C, A | R | A>G, T | 35 | (T)11GAAAGTTGAGCCAATAATGACGTG |
| 9 | U | m.12308A>G | R | T>C | 39 | (T)12ACTTTTATTTGGAGTTGCACCAAAATT |
| 10 | B4c | m.15346G>A | R | C>T | 39 | (T)21GTTTGATCCCGTTTCGTG |
| Panel | HT2 | |||||
| 1 | A | m.1736A>G | 22 | CCTTAGCCAAACCATTTACCCA | ||
| 2 | L1 | m.13789T>C | 21 | TTCCAAACAACAATCCCCCTC | ||
| 3 | E/M9 | m.4491G>A | R | C>T | 24 | CTGCAAAGATGGTAGAGTAGATGA |
| 4 | B4a | m.5465T>C | R | A>G | 27 | (T)7ATAGGTAGGAGTAGCGTGGT |
| 5 | J/T | m.4216T>C | 27 | CCACTCACCCTAGCATTACTTATATGA | ||
| 6 | R | m.12705T>C | R | A>G | 33 | AGCGGTAACTAAGATTAGTATGGTAATTAGGAA |
| 7 | N1 | m.10238T>C | 33 | CCCTTTCTCCATAAAATTCTTCTTAGTAGCTAT | ||
| 8 | M8/R30/B5 | m.8584G>A | 40 | (T)19ACAATCCTAGGCCTACCCGCC | ||
| 9 | L3/M/N | m.3594T>C | 39 | (T)24ACCCAACCCCCTGGT | ||
| 10 | D | m.5178C>A | R | G>T | 39 | (T)11GATGGAATTAAGGGTGTTAGTCATGTTA |
| 11 | H | m.7028T>C | 51 | (T)30CGACACGTACTACGTTGTAGC |
Haplogroups correspond to multiplex panels HT1 and HT2 in Figure 7.
The classification of mtDNAs of European ancestry using the ‘low resolution’ HT1 and HT2 SNV panels (yellow in Figure 6) is shown in Figure 7. The peak patterns for a haplogroup H individual are shown in Figure 7A (HT1 primer set) and B (HT2 primer set). Examples of other mtDNA haplotype results are presented in the four pairs of panels below Figure 7A and B. These include mtDNAs that belong to haplogroup clusters JT, U1811, U, and L3.
Figure 7.
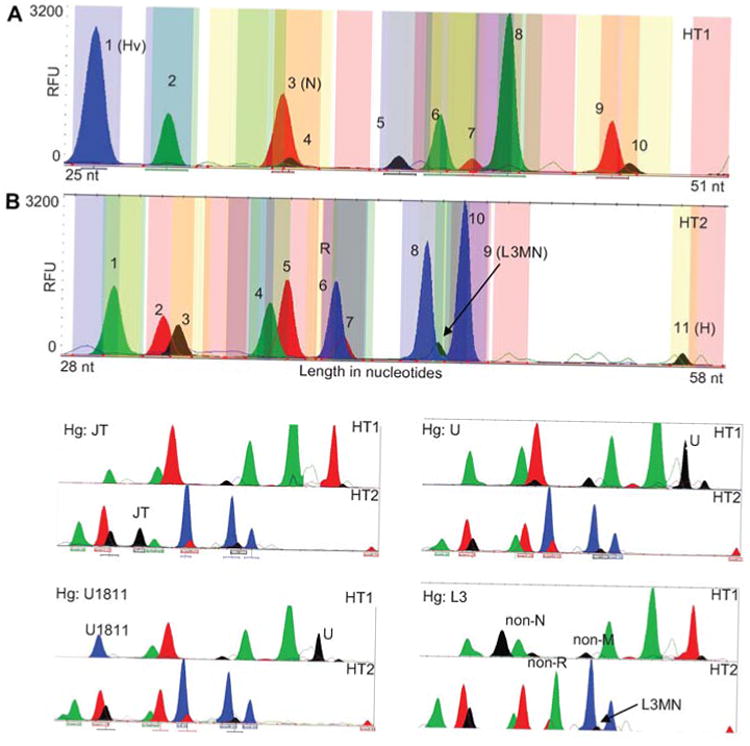
SNaPshot classification of European mtDNA haplogroups.
Two SNaPshot multiplex panels, HT1 and HT2, which interrogate the ‘low resolution’ SNVs (yellow in Figure 6) were applied to various European mtDNAs. Peak numbers correspond to SNVs listed in Table 3. Panels (A) and (B) show the peak distributions for an individual similar to the rCRS of haplogroup H. Peaks that identify a haplogroup in each panel are labeled with the appropriate marker. Panel (A), HT1: haplogroup (position): 1:Hv (11719), 2:U (1811), 3:N (9540), 4:B4bd (15535), 5:M (10400), 6:L0 (13276), 7:L2 (7146), 8:M7-D4 (9824), 9:U (12308), 10:B4c (15346). Panel (B), HT2: haplogroup (position): 1:A (1736), 2:L1-B4b (13789), 3:EM9 (4491), 4:B4a (5465), 5:JT (4216), 6:R (12705), 7:N1 (10238), 8:M8-B5-R30 (8584), 9:L3MN (3594), 10:D (5178), 11:H (7028). The lower four pairs of electropherograms demonstrate the HT1 and two peak patterns for a mtDNA of haplogroup cluster JT (upper left pair), U1811 (lower left pair), U (upper right pair), and L3 (lower right pair). Haplogroup L3 must be inferred using a combination of peaks: the L3MN SNV plus the non-M and non-N SNVs.
Discussion
Interest in human mtDNA variation in clinical medicine, forensic identification, and population genetics is growing rapidly. Although the mtDNA is much smaller than the nDNA, its high mutation rate and crucial function in energy metabolism mean that recent pathogenic and ancient functional mtDNA polymorphisms are both common and of great importance to medicine and human biology.
Because of uniparental inheritance of the mtDNA and its resulting lack of recombination, pathogenic, functional, and neutral variants are often linked and can interact. For example, the penetrance of mild pathogenic mutations can be modified by the background mtDNA haplogroup. Several SNVs can accumulate along the branches of a particular haplogroup, potentially altering the physiological significance of the different sub-haplogroups. Likewise, neutral variants can become enriched in a population along with adaptive variants by hitchhiking. Therefore, testing for individual SNVs often gives an inadequate picture of the functional or evolutionary significance of an individual's mtDNA haplotype. By permitting the testing of multiple SNVs, the multiplex capacity of SNaPshot permits a much more integrated perspective of mtDNA variation than that achieved by a single SNV analysis.
In additional to its multiplex capability, SNaPshot permits addressing two unique challenges of mtDNA genetics: multiple allelism and heteroplasmy. Because all four different colored bases can be incorporated into the 3′ end of the primer, SNaPshot naturally detects all possible alleles at a single SNV site. Moreover, if the SNV is heteroplasmic, then both the normal and mutant allele will be incorporated generating two colors in that primer's bin. The relative levels of the two colors then reflect the percentage heteroplasmy. Therefore, SNaPshot provides a uniquely appropriate platform for evaluating mtDNA variation for the individual clinical or population genetics laboratory, allowing one to focus on key changes in mtDNA, reducing analysis time and increasing throughput. It is also locus neutral with regard to being equally capable to interrogate any base found within mt-DNA. This presents significant advantages over traditional sequencing which requires the analysis of hundreds or thousands of bases.
Although SNaPshot permits the concurrent interrogation and quantification of multiple different SNVs, it does not tell the investigator the significance of the different SNV alleles or indicate the potential importance of their interaction. To provide this type of information, we have developed MITO-MASTER (Brandon et al., 2009). MITOMASTER provides information on the nature of the various SNV alleles, their conservation, population frequency, haplogroup associations, known relevance to pathogenicity, etc. Therefore, the combination of the current SNaPshot SNV detection system with our MITOMASTER computer interpretation system provides an integrated and inexpensive tool by which clinical and research laboratories can screen large numbers of samples and these address a broad range of questions.
Materials and methods
SNV site selection
In total, 20 of the most common pathogenic mtDNA mutations (http://www.mitomap.org/rimtab2.html) were chosen for development of primer extension assays using the SNaPshot system (Table 2). These were clustered into four groups: mitochondrial disease (MD), MD1, MD2, and LHON disease (LD), LD1 and LD2. The four most common pathogenic LHON mutations were duplicated in MD and LD panels 1 and 2, respectively. Three haplogroup-specific markers for haplogroups J and T were also incorporated into panel LD2 for their stated prevalence in LHON disease. The designation of all SNVs follows the numbering system of the revised Cambridge Reference Sequence (rCRS) (GenBank Accession NC_012920) (Andrews et al., 1999). A SNaPshot panel for mtDNA deafness mutations has also been reported (Bardien et al., 2009).
Extension primer web tool
To rapidly design extension primers for multiplex reactions, we developed a novel, previously unpublished computer program that takes into account the DNA sequence surrounding the SNV and then varies the primer length and primer location to generate primers of comparable melting temperatures. This permits the concurrent amplification and subsequent resolution of all of the multiplex extended primers on a single ABI 3130×1 automated sequencer column. The primer selection tool was written in Perl v5.8.8 and packaged into an object library. This new program was added to our existing suite of bioinformatic tools and has now been included as part of MITOMASTER (Brandon et al., 2009). To run the program, the user inputs a list of comma separated SNV locations corresponding to the rCRS, a range of permissible primer melting temperatures, and the reaction concentrations for salt and G-C ratios. The program then proposes primers that meet the specification by iteratively computing the melting temperature for a sliding window of nucleotides across the rCRS according to previously identified equations (Breslauer et al., 1986; Rychlik et al., 1990; SantaLucia, 1998). The program calculates and prints both upstream and downstream primer sequences potentially applicable for the proposed SNV in the standard 5′ to 3′ format. It specifies the reference nucleotide at that SNV location (RefNuc), the primer length (Length), melting temperature (MeltTemp), the forward or reverse strand sequence used (For/Rev), and the primer sequence (Primer Sequence) (Figure 1A). The results are output in a tabular format from which the user can perform the final primer selection based on desirable experimental criteria (Figure 1B).
A Perl::CGI module is used to extract the submitted parameters and the web interface is implemented in the TWiki software (Twiki.Net, Inc., Sunnyvale, CA, USA). All processing occurs on a Linux Apache server utilizing the mod_perl2 embedded Perl interpreter. An example of the program output is provided in Figure 1 and can be found at URL: http://mammag.web.uci.edu/bin/view/Mitomaster/AnalysisPrimerSelection.
Primers for SNV analysis by primer extension
The primers proposed for the current analysis system were prepared in lengths ranging from 18 to 70 nts, and differing by 3–5 nts each to prevent spatial overlap when resolved on the capillary columns of our ABI 3130xl sequencer. We emphasize the use of primers under 40 base pairs in length to cut costs, but more expensive PAGE purification allows the design of primers up to 120 bp in length and allows for higher multiplexing of the extension primers. This makes sense especially in cases where the panel will be run with a high number of samples so that the cost savings of SNaPshot reagents outweighs the cost of the longer PAGE purified primer. When primers were found to overlap, they were further modified by adding poly(T) tails to adjust their electrophoretic elution point. This permits the creation of a series of primers that do not overlap when extended with either the normal or mutant base, thus creating non-overlapping ‘bins’ for use in automated base calls with the GeneMapper software v4.0 (Life Technologies Inc.) (Table 2). Primers were synthesized by Integrated DNA Technologies Inc. (Iowa City, IA, USA), and those greater than 38 nts in length were PAGE purified. All primers were stored in 100 μM stocks in 0.5 mM EDTA, 10 mM Tris, pH 8.0 at −20°C.
Certain SNVs proved to be particularly difficult to extend with the correct base using cloned templates. This difficulty was most pronounced for the MELAS m.3243A>G and the NARP-LS m.8993T>G SNVs. These difficulties had to be resolved by empirically testing a number of alternative primer designs until the required specificity was obtained. Also, certain primers overlap polymorphic bases in certain mtDNA haplogroups, creating the possibility of amplification interference. The primers potentially affected by these interfering haplogroups and SNVs are listed in Table 4.
Table 4.
Potential haplotype interference for disease primers.
| Mutation | SNP | Frequency % (#) | Conflicting haplotype |
|---|---|---|---|
| Panel MD1 | |||
| m.8356T>C | None | ||
| m.11778G>A | None | ||
| m.3271T>C | None | ||
| m.1555A>G | None | ||
| m.8344A>G | None | ||
| m.3460G>A | None | ||
| m.7445A>G, C | 7444 | 0.47% (12) | H*, H6776, H* |
| Panel MD2 | |||
| m.14484T>C | 14 470 | 3.12% (79) | F1 |
| 14 476 | 0.91% (23) | X, H10 | |
| m.3243A>G, T | None | ||
| m.14487T>C | 14 502 | 0.59% (15) | M10, N*, X |
| m.8993T>G, C | 8994 | 2.49% (63) | L0, W, Hv |
| m.10191T>C | 10 181 | 0.83% (21) | D4b |
| m.14459G>A | None | ||
| Panel LD1 | |||
| m.11778G>A | None | ||
| m.3733G>A | None | ||
| m.10663T>C | 10 646 | 0.55% (14) | M10, M8, D, H6776, H* |
| m.14482C>G, A | 14 476 | 0.91% (23) | F1 |
| 14 470 | 0.59% (79) | X, H10 | |
| m.14495A>G | 14 502 | 0.59% (15) | |
| m.14568C>T | 14 569 | 3.63% (92) | Various |
| 14 582 | 1.03% (26) | N9a, H4 | |
| m.3460G>A | None | ||
| Panel LD2 | |||
| m.14484T>C | 14 476 | 0.91% (23) | F1 |
| 14 470 | 3.12% (79) | X, H10 | |
| m.4917A>G | 4907 | 0.55% (14) | L0, L3 |
| m.13708G>A | None | ||
| m.4216T>C | None | ||
| m.4171C>A | 4164 | 1.54% (39) | M7b |
| m.14459G>A | None |
As tested column, primer used was on the reverse strand.
Sample preparation
DNA was obtained from buccal swab, muscle, lymphoblast, or 143B osteosarcoma cell lines depending on the experiment. Genomic DNA was isolated from blood and lymphoblast cells using the puregene DNA isolation kit (Gentra Systems, Qiagen, Germantown, MA, USA). In some cases, the mtDNA was partially purified from human cell lines and tissue or their mitochondria using an mtDNA isolation kit (Biovision Research Products Inc., Mountain View, CA, USA). This method used a dounce homogenizer to release mitochondria, followed by enzyme treatment to obtain mtDNA from mitochondria.
mtDNA amplification
Whole mtDNA amplification was accomplished through PCR in overlapping fragments ranging in size from 1500 to 2500 bp involving seven fragments covering the coding region plus one fragment covering the control region. Following amplification, fragments were ExoSAP-IT treated and then added to the primer extension reaction mix. Because the entire mtDNA coding region is generated, the same mtDNA products can be used to interrogate any coding region SNV of interest. All mtDNA amplification primer sets were tested on genomic DNA isolated from cells lacking cytoplasmic mtDNA (ρ° cells) to confirm that none of the primer pairs amplified a nDNA-encoded mtDNA pseudogene sequence (Mishmar et al., 2004). Primer sequences for amplification of mtDNA templates are listed in Table 1.
To amplify the template mtDNA, we used between 25 and 50 ng of genomic DNA, 0.4 μmol of each primer, 50 μM dNTPs, and 1.25 U Taq polymerase (Roche, Indianapolis, IN, USA). Amplification conditions for mtDNA fragments were as follows: 94°C for 5 min, 35 cycles of 94°C 45 s/56°C 30 s/72°C 3 min, followed by 72°C for 5 min and hold at 4°C.
Multiplex PCR conditions
The template mtDNA coding region fragments were assayed in two multiplex reactions. Odd numbered fragments were added to multiplex 1 and even numbered fragments to multiplex 2 (Table 1). Other optimizations included increasing the polymerase to 3U/reaction and the Mg2+ concentration to 2.5 mM, doubling the DNA concentration, and reducing reaction volume from 50 to 15 μl. The control region fragment was amplified independently if needed.
Following PCR amplification, products were combined and treated with ExoSAP-IT (USB, Cleveland, OH, USA) for 15 min at 37°C followed by inactivation for 20 min at 80°C. This step removes any remaining dNTPs that might otherwise cause a multibase extension as opposed to a single base incorporation desired in the subsequent primer extension steps.
Primer extension assays for SNVs
For the concurrent interrogation of 6–11 SNVs, the reaction mixture includes a mixture of the required primers at 0.05–1 μM each, the ExoSAP-IT purified mtDNA template PCR products, 5 μl of 2× SNaPshot mastermix (Life Technologies, Inc.) including polymerase, labeled ddNTPs (adenine, dR6G, green; cytosine, dTAMRA, black; guanine, dR110, blue; thymidine, dROX, red), 2× reaction buffer, in a total volume of 10 μl. The mixture was denatured at 94°C for 1 min, and the primers repeatedly extended for 25–40 cycles of 94°C for 10 s, 53°C for 5 s, and 60°C for 10 min, followed by a final denaturation at 94°C for 30 s. Following the primer extension reactions, unincorporated ddNTPs were removed by digestion with shrimp alkaline phosphatase for 60 min at 37°C and the primer-template pairs denatured at 70°C for 15 min. The primer extension products were combined with GeneLiz120 size standards, diluted 1:10 in Hi-Di formamide, denatured for 5 min at 95°C, immediately chilled on ice, and resolved on the ABI 3130×1.
For heteroplasmy assessment, the relative fluorescent peak levels were determined. However, to avoid fluorescent signal saturation, the extension reaction often needed to be diluted before sequence analysis. Increasing the number of extension cycles increased the sensitivity for low allele levels as did altering primer sequences to improve binding efficiency and specificity and increasing or decreasing primer concentrations.
DNA collection, purification, and quantification
Experiments used to determine assay sensitivity were performed using serial dilutions of DNA quantified by spectrophotometry at 260 nm and confirmed through agarose gel electrophoreses alongside a standard DNA ladder of known DNA quantity (typically the O-gene Ruler plus Fermentas, Hanover, MD, USA). The products amplified were control products known to have either a WT or variant base at the intended interrogation site. The gel run PCR bands were resolved and quantified using a Bio-Rad gel GelDocXR imager (Bio-Rad Inc., Hercules, CA, USA) and image analysis was performed using Quantity One™ software from Bio-Rad and normalized over several iterations of gels by changing the loading volumes. The consistency of the gel quantification results was confirmed using pico green dye staining (Life Technologies Inc.) measured against a DNA standard curve and read using a NOVOstar fluorescent plate reader at 480 nm excitation and 520 nm emission spectra.
Analysis of extension products with GeneMapper
Because the primers, when 3′-extended by different fluorescent-labeled dideoxynucleotides, have slightly different mobilities on the ABI 3130×l capillaries, we used GeneMapper 4.0 to set up capillary elution regions, ‘bins’. These bins encompass the mobility of all four possible products of the primer extension assay. Because image analysis for many samples was impractical, result fields such as peak height, allele calls, and peak areas were collected in text format using the GeneMapper report manager and exported into Excel where they were scored for the variants. Extension peaks were only referenced in cases of low signal strength, when heteroplasmy was present, or there was a discrepancy in the data. Along with the multiplex amplifications and extension reactions, this greatly facilitated the high sample throughput and allowed for fast typing of pathogenic mutations or haplogroup-specific SNVs.
Haplogroup classification by identifying SNVs
A SNaPshot haplogroup determination strategy was developed based on selecting nodal SNVs that separate major branches of our mtDNA mutational tree (Ruiz-Pesini et al., 2007). These were grouped into a hierarchical series of SNaPshot multiplex reactions, thus permitting the sequential subdivision of the branches of the tree. The primers used for separating mtDNAs into the macro- and major haplogroups are presented in Table 3. Polymorphic bases associated with specific haplogroups that can interfere by causing a primer/template mismatch are listed in Table 5.
Table 5.
Potential SNV interference with haplogroup classification primers.
| Polymorphism | Conflicting position(s) | Frequency % (#) | Conflicting haplotype (s) |
|---|---|---|---|
| Panel HT1 | |||
| m.12308A>G | 12 311 | 0.39% (10) | H3010 |
| m.9540C>T | 9536 | 1.86% (47) | D, D2 |
| 9545 | 1.74% (44) | N, M1, C, A, U, H* | |
| 9548 | 0.99% (25) | F, U, J | |
| m.15535C>T | None | ||
| m.11719A>G | None | ||
| m.1811A>G | None | ||
| m.15346T>C | 15 355 | 0.55% (14) | M8, U, H3010 |
| m.10400C>T | 10 397 | 1.5% (10) | M7, C, D |
| 10 398 | 44.61% (1130) | N, Y, I, L1, P, B, U, J, | |
| 10 410 | 1.74% (44) | D, W, J, H6776 | |
| m.7146A>G | None | ||
| m.13276A>G | 13 281 | 0.39% (10) | L0, L2 |
| m.9824T>C, A | 9818 | 0.71% (18) | L0 |
| Panel HT2 | |||
| m.1736A>G | 1738 | 0.63% (16) | L1 |
| m.3594T>C | 3591 | 0.51% (13) | L2, A, U, T, H* |
| m.10238T>C | None | ||
| m.4216T>C | None | ||
| m.7028T>C | 7022 | 0.39% (10) | T |
| m.12705T>C | None | ||
| m.5465T>C | 5460 | 6.24% (158) | L0-2, Q, M7, D, W, P, U, |
| 5471 | 0.67% (17) | J, V, H3010, H3915 L3, N1b, U, J, Hv, H* | |
| m.8584G>A | None | ||
| m.5178C>A | None | ||
| m.13789T>C | None | ||
| m.4491G>A | None |
Acknowledgments
We would like to acknowledge Mehrdad Zoleikhaien for help with PCR fragment amplification and Jim Kreuziger for efforts related to the primer generation program. This research was supported by National Institutes of Health (NIH) grants NS21328, AG24373, DK73691, and AG16573, California Institute for Regenerative Medicine Comprehensive grant RC1-00353, and 2005 Doris Duke Clinical Interfaces Award 2005057 awarded to D.C.W. and NIH postdoctoral fellowship AG25638 awarded to J.C.P.
References
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- Bai RK, Leal SM, Covarrubias D, Liu A, Wong LJ. Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 2007;67:4687–4694. doi: 10.1158/0008-5472.CAN-06-3554. [DOI] [PubMed] [Google Scholar]
- Bannwarth S, Procaccio V, Paquis-Flucklinger V. Surveyor nuclease: a new strategy for a rapid identification of heteroplasmic mitochondrial DNA mutations in patients with respiratory chain defects. Hum Mutat. 2005;25:575–582. doi: 10.1002/humu.20177. [DOI] [PubMed] [Google Scholar]
- Bardien S, Human H, Harris T, Hefke G, Veikondis R, Schaaf HS, van der Merwe L, Greinwald JH, Fagan J, de Jong G. A rapid method for detection of five known mutations associated with aminoglycoside-induced deafness. BMC Med Genet. 2009;10:2. doi: 10.1186/1471-2350-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudouin SV, Saunders D, Tiangyou W, Elson JL, Poynter J, Pyle A, Keers S, Turnbull DM, Howell N, Chinnery PF. Mitochondrial DNA and survival after sepsis: a prospective study. Lancet. 2005;366:2118–2121. doi: 10.1016/S0140-6736(05)67890-7. [DOI] [PubMed] [Google Scholar]
- Besch D, Leo-Kottler B, Zrenner E, Wissinger B. Leber's hereditary optic neuropathy: clinical and molecular genetic findings in a patient with a new mutation in the ND6 gene. Graefes Arch Clin Exp Ophthalmol. 1999;237:745–752. doi: 10.1007/s004170050307. [DOI] [PubMed] [Google Scholar]
- Booker LM, Habermacher GM, Jessie BC, Sun QC, Baumann AK, Amin M, Lim SD, Fernandez-Golarz C, Lyles RH, Brown MD, et al. North American white mitochondrial haplogroups in prostate and renal cancer. J Urol. 2006;175:468–472. doi: 10.1016/S0022-5347(05)00163-1. discussion 472–473. [DOI] [PubMed] [Google Scholar]
- Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–4662. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- Brandon MC, Ruiz-Pesini E, Mishmar D, Procaccio V, Lott MT, Nguyen KC, Spolim S, Patil U, Baldi P, Wallace DC. MITOMASTER: a bioinformatics tool for the analysis of mitochondrial DNA sequences. Hum Mutat. 2009;30:1–6. doi: 10.1002/humu.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandstatter A, Parsons TJ, Parson W. Rapid screening of mtDNA coding region SNPs for the identification of west European Caucasian haplogroups. Int J Legal Med. 2003;117:291–298. doi: 10.1007/s00414-003-0395-2. [DOI] [PubMed] [Google Scholar]
- Breslauer KJ, Frank R, Blocker H, Marky LA. Predicting DNA duplex stability from the base sequence. Proc Natl Acad Sci USA. 1986;83:3746–3750. doi: 10.1073/pnas.83.11.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MD, Torroni A, Reckord CL, Wallace DC. Phylogenetic analysis of Leber's hereditary optic neuropathy mitochondrial DNA's indicates multiple independent occurrences of the common mutations. Hum Mutat. 1995;6:311–325. doi: 10.1002/humu.1380060405. [DOI] [PubMed] [Google Scholar]
- Brown MD, Sun F, Wallace DC. Clustering of Caucasian Leber hereditary optic neuropathy patients containing the 11778 or 14484 mutations on an mtDNA lineage. Am J Hum Genet. 1997;60:381–387. [PMC free article] [PubMed] [Google Scholar]
- Brown MD, Starikovskaya E, Derbeneva O, Hosseini S, Allen JC, Mikhailovskaya IE, Sukernik RI, Wallace DC. The role of mtDNA background in disease expression: a new primary LHON mutation associated with Western Eurasian haplogroup. J Hum Genet. 2002;110:130–138. doi: 10.1007/s00439-001-0660-8. [DOI] [PubMed] [Google Scholar]
- Carrieri G, Bonafe M, De Luca M, Rose G, Varcasia O, Bruni A, Maletta R, Nacmias B, Sorbi S, Corsonello F, et al. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer's disease. Hum Genet. 2001;108:194–198. doi: 10.1007/s004390100463. [DOI] [PubMed] [Google Scholar]
- Cassandrini D, Calevo MG, Tessa A, Manfredi G, Fattori F, Meschini MC, Carrozzo R, Tonoli E, Pedemonte M, Minetti C, et al. A new method for analysis of mitochondrial DNA point mutations and assess levels of heteroplasmy. Biochem Biophys Res Commun. 2006;342:387–393. doi: 10.1016/j.bbrc.2006.01.152. [DOI] [PubMed] [Google Scholar]
- Chagnon P, Gee M, Filion M, Robitaille Y, Belouchi M, Gauvreau D. Phylogenetic analysis of the mitochondrial genome indicates significant differences between patients with Alzheimer disease and controls in a French-Canadian founder population. Am J Med Genet. 1999;85:20–30. doi: 10.1002/(sici)1096-8628(19990702)85:1<20::aid-ajmg6>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Crespillo M, Paredes MR, Prieto L, Montesino M, Salas A, Albarran C, Alvarez-Iglesias V, Amorin A, Berniell-Lee G, Brehm A, et al. Results of the 2003–2004 GEP-ISFG collaborative study on mitochondrial DNA: focus on the mtDNA profile of a mixed semen-saliva stain. Forensic Sci Int. 2005;160:157–167. doi: 10.1016/j.forsciint.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Crispim D, Canani LH, Gross JL, Tschiedel B, Souto KE, Roisenberg I. The European-specific mitochondrial cluster J/T could confer an increased risk of insulin-resistance and type 2 diabetes: an analysis of the m.4216T>C and m.4917A>G variants. Ann Hum Genet. 2006;70:488–495. doi: 10.1111/j.1469-1809.2005.00249.x. [DOI] [PubMed] [Google Scholar]
- Darvishi K, Sharma S, Bhat AK, Rai E, Bamezai RN. Mitochondrial DNA G10398A polymorphism imparts maternal Haplogroup N a risk for breast and esophageal cancer. Cancer Lett. 2007;249:249–255. doi: 10.1016/j.canlet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- De Benedictis G, Rose G, Carrieri G, De Luca M, Falcone E, Passarino G, Bonafe M, Monti D, Baggio G, Bertolini S, et al. Mitochondrial DNA inherited variants are associated with successful aging and longevity in humans. FASEB J. 1999;13:1532–1536. doi: 10.1096/fasebj.13.12.1532. [DOI] [PubMed] [Google Scholar]
- Filippini S, Blanco A, Fernandez-Marmiesse A, Alvarez-Iglesias V, Ruiz-Ponte C, Carracedo A, Vega A. Multiplex SNaPshot for detection of BRCA1/2 common mutations in Spanish and Spanish related breast/ovarian cancer families. BMC Med Genet. 2007;8:40. doi: 10.1186/1471-2350-8-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischel-Ghodsian N, Prezant TR, Chaltraw WE, Wendt KA, Nelson RA, Arnos KS, Falk RE. Mitochondrial gene mutation is a significant predisposing factor in aminoglycoside ototoxicity. Am J Otolaryngol. 1997;18:173–178. doi: 10.1016/s0196-0709(97)90078-8. [DOI] [PubMed] [Google Scholar]
- Fuku N, Park KS, Yamada Y, Nishigaki Y, Cho YM, Matsuo H, Segawa T, Watanabe S, Kato K, Yokoi K, et al. Mitochondrial haplogroup N9a confers resistance against type 2 diabetes in Asians. Am J Hum Genet. 2007;80:407–415. doi: 10.1086/512202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genasetti A, Valentino ML, Carelli V, Vigetti D, Viola M, Karousou EG, Melzi d'Eril GV, De Luca G, Passi A, Pallotti F. Assessing heteroplasmic load in Leber's hereditary optic neuropathy mutation 3460G->A/MT-ND1 with a real-time PCR quantitative approach. J Mol Diagn. 2007;9:538–545. doi: 10.2353/jmoldx.2007.060183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D, Marelli C, Achilli A, Goldwurm S, Pezzoli G, Barone P, Pellecchia MT, Stanzione P, Brusa L, Bentivoglio AR, et al. Mitochondrial DNA haplogroup K is associated with a lower risk of Parkinson's disease in Italians. Eur J Hum Genet. 2005;13:748–752. doi: 10.1038/sj.ejhg.5201425. [DOI] [PubMed] [Google Scholar]
- Goto Y, Nonaka I, Horai S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5:857–866. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- Hendrickson SL, Hutcheson HB, Ruiz-Pesini E, Poole JC, Lautenberger J, Sezgin E, Kingsley L, Goedert JJ, Vlahov D, Donfield S, et al. Mitochondrial DNA haplogroups influence AIDS progression. AIDS. 2008;22:2429–2439. doi: 10.1097/QAD.0b013e32831940bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson SL, Kingsley LA, Ruiz-Pesini E, Poole JC, Jacobson LP, Palella FJ, Bream JH, Wallace DC, O'Brien SJ. Mitochondrial DNA haplogroups influence lipoatrophy after highly active anti-retroviral therapy. J Acquir Immune Defic Syndr. 2009;51:111–116. doi: 10.1097/QAI.0b013e3181a324d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428–433. [PMC free article] [PubMed] [Google Scholar]
- Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus ML. A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am J Hum Genet. 1991;48:1147–1153. [PMC free article] [PubMed] [Google Scholar]
- Ivanova R, Lepage V, Charron D, Schachter F. Mitochondrial genotype associated with French Caucasian centenarians. Gerontology. 1998;44:349. doi: 10.1159/000022041. [DOI] [PubMed] [Google Scholar]
- Johns DR, Neufeld MJ, Park RD. An ND-6 mito-chondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem Biophys Res Commun. 1992;187:1551–1557. doi: 10.1016/0006-291x(92)90479-5. [DOI] [PubMed] [Google Scholar]
- Jones MM, Manwaring N, Wang JJ, Rochtchina E, Mitchell P, Sue CM. Mitochondrial DNA haplogroups and age-related maculopathy. Arch Ophthalmol. 2007;125:1235–1240. doi: 10.1001/archopht.125.9.1235. [DOI] [PubMed] [Google Scholar]
- Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at np 14459 of the ND6 gene associated with maternally inherited Leber's hereditary optic neuropathy and dystonia. Proc Natl Acad Sci USA. 1994;91:6206–6210. doi: 10.1073/pnas.91.13.6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khusnutdinova E, Gilyazova I, Ruiz-Pesini E, Derbeneva O, Khusainova R, Khidiyatova I, Magzhanov R, Wallace DC. A mitochondrial etiology of neurodegenerative diseases: evidence from Parkinson's disease. Ann N Y Acad Sci. 2008;1147:1–20. doi: 10.1196/annals.1427.001. [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Mishmar D, Ruiz-Pesini E, Brandon M, Wallace DC. Mitochondrial DNA-like sequences in the nucleus (NUMTs): insights into our African origins and the mechanism of foreign DNA integration. Hum Mutat. 2004;23:125–133. doi: 10.1002/humu.10304. [DOI] [PubMed] [Google Scholar]
- Mohlke KL, Jackson AU, Scott LJ, Peck EC, Suh YD, Chines PS, Watanabe RM, Buchanan TA, Conneely KN, Erdos MR, et al. Mitochondrial polymorphisms and susceptibility to type 2 diabetes-related traits in Finns. Hum Genet. 2005;118:245–254. doi: 10.1007/s00439-005-0046-4. [DOI] [PubMed] [Google Scholar]
- Nelson TM, Just RS, Loreille O, Schanfield MS, Podini D. Development of a multiplex single base extension assay for mitochondrial DNA haplogroup typing. Croat Med J. 2007;48:460–472. [PMC free article] [PubMed] [Google Scholar]
- Niemi AK, Hervonen A, Hurme M, Karhunen PJ, Jylha M, Majamaa K. Mitochondrial DNA polymorphisms associated with longevity in a Finnish population. Hum Genet. 2003;112:29–33. doi: 10.1007/s00439-002-0843-y. [DOI] [PubMed] [Google Scholar]
- Nishigaki Y, Yamada Y, Fuku N, Matsuo H, Segawa T, Watanabe S, Kato K, Yokoi K, Yamaguchi S, Nozawa Y, et al. Mitochondrial haplogroup N9b is protective against myocardial infarction in Japanese males. Hum Genet. 2007;120:827–836. doi: 10.1007/s00439-006-0269-z. [DOI] [PubMed] [Google Scholar]
- Parfait B, Rustin P, Munnich A, Rotig A. Co-amplification of nuclear pseudogenes and assessment of heteroplasmy of mitochondrial DNA mutations. Biochem Biophys Res Commun. 1998;247:57–59. doi: 10.1006/bbrc.1998.8666. [DOI] [PubMed] [Google Scholar]
- Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. 1993;4:289–294. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- Procaccio V, Neckelmann N, Paquis-Flucklinger V, Bannwarth S, Jimenez R, Davila A, Poole J, Wallace DC. Detection of low levels of the m.3243A>G mutation in mitochondrial DNA in blood derived from diabetic patients. Mol Diagn Ther. 2006;10:381–389. doi: 10.1007/BF03256215. [DOI] [PubMed] [Google Scholar]
- Quintans B, Alvarez-Iglesias V, Salas A, Phillips C, Lareu MV, Carracedo A. Typing of mitochondrial DNA coding region SNPs of forensic and anthropological interest using SNaPshot minisequencing. Forensic Sci Int. 2004;140:251–257. doi: 10.1016/j.forsciint.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Raby BA, Klanderman B, Murphy A, Mazza S, Camargo CA, Jr, Silverman EK, Weiss ST. A common mitochondrial haplogroup is associated with elevated total serum IgE levels. J Allergy Clin Immunol. 2007;120:351–358. doi: 10.1016/j.jaci.2007.05.029. [DOI] [PubMed] [Google Scholar]
- Rose G, Passarino G, Carrieri G, Altomare K, Greco V, Bertolini S, Bonafe M, Franceschi C, De Benedictis G. Paradoxes in longevity: sequence analysis of mtDNA haplogroup J in centenarians. Eur J Hum Genet. 2001;9:701–707. doi: 10.1038/sj.ejhg.5200703. [DOI] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Wallace DC. Evidence for adaptive selection acting on the tRNA and rRNA genes of the human mitochondrial DNA. Hum Mutat. 2006;27:1072–1081. doi: 10.1002/humu.20378. [DOI] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 2004;303:223–226. doi: 10.1126/science.1088434. [DOI] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Lott MT, Procaccio V, Poole J, Brandon MC, Mishmar D, Yi C, Kreuziger J, Baldi P, Wallace DC. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007;35:D823–D828. doi: 10.1093/nar/gkl927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychlik W, Spencer WJ, Rhoads RE. Optimization of the annealing temperature for DNA amplification in vitro. Nucleic Acids Res. 1990;18:6409–6412. doi: 10.1093/nar/18.21.6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas A, Quintans B, Alvarez-Iglesias V. SNaPshot typing of mitochondrial DNA coding region variants. Methods Mol Biol. 2005;297:197–208. doi: 10.1385/1-59259-867-6:197. [DOI] [PubMed] [Google Scholar]
- SantaLucia J., Jr A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarzi E, Brown M, Lebon S, Chretien D, Munnich A, Rotig A, Procaccio V. A novel recurrent mitochondrial DNA mutation in ND3 gene is associated with isolated complex I deficiency causing Leigh syndrome and dystonia. Am J Med Genet. 2007;143A:33–41. doi: 10.1002/ajmg.a.31565. [DOI] [PubMed] [Google Scholar]
- Saxena R, de Bakker PI, Singer K, Mootha V, Burtt N, Hirschhorn JN, Gaudet D, Isomaa B, Daly MJ, Groop L, et al. Comprehensive association testing of common mitochondrial DNA variation in metabolic disease. Am J Hum Genet. 2006;79:54–61. doi: 10.1086/504926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, Wallace DC. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- Shoffner JM, Brown MD, Torroni A, Lott MT, Cabell MR, Mirra SS, Beal MF, Yang C, Gearing M, Salvo R, et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics. 1993;17:171–184. doi: 10.1006/geno.1993.1299. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Gong JS, Zhang J, Yoneda M, Yagi K. Mitochondrial genotype associated with longevity. Lancet. 1998;351:185–186. doi: 10.1016/S0140-6736(05)78211-8. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Gong J, Zhang J, Yamada Y, Borgeld HJ, Yagi K. Mitochondrial genotype associated with longevity and its inhibitory effect on mutagenesis. Mech Ageing Dev. 2000;116:65–76. doi: 10.1016/s0047-6374(00)00149-4. [DOI] [PubMed] [Google Scholar]
- Torroni A, Petrozzi M, D'Urbano L, Sellitto D, Zeviani M, Carrara F, Carducci C, Leuzzi V, Carelli V, Barboni P, et al. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet. 1997;60:1107–1121. [PMC free article] [PubMed] [Google Scholar]
- Udar N, Atilano SR, Memarzadeh M, Boyer DS, Chwa M, Lu S, Maguen B, Langberg J, Coskun P, Wallace DC, et al. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:2966–2974. doi: 10.1167/iovs.08-2646. [DOI] [PubMed] [Google Scholar]
- Vallone PM, Just RS, Coble MD, Butler JM, Parsons TJ. A multiplex allele-specific primer extension assay for forensically informative SNPs distributed throughout the mitochondrial genome. Int J Legal Med. 2004;118:147–157. doi: 10.1007/s00414-004-0428-5. [DOI] [PubMed] [Google Scholar]
- van den Bosch BJ, de Coo RF, Scholte HR, Nijland JG, van Den Bogaard R, de Visser M, de Die-Smulders CE, Smeets HJ. Mutation analysis of the entire mitochondrial genome using denaturing high performance liquid chromatography. Nucleic Acids Res. 2000;28:E89. doi: 10.1093/nar/28.20.e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt JM, Nicodemus KK, Martin ER, Scott WK, Nance MA, Watts RL, Hubble JP, Haines JL, Koller WC, Lyons K, et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am J Hum Genet. 2003;72:804–811. doi: 10.1086/373937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt JM, Dementieva YA, Martin ER, Scott WK, Nicodemus KK, Kroner CC, Welsh-Bohmer KA, Saunders AM, Roses AD, Small GW, et al. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci Lett. 2004;365:28–32. doi: 10.1016/j.neulet.2004.04.051. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005a;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondria and cancer: Warburg address. Cold Spring Harb Symp Quant Biol. 2005b;70:363–374. doi: 10.1101/sqb.2005.70.035. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Why do we have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem. 2007;76:781–821. doi: 10.1146/annurev.biochem.76.081205.150955. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, Nikoskelainen EK. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988a;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Zheng X, Lott MT, Shoffner JM, Hodge JA, Kelley RI, Epstein CM, Hopkins LC. Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell. 1988b;55:601–610. doi: 10.1016/0092-8674(88)90218-8. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Stugard C, Murdock D, Schurr T, Brown MD. Ancient mtDNA sequences in the human nuclear genome: a potential source of errors in identifying pathogenic mutations. Proc Natl Acad Sci USA. 1997;94:14900–14905. doi: 10.1073/pnas.94.26.14900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Brown MD, Lott MT. Mitochondrial DNA variation in human evolution and disease. Gene. 1999;238:211–230. doi: 10.1016/s0378-1119(99)00295-4. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Lott MT, Procaccio V. Mitochondrial genes in degenerative diseases, cancer and aging. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Emery and Rimoin's Principles and Practice of Medical Genetics. 5th. Philadelphia, PA, USA: Churchill Livingstone Elsevier; 2007. pp. 194–298. [Google Scholar]