

Abstract
Since 2011, outbreaks caused by influenza A(H3N2) variant [A(H3N2)v] viruses have become a public health concern in the United States. The A(H3N2)v viruses share the A(H1N1)pdm09 M gene containing the marker of M2 blocker resistance, S31N, but do not contain any known molecular markers associated with resistance to neuraminidase (NA) inhibitors (NAIs). Using a fluorescent NA inhibition (NI) assay, the susceptibilities of recovered A(H3N2)v viruses (n = 168) to FDA-approved (oseltamivir and zanamivir) and other (peramivir, laninamivir, and A-315675) NAIs were assessed. All A(H3N2)v viruses tested, with the exception of a single virus strain, A/Ohio/88/2012, isolated from an untreated patient, were susceptible to the NAIs tested. The A/Ohio/88/2012 virus contained two rare substitutions, S245N and S247P, in the NA and demonstrated reduced inhibition by oseltamivir (31-fold) and zanamivir (66-fold) in the NI assay. Using recombinant NA (recNA) proteins, S247P was shown to be responsible for the observed altered NAI susceptibility, in addition to an approximately 60% reduction in NA enzymatic activity. The S247P substitution has not been previously reported as a molecular marker of reduced susceptibility to the NAIs. Using cell culture assays, the investigational antiviral drugs nitazoxanide, favipiravir, and fludase were shown to inhibit the replication of A(H3N2)v viruses, including the virus with the S247P substitution in the NA. This report demonstrates the importance of continuous monitoring of susceptibility of zoonotic influenza viruses to available and investigational antiviral drugs.
INTRODUCTION
Influenza A viruses are highly contagious respiratory pathogens responsible for annual outbreaks and epidemics and, less frequently, pandemics. Influenza A viruses have several natural hosts, including swine. Swine are susceptible to a wide range of influenza viruses and shed naturally occurring reassortant influenza A viruses when infected with more than one subtype (1). So-called swine triple reassortants containing genes of influenza viruses from three different species (avian, human, and swine) became widespread in North American swine populations circa 1998 and pose a constant threat to human health.
Human infection with a novel influenza type A virus is a disease reportable to the World Health Organization (WHO) under International Health Regulations (2) and has been a nationally notifiable condition in the United States since 2007 (3). Human infections resulting from swine influenza viruses are relatively rare; between 1990 and 2010, only 27 such infections were documented in the United States (4). Influenza A viruses that circulate in swine but infect humans are called variant viruses (5).
In July 2011, a new reassortant virus was isolated from humans in the United States. This virus, named A(H3N2)v, differed from previously reported triple reassortants due to the replacement of the classic swine virus M gene with that of the A(H1N1)pdm09 virus (6), which is associated with improved virus transmissibility (7, 8).
There were 12 confirmed cases of A(H3N2)v infection in the United States in 2011 and 309 in 2012, with additional cases occurring in 2013 (9); nearly all infections have resulted from exposure to swine at state and local fairs (10). While no sustained community spread of A(H3N2)v has been detected, evidence of limited human-to-human transmission was reported (5, 10, 11).
Individuals infected with A(H3N2)v have mostly had mild illness with symptoms similar to that of seasonal influenza, which may result in the underreporting of A(H3N2)v cases (10). In some patients with underlying risk factors, symptoms have been more severe, and in 2012 there were 16 hospitalizations and a single death resulting from A(H3N2)v infections (12). The majority of reported A(H3N2)v cases have occurred in people aged <18 years, with the median age of all reported human cases being 7 years (13).
Due to antigenic differences with contemporary seasonal A(H3N2) viruses, the seasonal influenza vaccine is unlikely to be cross-protective against A(H3N2)v viruses (6). Indeed, the seasonal influenza vaccine has been shown to offer little to no protection against these A(H3N2)v viruses in the ferret model (14). Antiviral treatment is recommended for hospitalized patients and for high-risk patients with suspected variant virus infection (10). However, since A(H3N2)v viruses contain the A(H1N1)pdm09 M gene, they are resistant to M2 blockers (6), leaving the neuraminidase (NA) inhibitors (NAIs) oral oseltamivir and inhaled zanamivir as the only antiviral agents available for A(H3N2)v infections. Additional NAIs licensed outside the United States include the intravenous peramivir, which is approved in Japan, South Korea, and China, while inhaled laninamivir is licensed only in Japan; the oral prodrug of A-315675 (A-322278) was dropped from development (15).
Due to the limited therapeutic options, it was essential to monitor susceptibilities of the A(H3N2)v viruses to available and investigational antiviral drugs. Using a fluorescent NA inhibition (NI) assay, a single variant virus that carried a naturally occurring change affecting inhibition of the NA activity by oseltamivir and zanamivir, but not by other NAIs, was identified. The susceptibility of this virus to the antiviral drugs nitazoxanide, favipiravir, and fludase, which are currently undergoing clinical stage II/III trials (15), was assessed using cell culture-based assays.
MATERIALS AND METHODS
Antivirals.
NAIs zanamivir (GlaxoSmithKline, Uxbridge, United Kingdom), oseltamivir (carboxylate) (Hoffman-La Roche, Basel, Switzerland), peramivir (BioCryst Pharmaceuticals, Birmingham, AL), laninamivir (R-125489, the active metabolite of prodrug CS-8958; Biota, Melbourne, Australia), and A-315675 (the active metabolite of prodrug A-322278; Abbott Laboratories, Abbott Park, IL) were each dissolved in sterile distilled water and stored at −30°C.
The investigational antiviral drugs nitazoxanide (NTZ; Romark Laboratories, Tampa, FL), favipiravir (T-705; Toyama, Tokyo, Japan), and fludase (DAS181; NexBio, San Diego, CA) (15) were used to assess their effect on virus replication in cell culture; ribavirin (Sigma-Aldrich, St. Louis, MO) was used as a control. The nitazoxanide stock solution was prepared by dissolving 20 mg of nitazoxanide in 0.2 ml of dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO) and was then aliquoted and stored at −80°C until required. The working solutions of nitazoxanide, favipiravir, and ribavirin were prepared at the time of each experiment in virus growth medium (Dulbecco's modified eagles medium [DMEM]; Gibco, Carlsbad, CA) supplemented with 100 IU penicillin/streptomycin (Gibco, Carlsbad, CA), 0.5% bovine serum albumin (BSA; Gibco, Carlsbad, CA), and 2 μg/ml l-tosylamide-2-phenylethyl chloromethyl ketone (TPCK)-treated trypsin (Sigma-Aldrich, St. Louis, MO). The stock of fludase was stored at −80°C, and dilutions were made in EDB-BSA buffer (10 mM sodium acetate buffer, pH 6.0, supplemented with 0.1 M NaCl, 10 mM CaCl2, 0.5 mM MgCl2, and 0.5% BSA) prior to testing in cell culture (16).
Viruses.
Influenza A(H3N2)v viruses were submitted by U.S. public health laboratories to the WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza at the CDC, Atlanta, GA. One hundred sixty-eight viruses were isolated and propagated according to standard procedures (17) prior to testing in a biosafety level 2 (BSL2)-enhanced laboratory.
NI assays.
Drug susceptibilities were assessed in a fluorescent NI assay using the NA-Fluor influenza neuraminidase assay kit (Applied Biosystems, Carlsbad, CA) (18). Fifty percent inhibitory concentrations (IC50s), which are the NAI concentrations required to inhibit enzyme activity by 50%, were calculated using JASPR v1.2 curve-fitting software (18). Interpretation of IC50s was performed using the WHO antiviral working group (AVWG) criteria for influenza A viruses (a <10-fold increase in IC50 represents normal inhibition, and a 10- to 100-fold increase represents reduced inhibition, while a >100-fold increase is highly reduced inhibition) (19, 20).
Sequence analysis.
Sanger sequencing of NA genes was performed, and sequences were analyzed using BioEdit software (21), as previously described (4).
NA sequences at amino acid residues 245 and 247 were determined by pyrosequencing using the PyroMark ID platform (Qiagen, Valencia, CA) (22). RT-PCR primer N2-F670, 5′-AGA ACC CAG GAG TCG GAA TGC-3′, and the biotinylated N2-R871-Bio primer, 5′-AG ACG CAT CTG ACA CCA GGA TAT-3′, were designed and utilized at a concentration of 20 μM. The sequencing primer, N2-245-F715-seq, GTA GTC ATG ACT GAT GGG, was used at a concentration of 100 μM. The quantification of NA variants was performed using single nucleotide polymorphism (SNP) analysis with the target sequence A AG/AC GCT T/C CC GGA AAA GCT GAT ACT AAA ATA TTA TTC, where underlining indicates triplets encoding the amino acid of interest.
recNA expression and quantitation.
A codon-optimized cDNA encoding the ectodomain (residues 82 to 469) of the NA gene of A/Indiana/12/2012 A(H3N2)v was synthesized (GenScript USA Inc., Piscataway, NJ) and subcloned into pIEx-4 vector (EMD Millipore, Billerica, MA) using the In-Fusion HD cloning system (Clontech, Mountain View, CA). All NA variants were generated from this wild-type clone using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies Inc., Santa Clara, CA). Resulting constructs were transiently transfected into sf9 cells (EMD Millipore, Billerica, MA) using the Cellfectin II transfection reagent (Gibco, Grand Island, NY) and grown on a shaker at 27°C for 5 days. All recombinant NA (recNA) proteins contained a His tag at the N terminus followed by a tetramerization domain, from human vasodilator-stimulated phosphoprotein, and a thrombin site (23). The secreted recNA was quantified by Western blotting, using purified NA of A/Perth/16/2009 A(H3N2) as a standard. Supernatants were assessed for functional NA activity using MUNANA [2′-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid] as a substrate (18) and analyzed without further purification in the NI assay.
Inhibition of virus replication.
Madin-Darby canine kidney cells (MDCK) and MDCK cells modified to overexpress α2,6-linked neuraminic acid (NeuAc) containing glycan receptors, MDCK-SIAT1 (24), were used to assess susceptibility to non-NAI antiviral drugs. MDCK cells were used for experiments using nitazoxanide according to previously published protocols (25) and as recommended by the manufacturer. For all other antiviral compounds, experiments were carried out using MDCK-SIAT1 cells.
The reduction in the number of focus-forming units (FFU) was determined as described previously (16). Briefly, cell monolayers were pretreated for 2 h with fludase at concentrations ranging from 0.02 μM to 4 μM. Any unbound fludase was removed by washing cells three times with phosphate-buffered saline (PBS; Gibco, Grand Island, NY). Virus inoculum at a multiplicity of infection (MOI) of 0.0001 to 0.0003 was added, and cells were washed again after 1 h of incubation at 37°C with 5% CO2. Virus growth medium was added to the monolayers, and cells were incubated for 24 h, at which point cell monolayers were fixed and immunostained; FFU were counted under a microscope (16).
Reduction of infectious virus yield in the presence of nitazoxanide (25), favipiravir (26), and ribavirin (26) was assessed essentially as previously described. Virus inoculation was carried out at an MOI of 0.001 for 1 h at 37°C with 5% CO2. Unadsorbed virus was removed and nitazoxanide added at final concentrations of 0.5 μM to 10 μM. For nitazoxanide, cell culture supernatants were collected at 48 h postinfection (hpi) and viral titers were determined in MDCK cells and expressed as log10 of tissue culture infectious dose (TCID90) per ml. Favipiravir and ribavirin were each diluted to the final concentrations of 0.5 μM to 128 μM and 10 μM to 820 μM, respectively. Cell culture supernatants were collected at 24 hpi for both favipiravir and ribavirin. Viral titers were determined in MDCK cells and expressed as FFU per ml, as previously described (26). The IC90 value, the drug concentration at which the infectious virus replication (number of foci or yield) was reduced by 90%, was determined by nonlinear regression analysis using GraphPad Prism software v5.0 (GraphPad Software, San Diego, CA).
Structure modeling.
The model of the A/Indiana/12/2012 NA bound to zanamivir was generated by homology modeling (27) using the N2/zanamivir cocrystal structure (Protein Data Bank accession number 3TIC) as a template.
Nucleotide sequence accession numbers.
Accession numbers for the NA gene sequences determined in this study were deposited in GISAID (Global Initiative on Sharing All Influenza Data) as follows: EPI397960 for A/Ohio/88/2012, EPI397547 for A/Ohio/83/2012, EPI381848 for A/Indiana/12/2012.
RESULTS
Susceptibilities to NAIs.
Sequence analysis of NA genes (n = 338) of A(H3N2)v viruses collected in 2011 to 2013 revealed no known molecular markers of resistance to NAIs (28). In the NI assay, 168 A(H3N2)v viruses were tested with five NAIs (Table 1). Calculated IC50s were in the subnanomolar range (Table 1) with the exception of that of a single virus, A/Ohio/88/2012. This virus exhibited 31-, 66-, and 7-fold-elevated IC50s for oseltamivir, zanamivir, and laninamivir, respectively, compared to the respective median IC50 determined for the H3N2v subtype (Table 1). According to the WHO AVWG criteria (19), this result is interpreted as reduced inhibition by oseltamivir and zanamivir and normal inhibition by the remaining NAIs. A/Ohio/88/2012 was isolated from a patient <18 years old who had received no antiviral treatment, had no underlying medical conditions, was not hospitalized, and had symptoms lasting approximately a week.
TABLE 1.
Assessment of influenza A(H3N2)v virus susceptibilities to NAIs in the fluorescent NI assay
| NA source | Subtype | NA substitution(s)a | IC50, nM (fold)b |
||||
|---|---|---|---|---|---|---|---|
| Oseltamivir | Zanamivir | Peramivir | Laninamivir | A-315675 | |||
| Test viruses | |||||||
| Test viruses (n = 165) | H3N2v | — | 0.16 ± 0.11 [0.15] | 0.52 ± 0.09 [0.51] | 0.17 ± 0.04 [0.16] | 0.77 ± 0.19 [0.75] | 0.29 ± 0.05 [0.28] |
| A/Ohio/88/2012 | H3N2v | S245N, S247P | 4.69 ± 0.25 (31) | 33.86 ± 3.73 (66) | 0.20 ± 0.01 (1) | 4.85 ± 0.39 (7) | 0.51 ± 0.06 (2) |
| A/Ohio/83/2012 | H3N2v | S245N | 0.13 ± 0.01 (1) | 0.51 ± 0.02 (1) | 0.15 ± 0.01 (1) | 0.84 ± 0.06 (1) | 0.29 ± 0.04 (1) |
| A/Indiana/12/2012 | H3N2v | — | 0.15 ± 0.01 (1) | 0.42 ± 0.06 (1) | 0.13 ± 0.02 (1) | 0.61 ± 0.08 (1) | 0.22 ± 0.04 (1) |
| Recombinant NAs | |||||||
| recNA-1 | — | 0.09 ± 0.01 | 0.15 ± 0.01 | 0.11 ± 0.01 | 0.44 ± 0.04 | 0.15 ± 0.01 | |
| recNA-2 (A/Ohio/88/2012-like) | S245N, S247P | 3.60 ± 0.13 (40) | 35.32 ± 1.66 (235) | 0.17 ± 0.02 (2) | 3.84 ± 0.37 (9) | 0.32 ± 0.05 (2) | |
| recNA-3c | S247P | 4.23 ± 0.12 (42) | 39.91 ± 3.84 (266) | 0.21 ± 0.02 (2) | 3.79 ± 0.16 (9) | 0.62 ± 0.01 (4) | |
| recNA-4 (A/Ohio/83/2012-like) | S245N | 0.09 ± 0.01 (1) | 0.17 ± 0.03 (1) | 0.13 ± 0.01 (1) | 0.50 ± 0.02 (1) | 0.17 ± 0.01 (1) | |
| Reference viruses | |||||||
| A/Washington/01/2007, oseltamivir sensitive | H3N2 | — | 0.06 ± 0.01 | 0.46 ± 0.15 | 0.08 ± 0.01 | 0.33 ± 0.06 | 0.23 ± 0.16 |
| A/Texas/12/2007, oseltamivir resistant | H3N2 | E119V | 38.10 ± 2.35 (635) | 0.36 ± 0.06 (1) | 0.10 ± 0.01 (1) | 0.34 ± 0.03 (1) | 0.23 ± 0.15 (1) |
Amino acid substitution in the NA known or suspected to affect susceptibility to NAIs; —, absence of known or suspected markers of NAI resistance.
Values are means ± SD, based on at least three independent experiments. IC50 for A/Ohio/88/2012 virus was excluded from calculation of the mean and median IC50s. Fold values (in parentheses): for A(H3N2)v viruses, compared to a median IC50; for recNA, compared to an IC50 of recNA lacking changes at aa 245 and 247 in the NA; for the reference A(H3N2) viruses, compared to an IC50 for A/Washington/01/2007. Median IC50s for A(H3N2)v viruses are shown in square brackets.
No corresponding virus available.
The NA of A/Ohio/88/2012 virus isolate contained two rare and previously unreported substitutions, S245N and S247P; pyrosequencing confirmed the presence of both substitutions in the corresponding clinical sample (100% by SNP pyrosequencing). Therefore, these substitutions were not an artifact of virus propagation in cell culture, as has been seen with substitutions at D151 and at Q136 in the NA of seasonal influenza A viruses (29, 30, 31). Of note, only a single other virus, A/Ohio/83/2012, was shown to contain the NA substitution S245N. Of the 156 A(H3N2)v NAs sequenced, no other A(H3N2)v contained the S247P change either alone or in a combination with S245N. Furthermore, when the NA sequences of seasonal A(H3N2) viruses circulating in the United States from 2011 and 2012 were analyzed (n = 508), only a single virus contained the S245N substitution and none contained S247P (data not shown).
The IC50s of three A(H3N2)v viruses with identical NA sequences, with the exception of sequences at positions 245 and 247, were compared. A/Indiana/12/2012, the wild-type strain lacking the two changes, and A/Ohio/83/2012, carrying only the single substitution S245N, shared similar IC50s (Table 1), indicating that S247P alone was causing the elevated IC50s.
To confirm this finding, recNA proteins with and without the S245N and S247P changes were generated and tested in the NI assay. As anticipated, recNAs carrying S247P either alone or in combination with S245N showed elevated IC50s for oseltamivir and zanamivir (Table 1). Notably, the zanamivir IC50s of the two recNAs carrying S247P were similar to that of virus A/Ohio/88/2012. However, due to a low IC50 of the wild-type recNA compared to that of the wild-type virus, the difference associated with S247P was 235- to 266-fold, which would be interpreted as highly reduced inhibition by zanamivir. The reason for the somewhat low IC50 of the wild-type recNA compared to that of the wild-type virus is unknown.
The effect of S247P on the enzymatic activity on the recNA was next investigated as previously described (32). When expressed either alone or together with S245N, S247P reduced by 60% the recNA's ability to cleave a small synthetic substrate MUNANA, compared to the wild-type recNA (data not shown). When present alone, S245N resulted in a slight increase (∼15%) in the recNA's enzymatic activity.
Inhibition of A(H3N2)v replication by investigational antivirals.
The investigational antiviral drugs nitazoxanide, favipiravir, and fludase were evaluated for their efficacy against A(H3N2)v viruses. Each of these compounds has a different mechanism of action targeting various components of the influenza virus life cycle (15). Nitazoxanide was reported to reduce infectious viral yields via interference with hemagglutinin maturation and may also have an immunomodulatory effect. Favipiravir is a broad-spectrum antiviral drug that interferes with the viral RNA polymerase function, resulting in reduced viral titers. Fludase destroys virus receptors by cleaving sialyl moieties from receptors on the cell surface and thus preventing the attachment of the virus and preventing the spread of virus progeny to neighboring cells (15).
Antiviral potencies of nitazoxanide and favipiravir were assessed using an infectious virus yield reduction assay. The two A(H3N2)v viruses, with and without S247P, grew to titers comparable to those of the seasonal A(H3N2) viruses used as controls (Tables 2 and 3). Nitazoxanide was tested at concentrations ranging from 0.5 μM to 10 μM, and a reduction of ≥1 log in the A(H3N2)v virus titer was seen at concentrations 2 μM and higher (Table 2). At the highest concentration tested, nitazoxanide reduced A(H3N2)v titers by 3 to 6 log, which was better than, or similar to, that of the seasonal control viruses (Table 2). Favipiravir was used at concentrations ranging from 0.5 μM to 128 μM. At 8 μM and 32 μM there was a >1 log reduction in titer for all viruses, and at 128 μM there was no detectable virus (Table 3). The antiviral drug ribavirin was used as a control and was shown to reduce infectious yields by >2 log at a concentration of 10 μM (results not shown), consistent with previous data (27).
TABLE 2.
Reduction of A(H3N2)v infectious yields in MDCK cells in the presence of nitazoxanide
| Virus name | Subtype | NA substitution(s) | Mean virus titers ± SDa in the presence of nitazoxanide at concn of: |
|||||
|---|---|---|---|---|---|---|---|---|
| 0 μM | 0.5 μM | 1 μM | 2 μM | 4 μM | 10 μM | |||
| Test viruses | ||||||||
| A/Ohio/88/2012 | H3N2v | S245N, S247P | 8.3 ± 0.3 | 7.8 ± 0.9 | 7.3 ± 0.8 | 6.4 ± 1.3 | 4.6 ± 1.5 | 2.1 ± 0.9 |
| A/Ohio/83/2012 | H3N2v | S245N | 8.9 ± 0.4 | 8.6 ± 0.2 | 8.6 ± 0.1 | 7.8 ± 0.3 | 7.0 ± 1.0 | 5.9 ± 0.4 |
| Reference viruses | ||||||||
| A/Washington/01/2007 | H3N2 | —b | 8.6 ± 0.4 | 8.9 ± 0.2 | 8.6 ± 0.1 | 8.3 ± 0.1 | 8.3 ± 0.3 | 7.6 ± 0.2 |
| A/Texas/12/2007 | H3N2 | E119V | 8.1 ± 0.3 | 7.9 ± 0.4 | 7.8 ± 0.5 | 7.2 ± 0.4 | 5.4 ± 0.4 | 3.6 ± 0.5 |
| A/California/04/2009 | H1N1pdm09 | — | 6.4 ± 0.5 | 6.3 ± 0.5 | 6.0 ± 0.6 | 5.4 ± 0.5 | 4.8 ± 0.3 | 3.0 ± 0.8 |
Results are from three independently performed experiments.
—, absence of known or suspected markers of NAI resistance.
TABLE 3.
Reduction of influenza A(H3N2)v infectious yields in MDCK-SIAT1 cells in the presence of favipiravir
| Virus name | Subtype | NA substitution(s) | Mean virus titers ± SDa in the presence of favipiravir at concn of: |
|||||
|---|---|---|---|---|---|---|---|---|
| 0 μM | 0.5 μM | 2 μM | 8 μM | 32 μM | 128 μM | |||
| Test viruses | ||||||||
| A/Ohio/88/2012 | H3N2v | S245N, S247P | 5.9 ± 0.3 | 5.5 ± 0.3 | 5.1 ± 0.2 | 4.7 ± 0.2 | 2.6 ± 0.6 | <1 |
| A/Ohio/83/2012 | H3N2v | S245N | 6.6 ± 0.3 | 6.5 ± 0.1 | 5.9 ± 0.3 | 5.5 ± 0.6 | 3.3 ± 0.4 | <1 |
| Reference viruses | ||||||||
| A/Washington/01/2007 | H3N2 | —b | 6.5 ± 0.5 | 6.0 ± 0.1 | 6.2 ± 0.1 | 5.2 ± 0.4 | 2.3 ± 0.6 | <1 |
| A/Texas/12/2007 | H3N2 | E119V | 7.0 ± 0.1 | 6.7 ± 0.2 | 6.4 ± 0.2 | 5.6 ± 0.4 | 2.8 ± 0.2 | <1 |
| A/California/04/2009 | H1N1pdm09 | — | 5.5 ± 0.1 | 5.1 ± 0.4 | 4.3 ± 0.7 | 3.9 ± 0.5 | 2.2 ± 0.3 | <1 |
Results are from three independently performed experiments.
—, absence of known or suspected markers of NAI resistance.
Since fludase acts by removing virus receptors, MDCK-SIAT1 cells were pretreated with this antiviral drug prior to the addition of virus and the focus formation inhibition assay (16) was used to assess the inhibitory effect. Pretreatment with the lowest concentration of fludase, 0.02 μM, resulted in the number of foci being reduced by more than 50% for both the seasonal viruses and the A(H3N2)v viruses (Table 4).
TABLE 4.
Reduction in focus formation in MDCK-SIAT1 cell monolayers pretreated with fludase
| Virus name | Subtype | NA substitution(s) | Mean FFU ± SD (no. of foci in each expt)a at fludase concn of: |
|||||
|---|---|---|---|---|---|---|---|---|
| 0 μM | 0.02 μM | 0.06 μM | 0.25 μM | 1 μM | 4 μM | |||
| Test viruses | ||||||||
| A/Ohio/88/2012 | H3N2v | S245N, S247P | 18.5 ± 1.8 | 1.8 ± 0.8 | 2.0 ± 1.2 | (2;2;0) | (2;3;0) | (2;1;0) |
| A/Ohio/83/2012 | H3N2v | S245N | 24.3 ± 3.4 | 4.3 ± 0.8 | 2.5 ± 1.0 | 0.8 ± 0.4 | 0.5 ± 0.5 | 1.3 ± 0.8 |
| Reference viruses | ||||||||
| A/Washington/01/2007 | H3N2 | —b | 31.5 ± 6.6 | 5.8 ± 1.5 | 2.0 ± 0.7 | 3.5 ± 1.7 | 1.8 ± 0.4 | 2.5 ± 1.1 |
| A/Texas/12/2007 | H3N2 | E119V | 27.5 ± 3.2 | 4.0 ± 0.7 | 2.5 ± 0.9 | 2.0 ± 1.2 | 1.3 ± 0.4 | 1.8 ± 0.4 |
| A/California/04/2009 | H1N1pdm09 | — | 15.0 ± 2.3 | 2.7 ± 0.8 | 1.3 ± 0.4 | (1;1;0) | (1;0;0) | (1;0;0) |
Values are from three independently performed experiments; values in parentheses indicate the number of foci present in each of the independently performed experiments; FFU, focus-forming units.
—, absence of known or suspected markers of NAI resistance.
When IC90 values were determined for nitazoxanide, favipiravir, and fludase, each antiviral was shown to inhibit the replication of the A(H3N2)v viruses at least as well as that of the seasonal control viruses, irrespective of the presence of S247P (Table 5).
TABLE 5.
IC90a values determined in cell culture-based assays
| Virus name | Subtype | NA substitution(s) | Mean IC90 ± SD |
|||||
|---|---|---|---|---|---|---|---|---|
| Nitazoxanide |
Favipiravir |
Fludase |
||||||
| μM | μg/ml | μM | μg/ml | μM | μg/ml | |||
| Test viruses | ||||||||
| A/Ohio/88/2012 | H3N2v | S245N, S247P | 1.09 ± 0.54 | 0.34 ± 0.17 | 2.85 ± 0.77 | 0.45 ± 0.12 | 0.03 ± 0.02 | 1.41 ± 1.09 |
| A/Ohio/83/2012 | H3N2v | S245N | 0.86 ± 0.32 | 0.27 ± 0.10 | 2.77 ± 0.49 | 0.43 ± 0.08 | 0.05 ± 0.01 | 2.36 ± 0.67 |
| Reference viruses | ||||||||
| A/Washington/01/2007 | H3N2 | —b | 5.63 ± 0.15 | 1.75 ± 0.05 | 8.99 ± 1.56 | 1.40 ± 0.24 | 0.06 ± 0.05 | 2.63 ± 2.33 |
| A/Texas/12/2007 | H3N2 | E119V | 1.61 ± 0.25 | 0.50 ± 0.08 | 2.88 ± 0.19 | 0.45 ± 0.03 | 0.03 ± 0.01 | 1.43 ± 0.65 |
| A/California/04/2009 | H1N1pdm09 | — | 2.33 ± 0.21 | 0.72 ± 0.07 | 2.13 ± 0.44 | 0.33 ± 0.07 | 0.05 ± 0.03 | 2.58 ± 1.54 |
IC90 (the concentration of antiviral agent reducing the virus replication by 90%) values were determined in the infectious viral yield reduction (nitazoxanide and favipiravir) and the focus-forming reduction (fludase) assays (see also Tables 2 to 4). Results are from three independently performed experiments.
—, absence of known or suspected markers of NAI resistance.
DISCUSSION
The A(H3N2)v viruses have resulted in multistate outbreaks and continue to cause infections in humans on an annual basis. Although human-to-human transmission has been rare (5, 10, 11), the A(H3N2)v viruses transmit as efficiently as seasonal influenza viruses in a ferret model and replicate to significantly higher titers than seasonal influenza A viruses in human airway epithelial cell cultures (33). In this study, A(H3N2)v viruses were shown to replicate at least as well as seasonal viruses in two commonly used laboratory cell lines. The emergence of A(H3N2)v viruses is a public health concern, since vaccination with seasonal vaccines does not effectively protect against these zoonotic viruses, leaving antivirals as the main line of defense.
A(H3N2)v viruses recovered from individuals in the United States during 2011 to 2013 were tested for susceptibility to two FDA-approved and three investigational NAIs. In an NI assay, IC50s for A(H3N2)v viruses were similar to those of the seasonal A(H3N2) virus. The exception was one virus, A/Ohio/88/2012, which exhibited reduced inhibition by oseltamivir and zanamivir but normal inhibition by the three other NAIs, according to the established criteria (19). Presently, the clinical relevance of this laboratory finding is unknown.
The NA of A/Ohio/88/2012 virus contained two rare substitutions, S245N and S247P, but only the latter one was responsible for the elevated IC50s. Peculiarly, a deletion encompassing the region coding for amino acids 245 to 248 has been described in A(H3N2) viruses recovered from patients treated with an NAI (28). The proline that replaced serine at position 247 is likely to provide more rigidity to the 240-amino-acid loop by imposing strict torsion angles on this segment of the NA. The result may be an adverse effect in the flexibility and positioning of this loop and its contribution in the hydrogen bonding network involved in NAI binding, as shown in the NA/zanamivir model (Fig. 1). However, the S247P substitution did not affect IC50s for all NAIs used in the study. The greatest fold increases in IC50 were observed for oseltamivir and zanamivir. Laninamivir has a structure that is very similar to that of zanamivir (28, 34); however, only a minor change (7-fold) in its IC50 was observed. No changes were detected for the IC50s of either peramivir or A-315675. These data provide a rationale for having additional NAIs available for influenza treatment.
FIG 1.
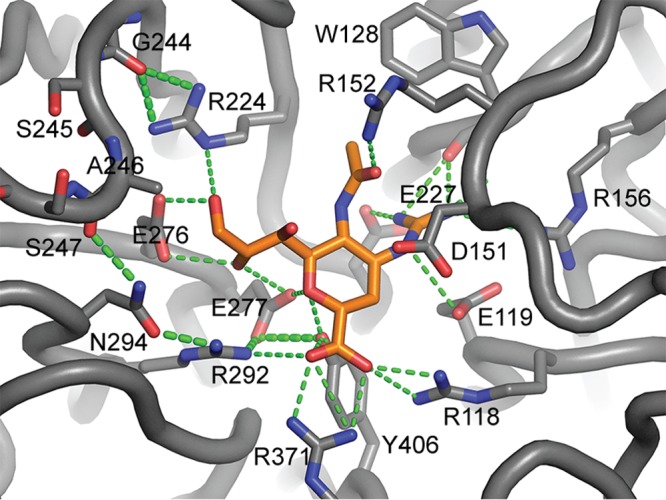
Structural model of zanamivir bound to the NA of A/Indiana/12/2012 A(H3N2)v virus. The 240-aa loop does not interact directly with the NAI but instead contributes to a hydrogen bond network that involves R224, R292, and N294 in binding zanamivir.
S247P caused a substantial loss in NA catalytic activity and reduction of infectious virus titer in cell culture (Tables 2 and 3), indications that A/Ohio/88/2012 may be less fit than the A/Ohio/83/2012 A(H3N2)v virus. These characteristics can potentially result in reduced virus transmissibility and pathogenicity.
Influenza viruses resistant to one or more antiviral drugs can emerge and spread rapidly (28, 34, 35), and active virological surveillance is essential to accurately and timely identify such changes in circulating viruses. To combat antiviral resistance acquired either naturally or under drug selection, new antivirals that target different molecular and replicative aspects of influenza virus are needed. The investigational antiviral compounds assessed in this study showed promise against the A(H3N2)v viruses tested, including the virus with the S247P substitution. In cell culture-based assays, replication of A(H3N2)v viruses was as effectively controlled by the investigational antivirals nitazoxanide, favipiravir, and fludase (15) as that of seasonal viruses. The results obtained for the seasonal viruses included in the study were consistent with previous findings for nitazoxanide (26, 36, 37), favipiravir (27), and fludase (16).
Influenza virus is an emerging and reemerging pathogen with a wide natural reservoir. Control of infections caused by zoonotic viruses requires active global surveillance and an arsenal of therapeutic options.
ACKNOWLEDGMENTS
We thank the U.S. public health laboratories for submission of specimens and Scott Epperson of the Epidemiology and Prevention Branch and other members of the Influenza Division at the CDC for their valuable contributions to this study. We also thank the pharmaceutical companies Abbott Laboratories, BioCryst Pharmaceuticals, Biota, GlaxoSmithKline, Hoffmann-La Roche Ltd., NexBio, Romark Laboratories, and Toyama Chemical Co. for providing the antiviral drugs used in this study.
We declare that we have no conflicts of interest.
The findings and conclusions of this report are those of the authors and do not necessarily represent the views of the funding agency, the U.S. Centers for Disease Control and Prevention.
Footnotes
Published ahead of print 21 January 2014
REFERENCES
- 1.Castrucci MR, Donatelli I, Sidoli L, Barigazzi G, Kawaoka Y, Webster RG. 1993. Genetic reassortment between avian and human influenza A viruses in Italian pigs. Virology 193:503–506. 10.1006/viro.1993.1155 [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. 2005. International health regulations (2005), 2nd ed. World Health Organization, Geneva, Switzerland [Google Scholar]
- 3.Council of State and Territorial Epidemiologists. 2007. National reporting for initial detections of novel influenza A viruses. Position statement 07-ID01 http://c.ymcdn.com/sites/www.cste.org/resource/resmgr/PS/07-ID-01.pdf Accessed 12 December 2012
- 4.Shu B, Garten RJ, Emery S, Balish A, Cooper L, Sessions W, Deyde V, Smith C, Berman L, Klimov A, Lindstrom S, Xu X. 2012. Genetic analysis and antigenic characterization of swine origin influenza viruses isolated from humans in the United States, 1990-2010. Virology 422:151–160. 10.1016/j.virol.2011.10.016 [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2012. Update: influenza A (H3N2)v transmission and guidelines—five states, 2011. MMWR Morb. Mortal. Wkly. Rep. 60:1741–1744 http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6051a4.htm [PubMed] [Google Scholar]
- 6.Lindstrom S, Garten R, Balish A, Shu B, Emery S, Berman L, Barnes N, Sleeman K, Gubareva L, Villanueva J, Klimov A. 2012. Human infections with novel reassortant influenza A(H3N2)v viruses, United States, 2011. Emerg. Infect. Dis. 18:834–837. 10.3201/eid1805.111922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou YY, Albrecht RA, Pica N, Lowen AC, Richt JA, Garcia-Sastre A, Palese P, Hai R. 2011. The M segment of the 2009 new pandemic H1N1 influenza virus is critical for its high transmission efficiency in the guinea pig model. J. Virol. 85:11235–11241. 10.1128/JVI.05794-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lakdawala SS, Lamirande EW, Suguitan AL, Jr, Wang W, Santos CP, Vogel L, Matsuoka Y, Lindsley WG, Jin H, Subbarao K. 2011. Eurasian-origin gene segments contribute to the transmissibility, aerosol release, and morphology of the 2009 pandemic H1N1 influenza virus. PLoS Pathog. 7:e1002443. 10.1371/journal.ppat.1002443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention. 2013. Case count: detected U.S. human infections with H3N2v by state since August 2011. http://www.cdc.gov/flu/swineflu/h3n2v-case-count.htm Accessed 5 June 2013
- 10.Jhung MA, Epperson S, Biggerstaff M, Allen D, Balish A, Barnes N, Beaudoin A, Berman L, Bidol S, Blanton L, Blythe D, Brammer L, D'Mello T, Danila R, Davis W, de Fijter S, DiOrio M, Durand LO, Emery S, Fowler B, Garten R, Grant Y, Greenbaum A, Gubareva L, Havers F, Haupt T, House J, Ibrahim S, Jiang V, Jain S, Jernigan D, Kazmierczak J, Klimov A, Lindstrom S, Longenberger A, Lucas P, Lynfield R, McMorrow M, Moll M, Morin C, Ostroff S, Page SL, Park SY, Peters S, Quinn C, Reed C, Richards S, Scheftel J, Simwale O, Shu B, Soyemi K, Stauffer J, Steffens C, Su S, Torso L, Uyeki TM, Vetter S, Villanueva J, Wong KK, Shaw M, Bresee J, Cox N, Finelli L. Outbreak of variant influenza A(H3N2) virus in the United States.Clin. Infect. Dis. 057:01703–1712. 10.1093/cid/cit649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention. 2011. Limited human-to-human transmission of novel influenza A (H3N2) virus—Iowa, November 2011. MMWR Morb. Mortal. Wkly. Rep. 60:1615–1617 http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6047a3.htm [PubMed] [Google Scholar]
- 12.Centers for Disease Control and Prevention. 2012. Influenza A (H3N2) variant virus-related hospitalizations—Ohio, 2012. MMWR Morb. Mortal. Wkly. Rep. 61:764–767 http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6138a3.htm [PubMed] [Google Scholar]
- 13.Centers for Disease Control and Prevention. 2012. Evaluation of rapid influenza diagnostic tests for influenza A (H3N2)v virus and updated case count—United States, 2012. MMWR Morb. Mortal. Wkly. Rep. 61:619–621 http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6132a4.htm [PubMed] [Google Scholar]
- 14.Houser KV, Katz JM, Tumpey TM. 2013. Seasonal trivalent inactivated influenza vaccine does not protect against newly emerging variants of influenza A (H3N2v) virus in ferrets. J. Virol. 87:1261–1263. 10.1128/JVI.02625-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wathen MW, Barro M, Bright RA. 2013. Antivirals in seasonal and pandemic influenza—future perspectives. Influenza Other Respir. Viruses 7(Suppl 1):76–80. 10.1111/irv.12049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Triana-Baltzer GB, Gubareva LV, Nicholls JM, Pearce MB, Mishin VP, Belser JA, Chen LM, Chan RW, Chan MC, Hedlund M, Larson JL, Moss RB, Katz JM, Tumpey TM, Fang F. 2009. Novel pandemic influenza A(H1N1) viruses are potently inhibited by DAS181, a sialidase fusion protein. PLoS One 4:e7788. 10.1371/journal.pone.0007788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.World Health Organization Global Influenza Surveillance Network. 2011. Manual for the laboratory diagnosis and virological surveillance of influenza. WHO Press, Geneva, Switzerland: http://whqlibdoc.who.int/publications/2011/9789241548090_eng.pdf [Google Scholar]
- 18.Okomo-Adhiambo M, Sleeman K, Lysen C, Nguyen HT, Xu X, Li Y, Klimov AI, Gubareva LV. 2013. Neuraminidase inhibitor susceptibility surveillance of influenza viruses circulating worldwide during the 2011 Southern Hemisphere season. Influenza Other Respir. Viruses 10.1111/irv.12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.World Health Organization. 2012. Meetings of the WHO working group on surveillance of influenza antiviral susceptibility—Geneva, November 2011 and June 2012. Wkly. Epidemiol. Rec. 87:369–380 http://www.who.int/wer/2012/wer8739/en/index.html [PubMed] [Google Scholar]
- 20.World Health Organization. 2013. Laboratory methodologies for testing the antiviral susceptibility of influenza viruses: neuraminidase inhibitor (NAI). http://www.who.int/influenza/gisrs_laboratory/antiviral_susceptibility/nai_overview/en/index.html Accessed 6 November 2013
- 21.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95–98 [Google Scholar]
- 22.Deyde VM, Okomo-Adhiambo M, Sheu TG, Wallis TR, Fry A, Dharan N, Klimov AI, Gubareva LV. 2009. Pyrosequencing as a tool to detect molecular markers of resistance to neuraminidase inhibitors in seasonal influenza A viruses. Antiviral Res. 81:16–24. 10.1016/j.antiviral.2008.08.008 [DOI] [PubMed] [Google Scholar]
- 23.Xu X, Zhu X, Dwek RA, Stevens J, Wilson IA. 2008. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J. Virol. 82:10493–10501. 10.1128/JVI.00959-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matrosovich M, Matrosovich T, Carr J, Roberts NA, Klenk HD. 2003. Overexpression of the alpha-2,6-sialyltransferase in MDCK cells increases influenza virus sensitivity to neuraminidase inhibitors. J. Virol. 77:8418–8425. 10.1128/JVI.77.15.8418-8425.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ashton LV, Callan RL, Rao S, Landolt GA. 2010. In vitro susceptibility of canine influenza A (H3N8) virus to nitazoxanide and tizoxanide. Vet. Med. Int. 10.4061/2010/891010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sleeman K, Mishin VP, Deyde VM, Furuta Y, Klimov AI, Gubareva LV. 2010. In vitro antiviral activity of favipiravir (T-705) against drug-resistant influenza and 2009 A(H1N1) viruses. Antimicrob. Agents Chemother. 54:2517–2524. 10.1128/AAC.01739-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwede T, Kopp J, Guex N, Peitsch MC. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385. 10.1093/nar/gkg520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samson M, Pizzorno A, Abed Y, Boivin G. 2013. Influenza virus resistance to neuraminidase inhibitors. Antiviral Res. 98:174–185. 10.1016/j.antiviral.2013.03.014 [DOI] [PubMed] [Google Scholar]
- 29.Hurt AC, Holien JK, Parker M, Kelso A, Barr IG. 2009. Zanamivir-resistant influenza viruses with a novel neuraminidase mutation. J. Virol. 83:10366–10373. 10.1128/JVI.01200-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okomo-Adhiambo M, Nguyen HT, Sleeman K, Sheu TG, Deyde VM, Garten RJ, Xu X, Shaw MW, Klimov AI, Gubareva LV. 2010. Host cell selection of influenza neuraminidase variants: implications for drug resistance monitoring in A(H1N1) viruses. Antiviral Res. 85:381–388. 10.1016/j.antiviral.2009.11.005 [DOI] [PubMed] [Google Scholar]
- 31.Sheu TG, Deyde VM, Okomo-Adhiambo M, Garten RJ, Xu X, Bright RA, Butler EN, Wallis TR, Klimov AI, Gubareva LV. 2008. Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob. Agents Chemother. 52:3284–3292. 10.1128/AAC.00555-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamura D, Nguyen HT, Sleeman K, Levine M, Mishin VP, Yang H, Guo Z, Okomo-Adhiambo M, Xu X, Stevens J, Gubareva LV. 2013. Cell culture-selected substitutions in influenza A(H3N2) neuraminidase affect drug susceptibility assessment. Antimicrob. Agents Chemother. 57:6141–6146. 10.1128/AAC.01364-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pearce MB, Jayaraman A, Pappas C, Belser JA, Zeng H, Gustin KM, Maines TR, Sun X, Raman R, Cox NJ, Sasisekharan R, Katz JM, Tumpey TM. 2012. Pathogenesis and transmission of swine origin A(H3N2)v influenza viruses in ferrets. Proc. Natl. Acad. Sci. U. S. A. 109:3944–3949. 10.1073/pnas.1119945109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen HT, Fry AM, Gubareva LV. 2012. Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir. Ther. 17:159–173. 10.3851/IMP2067 [DOI] [PubMed] [Google Scholar]
- 35.Ison MG. 2012. Expanding the armamentarium against respiratory viral infections: DAS181. J. Infect. Dis. 206:1806–1808. 10.1093/infdis/jis623 [DOI] [PubMed] [Google Scholar]
- 36.Belardo G, La Frazia S, Cenciarelli O, Carta S, Rossignol J-F, Santoro MG. 2011. Nitazoxanide, a novel potential anti-influenza drug, acting in synergism with neuraminidase inhibitors. IDSA%202011%20NTZ%20poster%20Belardo%20et%20al%20Final%20100611.pdf. Accessed 18 November 2013
- 37.Rossignol JF, La FS, Chiappa L, Ciucci A, Santoro MG. 2009. Thiazolides, a new class of anti-influenza molecules targeting viral hemagglutinin at the post-translational level. J. Biol. Chem. 284:29798–29808. 10.1074/jbc.M109.029470 [DOI] [PMC free article] [PubMed] [Google Scholar]