

Abstract

In this study the μ opioid receptor (MOR) ligands DALDA (Tyr-d-Arg-Phe-Lys-NH2) and Dmt1-DALDA (Dmt-d-Arg-Phe-Lys-NH2, Dmt = 2′,6′-dimethyltyrosine) were glycosylated at the N- or C-terminus. Subsequently, the modified peptides were subjected to in vitro and in vivo evaluation. In contrast to the N-terminally modified peptide (3), all peptide analogues derivatized at the C-terminus (4–7) proved to possess high affinity and agonist potency at both MOR and DOR (δ opioid receptor). Results of the Caco-2 monolayer permeation, as well as in vitro blood–brain barrier model experiments, showed that, in the case of compound 4, the glycosylation only slightly diminished the lumen-to-blood and blood-to-lumen transport. Altogether, these experiments were indicative of transcellular transport but not active transport. In vivo assays demonstrated that the peptides were capable of (i) crossing the blood–brain barrier (BBB) and (ii) activating both the spinal ascending as well as the descending opioid pathways, as determined by the tail-flick and hot-plate assays, respectively. In contrast to the highly selective MOR agonist Dmt1-DALDA 1, compounds 4–7 are mixed MOR/DOR agonists, expected to produce reduced opioid-related side effects.
Keywords: Opioid peptides, glycosylation, in vivo antinociception, Dmt1-DALDA
One strategy to improve the permeation of bioactive opioid peptides through several biological barriers consists of the conjugation of one or several sugar units.1,2 This strategy, termed glycosylation, can potentially (i) improve the intrinsically poor metabolic stability of peptides, (ii) enhance their systemic bioavailability, or can even (iii) target carriers systems located at physiological barriers, such as the blood–brain barrier (BBB). The metabolic stabilization of peptides is necessary to enhance in vivo half-life in order to maintain the targeted physiologic effect. In the opioid peptide field, glycosylation has been used as a method to increase enzymatic stability2,3 and to improve the analgesic profile of several opioid peptides.4
In previous studies, the synthesis of opioid glycopeptides strongly relied on the use of Fmoc-amino acid O-linked glycosides.4 This methodology, using, for example, O-glycosylated Fmoc-Ser, -Thr, or -Hyp residues, may however require heating for coupling of both the glycosidic residue and the subsequent amino acid.4,5 Alternatively, C-terminal amide-linked carbohydrate-containing opioid peptides6 and glucosyl and galactosyl peptide esters have been reported.7,8 The in vitro pharmacological evaluation of these peptides revealed that bioactivity was dependent on the linker type, the identity of the sugar unit, and the position of conjugation.4 With respect to the anchorage site and on the basis of previous research, two different conjugation positions were considered in this work. In the past, numerous glycosylations have been carried out on the enkephalin sequences.1−9 When the glycosylated residue was introduced at the C-terminus (e.g., 2 in Figure 1) of the sequence, high affinity for the μ opioid receptor was maintained.4 In contrast, when the sugar was attached in the proximity of the N-terminus in an enkephalin-based sequence, a substantial loss in potency was apparent.9 In addition, early SAR studies prematurely concluded that structural modifications at the phenolic function of aromatic amino acids in position 1 of opioid peptides would be detrimental for analgesic activity.10 It turned out that the replacement of Tyr1 by p-carboxamidoPhe (p-CONH2-Phe) gave compounds with retained activity.10−12
Figure 1.

Structures of Dmt1-DALDA 1, Enkephalin[1–4]-Ser(OGlu)-NH22 and glycosylated analogues 3 to 7.
Herein, the phenolic function of Tyr1 also served as a modification site. Because the Huisgen 1,3-dipolar cycloaddition has been shown to be a facile, robust, and bioorthogonal conjugation method;13,14 it was employed to anchor a glucose or galactose unit to an opioid tetrapeptide sequence, based on the structure of the μ-opioid receptor (MOR) peptide lead Dmt1-DALDA, Dmt-d-Arg-Phe-Lys-NH21 (Figure 1).15 Earlier studies identified this peptide as a highly potent and selective MOR agonist with a high binding affinity (Kiμ = 0.143 nM) and high metabolic stability.16 Moreover, [Dmt1]DALDA is able to cross the BBB.16 Despite these favorable pharmacodynamic and -kinetic properties and in order to optimize the structure of Dmt1-DALDA 1 for oral administration, the glycoconjugation strategy was employed in this work. Anchorage of the azido-sugars was performed on solid support and in solution phase synthesis. The peptides were pharmacologically characterized in vitro using opioid receptor binding assays and the functional guinea pig ileum (GPI) and mouse vas deferens (MVD) assays, and in vivo using the tail-flick and hot plate assays (vide infra).
The glycosylated peptides 3–7 can be obtained by introducing the protected glycosyl building blocks during the synthesis of the peptide sequence or by conjugation of the sugar once the peptide is synthesized. Since this latter method is the most convergent, it was selected in this study. The peptides 1 and the precursors of the glycosylated peptides 3–7, i.e., H-Tyr(propargyl)-d-Arg-Phe-Lys-NH2, H-Dmt-d-Arg-Phe-Pra-NH2, H-Dmt-d-Arg-Phe-Ser(propargyl)-NH2 , and H-Dmt-d-Arg-Phe-Tyr(propargyl)-NH2, respectively, were synthesized via standard solid phase peptide synthesis using Rink amide resin as the solid support and Nα-Fmoc (or Nα-Boc for terminal residue) protected amino acids. Boc-Dmt-OH or Boc-Tyr(propargyl)-OH were used as final amino acids in the peptide assembly in order to obtain fully deprotected peptides after cleavage from the resin under acidolytic conditions. The coupling of Boc-Dmt-OH was performed using 3 equiv of DIC/3 equiv of HOBt as the coupling mixture to avoid side reactions that occur when TBTU/DIPEA is used. Cleavage of the peptide from the resin and removal of the side chain protecting groups was achieved using a mixture of TFA/TES/H2O 95:2.5:2.5 v/v/v for 3 h. After evaporation of the cleavage mixture, precipitation in cold Et2O, and purification by preparative-HPLC, all compounds were obtained in high purity (>95%).
The conjugation of the galactose or glucose unit in peptides 3 to 7 was performed both on solid phase and in solution using unprotected azido-sugar (Scheme 1). For the reaction on solid phase, 8 equiv of the azido-sugar, 0.5 equiv of of CuBr, and 4 equiv of DIPEA were added to the resin-bound precursor swollen in DMF and shaken for 5 days. The cleavage of the peptide from the resin, the removal of side chain protecting groups, and the purification of the final compounds were carried out as described above. The conjugation of the monosaccharide in solution was performed by the addition of 6 equiv of sodium ascorbate, 4 equiv of CuSO4·5H2O, and 1.5 equiv of the azidosugar to a solution of the fully deprotected precursor in tBuOH/water 1:2 v/v. After overnight reaction at room temperature, the crude peptide was purified as described above. The reaction in solution is preferred, given the smaller amount of azido-sugar needed, the shorter reaction time and the increased yields after purification.
Scheme 1. Glycosylation by Huisgen Cycloaddition on Solid Phase (A) and in Solution (B).

Reference compound 1 and the glycosylated analogues (3 to 7) were subjected to in vitro binding and tissue functional assays. Despite a major loss in binding affinity, as compared to all other analogues, the data of compound 3 indicate that the derivatization of the phenolic position interferes with the ligand–receptor interaction but still allows for significant binding to the MOR. Despite the bulky sugar unit in 3, moderate binding (Kiμ 38 nM) is still observed. One needs to point out that in this specific case a Tyr was derivatized out of economic consideration, not the 2′,6′-dimethyltyrosine (Dmt), a Tyr analogue known to boost opioid agonist potency and receptor binding affinity.16,17 In terms of binding affinity and functional activity, DALDA (Tyr-d-Arg-Phe-Lys-NH2) is characterized by a Kiμ of 1.69 nM and GPI IC50 of 254 nM. When taking this into consideration, only a 22-fold loss in binding and 4.6-fold loss in functional activity resulted. Nonetheless, this anchorage site was not further pursued. When anchoring the sugar unit through Pra, Ser, or Tyr residues in position 4, MOR binding and activation (GPI assay) were preserved, as can be seen from comparison of 1 with compounds 4–7. In the case of compounds 4 and 5, the conjugated galactose and glucose subunits were found to be compatible with MOR activity (Kiμ, 0.222 and 0.203 nM, and GPI IC50, 0.66 and 1.46 nM, respectively). However, upon replacement of the aminoalkyl side chain of Lys4 in 1 with a triazolyl-Glu/Gal functionality, the μ/δ receptor binding selectivity ratio was dramatically lowered (Ki μ/δ ratio 11 400 (1) → 70 (4) and 58 (5)). The high Kiδ value of 1 (2100 nM) shifted to 15.6 and 11.9 nM for 4 and 5, respectively. Galactose-derivatized compounds 6 and 7 behaved similarly, showing extremely high, subnanomolar affinity for MOR and high DOR binding affinity (low nanomolar) (Table 1). These values are in qualitative agreement with the functional GPI and MVD assay data.
Table 1. Receptor MOR/DOR Binding Affinities and in Vitro Potency Dataa.
| no. | MOR Ki (nM) | DOR Ki (nM) | Ki ratio μ/δ | GPI(μ) IC50(nM) | MVD(δ) IC50 (nM) |
|---|---|---|---|---|---|
| 1 | 0.143 | 2100 | 11400 | 1.41 | 23.1 |
| 3 | 38.4 | >5000 | >130 | 1180 | 1990 |
| 4 | 0.222 | 15.6 | 70.3 | 0.66 | 1.98 |
| 5 | 0.203 | 11.9 | 58.6 | 1.46 | 2.75 |
| 6 | 0.112 | 6.36 | 56.8 | 0.35 | 4.9 |
| 7 | 0.286 | 22.2 | 77.6 | 2.3 | 6.00 |
Values represent means of 3–4 experiments. The GPI functional assay is representative of MOR activation, whereas the MVD is a DOR receptor-representative assay. Binding affinities of compounds for MOR and DOR opioid receptors were determined by displacement of [3H]DAMGO ([d-Ala2,NMePhe4,Gly-ol5]enkephalin) and [3H]DSLET ([d-Ser2,Leu5]enkephalin-Thr6) from rat brain membrane binding sites, respectively.
On the basis of the in vitro profile of compounds 4–7, peptide 4 was selected as a mixed MOR/DOR opioid agonist for evaluation in the Caco-2 cell monolayer translocation assay in comparison with [Dmt1]DALDA 1. Several studies provided evidence that compounds with dual MOR/DOR activity may have beneficial pharmacological effects in comparison to highly selective MOR agonists.18
The Caco-2 assay is used to perform an in vitro evaluation of intestinal uptake mechanisms. To allow for quantification, both peptides were labeled with 3H as previously described (see Supporting Information). The permeabilities of reference peptide 1 and glycopeptide 4 were measured in both gut–blood and blood–gut directions, across 21 day old Caco-2 cell monolayers (Figure 2) using standard protocols.19 The lumen–blood (A–B) and blood–lumen (B–A) permeabilities of Dmt-DALDA 1 and compound 4 were slightly higher than the permeability of the passive flux marker mannitol. For both peptides there was a slightly (but significantly) higher B–A permeability, as compared to A–B permeability, which possibly could be due to efflux transporters. The A–B and B–A fluxes were, however, of similar magnitude. The permeability of 4 was slightly, but significantly, lower than that of 1. There was no evidence for uptake transporter interactions for the substrates, as the A–B transport could not be inhibited by excess unlabeled substrate (Table 2). The slightly lower permeability of 4, as compared to 1, indicates that molecular mass is rate-limiting, which in turn suggests paracellular or passive transcellular transport. Both peptides displayed a permeability slightly higher than that of mannitol, although they also have a higher molecular weight. If transepithelial transport occurred via paracellular passage only, it would be expected that both peptides displayed a markedly lower permeability than that of mannitol, due to their much larger hydrodynamic radius. This indicates that a large fraction of the transepithelial transport of the peptides occurs via a transcellular route, possibly due to the cationic nature of the peptides. The permeabilities of mannitol in either direction are identical. The mannitol fluxes do not vary significantly in the presence of 1 and 4 (not shown), indicating that neither Dmt-DALDA 1 nor compound 4 increase the paracellular permeability (which can be the case if compounds show cell toxicity). The bioavailability of the two peptides cannot be directly calculated from the in vitro experiments; however, the permeability in Caco-2 cell monolayers of stable passively permeating compounds has been found to correlate with bioavailability.20 Given that the peptides have permeabilities in the range of the mannitol permeability, one would expect oral bioavailability similar to that of mannitol, i.e., ∼20%.21 This is comparable to the oral bioavailability of morphine22 and may be sufficient for oral dosing, taking into account the high potency of the peptides.
Figure 2.
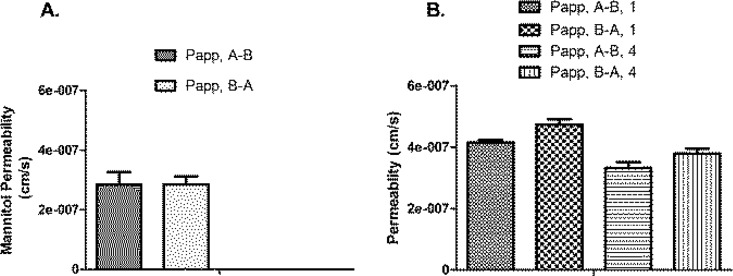
Steady-state permeabilities of (A) 14C-mannitol (1.7 μM) and (B) 3H-Dmt-DALDA 1* (13 nM) and 3H-labeled 4 (4*) (13 nM) across Caco-2 monolayers cultured for 21 days. Three individual experiments were performed (n = 3), and within each experiment, measurements were performed in triplicate (N = 3), except for the pooled mannitol data (n = 3, N = 12). Data are shown as means ± SEM (all values were calculated from the flux values, using Ficks law, i.e., permeability = flux × concentration gradient–1).
Table 2. Fluxes of Radiolabelled 1* and 4* at cis-Concentration of 13 nM, Across Caco-2 Cell Monolayers in the Lumen–Blood (A–B) and Blood–Lumen (B–A) Direction, in the Absence and Presence of 100 μM of Unlabelled (Cold) Compounds 1 and 4.
| 1* | 1* + cold 1 | p | 4* | 4* + cold 4 | p | |
|---|---|---|---|---|---|---|
| A–B | 3.24 ± 0.06 | 3.13 ± 0.13 | 0.62 | 2.59 ± 0.15 | 2.51 ± 0.12 | 0.24 |
| B–A | 3.7 ± 0.14 | 3.64 ± 0.13 | 0.66 | 2.96 ± 0.13 | 2.87 ± 0.09 | 0.7 |
*Labeled ligands. Fluxes are in nmol·cm–2·min–1 × 10–7. Flux values are shown as means ± SE, n = 3 cell passages for all experiments, and triplicate determinations were performed within each cell passage.
The permeability of 1 and 4 was further investigated in a bovine endothelial/rat astrocyte in vitro blood–brain barrier model.23 The transcellular electrical resistance in the model was 1276 ± 261 Ω cm2 (N = 12) confirming a tight monolayer with minimal space for paracellular diffusion (Figure 3).
Figure 3.

Steady-state transport of 1 (13 nM) and compound 4 (13 nM) across blood–brain barrier endothelial cells cocultured with astrocytes. Measurements were performed in triplicate (N = 3). Data are shown as means ± SEM.
As observed in the Caco-2 cell monolayers, the permeabilities of both compounds were similar in both directions, indicative of passive diffusion. Both peptides showed significantly higher permeability values than mannitol indicative of a transcellular transport route for the peptides. Previous in vivo data have shown the same tendencies with a relatively high brain influx of 1,24 compared to that of mannitol.25
Finally, the in vivo antinociceptive potency of the most potent compounds 4 and 5, as well as the reference peptide Dmt1-DALDA 1, were determined in the mouse tail-flick assay (Figure 4) and the mouse hot plate assay (Figure 5).
Figure 4.

Antinociceptive effect induced by intravenously administered compounds 1, 4, and 5 at a dose of 0.5 mg/kg in the tail-flick test (C57Bl6 mice; 23–25 g of body weight). *p ≤ 0.05, **p ≤ 0.01 (1 vs 5); (**)p ≤ 0.01 (1 vs 4).
Figure 5.

Antinociceptive effect induced by intravenously administered compounds 1, 4, and 5 at a dose of 0.5 mg/kg in the hot plate test (C57Bl6 mice; 23–25 g of body weight). (**)p ≤ 0.01 (1 and 4).
A systemically administered dose of 0.5 mg/kg of all three compounds produced a strong time- and dose-dependent analgesic response both at the spinal (tail-flick assay, Figure 4) and supraspinal (hot plate assay, Figure 5) level. In the tail-flick test, compounds 4 and 5 showed similar antinociceptive responses, as compared to the reference compound 1. All compounds exerted a peak pain relieving effect 60 min after administration. However, the observed maximal possible effect (%MPE) produced by compounds 4 and 5 was lower than that of reference peptide 1 (Figure 4). The dose–response curves result in higher ED50 values for both peptides 4 and 5 (Table 3) indicating a lower potency in this assay. Within the next two hours the analgesia produced by Dmt-DALDA 1 decreased and leveled with that of 4 and 5.
Table 3. ED50 Values Determined for Peptides 1, 4, and 5, Respectively, as Calculated Using Standard Nonlinear Regression Analysis of the Log Dose–Response Curvea.
| ED50 values (mg/kg) |
||
|---|---|---|
| no. | hot-plate test | tail-flick test |
| 1 | 0.25++ | 0.17 ** ,+, ### |
| 4 | 0.40 | 0.27* |
| 5 | 0.31 | 0.34 |
**hot-plate vs. tail-flick; ++p < 0.01; +p < 0.05 (1 vs. 4); ###p < 0.001 (1 vs. 5).
In the hot-plate test, that primarily determines analgesia resulting from the activation of supraspinal sites, compound 5 showed both a similar time course and magnitude of analgesia as the reference compound 1. The peak analgesic effect of compound 1 was observed at 30 min, while compound 4 exerted its maximum response after 60 min (Figure 5). The ED50 values for compounds 1 and 5 were similar (Table 3), whereas the ED50 for compound 4 was slightly higher (0.40 mg/kg, Table 3). Importantly, in both analgesic tests all three compounds showed a sustained antinociceptive effect at the level of 40–60% MPE even 180 min after administration. These results are in agreement with a performed stability assay. The metabolic plasma stability of compounds 4 and 5 was compared with the one of Dmt-Pro-Phe-Phe-NH2, an opioid peptide with a reported t1/2 of 39.55 min in rat brain homogenate (see Supporting Information). In our study this peptide presented a t1/2 of 53 min in human plasma, whereas no degradation products could be observed for peptides 4 and 5 up to 20 h.
As shown, the glycosylated compounds 4 and 5 acted via the spinal ascending as well as the descending pathway, which is a circuitry of the supraspinal CNS. Opioids manifest their analgesic action by modulation of both pathways.26 Populations of different types of opioid receptors were discovered to be expressed in the dorsal root ganglia, spinal cord, and the trigeminal nucleus constituting the ascending pain pathway. Opioid receptors are also expressed in the rostal vental medulla (RVM), the periaqueductal gray matter (PAG), and nucleus locus coeruleus (NLC), the main structures involved in descending pain control.
Opioid action via activation of the antinociceptive descending pathway is particularly important in chronic inflammatory or neuropathic pain relief by modulating, for example, GABA-ergic and adrenergic inputs.27,28 These observations emphasize the importance of the supraspinal action of potential new drugs for persistent pain relief. Hence, the discovery and design of compounds counteracting chronic pain should focus on those with both strong supraspinal action and long duration of action, properties exhibited by compounds 4 and 5. Although these glycosylated compounds did not show a net improvement in antinociceptive potency, as compared to the parent structure 1, they do represent alternative peptide lead molecules with a potentially favorable dual MOR/DOR agonist profile.18 Given that these peptides have permeabilities comparable to that of mannitol, an oral bioavailability similar to that of morphine is expected22 and may be sufficient for oral dosing, taking into account the high potency of the peptides.
Glossary
ABBREVIATIONS
- BBB
blood–brain barrier
- CNS
central nervous system
- DAMGO
[d-Ala2,NMePhe4,Gly-ol5]enkephalin
- DIC
N,N-diisopropylcarbodiimide
- DIPEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylformamide
- Dmt
2′,6′-dimethyl-(S)-tyrosine
- DOR
δ-opioid receptor
- DSLET
[d-Ser2,Leu5]enkephalin-Thr6
- GPI
guinea pig ileum
- HOBt
1-hydroxybenzotriazole
- Kin
unidirectional brain influx or uptake transfer constant
- KOR
κ-opioid receptor
- kout
brain efflux rate coefficient
- MOR
μ-opioid receptor
- MPE
maximal possible effect
- MVD
mouse vas deferens
- Pra
propargylglycine
- TBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate
- Pbf
2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl
- Vbr
brain distribution volume
Supporting Information Available
Experimental procedures as well as compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
¶ S.B. and C.B. contributed equally. The manuscript was written through contributions of all authors. S.B., C.B., A.N., and D.T. were in charge of the peptide synthesis and writing the manuscript. A.L., P.K., and A.W.L. have provided the experimental in vivo data. C.T. performed the H-3 labeling. C.U.N. and B.B. performed the Caco-2 cell monolayer permeability assay. H.C.H. performed the in vitro blood-brain barrier study. N.N.C. and P.W.S. performed the in vitro binding assays and the functional bioassays.
The work of S.B., C.B., A.N., D.T., and P.W.S. was supported by a collaboration convention between the Ministère du Développement Economique, de l′Innovation et de l′Exportation du Québec and the Research Foundation–Flanders (FWO Vlaanderen) (PSR-SIIRI-417), and by grants from the CIHR (MOP-89716) and the NIH (DA-004443). Financial support from the Hungarian Scientific Research Fund (K77783, CT) and the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (CT) are also acknowledged.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Egleton R. D.; Mitchell S. A.; Huber J. D.; Janders J.; Stropova D.; Polt R.; Yamamura H. I.; Hruby V. J.; Davis T. P. Improved bioavailability to the brain of glycosylated Met-enkephalin analogs. Brain Res. 2000, 881, 37–46. [DOI] [PubMed] [Google Scholar]
- Polt R.; Dhanasekaran M.; Keyari C. M. Glycosylated neuropeptides: a new vista for neuropsychopharmacology?. Med. Res. Rev. 2005, 25, 557–585. [DOI] [PubMed] [Google Scholar]
- Yamamoto T.; Nair P.; Ma S.; Davis P.; Yamamura H. I.; Vanderah T. W.; Porreca F.; Lai J.; Hruby V. J. The biological activity and metabolic stability of peptidic bifunctional compounds that are opioid receptor agonists and neurokinin-1 receptor antagonists with a cystine moiety. Bioorg. Med. Chem. 2009, 17, 7337–7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Lefever M. R.; Muthu D.; Bidlack J. M.; Bilsky E. J.; Polt R. Opioid glycopeptide analgesics derived from endogenous enkephalins and endorphins. Future Med. Chem. 2012, 4, 205–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R. E.; Rodriguez F. D.; Sacristán M. P.; Torres J. L.; Valencia G.; Garcia Antón J. M. New glycosylpeptides with high antinociceptive activity. Neurosci. Lett. 1989, 101, 89–94. [DOI] [PubMed] [Google Scholar]
- Torres J. L.; Reig F.; Valencia G.; Rodriguez R. E.; Garcia Antón J. M. [d-Met2, Pro5] enkephalin [N1.5-β-d-glucopyranosyl] amide: a glycosylpeptide with high antinociceptive activity. Int. J. Pept. Protein Res. 1988, 31, 474–480. [DOI] [PubMed] [Google Scholar]
- Horvat Š.; Horvat J.; Varga-Defterdarović L.; Pavelić K.; Chung N. N.; Schiller P. W. Methionine–enkephalin related glycoconjugates. Int. J. Pept. Protein Res. 1993, 41, 399–404. [PubMed] [Google Scholar]
- Skcrić M.; Horvat J.; Horvat Š.; Chung N. N.; Schiller P. W. Acetylated glucopyranosyl esters of enkephalins. Int. J. Pept. Protein Res. 1994, 43, 402–409. [DOI] [PubMed] [Google Scholar]
- Polt R.; Porreca F.; Szabò L. Z.; Bilsky E. J.; Davis P.; Abbruscato T. J.; Davis T. P.; Horvat R.; Yamamura H. I.; Hruby V. J. Glycopeptide enkephalin analogues produce analgesia in mice: evidence for penetration of the blood–brain barrier. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 7114–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezowska I.; Chung N. N.; Lemieux C.; Wilkes B. C.; Schiller P. W. Agonist vs antagonist behavior of δ opioid peptides containing novel phenylalanine analogues in place of Tyr1. J. Med. Chem. 2009, 52, 6941–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolle R. E.; Machaut M.; Martinez-Teipel B.; Belanger S.; Cassel J. A.; Stabley G. J.; Graczyk T. M.; DeHaven R. N. (4-Carboxamido)phenylalanine is a surrogate for tyrosine in opioid receptor peptide ligands. Bioorg. Med. Chem. Lett. 2004, 14, 3545–3548. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Luo J.; Liu C.; Weltrowska G.; Lemieux C.; Chung N. N.; Lu Y.; Schiller P. W. Novel opioid peptide derived antagonists containing (2S)-2-methyl-3-(2,6-dimethyl-4-carbamoylphenyl)propanoic acid [(2S)-Mdcp]. J. Med. Chem. 2008, 51, 5866–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumurugan P.; Matosiuk D.; Jozwiak K. Click Chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 2013, 113, 4905–4979. [DOI] [PubMed] [Google Scholar]
- Meldal M.; Tornøe C. W. Cu-catalyzed azide–alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [DOI] [PubMed] [Google Scholar]
- Schiller P. W.; Nguyen T. M.; Berezowska I.; Dupuis S.; Weltrowska G.; Chung N. N.; Lemieux C. Synthesis and in vitro opioid activity profiles of DALDA analogues. Eur. J. Med. Chem. 2000, 35, 895–901. [DOI] [PubMed] [Google Scholar]
- Schiller P. W. Bi- or multifunctional opioid peptide drugs. Life Sci. 2010, 86, 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D. W. Jr.; Stapelfeld A.; Savage M. A.; Reichman M.; Hammond D. L.; Haaseth R. C.; Mosberg H. I. Systemic analgesic activity and delta-opioid selectivity in [2,6-dimethyl-Tyr1,d-Pen2,d-Pen5]enkephalin. J. Med. Chem. 1992, 35, 684–687. [DOI] [PubMed] [Google Scholar]
- Ananthan S. Opioid Ligands with Mixed μ/δ Opioid Receptor Interactions: An Emerging Approach to Novel Analgesics. AAPS J. 2006, 8, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo S. A.; Nielsen C. U.; Amstrup J.; Froekjaer S.; Brodin B. In-depth evaluation of Gly-Sar transport parameters as a function of culture time in the Caco-2 cell model. Eur. J. Pharm. Sci. 2004, 21, 77–86. [DOI] [PubMed] [Google Scholar]
- Artursson P.; Palm K.; Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Delivery Rev. 2001, 46, 27–43. [DOI] [PubMed] [Google Scholar]
- Laker M. F.; Bull H. J.; Menzies I. S. Evaluation of mannitol for use as a probe marker of gastrointestinal permeability in man. Eur. J. Clin. Invest. 1982, 12, 485–91. [DOI] [PubMed] [Google Scholar]
- Hoskin P. J.; Hanks G. W.; Aherne G. W.; Chapman D.; Littleton P.; Filshie J. The bioavailability and pharmacokinetics of morphine after intravenous, oral and buccal administration in healthy volunteers. Br. J. Clin. Pharmacol. 1989, 27, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms H. C.; Waagepetersen H. S.; Nielsen C. U.; Brodin B. Para-cellular tightness and claudin-5 expression is increased in the BCEC/astrocyte blood–brain barrier model by increasing media buffer capacity during growth. AAPS J. 2010, 12, 759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa A.; Van Dorpe S.; Wynendaele E.; Spetea M.; Bracke N.; Stalmans S.; Betti C.; Chung N. N.; Lemieux C.; Zuegg J.; Cooper M. A.; Tourwé D.; De Spiegeleer B.; Schiller P. W.; Ballet S. Variation of the net charge, lipophilicity, and side chain flexibility in Dmt1-DALDA: Effect on opioid activity and biodistribution. J. Med. Chem. 2012, 55, 9549–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston J. E.; Al-Sarraf H.; Segal M. B. Permeability of the developing blood-brain barrier to 14C-mannitol using the rat in situ brain perfusion technique. Dev. Brain Res. 1995, 87, 69–76. [DOI] [PubMed] [Google Scholar]
- Mansour A.; Khachaturian H.; Lewis M. E.; Akil H.; Watson S. J. Review anatomy of CNS opioid receptors. Trends Neurosci. 1988, 11, 308–314. [DOI] [PubMed] [Google Scholar]
- Vaughan C. W.; Ingram S. L.; Connor M. A.; Christie M. J. How opioids inhibit GABA-mediated neuro-transmission. Nature 1997, 390, 611–614. [DOI] [PubMed] [Google Scholar]
- Hurley R. W.; Hammond D. L. The analgesic effects of supraspinal mu and delta opioid receptor agonists are potentiated during persistent inflammation. J. Neurosci. 2000, 20, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.