

Abstract
The aims of this study were to describe emtricitabine concentration-time courses in a large population of HIV-1-infected adults, to evaluate the influence of renal function on emtricitabine disposition, and to assess current dosing adjustment recommendations. Emtricitabine blood plasma concentrations were determined from samples collected from 161 adult patients during therapeutic drug monitoring and measured by liquid chromatography coupled to tandem mass spectrometry. The data were analyzed by a population approach. Emtricitabine pharmacokinetics was best described by a two-compartment model in which the absorption and distribution rate constants were assumed to be equal. Typical population parameter estimates (interindividual variability) were apparent elimination and intercompartmental clearances of 15.1 liters/h (17.4%) and 5.75 liters/h, respectively, and apparent central and peripheral volumes of distribution of 42.3 liters and 55.4 liters, respectively. The apparent elimination clearance was significantly related to creatinine clearance (CLCR), reflecting renal function. For 200 mg once a day (QD), the median area under the concentration-time curve over 24 h (AUC0-24) was 12.5 mg · h/liter for patients with normal renal function (CLCR, >80 ml/min), 14.7 mg · h/liter for patients with mild renal impairment (CLCR, 79 to 50 ml/min), and 17.9 mg · h/liter for patients with moderate renal impairment (CLCR, 49 to 30 ml/min). Simulations of the recommended dosing schemes for the oral solid form of emtricitabine (i.e., 200 mg per 48 h according to renal function) led to lower emtricitabine exposures for patients with moderate renal impairment (median AUC0-48, 17.2 mg · h/liter) than for patients with normal renal function (median AUC0-48, 25.6 mg · h/liter). Administering 18 ml of emtricitabine oral solution (10 mg/ml) QD to patients with moderate renal impairment should yield emtricitabine exposures similar to those in patients with normal renal function.
INTRODUCTION
Emtricitabine (FTC) is a potent nucleoside reverse transcriptase inhibitor (NRTI) widely used as part of first-line regimens for the combined treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults (1). While the pharmacokinetics of other NRTI, such as tenofovir or lamivudine, has been well documented, the pharmacokinetics of FTC has been much less studied. The pharmacokinetics of a 200-mg FTC dose given once daily (QD) has been described by noncompartmental analysis in studies performed in 6, 17, or 21 healthy volunteers (2–4). It has also been described in two very small studies that included HIV-1 infected adults (n = 20) (5, 6).
FTC is mainly eliminated by renal excretion, with 86% of an oral dose recovered unchanged in urine (5). FTC renal elimination combines glomerular filtration and active tubular secretion. Following the administration of one 200-mg FTC capsule QD, a significant increase in FTC exposure (area under the concentration-time curve over 24 h [AUC0-24]) has been shown in healthy volunteers with moderate renal impairment (creatinine clearance [CLCR], 49 to 30 ml/min). Thus, a dosing interval adjustment of one 200-mg capsule every 48 h has been recommended in this subpopulation (5). However, this study has some limitations: the influence of renal function on FTC pharmacokinetics has not been determined in HIV-1 infected patients, a small number of healthy volunteers with moderate renal impairment (n = 6) have been included, and the safety and efficacy of the recommended dosing adjustment (200 mg every 48 h) have not been clinically evaluated (5).
The aims of this study were (i) to describe the pharmacokinetics of FTC in a large population of HIV-1-infected adults, (ii) to evaluate the influence of renal function on FTC pharmacokinetics, and (iii) to assess recommended dosing adjustments according to the degree of renal impairment.
MATERIALS AND METHODS
Patients and treatment.
The study population included HIV-1-infected adult patients treated at Cochin Hospital, Paris, France, receiving an FTC dose of 200 mg QD. FTC was part of their antiretroviral regimen and was taken in oral solid forms (Emtriva, Truvada, or Atripla). FTC blood plasma concentrations at steady-state were measured at random times for therapeutic drug monitoring. For each patient of this retrospective study, the time elapsed between FTC intake and sampling times, gender, body weight (BW), age, serum creatinine (SCR), and associated antiretroviral drugs were recorded. Creatinine clearance (CLCR) was calculated for each occasion using the Cockroft-Gault formula (7).
Analytical method.
The FTC assay was performed according to a previously published method of liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) (8). The limit of quantification (LOQ) was 0.01 mg/liter, intra-assay precision was 3.6%, and interassay precision was 7.9%.
Population pharmacokinetic modeling strategy.
The data were analyzed with a nonlinear mixed-effect modeling approach using the Monolix software program version 4.1.3 (available at www.lixoft.eu) (9). The parameters were estimated by computing the maximum likelihood estimator of the parameters without any linearization of the model, using the stochastic approximation expectation maximization (SAEM) algorithm combined with a Markov chain Monte Carlo (MCMC) procedure. The number of MCMC chains was fixed at 5 for all estimations.
Several structural pharmacokinetic models were investigated using one or two compartments with linear absorption and elimination. Since very few samples were recorded in the absorption phase (Fig. 1), we could not estimate the absorption rate constant (ka). Models with fixed ka values (10, 11) were tested. However, these ka values have only been estimated in pregnant women. Thus, as the effects of pregnancy on absorption are not well understood and pregnancy might alter drug absorption (12), we also considered a model with an absorption rate constant (ka) equal to the distribution rate constant (α). This model was tested according to the following equation (13):
where α is the distribution rate constant, k21 is the constant of transfer from the peripheral to the central compartment, β is the elimination rate constant, Vc is the central volume of distribution, F is the bioavailability, and τ is the dosing interval.
FIG 1.
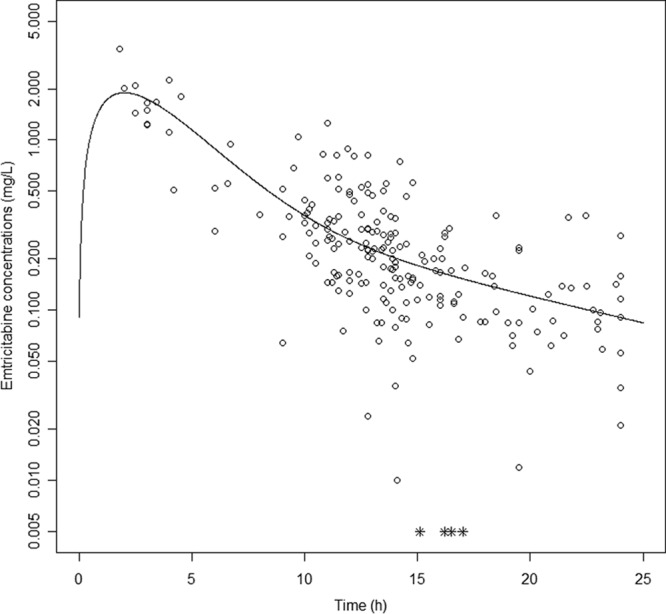
Observed (circles) and population-predicted (line) FTC concentrations versus time. Asterisks represent FTC concentrations below the LOQ.
Several error models (proportional, additive, and mixed) were investigated to describe the residual variability (ϵ). Interindividual variabilities (IIV or η) were tested using an exponential model. The influence of continuous covariates (CO) on pharmacokinetic parameters was tested according to the following equation, using apparent elimination clearance (CL/F), for example: CL/F = θCL/F · (CO/[median (CO)])θCO, where θCL/F is the typical value of apparent elimination clearance for a subject with the median covariate value, and θCO is the estimated influential factor for the continuous covariate. The main continuous covariates were BW, age, SCR, BW/SCR, and CLCR. The influence of categorical covariates (CA) was tested according to the equation CL/F = θCL/F · θCACA, where CA is 0 for the reference value of CL/F, CA is 1 for the value of CL/F with the covariate, and θCA is the estimated influential factor of the CA. The main CA were gender and associated antiretroviral treatments.
The Akaike information criterion (AIC) and the Bayesian information criterion (BIC) were used to test hypotheses for nonembedded models. The objective function value (OFV) was used to test hypotheses regarding the structural model, structure of the variance-covariance matrix for IIV, and covariate effect(s) on pharmacokinetic parameters. A covariate was retained in the model if its effect was biologically plausible, if the OFV was decreased by at least 3.84, and if it produced a reduction in the variability of the pharmacokinetic parameter (IIV). Individual Bayesian estimates of the pharmacokinetic parameters were used to calculate the individual area under the concentration-time curve (AUC) and the minimal concentration (Cmin) of FTC.
Model evaluation.
Graphical evaluation of the goodness of fit was performed with graphs of observed concentrations versus population predictions (PRED) or individual predictions (IPRED), weighted residuals versus time, and weighted residuals versus PRED.
In order to evaluate the model, simulated FTC concentrations and observed data were compared by a prediction-corrected visual predictive check (PC-VPC) (14). The 5th, 50th, and 95th percentiles of the observed data were overlaid on the 90% confidence interval of the 5th, 50th, and 95th simulated percentiles, and a visual inspection was performed. The model was also evaluated by the normalized prediction distribution errors (NPDE) metrics (15). Diagnostic graphics and distribution statistics were performed using RfN (see http://wfn.sourceforge.net) of the R program (16).
Dosing adjustment simulations.
Using the final model, the recommended dosing schemes for the oral solid form of FTC were simulated (1,000 Monte Carlo simulations), using 200 mg QD for patients with normal renal function or mild renal impairment (CLCR, 79 to 50 ml/min) and 200 mg every 48 h for patients with moderate renal impairment (CLCR, 49 to 30 ml/min). The AUC0-48 values and minimal concentrations (Cmin) obtained from the simulations were compared according to renal function. The percentage of patients with a daily AUC of <10 mg · h/liter was calculated. This value (10 mg · h/liter) was associated with maximal anti-HIV activity in a previous study (6). Simulations of other FTC dosing schemes were also performed for patients with moderate renal impairment, such as an oral solid dose of 200 mg QD or different doses of a 10-mg/ml oral solution QD. A relative bioavailability of 83% between the oral solution and the solid oral form was used (5). Similarly, the AUC0-24 values and minimal concentrations (Cmin) obtained from the simulations were compared according to renal function.
RESULTS
Demographic data.
A total of 211 blood plasma concentrations, determined from samples collected from 161 adult patients (85 men, 76 women), were available for FTC pharmacokinetic evaluation. All patients received an FTC dose of 200 mg QD in capsule or tablet form. Table 1 summarizes the characteristics of the patients.
TABLE 1.
Patient characteristics (n = 161)
| Characteristic | Median | Mean | Range |
|---|---|---|---|
| Age (yr) | 41 | 41 | 18–72 |
| BW (kg) | 69 | 71 | 37–133 |
| SCR (μmol/liter) | 76 | 79 | 32–242 |
| BW/SCR | 0.897 | 0.972 | 0.421–2.250 |
| CLCR (ml/min) | 104.5 | 109.9 | 34.1–246.1 |
In this population, 78% of the FTC samples came from patients with normal renal function (median CLCR, 110.9 ml/min; range, 80.3 to 246.1 ml/min), 19% came from patients with mild renal impairment (median CLCR, 71.3 ml/min; range, 50.4 to 79.6 ml/min), and 3% came from patients with moderate renal impairment (median CLCR, 40.6 ml/min; range, 34.1 to 44.9 ml/min). The mean number of samples per patient was 1.3 (range, 1 to 5). No differences in the sampling schemes between patients with normal renal function and moderate renal impairment were observed.
FTC was combined with tenofovir disoproxil fumarate (TDF) in 99.5% of the samples. The combination of FTC and TDF was associated with only one protease inhibitor in 66.7% of the samples and with only one nonnucleoside reverse transcriptase inhibitor in 21.9% of the samples.
Population pharmacokinetics.
Four concentrations were below the LOQ (1.9% of samples) and were set to half of the LOQ (17). The data did not allow us to estimate an absorption rate constant (Fig. 1). Thus, our study data were best described by a 2-compartment model in which the ka and the slope of the distribution rate constant (α) were equal (13). The parameters of this model were apparent elimination clearance (CL/F), apparent intercompartmental clearance (Q/F), and apparent central (Vc/F) and peripheral (Vp/F) volumes of distribution, with F being the unknown bioavailability.
Residual variability was best described by a proportional error model. Interindividual variability was described by an exponential error model and was estimated only on CL/F. Gender and associated antiretroviral treatments had no effect on the objective function value (OFV). Age, BW, SCR, CLCR, and BW/SCR decreased the OFV significantly. The most significant decrease in OFV was obtained with CLCR, calculated by the Cockroft-Gault formula for each patient. After the inclusion of CLCR in the model, the addition of other covariates did not further improve the model. Thereby, the final covariate model was CL/F = θCL/F · (CLCR/104.5)0.278, where θCL/F is the typical value of CL/F for an adult, with a CLCR of 104.5 ml/min.
Table 2 summarizes the final population pharmacokinetic estimates. All the parameters were well estimated, with a relative standard error (RSE) of <40%.
TABLE 2.
Population pharmacokinetic parameters of emtricitabine in 161 HIV-1-infected adult patients
| Parametera | Estimated value | RSE (%)b |
|---|---|---|
| Structural model | ||
| CL/F (liters/h) | 15.1 | 6 |
| Vc/F (liters) | 42.3 | 12 |
| Q/F (liters/h) | 5.75 | 38 |
| Vp/F (liters) | 55.4 | 28 |
| Effect of creatinine clearance on CL/F | 0.278 | 23 |
| ka (h−1) | 0.53 | |
| Statistical model | ||
| IIVCL/F | 0.174 | 14 |
| Proportional error | 0.422 | 7 |
CL/F, apparent elimination clearance; Vc/F, apparent central volume of distribution; Q/F, apparent intercompartmental clearance; Vp/F, apparent peripheral volume of distribution; ka, absorption rate constant; IIVCL/F, interindividual variability estimate.
RSE, relative standard error (standard error of estimate/estimate × 100).
Model evaluation.
The prediction-corrected visual predictive check showed that the 5th, 50th, and 95th percentiles of the observed data are well included in the 90% confidence interval of the 5th, 50th, and 95th simulated percentiles (Fig. 2). The mean and variance of the normalized prediction distribution errors were not significantly different from 0 (P = 0.75, Wilcoxon signed rank test) and 1 (P = 0.64, Fisher variance test), respectively. Their distribution was not different from a normal one (P = 0.23, Shapiro-Wilk test of normality). The global adjusted P value was 0.69.
FIG 2.

Prediction-corrected visual predictive check. The lines represent the 5th, 50th, and 95th percentiles of observed data. The shaded areas represent the 90% confidence intervals of the simulated percentiles.
Individual exposure to FTC.
Individual Bayesian clearances were used to calculate individual AUC0–24 values. FTC exposure after a standard dose of 200 mg QD increased when the CLCR decreased: the median AUC0–24 (95% confidence interval) increased from 12.5 (12.3 to 12.8) mg · h/liter in patients with normal renal function (CLCR, >80 ml/min) to 14.7 (13.9 to 15.5) mg · h/liter in patients with mild renal impairment (CLCR, 79 to 50 ml/min), and to 17.9 (15.6 to 20.6) mg · h/liter in patients with moderate renal impairment (CLCR, 49 to 30 ml/min).
Dosing adjustment simulations.
Figure 3 displays FTC exposures derived from 1,000 simulations of the final model, according to the degree of renal impairment. Figure 3a shows that the recommended dosing schemes for the oral solid form of FTC (200 mg QD for patients with normal renal function and patients with a CLCR of 79 to 50 ml/min; 200 mg every 48 h for patients with a CLCR of 49 to 30 ml/min) led to lower AUC0–48 for patients, with a CLCR of 49 to 30 ml/min (median AUC0–48, 17.2 versus 25.6 mg · h/liter). Despite the median AUC0–24 being close (15.6 mg · h/liter in patients with moderate renal impairment versus 12.8 mg · h/liter in patients with normal renal function), 100% of the patients with moderate renal impairment had an AUC24–48 of <10 mg · h/liter (median AUC24–48, 1.6 mg · h/liter). The same trend was observed for minimal concentrations, with a mean Cmin of 0.032 mg/liter for patients with moderate renal impairment and 0.091 mg/liter for patients with normal renal function.
FIG 3.

Simulated FTC exposures (AUC0–48 or AUC0–24) versus degree of renal impairment (CLCR). (a) AUC0–48 for oral solid form recommended dosing schemes: 200 mg QD in patients with normal renal function or mild renal impairment, 200 mg every 48 h in patients with moderate renal impairment. (b and c) AUC0–24 for 200 mg oral solid form QD in patients with moderate renal impairment (b) and for 18 ml of FTC oral solution (10 mg/ml) QD in patients with moderate renal impairment (c). Boxes represent the median and interquartile range, and whisker plots represent values within 1.5 times the interquartile range.
Simulations of the 200-mg oral solid form dose QD in patients with a CLCR of 49 to 30 ml/min resulted in a slightly higher AUC0–24 than in patients with mild renal impairment (Fig. 3b). With this dosing scheme, only 0.1% of the patients with moderate renal impairment had a daily AUC of <10 mg · h/liter.
Figure 3c shows that similar FTC exposures were obtained in patients with moderate renal impairment receiving 18 ml of FTC oral solution QD and patients with normal renal function receiving 200 mg of the oral solid form QD (median AUC0–24, 12.9 versus 12.8 mg · h/liter). The 18 ml of FTC oral solution was considered equivalent to 150 mg of FTC in oral solid form, taking into account a relative bioavailability of 83% (5). The percentage of patients with moderate renal impairment having an AUC0–24 of <10 mg · h/liter was also close (9.6% for the oral solid form versus 7.2% for the oral solution).
DISCUSSION
This article describes the emtricitabine pharmacokinetics in 161 HIV-1-infected adults. FTC blood plasma concentrations were satisfactorily described by a two-compartment model. As the data did not allow us to estimate an absorption rate constant, a model with a ka equal to the slope of the distribution phase or models with fixed ka values were tested. The ka value estimated in our population (0.53 h−1) thanks to the model developed (i.e., with ka = α) was close to previously published values for pregnant women (0.54 h−1 and 0.709 h−1) (10, 11). However, even by fixing ka to these values, the best fit was obtained for the model with a common value estimated for ka and α.
Our typical CL/F estimate (15.1 liters/h) was slightly lower than the calculated values (dose-to-AUC ratios) in healthy volunteers (2–4) and HIV-1-infected patients (5, 6). However, most of these studies were performed in adults with normal renal function only. The mean half-life was 9.9 h and was in agreement with previously published values of 9.44 h (2) and 10.7 h (3). The mean Cmin was 0.094 mg/liter, which was close to reported Cmin values of 0.09 mg/liter in HIV-infected adults (5) and 0.075 mg/liter in healthy volunteers (3).
The population model was used to investigate whether demographic and biological characteristics could influence FTC pharmacokinetics. Our results confirmed that renal function was the most important factor influencing FTC pharmacokinetics. The BW/SCR ratio has been suggested to be a surrogate marker of renal function (18). However, in our study, the effect of BW/SCR on FTC CL/F was less pronounced than the effect of CLCR. As ethnicity data were not available, we could not evaluate renal function by the modification of diet in renal disease (MDRD) or chronic kidney disease epidemiology collaboration (CKD-EPI) formulas, so we used the Cockroft-Gault formula, despite its limitations. Thus, the only and most significant influential factor for CL/F was CLCR.
In our population, FTC exposure (AUC0-24) after a dose of 200 mg QD increased, while CLCR decreased. The FTC exposure increasing in accordance with the degree of renal impairment is in agreement with the FTC-107 study results (5). However, the increase in FTC exposure was more pronounced in the FTC-107 study (11.8 ± 2.9 mg · h/liter to 19.9 ± 1.2 mg · h/liter to 25.1 ± 5.7 mg · h/liter) compared to that in our study.
The current dosing recommendations for FTC in oral solid form are 200 mg QD for patients with normal renal function and patients with mild renal impairment and 200 mg every 48 h for patients with moderate renal impairment. However, for patients with moderate renal impairment, this dosing interval adjustment has never been evaluated. Thus, our population model was used to simulate this dosing adjustment recommendation. For patients with moderate renal impairment, the median AUC0-48 was 17.2 mg · h/liter. This median exposure was 33% lower than the one in patients with normal renal function receiving 200 mg QD (25.6 mg · h/liter). Even if FTC 5′-triphosphate is the active moiety of emtricitabine, a plasma FTC concentration-response relationship was previously demonstrated. A relationship between anti-HIV activity and plasma FTC AUC0-24 has been shown, with a plateau in the anti-HIV activity, for a daily AUC value of ≈10 mg · h/liter (6). Thus, as 100% of the simulated patients with moderate renal impairment receiving 200 mg every 48 h had an AUC24-48 of <10 mg · h/liter, we expected reduced anti-HIV activities for these patients.
Therefore, in order to guarantee the anti-HIV activity of FTC, we used our population model to simulate other dosing schemes for patients with moderate renal impairment to obtain exposures similar to what is found in patients with normal renal function receiving 200 mg FTC oral solid form QD. Simulated patients with moderate renal impairment receiving a 200-mg oral solid dose QD had a higher AUC0-24 than patients with normal renal function (17.2 versus 12.8 mg · h/liter). However, only 0.1% of patients with moderate renal impairment had a daily AUC lower than the threshold of maximal anti-HIV activity (10 mg · h/liter). As FTC is a well-tolerated drug, even at higher dosing levels (300 mg QD [19] or 200 mg twice a day (BID) [20]), an increase in FTC exposure for patients with moderate renal impairment receiving 200 mg QD is not expected to have major effects on the safety profile of FTC.
Simulated patients with moderate renal impairment receiving 18 ml of a 10 mg/ml oral solution of emtricitabine QD had a median AUC0-24 similar to that of patients with normal renal function receiving 200 mg QD in oral solid form (12.9 mg · h/liter versus 12.8 mg · h/liter L). Only 7.2% of patients with moderate renal impairment had an FTC AUC0-24 of <10 mg · h/liter. However, one limitation of this dosing recommendation may be the practicality of administering an oral solution, which is less convenient than an oral solid form.
These simulations may be regarded with caution since the number of HIV-1-infected patients with moderate renal impairment in our study was low. However, the current dosing recommendations derived from a study (FTC-107) that included only 6 healthy volunteers with moderate renal impairment.
In conclusion, this study reports FTC pharmacokinetics in a large population of HIV-1-infected patients with various degrees of renal impairment. FTC clearance was related to CLCR. In our study, the current dosing recommendations led to lower FTC exposures for patients with moderate renal impairment. In order to reach exposures similar to those obtained in HIV-1-positive patients with normal renal function, patients with moderate renal impairment should receive 18 ml of a 10 mg/ml FTC oral solution QD. If the switch from an oral solid form to an oral solution is not possible for practical reasons, we propose to give 200 mg oral solid form QD rather than every 48 h, as long as FTC is well tolerated. These proposed recommendations should be prospectively confirmed.
Footnotes
Published ahead of print 3 February 2014
REFERENCES
- 1.World Health Organization. 2010. Antiretroviral therapy for HIV infection in adults and adolescents: recommendations for a public health approach: 2010 revision. World Health Organization, Geneva, Switzerland: http://whqlibdoc.who.int/publications/2010/9789241599764_eng.pdf?ua=1 [PubMed] [Google Scholar]
- 2.Zong J, Chittick GE, Wang LH, Hui J, Begley JA, Blum MR. 2007. Pharmacokinetic evaluation of emtricitabine in combination with other nucleoside antivirals in healthy volunteers. J. Clin. Pharmacol. 47:877–889. 10.1177/0091270007300808 [DOI] [PubMed] [Google Scholar]
- 3.Blum MR, Chittick GE, Begley JA, Zong J. 2007. Steady-state pharmacokinetics of emtricitabine and tenofovir disoproxil fumarate administered alone and in combination in healthy volunteers. J. Clin. Pharmacol. 47:751–759. 10.1177/0091270007300951 [DOI] [PubMed] [Google Scholar]
- 4.Chittick GE, Zong J, Begley JA, Alianti JR, Sorbel JJ, Blum MR. 2008. Pharmacokinetics of emtricitabine/tenofovir disoproxil fumarate and tacrolimus at steady state when administered alone or in combination. Int. J. Clin. Pharmacol. Ther. 46:627–636. 10.5414/CPP46627 [DOI] [PubMed] [Google Scholar]
- 5.Gilead Sciences. 2012. Emtriva prescribing information. Gilead Sciences, Foster City, CA: http://www.gilead.com/pdf/emtriva_pi.pdf [Google Scholar]
- 6.Wang LH, Begley J, St. Claire RL, III, Harris J, Wakeford C, Rousseau FS. 2004. Pharmacokinetic and pharmacodynamic characteristics of emtricitabine support its once daily dosing for the treatment of HIV infection. AIDS Res. Hum. Retroviruses 20:1173–1182. 10.1089/aid.2004.20.1173 [DOI] [PubMed] [Google Scholar]
- 7.Cockcroft DW, Gault MH. 1976. Prediction of creatinine clearance from serum creatinine. Nephron 16:31–41. 10.1159/000180580 [DOI] [PubMed] [Google Scholar]
- 8.Le Saux T, Chhun S, Rey E, Launay O, Weiss L, Viard JP, Pons G, Jullien V. 2008. Quantification of seven nucleoside/nucleotide reverse transcriptase inhibitors in human plasma by high-performance liquid chromatography with tandem mass-spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 865:81–90. 10.1016/j.jchromb.2008.02.008 [DOI] [PubMed] [Google Scholar]
- 9.Lavielle M, Mentré F. 2007. Estimation of population pharmacokinetic parameters of saquinavir in HIV patients with the MONOLIX software. J. Pharmacokinet. Pharmacodyn. 34:229–249. 10.1007/s10928-006-9043-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirt D, Pruvost A, Ekouévi DK, Urien S, Arrivé E, Kone M, Nerrienet E, Nyati M, Gray G, Kruy LS, Blanche S, Dabis F, Tréluyer JM. 2011. Very high concentrations of active intracellular phosphorylated emtricitabine in neonates (ANRS 12109 trial, step 2). Antimicrob. Agents Chemother. 55:2953–2960. 10.1128/AAC.01376-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirt D, Urien S, Rey E, Arrivé E, Ekouévi DK, Coffié P, Leang SK, Lalsab S, Avit D, Nerrienet E, McIntyre J, Blanche S, Dabis F, Tréluyer JM. 2009. Population pharmacokinetics of emtricitabine in human immunodeficiency virus type 1-infected pregnant women and their neonates. Antimicrob. Agents Chemother. 53:1067–1073. 10.1128/AAC.00860-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson GD. 2005. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin. Pharmacokinet. 44:989–1008. 10.2165/00003088-200544100-00001 [DOI] [PubMed] [Google Scholar]
- 13.Wijnand HP. 1988. Pharmacokinetic model equations for the one- and two-compartment models with first-order processes in which the absorption and exponential elimination or distribution rate constants are equal. J. Pharmacokinet. Biopharm. 16:109–128. 10.1007/BF01061864 [DOI] [PubMed] [Google Scholar]
- 14.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. 2011. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 13:143–151. 10.1208/s12248-011-9255-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comets E, Brendel K, Mentré F. 2008. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput. Methods Programs Biomed. 90:154–166. 10.1016/j.cmpb.2007.12.002 [DOI] [PubMed] [Google Scholar]
- 16.Ihaka R, Gentleman R. 1996. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 5:299–314. 10.2307/1390807 [DOI] [Google Scholar]
- 17.Beal SL. 2001. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 28:481–504. 10.1023/A:1012299115260 [DOI] [PubMed] [Google Scholar]
- 18.Jullien V, Tréluyer JM, Rey E, Jaffray P, Krivine A, Moachon L, Lillo-Le Louet A, Lescoat A, Dupin N, Salmon D, Pons G, Urien S. 2005. Population pharmacokinetics of tenofovir in human immunodeficiency virus-infected patients taking highly active antiretroviral therapy. Antimicrob. Agents Chemother. 49:3361–3366. 10.1128/AAC.49.8.3361-3366.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gish RG, Leung NW, Wright TL, Trinh H, Lang W, Kessler HA, Fang L, Wang LH, Delehanty J, Rigney A, Mondou E, Snow A, Rousseau F. 2002. Dose range study of pharmacokinetics, safety, and preliminary antiviral activity of emtricitabine in adults with hepatitis B virus infection. Antimicrob. Agents Chemother. 46:1734–1740. 10.1128/AAC.46.6.1734-1740.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rousseau FS, Kahn JO, Thompson M, Mildvan D, Shepp D, Sommadossi JP, Delehanty J, Simpson JN, Wang LH, Quinn JB, Wakeford C, van der Horst C. 2001. Prototype trial design for rapid dose selection of antiretroviral drugs: an example using emtricitabine (Coviracil). J. Antimicrob. Chemother. 48:507–513. 10.1093/jac/48.4.507 [DOI] [PubMed] [Google Scholar]