

Abstract
Clostridium difficile is the causative agent of C. difficile-associated diarrhea (CDAD), with increased risk in elderly populations. Kibdelomycin, a novel natural-product inhibitor of type II topoisomerase enzymes, was evaluated for activity against C. difficile and gastrointestinal anaerobic organisms. Toxigenic C. difficile isolates (n = 168) from U.S. hospitals and anaerobic Gram-positive and Gram-negative organisms (n = 598) from Chicago-area hospitals were tested. Kibdelomycin showed potent activity against toxigenic C. difficile (MIC90 = 0.25 μg/ml) and most Gram-positive aerobic organisms but had little activity against Bacteroides species (MIC50 > 32 μg/ml; n = 270). Potent anti-C. difficile activity was also observed in the hamster model of C. difficile colitis. Dosing at 1.6 mg/kg (twice-daily oral dose) resulted in protection from a lethal infection and a 2-log reduction in C. difficile cecal counts. A 6.25-mg/kg twice-daily oral dose completely eliminated detectable C. difficile counts in cecal contents. A single 6.25-mg/kg oral dose showed that cecal contents were exposed to the drug at >2 μM (eightfold higher than the MIC), with no significant plasma exposure. These findings support further exploration of kibdelomycin for development of an anti-C. difficile agent.
INTRODUCTION
Clostridium difficile infection (CDI) is increasingly prevalent worldwide and is the leading cause of antibiotic-associated nosocomial C. difficile-associated diarrhea (CDAD) in community hospitals (1). This organism surpasses even methicillin-resistant Staphylococcus aureus (MRSA) as a leading cause of hospital-acquired infection in the United States, and the severity of CDAD has worsened over time (2, 3). The rise of CDAD is due, in part, to the emergence of a hypervirulent toxigenic strain (NAP-1, ribotype 027, REA type BI) which causes more severe disease that is more frequently fatal among susceptible and elderly patients (4). CDI is caused by an imbalance of bacterial species in the gut microbiota (5), generally due to treatment with broad-spectrum antibiotics. These treatments cause selective inhibition of commensal Gram-negative bacteria, leading to disproportional increased growth of Gram-positive C. difficile. Withdrawal of the offending antibiotic often improves the underlying symptoms, although many patients require treatment of CDI with a narrow-spectrum antibiotic. Metronidazole and vancomycin are the first-line treatment options for CDAD, and these agents are initially efficacious in eradicating symptoms. However, cessation of therapy results in disease recurrence in a significant number of patients (∼20%) and the recurrent disease becomes progressively worse after subsequent treatment rounds (6, 7). Fidaxomicin was approved recently as a new treatment for CDI, with significantly improved sustained response. Unfortunately, such improvement does not translate in patients infected with hypervirulent strains, which are causing CDAD in North America and Europe with increasing frequency (8–10). Fecal microbiota transplant is another approach that has been recently used for the treatment of recurrent CDI, with remarkable clinical success (11). A number of additional new CDAD agents are in clinical development (12–15), and there remains an unmet medical need for a new CDI agent with a new mechanism of action that could provide a beneficial effect to patients with all types of CDI.
Kibdelomycin (KBD) (Fig. 1) is a novel natural-product broad-spectrum Gram-positive antibiotic discovered using a genomics-based Staphylococcus aureus fitness test screening method (16, 17). It prevents DNA synthesis by inhibiting the ATPase activity of type II topoisomerase enzymes, DNA gyrase, and topoisomerase IV (17, 18). Kibdelomycin has a large molecular size, which limits systemic absorption following oral dosing, a desirable feature for a safe CDI agent. Our initial studies showed potent inhibition of C. difficile with a MIC of 0.12 μg/ml (19). This study reports an initial microbiological characterization of kibdelomycin, including the in vitro activity against a diverse collection of 168 C. difficile toxigenic clinical isolates, including the hypervirulent BI strains, and other gastrointestinal anaerobes that are generally commensal organisms. The in vivo efficacy and exposure (pharmacokinetics [PK]) were measured and correlated in a hamster model of infection to assess this compound's potential utility as a C. difficile agent.
FIG 1.
Chemical structure of kibdelomycin.
MATERIALS AND METHODS
Test agents.
Kibdelomycin (KBD) was obtained from Merck Research Laboratories (17), fidaxomicin was purified from DIFICID tablets (Optimer Pharmaceuticals, Inc., San Diego, CA), metronidazole was purchased from Sigma (Saint Louis, MO), vancomycin was purchased from ViroPharma Inc. (Exton, PA), and moxifloxacin was produced by Bayer Healthcare (Leverkusen, Germany).
Bacterial strains.
C. difficile ATCC 700057 and B. fragilis ATCC 25285 were used as the control strains, and all quality controls were within the range for approved drugs. All 598 non-C. difficile isolates were derived from patient samples collected from 2004 to 2009 at multiple medical centers (>10) in the Chicago area and stored (−70°C) at the Loyola Anaerobe Laboratory. The C. difficile isolates (n = 168) were obtained from geographically widely dispersed U.S. hospitals and were voluntarily submitted as part of identified stool specimens from CDI patients to the Loyola University reference laboratory for isolation and typing of the C. difficile organisms. The majority of the C. difficile strains were collected between 2006 and 2008 and were all verified as toxigenic. All isolates were DNA fingerprinted using restriction endonuclease analysis (REA). The so-called hypervirulent epidemic strains were designated by the letters “BI” for the REA group, which is the same designation as PFGE NAP1 and PCR ribotype 027. Isolates of the BI group constituted approximately 50% of the isolates tested during this study. Other commonly identified and epidemic or highly endemic C. difficile groups include the J group (PCR type 001), the K group, the Y group (PCR type 014), and the G group. The BK group is found in animals and humans (PCR ribotype 078). There is diversity within each of these groups, as defined by restriction endonuclease analysis.
Susceptibility testing.
The in vitro activity (MIC) was measured using the CLSI agar dilution method for anaerobe susceptibility testing (M11-A7) (20). Comparator antimicrobial agents used for the C. difficile susceptibility test were fidaxomicin (OPT-80), metronidazole (MET), and vancomycin (VAN). Comparator antimicrobial agents used for other anaerobic organisms were fidaxomicin and metronidazole.
Hamster C. difficile gastrointestinal infection model.
The in vivo efficacy of KBD was measured in a C. difficile hamster infection model with the toxigenic B1 strain (SM8-6865). Groups of five golden Syrian hamsters (average weight = 98 g) were rendered susceptible to C. difficile by administration of clindamycin hydrochloride by oral gavage (PO) at 30 mg/kg 5 days prior to infection. Animals were then infected with spores of the toxigenic B1 strain by oral gavage, and 53 spores (average value) were administered in 0.5 ml sterile saline. Vancomycin and kibdelomycin treatments were initiated 5 h postinfection by oral dosing. Vancomycin was dissolved in sterile saline and given once daily for a total of 4 days. Kibdelomycin was given once on the first day of infection and then twice daily for the next 3 days. Kibdelomycin (1.25 mg/ml) was dissolved in the dosing vehicle dimethyl sulfoxide (DMSO)-Tween 80-water (10:10:80 [vol/vol]), providing a clear solution. All surviving animals were euthanized 4 days postinfection, and the viable C. difficile burden in the cecum contents was measured by plating dilutions onto Clostridium difficile-selective agar supplemented with 10% sodium taurocholate.
Pharmacokinetic (PK) measurements.
Two fasting male C57BL/6 mice were used for PK measurements using intravenous (IV) or oral administration. Kibdelomycin, 1 mg/ml, was dissolved in ethyl alcohol (EtOH)–polyethylene glycol (PEG) 400–water (10/50/40), yielding a clear solution for intravenous dosing at 1 mg/kg. For oral dosing, KBD was suspended at 0.4 mg/ml in Tween 80–0.5% Methocel (10/90) and given at 2 mg/kg. Plasma concentrations at time points over 24 h were determined for IV and oral routes of administration using liquid chromatography-tandem mass spectrometry (LC-MS/MS) in which the lowest level of quantification (LLOQ) was 4.45 nM. Hamster PK analysis was performed with 15 golden Syrian hamsters (average weight = 99 g). Animals were dosed orally with kibdelomycin, 6.25 mg/kg, as a clear solution (1.25 mg/ml) in DMSO–Tween 80–water (10/10/80). Samples of plasma and cecum contents were collected at 1, 2, 4, 8, and 24 h, and 50 μl sample was combined with 300 μl acetonitrile spiked with a diclofenac internal standard. Samples were analyzed by LC-MS/MS in which the LLOQ was 10 nM. Analysis was performed with using a Thermo Scientific LX-2 LC instrument, an Acquity UPLC HSS T3 column (1.8-μm particle size; 50- by 2.1-mm inside diameter), and an API 4000 LC-MS/MS triple-quadrupole mass spectrometer equipped with a Turbo ion spray ionization source (Applied Biosystems/MDS Sciex, Concord, Ontario, Canada) for tandem mass spectrometry. Chromatography was performed with gradient elution in which mobile phase A was 0.1% formic acid in water, mobile phase B was 0.1% formic acid in acetonitrile, and the solvent composition was changed from 5% B to 95% B in 1.5 min and then held at 95% B for an additional 0.5 min. MS/MS analysis was performed in the SRM (selected reaction monitoring) negative ionization mode using the mass transitions m/z 937→762 (CE = −60eV) for kibdelomycin and m/z 294→250 (CE = −30eV) for diclofenac. The nebulization temperature was 550°C. Peak areas were integrated using Analyst software. Linear regression with a weighting of 1/x2 was used to construct the standard curves.
RESULTS
In vitro antibacterial activity.
The MICs for various C. difficile strains, categorized by the restriction endonuclease (REA) type and the clindamycin and moxifloxacin susceptibility, are shown in Table 1 (21). The cumulative MICs are presented in Fig. 2. Kibdelomycin demonstrated potent growth inhibition of C. difficile with MIC50 and MIC90s of 0.125 and 0.5 μg/ml, respectively. The MIC distribution range was narrow, 0.06 to 0.5 μg/ml, with over 75% of strains having an MIC of 0.125 μg/ml (Fig. 2). The kibdelomycin MIC50s and MIC90s were identical to those of fidaxomicin, although fidaxomicin had a somewhat lower MIC range, ≤0.03 to 0.5 μg/ml. Kibdelomycin was 2- to 4-fold more potent than metronidazole (MIC90 = 0.5 μg/ml) and vancomycin (MIC90 = 2 μg/ml). The potencies of kibdelomycin, fidaxomicin, metronidazole, and vancomycin were consistent among the different REA types, including the hypervirulent BI (NAP1 027) strains. These agents were also equally potent against clindamycin- and moxifloxacin-resistant strains.
TABLE 1.
Comparative in vitro activity of kibdelomycin, fidaxomicin, metronidazole, and vancomycin against Clostridium difficile strains
| Organism typea (no. of isolates) | Antimicrobial agent | MIC (μg/ml) |
||
|---|---|---|---|---|
| 50% | 90% | Range | ||
| All C. difficile strains (168)b | Kibdelomycin | 0.125 | 0.25 | 0.06–0.5 |
| Fidaxomicin | 0.125 | 0.25 | ≤ 0.03–0.5 | |
| Metronidazole | 0.25 | 0.5 | 0.125–2 | |
| Vancomycin | 0.5 | 2 | 0.5–4 | |
| REA BI type (70) | Kibdelomycin | 0.125 | 0.25 | 0.06–0.25 |
| Fidaxomicin | 0.25 | 0.25 | ≤0.03–0.25 | |
| Metronidazole | 0.25 | 0.5 | 0.125–2 | |
| Vancomycin | 0.5 | 2 | 0.5–4 | |
| REA Y type (21) | Kibdelomycin | 0.125 | 0.125 | 0.06–0.125 |
| Fidaxomicin | 0.125 | 0.25 | ≤0.03–0.25 | |
| Metronidazole | 0.125 | 0.25 | 0.125–0.25 | |
| Vancomycin | 0.5 | 1 | 0.5–2 | |
| REA DH type (11) | Kibdelomycin | 0.125 | 0.125 | 0.06–0.125 |
| Fidaxomicin | 0.125 | 0.25 | ≤0.03–0.25 | |
| Metronidazole | 0.25 | 0.25 | 0.25 | |
| Vancomycin | 0.5 | 1 | 0.5–1 | |
| REA J type (9) | Kibdelomycin | 0.06–0.5 | ||
| Fidaxomicin | ≤0.03–0.5 | |||
| Metronidazole | 0.125–0.5 | |||
| Vancomycin | 0.5–2 | |||
| CF (5) | Kibdelomycin | 0.125–0.25 | ||
| Fidaxomicin | 0.125–0.25 | |||
| Metronidazole | 0.125–0.25 | |||
| Vancomycin | 0.5–0.5 | |||
| K (5) | Kibdelomycin | 0.0.06–0.125 | ||
| Fidaxomicin | ≤0.03–0.25 | |||
| Metronidazole | 0.25–0.5 | |||
| Vancomycin | 1–2 | |||
| CLX susceptiblec (47) | Kibdelomycin | 0.125 | 0.25 | 0.06–0.25 |
| Fidaxomicin | 0.125 | 0.25 | ≤0.03–0.25 | |
| Metronidazole | 0.25 | 0.5 | 0.125–2 | |
| Vancomycin | 0.5 | 2 | 0.5–4 | |
| CLX resistantc (121) | Kibdelomycin | 0.125 | 0.25 | 0.06–0.5 |
| Fidaxomicin | 0.25 | 0.25 | ≤0.03–0.5 | |
| Metronidazole | 0.25 | 0.5 | 0.125–1 | |
| Vancomycin | 1 | 2 | 0.5–4 | |
| MFX susceptibled (77) | Kibdelomycin | 0.125 | 0.125 | 0.06–0.25 |
| Fidaxomicin | 0.125 | 0.25 | ≤0.03–0.25 | |
| Metronidazole | 0.125 | 0.25 | 0.125–0.25 | |
| Vancomycin | 0.5 | 1 | 0.5–2 | |
| MFX resistantd (91) | Kibdelomycin | 0.125 | 0.25 | 0.06–0.5 |
| Fidaxomicin | 0.25 | 0.25 | ≤0.03–0.5 | |
| Metronidazole | 0.25 | 0.5 | 0.125–2 | |
| Vancomycin | 1 | 2 | 0.5–4 | |
REA (restriction endonuclease) groups include multiple PCR ribotypes as described previously (21). The REA groups evaluated in this study and corresponding ribotypes are BI (ribotypes 016, 027, and 036), Y (ribotypes 014, 020, and 9 others), DH (106, 118, 147, and 174), J (ribotypes 001, 055, 072, and 115), CF (ribotypes 017, 047, and 066), and K (ribotypes 053, and 163).
Data from 47 organisms are represented in the set of all 163 strains but not in individual groups. The REA types of 43 isolates were not determined with certainty, and three groups (BK, G, and I) were identified infrequently (1 or 2 isolates).
Clindamycin (CLX) susceptible, MIC ≤ 4; clindamycin resistant, MIC ≥ 8 μg/ml.
Moxifloxacin (MFX) susceptible, MIC ≤ 4; moxifloxacin resistant, MIC ≥ 8 μg/ml.
FIG 2.
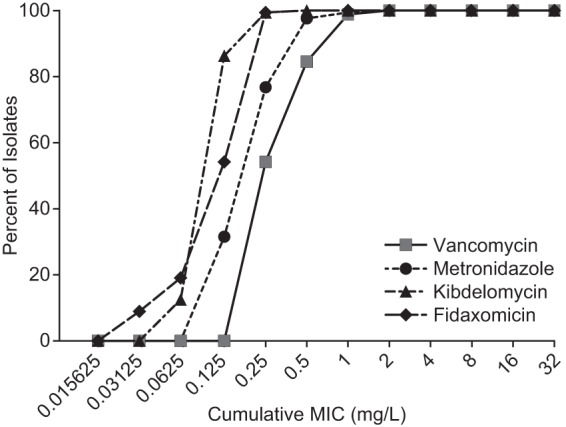
Cumulative MICs of kibdelomycin and comparators against C. difficile isolates (n = 168) (Table 1).
Kibdelomycin and the comparator antibiotics metronidazole and fidaxomicin were evaluated for activity against 598 strains (Tables 2 and 3) of anaerobic Gram-positive and -negative bacteria, including Bacteroides species (Tables 2 and 3), which predominate in the gastrointestinal flora (5). Like fidaxomicin, kibdelomycin is primarily active against Gram-positive anaerobic organisms, with less or no activity against most Gram-negative organisms. Kibdelomycin was active against most Clostridium species, with MIC90s of 0.06 to 0.25 μg/ml. Clostridium cadaveris was the Clostridium species which was insensitive (MIC > 32 μg/ml, n = 2). Kibdelomycin and fidaxomicin were relatively inactive against members of the Bacteroides fragilis group. More than 85% of the Bacteroides isolates were not susceptible to kibdelomycin (MIC > 32 μg/ml). In contrast, metronidazole was active against Gram-positive and -negative organisms.
TABLE 2.
Comparative in vitro activity of kibdelomycin, fidaxomicin, metronidazole, clindamycin, and moxifloxacin against anaerobic gut bacteria
| Organism (no. of isolates) | Antimicrobial agent | MIC (μg/ml) |
||
|---|---|---|---|---|
| 50% | 90% | Range | ||
| Gram positive | ||||
| Actinomyces meyer (15) | Kibdelomycin | 0.25 | 4 | ≤0.015 − 8 |
| Fidaxomicin | 0.5 | 4 | ≤0.015 − 8 | |
| Metronidazole | >16 | >16 | 16 − >16 | |
| Clindamycin | ≤0.125 | >64 | ≤0.125 − >64 | |
| Moxifloxacin | 0.5 | 1 | 0.125 − 4 | |
| Bifidobacterium spp. (6) | Kibdelomycin | ≤0.015 − 32 | ||
| Fidaxomicin | ≤0.015 − 0.06 | |||
| Metronidazole | 0.125 − 32 | |||
| Clindamycin | ≤0.125− 2 | |||
| Moxifloxacin | 0.125 − 4 | |||
| Clostridium spp.a (62) | Kibdelomycin | 0.06 | 8 | ≤0.015 − 32 |
| Fidaxomicin | ≤0.015 | >32 | ≤0.015 − >32 | |
| Metronidazole | 0. 5 | 1 | ≤0.015 − 2 | |
| Clindamycin | 1 | 4 | ≤0.125− >64 | |
| Moxifloxacin | 0.5 | 4 | 0.125 − >32 | |
| Gamella morbillorum (6) | Kibdelomycin | ≤0.015 − 32 | ||
| Fidaxomicin | ≤0.015 − 32 | |||
| Metronidazole | 0.25 − >16 | |||
| Clindamycin | ≤0.125 − 1 | |||
| Moxifloxacin | ≤0.06 − 8 | |||
| Mobiluncus spp. (3) | Kibdelomycin | 0.03 − 8 | ||
| Fidaxomicin | 0.5 − 1 | |||
| Metronidazole | 8 − 32 | |||
| Clindamycin | ≤0.125 − 4 | |||
| Moxifloxacin | 0.5 − 4 | |||
| Peptostreptococcus spp.b (101) | Kibdelomycin | 0.03 | 0.25 | ≤0.015 − 32 |
| Fidaxomicin | 0.25 | 1 | 0.25 − >32 | |
| Metronidazole | 0.5 | 1 | ≤0.06 − >16 | |
| Clindamycin | 0.5 | >64 | ≤0.125 − >64 | |
| Moxifloxacin | 0.5 | 8 | 0.015 − >32 | |
| Propionibacterium spp.c (19) | Kibdelomycin | 8 | 8 | 0.03 − 32 |
| Fidaxomicin | 4 | 8 | 0.03 − 32 | |
| Metronidazole | >16 | >16 | 0.5 − >16 | |
| Clindamycin | ≤0.125 | 4 | ≤0.125 − 32 | |
| Moxifloxacin | 0.25 | 1 | 0.25 − 8 | |
| Gram negative | ||||
| Bacteroides spp.d (308) | Kibdelomycin | >32 | >32 | 0.03 − >32 |
| Fidaxomicin | >32 | >32 | 32 − >32 | |
| Metronidazole | 0.5 | 1 | 0.125 − >16 | |
| Clindamycin | 4 | >64 | ≤0.125 − >64 | |
| Moxifloxacin | 4 | 32 | 0.25 − >32 | |
| Fusobacterium spp.e (17) | Kibdelomycin | 0.03 | 32 | ≤0.015 − >32 |
| Fidaxomicin | 0.03 | 64 | ≤0.015 − >32 | |
| Metronidazole | 0.125 | 0.5 | ≤0.06 − 1 | |
| Clindamycin | ≤0.125 | 8 | ≤0.125 − 64 | |
| Moxifloxacin | 0.25 | 8 | ≤0.06 − >32 | |
| Prevotella spp.f (50) | Kibdelomycin | 16 | >32 | ≤0.015 − >32 |
| Fidaxomicin | >32 | >32 | 0.03 − >32 | |
| Metronidazole | 0.5 | 1 | ≤0.06 − 2 | |
| Clindamycin | 2 | >64 | ≤0.125 − >64 | |
| Moxifloxacin | 2 | >32 | 0.5 − >32 | |
| Veillonella spp. (11) | Kibdelomycin | 0.125 | 32 | 0.03 − 32 |
| Fidaxomicin | 32 | 32 | 0.25 − 32 | |
| Metronidazole | 1 | 2 | 0.25 − 2 | |
| Clindamycin | 2 | >64 | ≤0.125 − >64 | |
| Moxifloxacin | 0.5 | 4 | 0.125 − 8 | |
Clostridium beijerinckii (1 isolate), C. butyricum (1), C. cadaverus (2), C. clostridioforme (3), C. hastiforme (4), C. innocuum (3), C. perfringens (40), C. ramosum (6), C. subterminale (2).
Peptostreptococcus anaerobius (15 isolates), P. asaccharolyticus (22), P. magnus (38), P. micros (16), P. prevotii (8), P. tetradius (2).
Propionibacterium acnes (16 isolates), P. granulosum (3).
Bacteroides caccae (10 isolates), B. distasonis (29), B. stercoris (4), B. thetaiotaomicron (59), B. eggerthii (30), B. fragilis (105), B. ovatus (10), B. uniformis (33), B. vulgatus (28). See Table 3 for the MIC50, MIC90, and MIC range for this group of organisms.
Fusobacterium necrophorum (10 isolates), F. nucleatum (2), F. varium (5).
Prevotella bivia (19 isolates), P. buccae (7), P. disiens (4), P. intermedia (3), P. loescheii (2), P. melaninogenica (10), P. oralis group (5).
TABLE 3.
Comparative in vitro activities of kibdelomycin, fidaxomicin, metronidazole, clindamycin, and moxifloxacin against members of the Bacteroides group
| Organism (no. of isolates) | Antimicrobial agent | MIC (μg/ml) |
||
|---|---|---|---|---|
| 50% | 90% | Range | ||
| Bacteroides caccae (10) | Kibdelomycin | 32 | >32 | 0.25–>32 |
| Fidaxomicin | >32 | >32 | >32–>32 | |
| Metronidazole | 0.5 | 1 | 0.25–1 | |
| Clindamycin | 8 | >64 | ≤0.125–>64 | |
| Moxifloxacin | 16 | >32 | 0.5–>32 | |
| Bacteroides distasonis (29) | Kibdelomycin | 0.125 | >32 | 0.03–>32 |
| Fidaxomicin | >32 | >32 | >32–>32 | |
| Metronidazole | 0.5 | 1 | 0.125–1 | |
| Clindamycin | 8 | >64 | 1–>64 | |
| Moxifloxacin | 1 | 16 | 0.25–>32 | |
| Bacteroides eggerthii (30) | Kibdelomycin | >32 | >32 | 0.125–>32 |
| Fidaxomicin | >32 | >32 | 32–>32 | |
| Metronidazole | 0.5 | 1 | 0.5–1 | |
| Clindamycin | 4 | >64 | 0.5–>64 | |
| Moxifloxacin | 4 | >32 | 0.5–>32 | |
| Bacteroides fragilis (105) | Kibdelomycin | >32 | >32 | 0.5–>32 |
| Fidaxomicin | >32 | >32 | 32–>32 | |
| Metronidazole | 0.5 | 1 | 0.125–2 | |
| Clindamycin | 2 | >64 | ≤0.125–>64 | |
| Moxifloxacin | 1 | 16 | 0.25–>32 | |
| Bacteroides ovatus (10) | Kibdelomycin | >32 | >32 | 0.25–>32 |
| Fidaxomicin | >32 | >32 | >32–>32 | |
| Metronidazole | 0.5 | 1 | 0.25–1 | |
| Clindamycin | 4 | >64 | 4–>64 | |
| Moxifloxacin | 4 | 16 | 2–32 | |
| Bacteroides thetaiotaomicron (59) | Kibdelomycin | >32 | >32 | 0.25–>32 |
| Fidaxomicin | >32 | >32 | 32–>32 | |
| Metronidazole | 1 | 1 | 0.015–2 | |
| Clindamycin | 8 | >64 | ≤0.125–>64 | |
| Moxifloxacin | 4 | 32 | 0.25–>32 | |
| Bacteriodes uniformis (33) | Kibdelomycin | >32 | >32 | 0.125–>32 |
| Fidaxomicin | >32 | >32 | >32–>32 | |
| Metronidazole | 1 | 1 | 0.25–1 | |
| Clindamycin | 8 | >64 | ≤0.125–>64 | |
| Moxifloxacin | 8 | >32 | 2–>32 | |
| Bacteriodes vulgatus (28) | Kibdelomycin | 1 | >32 | 0.125–>32 |
| Fidaxomicin | >32 | >32 | 32–>32 | |
| Metronidazole | 0.5 | 1 | 0.125–1 | |
| Clindamycin | 16 | >64 | ≤0.125–>64 | |
| Moxifloxacin | 32 | >32 | 0.5–>32 | |
In vivo efficacy.
The efficacy of kibdelomycin was observed in the hamster CDAD model, which monitored the 4-day survival of test animals and the C. difficile counts in the cecal contents (Fig. 3). The separate animal groups (n = 5) were orally dosed with vancomycin or kibdelomycin 5 h postinfection. Dosing was for a total of 4 days. Vancomycin was given once daily at 6.25 mg/kg, whereas kibdelomycin was given twice daily at 12.5, 6.25, and 1.56 mg/kg. Kibdelomycin demonstrated efficacy comparable to that of vancomycin, conferring 100% protection when given at 12.5 and 6.25 mg/kg and 80% protection at 1.56 mg/kg. A large reduction in C. difficile counts was observed following kibdelomycin treatment: a >2-log reduction was observed in the 1.56-mg/kg treatment group, and a >5-log reduction was seen in the 6.25- and 12.5-mg/kg treatment groups (counts below the limit of detection [LOD]).
FIG 3.
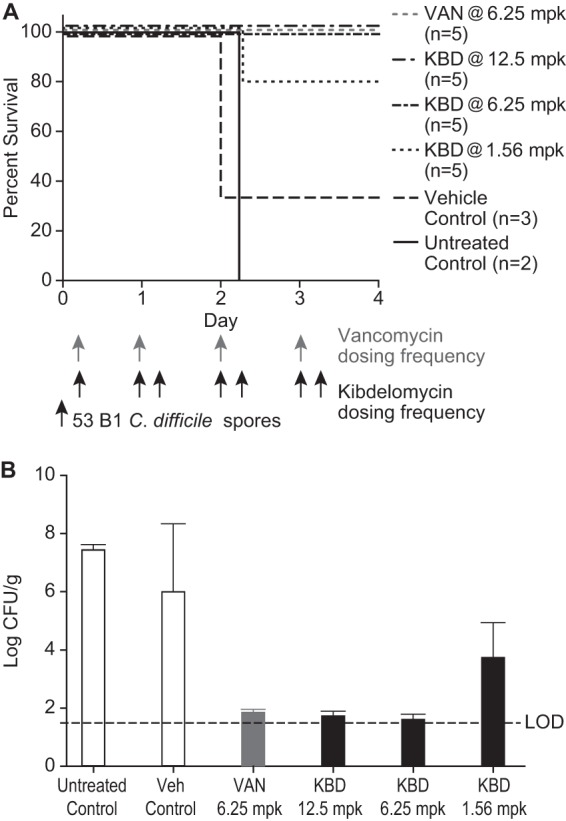
Efficacy of kibdelomycin and vancomycin in the hamster CDAD model. (A) Four-day survival of the lethal infection. (B) C. difficile counts in cecal contents recovered 4 days postinfection. LOD, lowest level of organism detection.
Pharmacokinetics.
Plasma concentration in C57BL/6 mice was measured following a single dose, 1 mg/kg (IV) or 2 mg/kg (oral). Kibdelomycin demonstrated no systemic exposure in plasma following oral dosing: plasma concentrations were below the LOQ at all of the time points (Fig. 4). KBD was reasonably stable in plasma following IV dosing: terminal half-life [t1/2], 3 h; normalized area under the concentration-time curve (AUC), 0.41 μM · h; clearance (CL), 43 ml/min/kg; steady-state volume of distribution (Vss), 6.08 liters/kg. Kibdelomycin was significantly protein bound, as demonstrated by a 32-fold shift in the S. aureus MIC (17).
FIG 4.
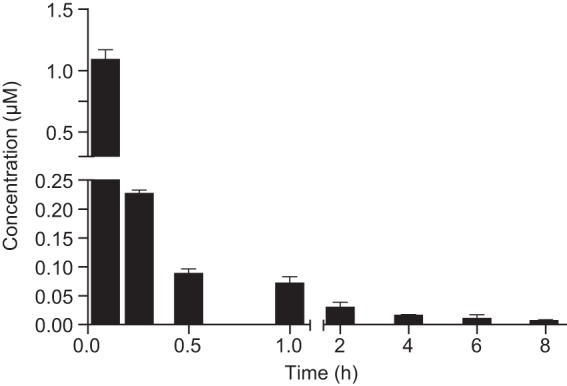
Plasma time-concentration profile of kibdelomycin at a 1-mg/kg intravenous dose in C57BL/6 mice (n = 2). Plasma levels after an oral dose of 2 mg/kg were below the level of detection (<10 nM).
PK was also evaluated in noninfected hamsters following a single oral dose, 6.25 mg/kg, after which kibdelomycin was measured in plasma samples and cecal contents (Fig. 5). Concentrations in cecal contents reached 35.5 μM at 1 h postdosing, with a low clearance, and more than 2 μM remained at 24 h. The remaining concentration at 24 h is at least 8-fold greater than the kibdelomycin MIC, 0.25 μg/ml (0.26 μM). The systemic exposure following oral dosing was insignificant: 60 to 70 nM was detected in plasma 1 to 2 h postdosing. The lack of significant systemic exposure indicates a strong potential for a large therapeutic index of safety.
FIG 5.
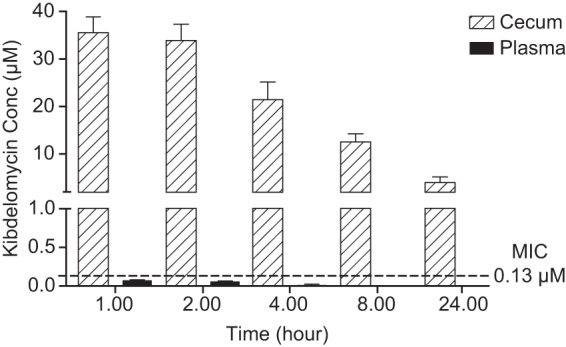
Time-concentration profile of kibdelomycin given at 6.25 mg/kg oral dose in the cecum contents and plasma of uninfected hamsters (n = 3).
DISCUSSION
Kibdelomycin is a novel natural product antibiotic with potent antibacterial activity against aerobic Gram-positive bacteria (17). It works by inhibiting the ATPase activity of bacterial topoisomerase enzymes, DNA gyrase and topoisomerase IV, a mechanism of action that is unexploited among C. difficile therapies. Kibdelomycin has a low frequency of resistance (<10−9) in S. aureus, and it is anticipated that resistance emergence will also be low in C. difficile. The lack of oral absorption in mice and the Gram-positive selectivity make this compound a compelling lead for the development of a C. difficile agent. Kibdelomycin demonstrated potent activity against a panel of 168 toxigenic C. difficile clinical strains, with a MIC90 of 0.25 μg/ml and a very narrow MIC range (0.06 to 0.5 μg/ml). Potency against C. difficile was comparable to that of fidaxomicin and exceeded those of metronidazole and vancomycin. Activity was similar among antibiotic-resistant and -susceptible strains and against the various REA types, including the hypervirulent BI (NAPI, 027) strains. Like fidaxomicin, kibdelomycin was active against other Gram-positive anaerobic organisms and was relatively inactive against most members of the Bacteroides fragilis group (MIC50 ≥ 32 μg/ml), a prevalent component of the gastrointestinal commensal flora (5). Protection of the prevalent Gram-negative intestinal flora may help to yield a more efficacious therapy with reduced disease recurrence.
Kibdelomycin was efficacious in the hamster CDAD model in which protection from a lethal infection and reduction in the C. difficile counts were measured. Efficacy was observed following oral dosing at 1.65 mg/kg per day and was correlated with gastrointestinal concentrations over a 24-h duration that exceeded the MIC. Significantly, KBD showed little to no oral absorption in mice and hamsters, suggesting the potential for a large safety margin. Kibdelomycin was not cytotoxic and was inactive in in vitro safety assays, including the Panlabs panel of 117 enzymes and receptors (10 μM), an assay using cytochrome P450s (3A4, 2D6, and 2C9; 10 μM), and the hERG Patch Express assay (60 μM) (data not shown). The lack of activity in the in vitro safety assays provides further confidence in the feasibility of developing a safe C. difficile therapy.
Kibdelomycin is a recently discovered novel natural product which has bactericidal activity against Gram-positive bacteria with a novel mechanism of action and a low frequency of resistance emergence. The potent in vitro and in vivo activity against C. difficile support the investigation of kibdelomycin for developing a new CDAD therapy.
Footnotes
Published ahead of print 10 February 2014
REFERENCES
- 1.Miller BA, Chen LF, Sexton DJ, Anderson DJ. 2011. Comparison of the burdens of hospital-onset, healthcare facility-associated Clostridium difficile infection and of healthcare-associated infection due to methicillin-resistant Staphylococcus aureus in community hospitals. Infect. Control Hosp. Epidemiol. 32:387–390. 10.1086/659156 [DOI] [PubMed] [Google Scholar]
- 2.Ananthakrishnan AN. 2011. Clostridium difficile infection: epidemiology, risk factors and management. Nat. Rev. Gastroenterol. Hepatol. 8:17–26. 10.1038/nrgastro.2010.190 [DOI] [PubMed] [Google Scholar]
- 3.Petrella LA, Sambol SP, Cheknis A, Nagaro K, Kean Y, Sears PS, Babakhani F, Johnson S, Gerding DN. 2012. Decreased cure and increased recurrence rates for Clostridium difficile infection caused by the epidemic C. difficile BI strain. Clin. Infect. Dis. 55:351–357. 10.1093/cid/cis430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerding DN. 2010. Global epidemiology of Clostridium difficile infection in 2010. Infect. Control Hosp. Epidemiol. 31(Suppl 1):S32–S34. 10.1086/655998 [DOI] [PubMed] [Google Scholar]
- 5.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Meta HITC, Bork P, Ehrlich SD, Wang J. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65. 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerding DN, Johnson S. 2010. Management of Clostridium difficile infection: thinking inside and outside the box. Clin. Infect. Dis. 51:1306–1313. 10.1086/657116 [DOI] [PubMed] [Google Scholar]
- 7.Surawicz CM, Alexander J. 2011. Treatment of refractory and recurrent Clostridium difficile infection. Nat. Rev. Gastroenterol. Hepatol. 8:330–339. 10.1038/nrgastro.2011.59 [DOI] [PubMed] [Google Scholar]
- 8.Mullane KM, Miller MA, Weiss K, Lentnek A, Golan Y, Sears PS, Shue YK, Louie TJ, Gorbach SL. 2011. Efficacy of fidaxomicin versus vancomycin as therapy for Clostridium difficile infection in individuals taking concomitant antibiotics for other concurrent infections. Clin. Infect. Dis. 53:440–447. 10.1093/cid/cir404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Louie TJ, Miller MA, Mullane KM, Weiss K, Lentnek A, Golan Y, Gorbach S, Sears P, Shue YK, OPT-80-003 Clinical Study Group 2011. Fidaxomicin versus vancomycin for Clostridium difficile infection. N. Engl. J. Med. 364:422–431. 10.1056/NEJMoa0910812 [DOI] [PubMed] [Google Scholar]
- 10.Johnson AP. 2010. New antibiotics for selective treatment of gastrointestinal infection caused by Clostridium difficile. Expt. Opin. Ther. Pat. 20:1389–1399. 10.1517/13543776.2010.511177 [DOI] [PubMed] [Google Scholar]
- 11.van Nood E, Dijkgraaf MG, Keller JJ. 2013. Duodenal infusion of feces for recurrent Clostridium difficile. N. Engl. J. Med. 368:2145. 10.1056/NEJMc1209761 [DOI] [PubMed] [Google Scholar]
- 12.Goldstein EJ, Citron DM, Tyrrell KL, Merriam CV. 2013. Comparative in vitro activities of SMT19969, a new antimicrobial agent, against Clostridium difficile and 350 gram-positive and gram-negative aerobic and anaerobic intestinal flora isolates. Antimicrob. Agents Chemother. 57:4872–4876. 10.1128/AAC.01136-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ting LS, Praestgaard J, Grunenberg N, Yang JC, Leeds JA, Pertel P. 2012. A first-in-human, randomized, double-blind, placebo-controlled, single- and multiple-ascending oral dose study to assess the safety and tolerability of LFF571 in healthy volunteers. Antimicrob. Agents Chemother. 56:5946–5951. 10.1128/AAC.00867-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mascio CT, Mortin LI, Howland KT, Van Praagh AD, Zhang S, Arya A, Chuong CL, Kang C, Li T, Silverman JA. 2012. In vitro and in vivo characterization of CB-183,315, a novel lipopeptide antibiotic for treatment of Clostridium difficile. Antimicrob. Agents Chemother. 56:5023–5030. 10.1128/AAC.00057-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pucci MJ, Bush K. 2013. Investigational antimicrobial agents of 2013. Clin. Microbiol. Rev. 26:792–821. 10.1128/CMR.00033-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donald RG, Skwish S, Forsyth RA, Anderson JW, Zhong T, Burns C, Lee S, Meng X, LoCastro L, Jarantow LW, Martin J, Lee SH, Taylor I, Robbins D, Malone C, Wang L, Zamudio CS, Youngman PJ, Phillips JW. 2009. A Staphylococcus aureus fitness test platform for mechanism-based profiling of antibacterial compounds. Chem. Biol. 16:826–836. 10.1016/j.chembiol.2009.07.004 [DOI] [PubMed] [Google Scholar]
- 17.Phillips JW, Goetz MA, Smith SK, Zink DL, Polishook J, Onishi R, Salowe S, Wiltsie J, Allocco J, Sigmund J, Dorso K, Lee S, Skwish S, de la Cruz M, Martin J, Vicente F, Genilloud O, Lu J, Painter RE, Young K, Overbye K, Donald RG, Singh SB. 2011. Discovery of kibdelomycin, a potent new class of bacterial type II topoisomerase inhibitor by chemical-genetic profiling in Staphylococcus aureus. Chem. Biol. 18:955–965. 10.1016/j.chembiol.2011.06.011 [DOI] [PubMed] [Google Scholar]
- 18.Singh SB, Goetz MA, Smith SK, Zink DL, Polishook J, Onishi R, Salowe S, Wiltsie J, Allocco J, Sigmund J, Dorso K, de la Cruz M, Martin J, Vicente F, Genilloud O, Donald RG, Phillips JW. 2012. Kibdelomycin A, a congener of kibdelomycin, derivatives and their antibacterial activities. Bioorg. Med. Chem. Lett. 22:7127–7130. 10.1016/j.bmcl.2012.09.071 [DOI] [PubMed] [Google Scholar]
- 19.Singh SB, Polishook JD, Zink DL, Genilloud O, Goetz MA, Vicente F. 2011. Extracts of Kibdelosporangium as antibacterial agents. WO2011/079034A1. [Google Scholar]
- 20.Clinical and Laboratory Standards Institute (CLSI). 2007. Methods antimicrobial susceptibility testing of anaerobic bacteria: approved standard, 7th ed. CLSI document M11-A7 CLSI, Wayne, PA [Google Scholar]
- 21.Perdue ER, Fawley W, Wilcox M, Gerding D, Johnson S. 2013. Correlation of PCR ribotyping and REA of Clostridium difficile, abstr. K-157 Abstr. 53rd Intersci. Conf. Antimicrob. Agents Chemother [Google Scholar]