

Abstract
As the incidence of Gram-negative bacterial infections for which few effective treatments remain increases, so does the contribution of drug-hydrolyzing β-lactamase enzymes to this serious clinical problem. This review highlights recent advances in β-lactamase inhibitors and focuses on agents with novel mechanisms of action against a wide range of enzymes. To this end, we review the β-lactamase inhibitors currently in clinical trials, select agents still in preclinical development, and older therapeutic approaches that are being revisited. Particular emphasis is placed on the activity of compounds at the forefront of the developmental pipeline, including the diazabicyclooctane inhibitors (avibactam and MK-7655) and the boronate RPX7009. With its novel reversible mechanism, avibactam stands to be the first new β-lactamase inhibitor brought into clinical use in the past 2 decades. Our discussion includes the importance of selecting the appropriate partner β-lactam and dosing regimens for these promising agents. This “renaissance” of β-lactamase inhibitors offers new hope in a world plagued by multidrug-resistant (MDR) Gram-negative bacteria.
INTRODUCTION
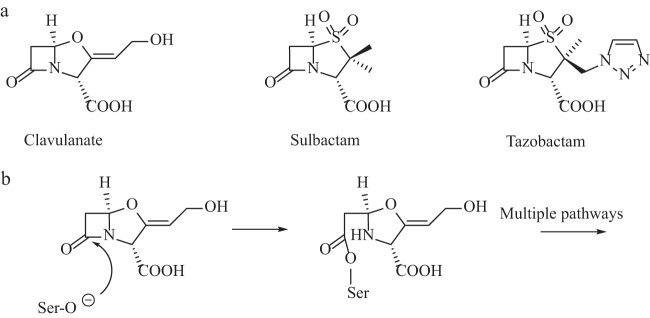
The production of β-lactam-hydrolyzing enzymes, i.e., β-lactamases, by Gram-negative and -positive bacteria remains one of the most significant threats to the efficacy of this life-saving class of antimicrobial agents (1). Drug discovery and development since the mid-to-late 1980s led to the introduction of β-lactamase inhibitors which provided “new approaches” for combating this clinical challenge. However, bacteria continue to evolve, as they are amazingly capable of responding to environmental pressure via selection of existing mutations and acquisition of new genes (2, 3). The currently available β-lactamase inhibitors, clavulanic acid, tazobactam, and sulbactam (Fig. 1a), are now met with an increasingly prevalent panel of inhibitor-resistant bacterial strains (4). Regrettably, we are faced with the daunting challenge of designing effective inhibitors for an ever-increasing number of diverse β-lactamases.
FIG 1.
(a) Chemical structures of current clinically available β-lactamase inhibitors. (b) Acylation step in general mechanism of inhibition of a class A β-lactamase by a β-lactamase inhibitor, illustrated here for clavulanic acid (4).
The clinically available inhibitors share a β-lactam backbone. Sulbactam and tazobactam are penicillanic acid sulfones, while clavulanic acid is a clavam. β-Lactamase inhibitors take advantage of conserved active-site residues to interact with their target (Fig. 1b). However, inhibitors differ from substrates in their abilities to assume long-lived, stable intermediates with β-lactamases, thus “tying up” the enzymes, while the partner β-lactam inhibits the penicillin binding protein target. As more catalytically versatile β-lactamases (e.g., Klebsiella pneumoniae carbapenemases [KPCs]) continue to emerge and acquire the ability to hydrolyze inhibitors faster, a new approach is required (5).
This review focuses on the recent studies illustrating the exceptional promise of agents with novel mechanisms of inhibition that are on the threshold of clinical application. Specifically, we summarize the growing body of data supporting the potential use of avibactam as the first clinically available β-lactamase inhibitor introduced in the United States since the piperacillin-tazobactam combination in 1993. We advance the claim that this novel inhibitor illustrates several important features which serve as lessons for improved therapeutic design. Next, we discuss a compound of the same chemical class as avibactam, MK-7655, as well as other non-β-lactams, such as the RPX7009 boronate which recently completed phase 1 trials and cyclobutanones that offer additional new approaches for Ambler class A and C β-lactamases. Lastly, we highlight some of the specific challenges of inhibiting class B and D enzymes. Although the quest for a “universal” β-lactamase inhibitor continues, it is becoming concerning that this notion may be unrealistic. Fundamental research is still required to decipher the basic mechanisms of catalysis of each β-lactamase class.
AVIBACTAM AND MK-7655: “NON-β-LACTAM INHIBITORS”
(i) Avibactam.
Avibactam, known formerly as both AVE1330A and NXL104, is a bridged diazabicyclo[3.2.1]octanone non-β-lactam inhibitor (Fig. 2a and c) (6–8). Compared to clavulanic acid, sulbactam, and tazobactam, this non-β-lactam achieved both lower 50% inhibitory concentrations (IC50s) (range, 3 to 170 nM) and decreased reactivation rates for the clinically relevant class A and C β-lactamases such as TEM-1, KPC-2, and P99 and the AmpC from Pseudomonas aeruginosa (7, 9, 10). Comparable IC50s were observed for the extended-spectrum β-lactamases (ESBLs) CTX-M-15 and SHV-4.
FIG 2.
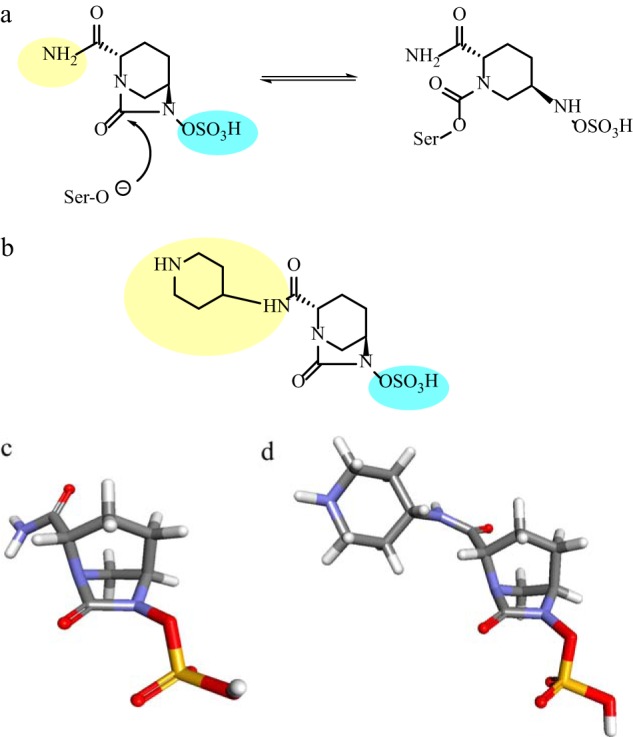
(a) Hypothesized mechanism of avibactam acylation and regeneration with a class A β-lactamase; the amine and sulfate are highlighted in yellow and blue, respectively (11). (b) Structure of MK-7655; the piperidine ring and sulfate are highlighted in yellow and blue, respectively (40). (c and d) Three-dimensional structures of avibactam (c) and MK-7655 (d), constructed using Fragment Builder tools and minimized using a Standard Dynamics Cascade protocol in Discovery Studio 3.1.
The success of avibactam may be owing first to its structural similarity to β-lactams at the electrophilic carbonyl group. This molecular mimicry is important for rapid recognition and formation of a stable adduct by β-lactamases, as indicated by rapid enzyme “on” rates (i.e., 370,000 M−1 s−1 for TEM-1) (9, 11).
The second defining feature of this promising compound is the stable acyl enzyme formed by the carbamoyl link between the inhibitor and the enzyme active-site serine residue. Current analyses reveal that the inhibitor comes off (koff) of class A, C, and D β-lactamases at a very low rate; measured for TEM-1, CTX-M-15, KPC-2, P99, P. aeruginosa AmpC, OXA-10, and OXA-48, values ranged from 0.0019 s−1 to <0.0000016 s−1, yielding enzyme reactivation half-lives of 6 to >7,200 min (11, 12). In contrast, the kinetic details of avibactam interactions with OXA-10 merit consideration (12). Against OXA-10, acylation (1.1 × 101 M−1 s−1) and deacylation (1.6 × 10−6 M−1 s−1) were significantly slowed, resulting in an enzyme that is relatively resistant to inactivation yet slow to reactivate. In contrast, the acylation rate for OXA-48 was 100-fold higher. Similarly to class D β-lactamases, BlaC, a class A β-lactamase from Mycobacterium tuberculosis, also demonstrated slow acylation and deacylation by avibactam (13).
Further, avibactam's inhibition is believed to be reversible and the active inhibitor is regenerated via deacylation and recyclization of the 5-membered urea ring. Notably, such cyclic regeneration is not observed with sulfones and clavulanic acid, presumably because the four-member β-lactam ring is too constrained (i.e., after inhibitors are hydrolyzed, the energy required to close and form the original β-lactam ring is too great). Detailed kinetic studies of TEM-1, combined with nuclear magnetic resonance (NMR) analysis and mass spectroscopy, did not yield evidence for irreversible deacylation pathways through hydrolysis or chemical rearrangements (11). Acyl enzyme transfer experiments added support to the idea of the reversible mechanism, where deacylated avibactam was released from a donor enzyme-avibactam mixture and acylated a second enzyme. The mixtures of these apo and acyl enzyme species showed proportions of acyl enzyme that reflected avibactam's affinity for each β-lactamase (11). However, with KPC-2, avibactam hydrolysis was observed after 24 h (only 10% of the enzyme remained acylated with avibactam, as shown by mass spectrometry) (12). Several intermediates that resulted from loss of SO3, loss of a water molecule, and imine hydrolysis were observed using mass spectrometry. The carbamate linkage was subsequently hydrolyzed, and a decarboxylation reaction regenerated free KPC-2.
The recently defined crystal structures of avibactam in complex with CTX-M-15, P. aeruginosa AmpC, and M. tuberculosis BlaC have offered important insight into the structural bases of the inhibitor's activity (13, 14). Avibactam adopts very similar active-site conformations in class A and C enzymes, making contact with key conserved residues with limited molecular flexibility. Additionally, the sulfate group has more polarity than the C3/C4 carboxylate β-lactams, forming multiple hydrogen bonds in the active site (14). The opened avibactam ring retains a conformation similar to that of the native form, which aids in the recyclization mechanism. Deacylation over hydrolysis is likely explained by the stability of the carbamoyl bond and the lack of an appropriately positioned and activated water molecule, i.e., the latter due to the charges created by the protonated glutamic acid at position 166 (Glu166) in CTX-M-15 (14). These mechanistic details have important implications not just for avibactam but also as possible strategies for additional inhibitor compounds.
As stated above, release of intact avibactam allows the compound to acylate another β-lactamase, in contrast to the β-lactam inhibitors which follow hydrolytic routes that yield molecules without inhibitory activity. Not only is the active inhibitor regenerated, but so is the active enzyme. As shown by acyl enzyme exchange experiments, this can result in “shuffling” of the inhibitor to higher-affinity enzymes (11). The outcome of this partitioning, and possible selective inhibition of certain β-lactamases within a strain producing multiple enzymes, is not clear and awaits further clinical data. The koff rates and the amount of enzymes present likely have some bearing on the proportion of enzymes inactivated.
The importance of complementing enzyme kinetic and structural studies with whole-cell and in vivo assays is central to drug development. Fortunately, the studies with avibactam are also promising. Avibactam has been studied primarily with two partner cephalosporins, ceftazidime and ceftaroline (the active metabolite of ceftaroline-fosamil). These combinations restored activity against Enterobacteriaceae, most strains of P. aeruginosa, and Burkholderia cepacia complex as well as isolates producing one or multiple KPCs (a serine carbapenemase) and ESBL and AmpC (cephalosporinases) enzymes (see Table 1 for representative MIC values) (10, 15–23). Ceftazidime-avibactam lacked activity against bacteria carrying class B metallo-β-lactamases (MBLs) as well as against Acinetobacter baumannii, which was most likely due to coexpression of OXA β-lactamases (i.e., OXA-23, -24/40, -51, and -58). Enterobacteriaceae expressing the carbapenemase OXA-48 were susceptible to ceftazidime with avibactam, while the alternative combination of aztreonam and avibactam was very effective in vitro against strains carrying MBLs (see MBL section below) (19, 24). The lack of activity versus the carbapenem-hydrolyzing OXA enzymes (i.e., OXA-23 and OXA 24/40) remains to be explained.
TABLE 1.
MICs of β-lactam and β-lactam-avibactam combinations against select pathogensa
| Pathogen | MIC (μg/ml)b |
|||||
|---|---|---|---|---|---|---|
| CAZ | CAZ-AVI | CPT | CPT-AVI | ATM | ATM-AVI | |
| K. pneumoniae with OXA-48 | 256/512 | 0.25/0.5 | ||||
| K. pneumoniae with CTX-M-15 | 8/64 | 0.06/0.25 | ||||
| K. pneumoniae with KPC-2 | ≥512/≥512 | 0.25/1 | ≥512/≥512 | ≤0.06/≤0.06 | ||
| E. coli with ESBL | 16/64 | 0.12/0.25 | ||||
| E. coli with AmpC | 16/64 | 0.12/0.5 | ||||
| E. coli with OXA-48 | 4 | <0.008 | ||||
| E. coli with IMP-1 | 256 | 64 | ||||
| Enterobacteriaceae with multiple β-lactamases, including KPC-2 | >64/>64 | 0.5/2 | ||||
| Enterobacteriaceae with multiple β-lactamases, including AmpC | 256/>256 | 0.5/2 | ||||
| Enterobacteriaceae with VIM | 64–512 | 64–512 | 0.25–256 | 0.12–0.5 | ||
| P. aeruginosa | 8/64 | 4/8 | >64/>64 | 16/>32 | 16/32 | 8/32 |
| P. aeruginosa with ESBL PER-1 | 128/128 | 4/16 | ||||
| A. baumannii | >64/>64 | 32/>32 | ||||
| A. baumannii with PER-1, OXA-51, and OXA-58 | 128/≥512 | 32/256 | ||||
| S. aureus | 1/2 | 1/2 | ||||
The addition of avibactam to ceftaroline may be particularly useful for treatment of infections with a presumptively high burden of Gram-positive organisms, such as Staphylococcus aureus in diabetic foot wounds (25). The potent activity of ceftaroline against most aerobic organisms is complemented by the ability of avibactam to inhibit β-lactamases from anaerobes, including Bacteroides fragilis. Murine sepsis models demonstrated that avibactam added to ceftazidime and ceftaroline regimens increased the susceptibility of highly resistant ESBL- and KPC-producing K. pneumoniae isolates (26, 27). Depending on the partner β-lactam, avibactam combinations have the potential to be highly effective against many multidrug-resistant (MDR) pathogens.
Optimal dosing regimens for these novel combinations have been studied in multiple hollow-fiber models, assays which exposed a bacterial suspension to clinically relevant and fluctuating drug concentrations (22, 23, 28, 29). The increasing sophistication of pharmacodynamic modeling may help refine regimens for potential inhibitors to prevent mislabeling a drug as ineffective due to dosing failures (30). Investigations of dosing schedules for both the ceftazidime-avibactam and ceftaroline-avibactam combinations argued that time above a critical concentration (time > MIC) was the essential parameter for suppressing bacterial growth, as has been shown for imipenem (23, 28, 29, 31). For example, for K. pneumoniae 27-908M, the ceftaroline-avibactam MIC was 0.75 µg/ml for ceftaroline and 4 µg/ml for avibactam. The ceftaroline-avibactam concentrations needed to be above the MICs for 62% to 80% of the dosing interval for treatment success with ceftaroline-avibactam (administered as ceftaroline at 600 mg every 8 h and avibactam at a daily dose as a continuous infusion or as divided doses every 8 h) (23). The concentrations differed depending on the organism, inoculum, partner antibiotic, and β-lactamase expression profile. Louie et al., who examined KPC-2-, CTX-M-15-producing K. pneumoniae, and AmpC-overproducing Enterobacter cloacae, concluded that avibactam trough levels should be at least 2.5 μg/ml and that patients should be dosed with ceftaroline every 8 h for infections with highly resistant pathogens (23). In contrast, Nichols et al. determined that the required avibactam trough concentration was closer to 0.3 μg/ml when the drug was partnered with ceftazidime in a model with E. cloacae and K. pneumoniae at lower inocula (29).
These hollow-fiber models and pharmacokinetic analyses have helped inform dosing regimens for clinical trials, and avibactam is well on its way in moving from “bench to bedside.” The first study to report avibactam data from a phase 2 trial showed that a ceftazidime-avibactam combination (500 mg/125 mg, every 8 h) is comparable in efficacy and safety to imipenem-cilastatin in hospitalized patients with complicated urinary tract infections (UTIs) (32). That study did not elucidate particular resistance mechanisms, but it is likely that the most common uropathogen isolated from these patients, Escherichia coli, harbored only class A enzymes (33). More recently published phase 2 results of complicated intra-abdominal infections (IAIs) treated with ceftazidime-avibactam-metronidazole (2,000 mg/500 mg/500 mg, every 8 h) versus meropenem demonstrated comparable clinical responses (91.2% and 93.4%, respectively) and similar rates of treatment-emergent adverse events (64.4% and 57.8%, respectively) (34). That investigation nicely translates in vitro data of the potency of the triple combination against anaerobes, making this formulation well-suited for polymicrobial IAIs (of note, the anaerobic activity of ceftazidime-avibactam was limited without the addition of metronidazole) (35, 36). With these promising results, multiple phase 3 trials are currently recruiting for ceftazidime-avibactam in complicated cases of UTIs and IAIs (www.clinicaltrials.gov), as well as for one for nosocomial pneumonia (registration no. NCT01808092). The ceftaroline-avibactam combination is not yet as thoroughly tested, although one recently completed phase 2 study compared the combination to doripenem for complicated UTIs (NCT01281462). Finally, a phase 1 trial of aztreonam-avibactam safety has suspended recruitment following a change in dosing regimens, totaling a second suspension for this study (NCT01689207). While it is unclear what types of problems underlie these suspensions, aztreonam-avibactam is likely to receive further attention. In latter half of 2013, the European Innovative Medicines Initiative called for proposals for development of phase IIa pharmacokinetic/pharmacodynamic and phase III efficacy and safety studies of Gram-negative pathogens, with particular attention to MBL producers (www.imi.europa.eu/sites/default/files/uploads/documents/9th_Call/Calll_9_Text.pdf) (37).
Clinicians and researchers need to be aware that, as with all agents, bacteria with mechanisms of resistance to these novel inhibitors can emerge. Passage of E. coli and E. cloacae in the presence of ceftaroline-avibactam selected for organisms harboring AmpC and CTX-M-15 enzymes with deletions or mutations that conferred resistance (38). In addition, CMY-2 variants (i.e., enzymes with amino acid substitutions of Asn152 to alanine, threonine, and serine) demonstrated higher IC50s for avibactam (i.e., 0.5 to 1.0 μM) than their wild-type counterpart (i.e., 0.06 μM) (39). The clinical impact of these findings is being monitored. Nevertheless, we can optimistically project that with appropriate antibiotic stewardship, any one β-lactamase conferring resistance to a particular inhibitor may remain susceptible to inactivators that work by different mechanisms. As such, introduction of agents employing diverse mechanisms may allow successful therapy. For example, those strains selected by ceftaroline-avibactam pressure were relatively “unstable,” lost significant ESBL activity, and were susceptible to tazobactam (38).
(ii) MK-7655.
MK-7655 is similar to avibactam's diazabicyclooctane core with the addition of a piperidine ring (Fig. 2b and d). MK-7655 is predicted to function through a similar mechanism (40). MK-7655 exhibits synergistic activity in combination with imipenem against carbapenem-resistant Enterobacteriaceae and P. aeruginosa with diverse resistance mechanisms, including KPC production and impermeability due to porin loss and class A and C β-lactamase production (41–43). For example, imipenem MICs for KPC-producing K. pneumoniae and Enterobacter spp. fell from a range of 16 to 64 μg/ml to a range of 0.12 to 1 μg/ml with the addition of 4 μg/ml of MK-7655 (42). Additional data from another study showed that imipenem and MK-7655 were synergistic for K. pneumoniae and Enterobacter strains with imipenem resistance due to both impermeability and ESBL or AmpC activity, lowering MICs from a range of 2 to 16 μg/ml to ≤1 μg/ml. The combination was not effective in restoring susceptibility to the Enterobacteriaceae expressing OXA-48 carbapenemase or IMP, NDM, or VIM MBLs, and similarly, MK-7655 did not lower MICs for imipenem-susceptible isolates with ESBLs or AmpCs. Against P. aeruginosa, MK-7655 was successful in lowering imipenem MICs for imipenem-susceptible strains, likely by inhibiting endogenous AmpC-mediated imipenem protection (42). OprD porin-deficient P. aeruginosa required 8 μg/ml of MK-7655 to lower imipenem MICs from a range of 16 to 64 μg/ml to ≤2 μg/ml for most (7/8) isolates. MK-7655 was unable to lower imipenem MICs for MBL-producing P. aeruginosa and lowered them only to 4 to 8 μg/ml for MDR strains, including those from patients with cystic fibrosis.
MK-7655 dosing regimens were examined in a hollow-fiber model (41). Five hundred milligrams of both imipenem and MK-7655 suppressed growth of a KPC-2-producing K. pneumoniae strain and an OprD-deficient, AmpC-overexpressing P. aeruginosa strain at 72 h. Increasing MK-7655 to 1,000 mg achieved growth suppression in an additional P. aeruginosa strain. In a related hollow-fiber model, a unique parameter, time above instantaneous MIC (T>MICi), was derived to reflect changing in vivo susceptibility (44). Provided the MK-7655 T>MICi was greater than 69% in the presence of 500 mg imipenem, similar levels of 48-h killing were observed for the KPC-2-producing K. pneumoniae strain regardless of escalating inhibitor doses. These data suggest that time over a susceptibility threshold is essential to bacterial killing for this novel inhibitor as well.
A phase 1 trial of the pharmacokinetics of MK-7655 in patients with impaired renal function was completed in March 2012 (www.clinicaltrials.gov; NCT01275170), and reported data suggested that the required dosage reduction was unchanged by the addition of the β-lactamase inhibitor (45). Phase 2 clinical trials are presently examining the imipenem-cilastatin-MK-7655 combination for treatment of complicated UTIs and IAIs, with doses of 125 mg or 250 mg of MK-7655 combined with 500 mg of imipenem and cilastatin every 6 h (NCT01505634 and NCT01506271).
Taking a reductionist view, avibactam and MK-7655 are “similar.” Although the clinical development of avibactam is ahead of that of MK-7655 based upon the number of clinical trials, it is very likely that in controlled studies, both will perform well, especially as they are effective against ESBLs, AmpCs, and serine carbapenemases (KPCs). That property alone is a truly welcome addition to our therapeutic armamentarium and a significant advance with respect to what we currently have available to us. As a result, barring unforeseen complications or regulatory problems, avibactam and MK-7655 should proceed to market. Once in use, clinicians will witness a “natural experiment” and observe what ends up being the “better partner” β-lactam. Is a “carbapenem-sparing” combination less likely to foster more resistance? The cephaloporins have well-established clinical safety records, and yet carbapenems may be less susceptible to efflux in P. aeruginosa (46). Ultimately, clinical use and the emergence of resistance will answer this vexing question. We must keep in mind, however, that resistance is inevitable, and although these inhibitors promise “a battlefield victory” over certain MDR organisms, we have not yet “won the war” and the struggle between “bug and drug” will likely continue.
BORONIC ACID β-LACTAMASE INHIBITORS
Since the late 1970s, boronates have been documented as effective inhibitors of serine β-lactamases in vitro (47–49). Working via a novel mechanism compared to that of the clinically available β-lactamase inhibitors, boron forms a reversible dative bond with the β-lactamase, is not hydrolyzed by the enzyme, and serves as a competitive inhibitor (Fig. 3a) (48). Recently, the functional groups bound to the boronate core have been modified extensively based on hypothesized and structurally confirmed interactions with β-lactamase active-site residues (50–52).
FIG 3.

(a) General mechanism of reversible inhibition of a class A β-lactamase by a boronic acid compound. (b) Structure of β-lactam substrate analog boronic acid inhibitors (ampicillin, cephalothin, and cefoperazone mimics). (c) Structure of sulfonamide boronic acid (tested as described in reference 57); the sulfonamide boronic acid group is highlighted in yellow. (d) Compound 5 developed from FBLD optimization of sulfonamide boronate structure (59); the sulfonamide boronic acid group and tetrazole are highlighted in yellow and blue, respectively. (e) Structure of RPX7009 (65).
Glycylboronates are based on β-lactam substrate homology and contain side chains of penicillins and cephalosporins. Studies have shown analogs of ampicillin, cephalothin, and cefoperazone to be inhibitors of clinically relevant class A and C β-lactamases in the nM range (i.e., 9.3 nM, 420 nM, and 11 nM for K. pneumoniae SHV and for AmpCs from P. aeruginosa and Acinetobacter spp., respectively) (Fig. 3b) (53–56). These compounds are still preclinical.
More recently, new modifications to the β-lactam analog boronate were added by replacing the carboxamide group, conserved in all penicillin and cephalosporins, with a sulfonamide (Fig. 3c) (57, 58). These novel derivatives result in very potent E. coli AmpC inhibitors with affinities approaching 0.025 μM, 23 times more potent than their carboxamide analogs. Combined with ceftazidime, the lead compounds lowered MICs up to 64-fold (from 64 μg/ml to 1 μg/ml) against microorganisms expressing class A and class C β-lactamases.
Shortly thereafter, using a boronic acid scaffold, fragment-based lead discovery (FBLD) compounds were optimized to better target β-lactamases (59). FBLD is a powerful new method for building drug leads from “fragments of molecules” (this approach has recently yielded important therapeutic candidates for treatment of Alzheimer's disease and cancer) (60). The strategy here lies in the ability to identify multiple small molecules with favorable β-lactamase binding properties and then to apply structural and chemical information to combine the components into drug leads. In this regard, FBLD is used to refine the highest-affinity boronates with the aim of yielding compounds with significant in vivo antimicrobial activity, as previous generations of boronates have demonstrated favorable in vitro kinetics but failed to improve susceptibility (e.g., affinities in the nM range but poor MICs) (59). Examination of binding models using FBLD revealed subtle changes such as reorientation of functional groups and ring structures that improved affinity by 500-fold. Lead compound 5, in Eidam et al., when combined with cefotaxime, reduced E. coli MICs from a range of 8 to 128 μg/ml to a range of 0.5 to 1 μg/ml (Fig. 3d). Mice challenged with AmpC-overproducing E. coli had a significant 65% improved survival when treated with cefotaxime combined with compound 5 at 50 and 200 mg/kg of body weight compared to 50 mg/kg cefotaxime alone (P ≤ 0.0005). Despite these promising in vivo animal studies, the FBLD approach remains a work in progress that has not yet promoted any candidates into anti-infective preclinical assessment.
At the 2012 Interscience Conference on Antimicrobial Agents and Chemotherapy, a novel boronic acid-based β-lactamase inhibitor, RPX7009 (Fig. 3e), was presented in combination with a carbapenem antibiotic (RPX2003; biapenem) (61–64). Alone, RPX7009 did not exhibit antibacterial activity, but the combination showed strong potentiation of biapenem against class A carbapenemase-producing Enterobacteriaceae (e.g., KPC, SME, and IMI) (65). Of a panel of 145 KPC-positive isolates, 89.7% were inhibited by ≤1 μg/ml of biapenem with 2 μg/ml of RPX7009 and 94.4% were inhibited when RPX7009 was increased to 8 μg/ml. Biapenem with RPX7009 at 4 μg/ml demonstrated MIC50 values of 0.12 μg/ml for KPC-producing Enterobacteriaceae that coexpressed either one to four additional β-lactamases (including class A [narrow- and extended-spectrum], CTX-M, and noncarbapenemase OXA) or hyperexpressed chromosomal AmpC enzymes (62). However, against isolates with impermeability and either AmpC or ESBL expression (Enterobacter and Klebsiella spp., respectively), 8 μg/ml of RPX7009 showed only weak potentiation of biapenem (50% and 33% of isolates, respectively, had MICs > 1 μg/ml) (65). Potentiation was not seen with Enterobacteriaceae expressing IMP, VIM, or NDM MBLs or OXA-48.
Biapenem-RPX7009 activity for the nonfermenters P. aeruginosa and A. baumannii was similar to that of meropenem and imipenem (66). Testing of a panel of anaerobic pathogens, including more than 25 Bacteroides spp., demonstrated that biapenem and RPX7009 were comparable to meropenem alone. However, biapenem given without RPX7009 showed similar results, suggesting that RPX7009 lacked anaerobic activity (67). Animal models (neutropenic thigh models of infection) with KPC-producing strains also demonstrated promising results for this boronate inhibitor (63). Biapenem-RPX7009 recently completed a phase 1 clinical trial without current reports of toxicity (NCT01772836). Interestingly, an additional trial of RPX7009 in combination with a novel carbapenem, RPX2014, is currently in recruitment phase (NCT01897779). The structure of RPX2014 is unknown at this time, and insights as to efficacy are awaited.
CYCLOBUTANONE AND PENAM SULFONE β-LACTAMASE INHIBITORS
Cyclobutanone-based inhibitors, as mimetics of β-lactams, demonstrate activity against all four classes of β-lactamases; however, they appear most effective against class C enzymes. One lead compound (Fig. 4) inhibited KPC-2, IMP-1, E. cloacae GC1, and OXA-10 with IC50s of 26, 213, 4.5, and 370 μM, respectively (68). These compounds are reversible inhibitors with a bicyclic system, and particular substituents help force the 5-membered ring into conformations that enhance broad-spectrum-β-lactamase binding. While this lead inhibitor reduced meropenem MICs from 64 μg/ml to 16 μg/ml for MBL-producing strains of Chryseobacterium meningosepticum and Stenotrophomonas maltophilia, this limited potentiation may not be clinically useful.
FIG 4.
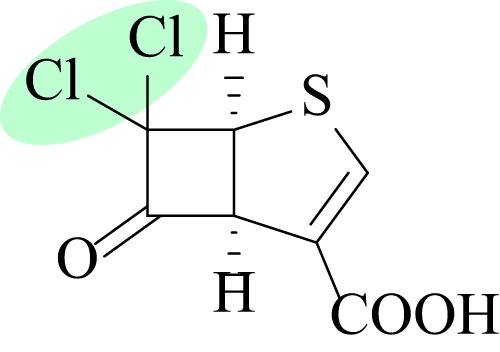
Structure of cyclobutanone inhibitor with high affinity for class C enzymes; the chlorine atoms are highlighted in green (68).
Derivativized penam sulfone inhibitors are another group of inhibitors with a β-lactam core that are being rationally designed to help address specific clinical problems. For example, functional groups taking advantage of active-site residues have led to compounds with high affinities for KPC-2 and P. aeruginosa AmpC (e.g., 3.9 μM and 180 nM, respectively) (69, 70). Despite what may be promising prototypes, there are no current reports suggesting the promotion of cyclobutanone and penam sulfone inhibitors beyond the preclinical stage.
THE UNIQUE CHALLENGE OF CLASS B MBLS; CLOSER THAN WE THINK?
This “class apart” of β-lactamases presents a particular challenge for clinicians and medicinal chemists (71). The spread of these metalloenzymes is often facilitated by mobile genetic elements, and the rapid emergence of the NDM-1 β-lactamase demonstrates how an MDR strain from one part of the world can quickly become a global problem (72). The hydrolytic mechanisms of MBLs are substantially different from those of the other classes, requiring one or two zinc atoms depending on subclass (i.e., B1, B2, or B3). The amino acid or primary sequence diversity is extremely broad, and the substrate profile includes all known β-lactams (including the available β-lactamase inhibitors), with the exception of monobactams (73).
While none of the promising inhibitors mentioned have significant activity against class B enzymes, there are two important exceptions, the combinations of cyclobutanone with meropenem (see above) and avibactam with aztreonam (19, 68). The aztreonam-avibactam combination, with MICs of ≤4 μg/ml for carbapenem-resistant Enterobacteriaceae, including MBL-containing pathogens, may offer great promise for this clinical challenge (19). Most likely, avibactam inhibits coproduced ESBL and AmpC enzymes as aztreonam evades the MBLs and exerts its antimicrobial effect. Aztreonam is hypothesized to bind poorly and unproductively to MBLs; molecular modeling reveals that the β-lactam moiety is too far from the nucleophile at the first Zn2+ binding site due to the interactions of the sulfonate group of aztreonam with the second Zn2+ binding site (74). As discussed above, the phase 1 trial of aztreonam-avibactam (NCT01689207) has been suspended for the second time in the study's history, but further work fostered by the European Innovative Medicines Initiative may provide important data for further therapeutic development.
This review focuses on the merits of inhibitor design which move beyond the β-lactam core structure for class A and C β-lactamases. Because of the special characteristics of the class B enzymes, this approach has already been applied and yielded candidate agents such as thiols and succinate derivatives, pyridine dicarboxylates, and tricyclic natural products (4). Of these, the thiol derivatives have shown the most promise for meeting the need for broad-spectrum activity against each of the MBL subclasses. Thiols, including the clinically available antihypertension agent l-captopril, have shown effective inhibition of NDM-1 and subclass B1, B2, and B3 enzymes (75–78). For example, synthesis informed by binding mode structural analyses led to a thiol compound with 0.019, 5.7, and 1.8 μM affinity values for MBLs from each class, IMP-1, CphA, and L1, respectively (Fig. 5a) (76). A single thiol compound can adopt unique binding conformations in different enzymes and yet still achieve inhibition through the common mechanisms of zinc chelation and/or displacement of the hydrolytic water.
FIG 5.
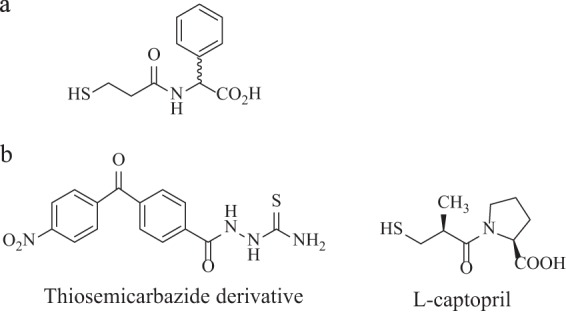
(a) Thiol compound with inhibitory activity for the three MBL classes (76). (b) Structure of thiosemicarbazide derivative and l-captopril, metallo-β-lactamase IMP-1 inhibitors (85, 87).
Much of the recent literature on identifying possible inhibitors for MBLs describes in vitro testing of compounds based on insights gained from solved crystal structures. This trend may be partly explained by our relatively rudimentary understanding of this group of enzymes compared to the better studied class A and C β-lactamases. Not until the last couple decades have MBLs presented in pathogenic Gram-negative organisms such as Enterobacteriaceae, P. aeruginosa, and Acinetobacter spp. (79–81). As these enzymes have “grown up” during this technological age, multiple computer-based approaches, such as computer modeling of target compounds, integrating protein shape and surface charge profiles into in silico screening, and compiling crystal structures in automated databases for future studies, have been described for drug discovery (82–84).
To illustrate, a modified form of fragment screening was employed to identify lead molecules for inhibition of the increasingly common IMP-1 enzyme, followed by in silico studies of the binding mechanisms of 1,2,4-triazole-3-thiols (85, 86). While optimization of the initial thiols yielded compounds with only mid-μM affinities, derivatives of thiosemicarbazides achieved low-μM affinity (range of 11 to 75 μM) (87). Interestingly, the final affinity value of the lead compound was comparable to that of l-captopril for IMP-1 (11 and 12.5 μM, respectively) (85) (Fig. 5b).
Few papers describing animal models of potential MBL inhibitors have been published. Two studies showed inhibitory effects of calcium-ethylenediamine-N,N,N′,N′-tetraacetic acid (CaEDTA) in whole-cell assays and murine models of MBL-producing P. aeruginosa and E. coli (Fig. 6a) (88, 89). In combination with imipenem, CaEDTA improved survival and decreased bacterial burden in cases of pneumonia caused by IMP- and VIM-producing P. aeruginosa (88). Furthermore, CaEDTA inhibited the activity of the elastase-type P. aeruginosa metalloprotease virulence factor. A murine neutropenic sepsis model of NDM-1-producing E. coli also showed reduced bacterial liver and blood counts after administration of imipenem-CaEDTA combinations, although the effects on overall survival were less clear (89).
FIG 6.
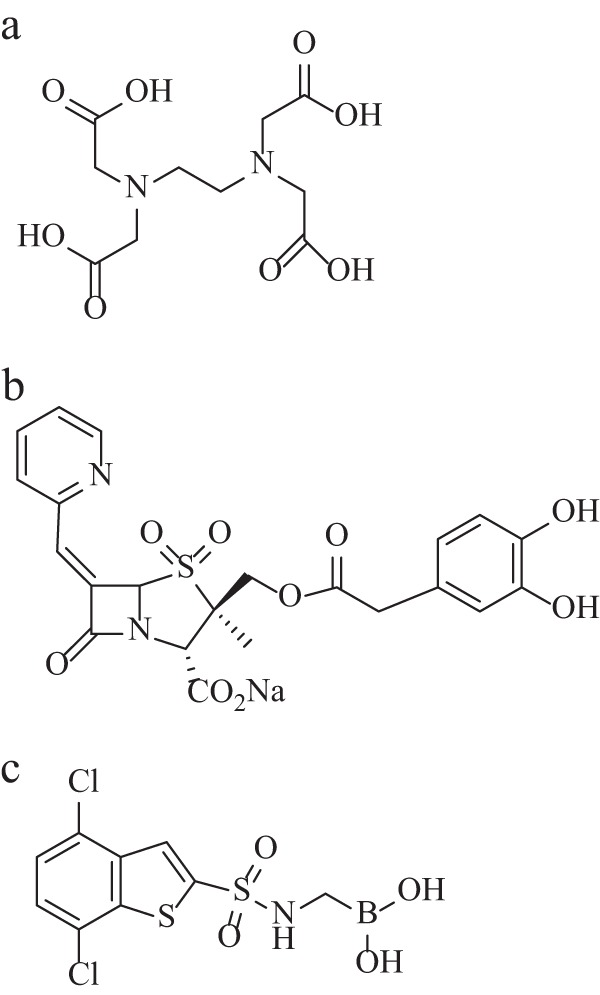
(a) Structure of EDTA. (b) Structure of LN-1-255 (108). (c) Structure of DSABA (109).
Whether these in vivo studies offer real clinical promise is unclear. The chelating properties of EDTA for metals present in MBLs have been known for decades such that the agent is used to screen for MBL-producing organisms in clinical laboratories (90–93). In addition to chelating the zinc ions required for MBL hydrolysis, ion chelation may disrupt bacterial cell membranes and help disperse bacterial biofilms (94, 95). Literature also exists supporting the potential benefits of Na2EDTA, including decalcifying atherosclerotic plaque; however, recently published data from a clinical trial (NCT00044213) found an only modestly reduced risk of adverse cardiovascular outcomes from Na2EDTA therapy in patients with a history of myocardial infarction (96).
Presently, clinicians often have to resort to treatment regimens that include relatively toxic antimicrobials, such as colistin, and/or those likely to foster further resistance, such as tigecycline, for MBL-producing organisms (97). However, serious concerns exist about the possible toxicities of EDTA therapy, including deaths after documented infusions of Na2EDTA (98). In fact, Na2EDTA was withdrawn from the market in 2008 because of safety concerns. Approximately one-third of human proteins are metalloproteins, and nonspecific chelation of essential cofactors could have significant biological effects (99). While pharmaceutical companies are unlikely to select EDTA for development as an MBL inhibitor, the concept of chelator fragments with improved specificity may still be worth attention. This notion of selective inhibition has been applied to the design of inhibitors for the metalloenzyme HIV integrase which inhibited viral replication in cell culture assays (100). Many important issues about a chelation strategy for MBLs, including whether these agents can penetrate bacterial cell walls to reach their targets and whether modification is possible to create safe therapeutic options, remain unresolved.
CLASS D β-LACTAMASES; ANTICIPATING A GROWING NEED
The class D β-lactamases, named the OXA type for their oxacillin-hydrolyzing properties, comprise a very diverse group of enzymes with substrate profiles ranging from narrow to extended spectrum and more recently including the carbapenems (101, 102). As a class, these OXA β-lactamases are not effectively inactivated by the available β-lactam-type inhibitors; however, in vitro, concentrations of NaCl > 100 mM inactivate most class D enzymes (103, 104). This inhibitory property is not well understood, although substitution of a specific active-site Tyr144 residue can produce a resistant phenotype (103); of note, this mechanistic insight may someday provide a focus for inhibitor design. The OXA enzymes can be spread through mobile elements such as insertion sequences and transposons and have recently been increasingly detected worldwide in Enterobacteriaceae, P. aeruginosa, and A. baumannii (105, 106).
There is a relative dearth of literature addressing the challenge of inhibiting class D enzymes. With regard to β-lactam derivatives, substituted penicillin sulfones have turnover numbers (i.e., partitioning of the initial enzyme inhibitor complex between hydrolysis and enzyme inactivation) ranging from 0 to 8 for narrow- and extended-spectrum and carbapenemase OXA enzymes (107, 108). Bou et al. showed that select compounds from this series demonstrate exceptional potency (nM affinity versus OXA-24/40) (108). In addition, LN-1-255, compound 1, lowered meropenem MICs for A. baumannii possessing OXA-24/40 from 32 μg/ml to 4 μg/ml (Fig. 6b).
While the studies mentioned above describe penicillin sulfones with promising inhibition of class A and C enzymes, boronates with activity against OXA β-lactamases had not been described until recently. In 2010, medicinal chemists reported on a 4,7-dichloro-1-benzothien-2-yl sulfonylaminomethyl (DSABA) compound with a 5.6 μM IC50 against OXA-24/40 (Fig. 6c) (109). DSABA also inhibits class A and C β-lactamases with low-μM IC50s (i.e., 0.57 and 1.1 μM for SHV-5 and TEM-1 and 0.62 and 1.2 μM for P99 and P. aeruginosa AmpC, respectively). The whole-cell data were less impressive, as 100 μM DSABA reduced imipenem MICs only from 256 μg/ml to 128 μg/ml for A. baumannii expressing class A, C, and D β-lactamases.
Phosphonates, which share functional similarities with boronates and have been similarly derivatized with side chains to screen for high-affinity active-site binding, may offer a better ability to bridge the gap to class D enzymes. The same medicinal chemistry group working on DBASA evaluated a panel of thiophenyl oxime-derived phosphonate compounds among which a lead compound showed strong nM IC50 data against class C β-lactamases (i.e., 7.5 to 32 nM for P. aeruginosa AmpC and P99) and 17 to 22 μM values against class A and D enzymes (Fig. 7) (110). This compound potentiated imipenem against clinical strains of AmpC-producing P. aeruginosa in a dose-dependent manner and, despite its relatively weaker class D activity, lowered imipenem MICs from 128 to 16 μg/ml in OXA-24/40-expressing A. baumannii.
FIG 7.
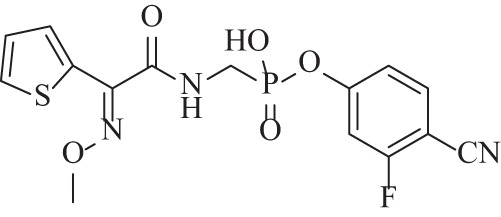
Structure of thiophenyl oxime-derived phosphonate compound with strong class C inhibitory activity (110).
While we recognize that the impact of OXA-23 and -48 is significant, the clinical contribution of other carbapenem-hydrolyzing members of the class has been debated (111, 112). The carbapenem inactivation rate is relatively low; however, the affinities are high, and recent in vitro data suggest that OXA-23, OXA-24/40, and OXA-58 add significantly to decreased carbapenem susceptibilities, particularly when combined with other resistance mechanisms such as decreased membrane permeability (113). Owing likely to the significant diversity in substrate spectra of this class of β-lactamases, laboratory detection of these enzymes is quite challenging (105). Specific class-wide phenotypic markers have not been identified, and this difficulty in detection may contribute to the spread of OXA-expressing organisms as their presence goes unidentified.
The search for novel mechanisms again presents promising data for avibactam combinations, specifically with ceftazidime or ceftaroline, restoring susceptibility in K. pneumoniae expressing OXA-48 (Table 1) (19). In contrast, ceftazidime-avibactam did not lower MICs for A. baumannii strains harboring carbapenemases OXA-23, -24/40, -51, and -58 (18). This failure may reflect problems with membrane permeability or an intrinsic lack of activity against these enzymes. The ceftazidime-avibactam combination is also inactive against the structurally distinct extended-spectrum-type enzymes in P. aeruginosa, such as those derived from OXA-2 and OXA-10 (18, 114). These extended-spectrum-type class D enzymes generally maintain susceptibility to carbapenems, although broad inhibition certainly remains a paramount goal in this era of antibiotic stewardship. The further dissemination of OXA-48-type enzyme expressers in Enterobacteriaceae was foreshadowed, coining the phrase “the phantom menace” for this β-lactamase (111). Treatment of Acinetobacter spp. expressing OXA carbapenemases may also become an increasingly relevant clinical problem (115, 116). None of the discussed inhibitors are likely solutions for carbapenem- and aztreonam-resistant strains; however, the novel siderophore monosulfactam BAL30072 restores susceptibility in many MDR strains (117).
PERSPECTIVE
This paper reviews the current state of research and development for β-lactamase inhibitors of novel or non-β-lactam-based mechanisms (for a full review of the existing patent literature, see reference 118). Since our last review on β-lactamase inhibitors 4 years ago (4), we have noted a growing focus on computation-based design methods. Such approaches are yielding inhibitors with low-nM affinities rarely before reported. We are eager to see how these agents will survive thorough in vivo testing and if they will continue to adapt as the boronates have.
However, the reader must appreciate that the majority of the compounds we discuss, with the exceptions of avibactam, MK-7756, and RPX7009, are years from clinical testing (116, 119). This “gap” is a call to action and, fortunately, one that has recently seen an increase in attention and momentum in a world with ever-rising rates of MDR pathogens. In much the same way that we need novel inhibition mechanisms, researchers, clinicians, and policy makers alike need to embrace a diverse and multifaceted approach to this problem. The Infectious Disease Society of America outlined in their 2012 white paper a new approach to streamline and expedite trials for treatment of highly resistant bacteria (120). Additionally, authorities have advocated focusing not just on incentivizing new drug development but also on interventions such as requiring transparency through public reporting of antibiotic use tied to reimbursement, harnessing molecular techniques for diagnostic confirmation of antibiotic indications, and exploring agents that modify host immune responses to pathogens to circumvent resistance selection (121). The role of regulatory agencies in this process is also highlighted (122). We need to pay closer attention to the variables over which clinicians do have control, such as dosing regimens. A recent double-blind randomized controlled trial showed improved clinical cure for critically ill septic patients receiving continuous infusion versus intermittent boluses of β-lactams (123). Why have we been reticent to embrace these approaches? A better understanding of the relationships between bacterial burden, patient factors, and pharmacodynamics has the potential to revitalize our β-lactam armamentarium (124). In this context, we can answer that novel mechanisms are indeed an advantage. The renaissance of interest is unlikely to provide a “magic bullet” that effectively inhibits all β-lactamase classes but will hopefully engender a collaborative solution.
In closing, a position that has some merit is to design optimal β-lactamase inhibitors for specific applications, based upon the pathogen detected. This would require a level of expertise beyond the scope of our current clinical microbiology laboratory. Yet, with the advent of molecular diagnostics, we may be able to define the β-lactamase background present and target appropriately. The current clinical threats should force decisions about antibiotic treatment to become more deliberate. We hope this prudence will allow agents with novel inhibitory mechanisms to succeed.
ACKNOWLEDGMENTS
We thank Andrea Hujer and the anonymous reviewers for their careful attention to the manuscript. Their suggestions have greatly improved the content and our message.
This work was supported by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, the Veterans Affairs Merit Review Program, and the Geriatric Research Education and Clinical Center VISN 10 to R.A.B. and the Veterans Affairs Career Development Program to K.M.P.-W. Research reported in this publication was also supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI100560 and R01AI063517 to R.A.B.
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 30 December 2013
REFERENCES
- 1.Bush K, Courvalin P, Dantas G, Davies J, Eisenstein B, Huovinen P, Jacoby GA, Kishony R, Kreiswirth BN, Kutter E, Lerner SA, Levy S, Lewis K, Lomovskaya O, Miller JH, Mobashery S, Piddock LJ, Projan S, Thomas CM, Tomasz A, Tulkens PM, Walsh TR, Watson JD, Witkowski J, Witte W, Wright G, Yeh P, Zgurskaya HI. 2011. Tackling antibiotic resistance. Nat. Rev. Microbiol. 9:894–896. 10.1038/nrmicro2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Livermore DM. 2012. Fourteen years in resistance. Int. J. Antimicrob. Agents 39:283–294. 10.1016/j.ijantimicag.2011.12.012 [DOI] [PubMed] [Google Scholar]
- 3.Fisher J, Mobashery S. 2010. Enzymology of antibacterial resistance, p 443–487 In Mander L, Liu H. (ed), Comprehensive natural products chemistry II. Elsevier, Oxford, United Kingdom [Google Scholar]
- 4.Drawz SM, Bonomo RA. 2010. Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 23:160–201. 10.1128/CMR.00037-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papp-Wallace KM, Bethel CR, Distler AM, Kasuboski C, Taracila M, Bonomo RA. 2010. Inhibitor resistance in the KPC-2 β-lactamase, a preeminent property of this class A beta-lactamase. Antimicrob. Agents Chemother. 54:890–897. 10.1128/AAC.00693-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coleman K. 2011. Diazabicyclooctanes (DBOs): a potent new class of non-β-lactam β-lactamase inhibitors. Curr. Opin. Microbiol. 14:550–555. 10.1016/j.mib.2011.07.026 [DOI] [PubMed] [Google Scholar]
- 7.Bonnefoy A, Dupuis-Hamelin C, Steier V, Delachaume C, Seys C, Stachyra T, Fairley M, Guitton M, Lampilas M. 2004. In vitro activity of AVE1330A, an innovative broad-spectrum non-β-lactam β-lactamase inhibitor. J. Antimicrob. Chemother. 54:410–417. 10.1093/jac/dkh358 [DOI] [PubMed] [Google Scholar]
- 8.Livermore DM, Mushtaq S, Warner M, Miossec C, Woodford N. 2008. NXL104 combinations versus Enterobacteriaceae with CTX-M extended-spectrum β-lactamases and carbapenemases. J. Antimicrob. Chemother. 62:1053–1056. 10.1093/jac/dkn320 [DOI] [PubMed] [Google Scholar]
- 9.Stachyra T, Péchereau MC, Bruneau JM, Claudon M, Frère JM, Miossec C, Coleman K, Black MT. 2010. Mechanistic studies of the inactivation of TEM-1 and P99 by NXL104, a novel non-β-lactam β-lactamase inhibitor. Antimicrob. Agents Chemother. 54:5132–5138. 10.1128/AAC.00568-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stachyra T, Levasseur P, Péchereau MC, Girard AM, Claudon M, Miossec C, Black MT. 2009. In vitro activity of the β-lactamase inhibitor NXL104 against KPC-2 carbapenemase and Enterobacteriaceae expressing KPC carbapenemases. J. Antimicrob. Chemother. 64:326–329. 10.1093/jac/dkp197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehmann DE, Jahić H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL. 2012. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. U. S. A. 109:11663–11668. 10.1073/pnas.1205073109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Durand-Réville TF, Lahiri S, Thresher J, Livchak S, Gao N, Palmer T, Walkup GK, Fisher SL. 2013. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J. Biol. Chem. 288:27960–27971. 10.1074/jbc.M113.485979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu H, Hazra S, Blanchard JS. 2012. NXL104 irreversibly inhibits the β-lactamase from Mycobacterium tuberculosis. Biochemistry 51:4551–4557. 10.1021/bi300508r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lahiri SD, Mangani S, Durand-Reville T, Benvenuti M, De Luca F, Sanyal G, Docquier JD. 2013. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-lactamases. Antimicrob. Agents Chemother. 57:2496–2505. 10.1128/AAC.02247-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castanheira M, Sader HS, Farrell DJ, Mendes RE, Jones RN. 2012. Activity of ceftaroline-avibactam tested against Gram-negative organism populations, including strains expressing one or more β-lactamases and methicillin-resistant Staphylococcus aureus carrying various staphylococcal cassette chromosome mec types. Antimicrob. Agents Chemother. 56:4779–4785. 10.1128/AAC.00817-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Endimiani A, Choudhary Y, Bonomo RA. 2009. In vitro activity of NXL104 in combination with β-lactams against Klebsiella pneumoniae isolates producing KPC carbapenemases. Antimicrob. Agents Chemother. 53:3599–3601. 10.1128/AAC.00641-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mushtaq S, Warner M, Williams G, Critchley I, Livermore DM. 2010. Activity of chequerboard combinations of ceftaroline and NXL104 versus β-lactamase-producing Enterobacteriaceae. J. Antimicrob. Chemother. 65:1428–1432. 10.1093/jac/dkq161 [DOI] [PubMed] [Google Scholar]
- 18.Mushtaq S, Warner M, Livermore DM. 2010. In vitro activity of ceftazidime+NXL104 against Pseudomonas aeruginosa and other non-fermenters. J. Antimicrob. Chemother. 65:2376–2381. 10.1093/jac/dkq306 [DOI] [PubMed] [Google Scholar]
- 19.Livermore DM, Mushtaq S, Warner M, Zhang J, Maharjan S, Doumith M, Woodford N. 2011. Activities of NXL104 combinations with ceftazidime and aztreonam against carbapenemase-producing Enterobacteriaceae. Antimicrob. Agents Chemother. 55:390–394. 10.1128/AAC.00756-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lagacé-Wiens PR, Tailor F, Simner P, DeCorby M, Karlowsky JA, Walkty A, Hoban DJ, Zhanel GG. 2011. Activity of NXL104 in combination with β-lactams against genetically characterized Escherichia coli and Klebsiella pneumoniae isolates producing class A extended-spectrum β-lactamases and class C β-lactamases. Antimicrob. Agents Chemother. 55:2434–2437. 10.1128/AAC.01722-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levasseur P, Girard AM, Claudon M, Goossens H, Black MT, Coleman K, Miossec C. 2012. In vitro antibacterial activity of the ceftazidime-avibactam (NXL104) combination against Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 56:1606–1608. 10.1128/AAC.06064-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crandon JL, Schuck VJ, Banevicius MA, Beaudoin ME, Nichols WW, Tanudra MA, Nicolau DP. 2012. Comparative in vitro and in vivo efficacies of human simulated doses of ceftazidime and ceftazidime-avibactam against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 56:6137-6146. 10.1128/AAC.00851-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louie A, Castanheira M, Liu W, Grasso C, Jones RN, Williams G, Critchley I, Thye D, Brown D, Vanscoy B, Kulawy R, Drusano GL. 2012. Pharmacodynamics of β-lactamase inhibition by NXL104 in combination with ceftaroline: examining organisms with multiple types of β-lactamases. Antimicrob. Agents Chemother. 56:258–270. 10.1128/AAC.05005-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aktaş Z, Kayacan C, Oncul O. 2012. In vitro activity of avibactam (NXL104) in combination with β-lactams against Gram-negative bacteria, including OXA-48 β-lactamase-producing Klebsiella pneumoniae. Int. J. Antimicrob. Agents 39:86–89. 10.1016/j.ijantimicag.2011.09.012 [DOI] [PubMed] [Google Scholar]
- 25.Goldstein EJ, Citron DM, Merriam CV, Tyrrell KL. 2013. Comparative in vitro activity of ceftaroline, ceftaroline-avibactam, and other antimicrobial agents against aerobic and anaerobic bacteria cultured from infected diabetic foot wounds. Diagn. Microbiol. Infect. Dis. 76:347–351. 10.1016/j.diagmicrobio.2013.03.019 [DOI] [PubMed] [Google Scholar]
- 26.Endimiani A, Hujer KM, Hujer AM, Pulse ME, Weiss WJ, Bonomo RA. 2011. Evaluation of ceftazidime and NXL104 in two murine models of infection due to KPC-producing Klebsiella pneumoniae. Antimicrob. Agents Chemother. 55:82–85. 10.1128/AAC.01198-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiskirchen DE, Crandon JL, Furtado GH, Williams G, Nicolau DP. 2011. In vivo efficacy of a human-simulated regimen of ceftaroline combined with NXL104 against extended-spectrum-beta-lactamase (ESBL)-producing and non-ESBL-producing Enterobacteriaceae. Antimicrob. Agents Chemother. 55:3220–3225. 10.1128/AAC.00024-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borgonovi M, Merdjan H, Girard A, Levasseur P, Quernin J, Lowther J, Miossec C, Shlaes D, Drusano G. 2008. Importance of NXL-104 pharmacokinetics in the pharmacodynamics of ceftazidime-NXL-104 combinations, abstr A-023 Abstr. Intersci. Conf. Antimicrob. Agents Chemother., Washington, DC [Google Scholar]
- 29.Nichols W, Levasseur P, Das S. 2012. A threshold concentration of avibactam (AVI) during the pharmacokinetic decline phase, below which β-lactamase inhibition in Enterobacteriaceae becomes ineffective, abstr A-1760 Abstr. Intersci. Conf. Antimicrob. Agents Chemother., San Francisco, CA [Google Scholar]
- 30.Bush K. 2012. Improving known classes of antibiotics: an optimistic approach for the future. Curr. Opin. Pharmacol. 12:527–534. 10.1016/j.coph.2012.06.003 [DOI] [PubMed] [Google Scholar]
- 31.Flückiger U, Segessenmann C, Gerber AU. 1991. Integration of pharmacokinetics and pharmacodynamics of imipenem in a human-adapted mouse model. Antimicrob. Agents Chemother. 35:1905–1910. 10.1128/AAC.35.9.1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vazquez JA, González Patzán LD, Stricklin D, Duttaroy DD, Kreidly Z, Lipka J, Sable C. 2012. Efficacy and safety of ceftazidime-avibactam versus imipenem-cilastatin in the treatment of complicated urinary tract infections, including acute pyelonephritis, in hospitalized adults: results of a prospective, investigator-blinded, randomized study. Curr. Med. Res. Opin. 28:1921–1931. 10.1185/03007995.2012.748653 [DOI] [PubMed] [Google Scholar]
- 33.Azap OK, Arslan H, Serefhanoğlu K, Colakoğlu S, Erdoğan H, Timurkaynak F, Senger SS. 2010. Risk factors for extended-spectrum β-lactamase positivity in uropathogenic Escherichia coli isolated from community-acquired urinary tract infections. Clin. Microbiol. Infect. 16:147–151. 10.1111/j.1469-0691.2009.02941.x [DOI] [PubMed] [Google Scholar]
- 34.Lucasti C, Popescu I, Ramesh MK, Lipka J, Sable C. 2013. Comparative study of the efficacy and safety of ceftazidime/avibactam plus metronidazole versus meropenem in the treatment of complicated intra-abdominal infections in hospitalized adults: results of a randomized, double-blind, phase II trial. J. Antimicrob. Chemother. 68:1183–1192. 10.1093/jac/dks523 [DOI] [PubMed] [Google Scholar]
- 35.Citron DM, Tyrrell KL, Merriam V, Goldstein EJ. 2011. In vitro activity of ceftazidime-NXL104 against 396 strains of β-lactamase-producing anaerobes. Antimicrob. Agents Chemother. 55:3616–3620. 10.1128/AAC.01682-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dubreuil LJ, Mahieux S, Neut C, Miossec C, Pace J. 2012. Anti-anaerobic activity of a new β-lactamase inhibitor NXL104 in combination with β-lactams and metronidazole. Int. J. Antimicrob. Agents 39:500–504. 10.1016/j.ijantimicag.2012.02.013 [DOI] [PubMed] [Google Scholar]
- 37.Pucci MJ, Bush K. 2013. Investigational antimicrobial agents of 2013. Clin. Microbiol. Rev. 26:792–821. 10.1128/CMR.00033-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Livermore DM, Mushtaq S, Barker K, Hope R, Warner M, Woodford N. 2012. Characterization of β-lactamase and porin mutants of Enterobacteriaceae selected with ceftaroline + avibactam (NXL104). J. Antimicrob. Chemother. 67:1354–1358. 10.1093/jac/dks079 [DOI] [PubMed] [Google Scholar]
- 39.Skalweit M, Li M, Conklin B, Taracila M. 2011. Effect of N152G, S and T substitution on CMY-2 β-lactamase activity and inhibition, abstr C1-601 Abstr. Intersci. Conf. Antimicrob. Agents Chemother., Chicago, IL [Google Scholar]
- 40.Mangion IK, Ruck RT, Rivera N, Huffman MA, Shevlin M. 2011. A concise synthesis of a β-lactamase inhibitor. Org. Lett. 13:5480–5483. 10.1021/ol202195n [DOI] [PubMed] [Google Scholar]
- 41.Hirsch EB, Ledesma KR, Chang KT, Schwartz MS, Motyl MR, Tam VH. 2012. In vitro activity of MK-7655, a novel β-lactamase inhibitor, in combination with imipenem against carbapenem-resistant Gram-negative bacteria. Antimicrob. Agents Chemother. 56:3753–3757. 10.1128/AAC.05927-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Livermore DM, Warner M, Mushtaq S. 2013. Activity of MK-7655 combined with imipenem against Enterobacteriaceae and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 68:2286–2290. 10.1093/jac/dkt178 [DOI] [PubMed] [Google Scholar]
- 43.Young K, Hackel M, Lascols C, Bouchillon S, Badal R, Martinez-Martinez L, Pillar C, Shinabarger D, Deane J, Sahm D. 2012. Response to imipenem plus MK-7655, a novel β-lactamase inhibitor, among 212 recent clinical isolates of P. aeruginosa, abstr 1620 Abstr. IDSA Week 2012, San Diego, CA [Google Scholar]
- 44.Bhagunde P, Chang KT, Hirsch EB, Ledesma KR, Nikolaou M, Tam VH. 2012. Novel modeling framework to guide design of optimal dosing strategies for β-lactamase inhibitors. Antimicrob. Agents Chemother. 56:2237–2240. 10.1128/AAC.06113-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rizk M, Ahmed G, Young K, Motyl M, Butterton J, Racine F, Wu J, Li C, Wenning L. 2012. A semi-mechanistic pharmacokinetic/pharmacodynamic (PK/PD) model for MK-7655, a novel β-lactamase inhibitor (BLI) for use in combination with imipenem/cilastatin, abstr A-1763 Abstr. Intersci. Conf. Antimicrob. Agents Chemother., San Francisco, CA [Google Scholar]
- 46.Köhler T, Michea-Hamzehpour M, Epp SF, Pechere JC. 1999. Carbapenem activities against Pseudomonas aeruginosa: respective contributions of OprD and efflux systems. Antimicrob. Agents Chemother. 43:424–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strynadka NC, Martin R, Jensen SE, Gold M, Jones JB. 1996. Structure-based design of a potent transition state analogue for TEM-1 β-lactamase. Nat. Struct. Biol. 3:688–695. 10.1038/nsb0896-688 [DOI] [PubMed] [Google Scholar]
- 48.Beesley T, Gascoyne N, Knott-Hunziker V, Petursson S, Waley SG, Jaurin B, Grundström T. 1983. The inhibition of class C β-lactamases by boronic acids. Biochem. J. 209:229–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kiener PA, Waley SG. 1978. Reversible inhibitors of penicillinases. Biochem. J. 169:197–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morandi S, Morandi F, Caselli E, Shoichet BK, Prati F. 2008. Structure-based optimization of cephalothin-analogue boronic acids as β-lactamase inhibitors. Bioorg. Med. Chem. 16:1195–1205. 10.1016/j.bmc.2007.10.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weston GS, Blázquez J, Baquero F, Shoichet BK. 1998. Structure-based enhancement of boronic acid-based inhibitors of AmpC β-lactamase. J. Med. Chem. 41:4577–4586. 10.1021/jm980343w [DOI] [PubMed] [Google Scholar]
- 52.Morandi F, Caselli E, Morandi S, Focia PJ, Blázquez J, Shoichet BK, Prati F. 2003. Nanomolar inhibitors of AmpC β-lactamase. J. Am. Chem. Soc. 125:685–695. 10.1021/ja0288338 [DOI] [PubMed] [Google Scholar]
- 53.Winkler ML, Rodkey EA, Taracila MA, Drawz SM, Bethel CR, Papp-Wallace KM, Smith KM, Xu Y, Dwulit-Smith JR, Romagnoli C, Caselli E, Prati F, van den Akker F, Bonomo RA. 2013. Design and exploration of novel boronic acid inhibitors reveals important interactions with a clavulanic acid-resistant sulfhydryl-variable (SHV) β-lactamase. J. Med. Chem. 56:1084–1097. 10.1021/jm301490d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ke W, Sampson JM, Ori C, Prati F, Drawz SM, Bethel CR, Bonomo RA, van den Akker F. 2011. Novel insights into the mode of inhibition of class A SHV-1 β-lactamases revealed by boronic acid transition state inhibitors. Antimicrob. Agents Chemother. 55:174–183. 10.1128/AAC.00930-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drawz SM, Babic M, Bethel CR, Taracila M, Distler AM, Ori C, Caselli E, Prati F, Bonomo RA. 2010. Inhibition of the class C β-lactamase from Acinetobacter spp.: insights into effective inhibitor design. Biochemistry 49:329–340. 10.1021/bi9015988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Drawz SM, Taracila M, Caselli E, Prati F, Bonomo RA. 2011. Exploring sequence requirements for C3/C4 carboxylate recognition in the Pseudomonas aeruginosa cephalosporinase: insights into plasticity of the AmpC β-lactamase. Protein Sci. 20:941–958. 10.1002/pro.612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eidam O, Romagnoli C, Caselli E, Babaoglu K, Pohlhaus DT, Karpiak J, Bonnet R, Shoichet BK, Prati F. 2010. Design, synthesis, crystal structures, and antimicrobial activity of sulfonamide boronic acids as β-lactamase inhibitors. J. Med. Chem. 53:7852–7863. 10.1021/jm101015z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tondi D, Calò S, Shoichet BK, Costi MP. 2010. Structural study of phenyl boronic acid derivatives as AmpC β-lactamase inhibitors. Bioorg. Med. Chem. Lett. 20:3416–3419. 10.1016/j.bmcl.2010.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eidam O, Romagnoli C, Dalmasso G, Barelier S, Caselli E, Bonnet R, Shoichet BK, Prati F. 2012. Fragment-guided design of subnanomolar β-lactamase inhibitors active in vivo. Proc. Natl. Acad. Sci. U. S. A. 109:17448–17453. 10.1073/pnas.1208337109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baker M. 2013. Fragment-based lead discovery grows up. Nat. Rev. Drug Discov. 12:5–7. 10.1038/nrd3926. [DOI] [PubMed] [Google Scholar]
- 61.Livermore D, Mushtaq S, Dhanji H. 2012. Activity of RPX2003 - RPX7009 combinations against carbapenem-resistant Enterobacteriaceae, abstr F-853 Abstr. Intersci. Conf. Antimicrob. Agents Chemother., San Francisco, CA [Google Scholar]
- 62.Castanheira M, Becker H, Rhomberg P, Jones R. 2012. Pre-clinical evaluation of a carbapenem/β-lactamase inhibitor combination (RPX2003/RPX7009) tested against serine-carbapenemase-producing pathogens, abstr F-856 Abstr. Intersci. Conf. Antimicrob. Agents Chemother., San Francisco, CA [Google Scholar]
- 63.Sabet M, Tarazi Z, Lomovskaya O, Hecke S, Dudley M, Griffith O. 2012. In vivo efficacy of the β-lactamase inhibitor RPX7009 combined with the carbapenem RPX2003 against KPC-producing K. pneumoniae, abstr F-858 Abstr. Intersci. Conf. Antimicrob. Agents Chemother., San Francisco, CA [Google Scholar]
- 64.Hecker S, Reddy K, Totrov M, Hirst G, Sabet M, Tarazi Z, Dudley M. 2012. Discovery of RPX7009, a broad-spectrum β-lactamase inhibitor with utility vs. class A serine carbapenemase, abstr F-848 Abstr. Intersci. Conf. Antimicrob. Agents Chemother., San Francisco, CA [Google Scholar]
- 65.Livermore DM, Mushtaq S. 2013. Activity of biapenem (RPX2003) combined with the boronate β-lactamase inhibitor RPX7009 against carbapenem-resistant Enterobacteriaceae. J. Antimicrob. Chemother. 68:1825–1831. 10.1093/jac/dkt118 [DOI] [PubMed] [Google Scholar]
- 66.Castanheira M, Konrardy M, Rhomberg P, Jones R. 2012. In vitro activity of a carbapenem and novel-β-lactamase inhibitor combination (RPX2003/RPX7009) tested against contemporary populations of Gram-negative organisms, abstr 1615 Abstr. ID Week 2012, San Diego, CA [Google Scholar]
- 67.Goldstein EJ, Citron DM, Tyrrell KL, Merriam CV. 2013. In vitro activity of biapenem plus RPX7009, a carbapenem combined with a serine β-lactamase inhibitor, against anaerobic bacteria. Antimicrob. Agents Chemother. 57:2620–2630. 10.1128/AAC.02418-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson JW, Gretes M, Goodfellow VJ, Marrone L, Heynen ML, Strynadka NC, Dmitrienko GI. 2010. Cyclobutanone analogues of β-lactams revisited: insights into conformational requirements for inhibition of serine- and metallo-β-lactamases. J. Am. Chem. Soc. 132:2558–2560. 10.1021/ja9086374 [DOI] [PubMed] [Google Scholar]
- 69.Ke W, Bethel CR, Papp-Wallace KM, Pagadala SR, Nottingham M, Fernandez D, Buynak JD, Bonomo RA, van den Akker F. 2012. Crystal structures of KPC-2 β-lactamase in complex with 3-nitrophenyl boronic acid and the penam sulfone PSR-3-226. Antimicrob. Agents Chemother. 56:2713–2718. 10.1128/AAC.06099-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Papp-Wallace KM, Bethel CR, Gootz TD, Shang W, Stroh J, Lau W, McLeod D, Price L, Marfat A, Distler A, Drawz SM, Chen H, Harry E, Nottingham M, Carey PR, Buynak JD, Bonomo RA. 2012. Inactivation of a class A and a class C β-lactamase by 6β-(hydroxymethyl)penicillanic acid sulfone. Biochem. Pharmacol. 83:462–471. 10.1016/j.bcp.2011.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bush K. 1998. Metallo-β-lactamases: a class apart. Clin. Infect. Dis. 27(Suppl 1):S48–S53. 10.1086/514922 [DOI] [PubMed] [Google Scholar]
- 72.Nordmann P, Poirel L, Walsh TR, Livermore DM. 2011. The emerging NDM carbapenemases. Trends Microbiol. 19:588–595. 10.1016/j.tim.2011.09.005 [DOI] [PubMed] [Google Scholar]
- 73.Palzkill T. 2013. Metallo-β-lactamase structure and function. Ann. N. Y. Acad. Sci. 1277:91–104. 10.1111/j.1749-6632.2012.06796.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Poeylaut-Palena AA, Tomatis PE, Karsisiotis AI, Damblon C, Mata EG, Vila AJ. 2007. A minimalistic approach to identify substrate binding features in B1 metallo-β-lactamases. Bioorg. Med. Chem. Lett. 17:5171–5174. 10.1016/j.bmcl.2007.06.089 [DOI] [PubMed] [Google Scholar]
- 75.García-Saez I, Hopkins J, Papamicael C, Franceschini N, Amicosante G, Rossolini GM, Galleni M, Frère JM, Dideberg O. 2003. The 1.5-A structure of Chryseobacterium meningosepticum zinc β-lactamase in complex with the inhibitor, D-captopril. J. Biol. Chem. 278:23868–23873. 10.1074/jbc.M301062200 [DOI] [PubMed] [Google Scholar]
- 76.Liénard BM, Garau G, Horsfall L, Karsisiotis AI, Damblon C, Lassaux P, Papamicael C, Roberts GC, Galleni M, Dideberg O, Frère JM, Schofield CJ. 2008. Structural basis for the broad-spectrum inhibition of metallo-β-lactamases by thiols. Org. Biomol. Chem. 6:2282–2294. 10.1039/b802311e [DOI] [PubMed] [Google Scholar]
- 77.King DT, Worrall LJ, Gruninger R, Strynadka NC. 2012. New Delhi metallo-β-lactamase: structural insights into β-lactam recognition and inhibition. J. Am. Chem. Soc. 134:11362–11365. 10.1021/ja303579d [DOI] [PubMed] [Google Scholar]
- 78.Heinz U, Bauer R, Wommer S, Meyer-Klaucke W, Papamichaels C, Bateson J, Adolph HW. 2003. Coordination geometries of metal ions in d- or l-captopril-inhibited metallo-β-lactamases. J. Biol. Chem. 278:20659–20666. 10.1074/jbc.M212581200 [DOI] [PubMed] [Google Scholar]
- 79.Maltezou HC. 2009. Metallo-β-lactamases in Gram-negative bacteria: introducing the era of pan-resistance?. Int. J. Antimicrob. Agents 33:405.e1–405.e7. 10.1016/j.ijantimicag.2008.09.003 [DOI] [PubMed] [Google Scholar]
- 80.Cornaglia G, Riccio ML, Mazzariol A, Lauretti L, Fontana R, Rossolini GM. 1999. Appearance of IMP-1 metallo-β-lactamase in Europe. Lancet 353:899–900. 10.1016/S0140-6736(98)05954-6 [DOI] [PubMed] [Google Scholar]
- 81.Lauretti L, Riccio ML, Mazzariol A, Cornaglia G, Amicosante G, Fontana R, Rossolini GM. 1999. Cloning and characterization of blaVIM, a new integron-borne metallo-β-lactamase gene from a Pseudomonas aeruginosa clinical isolate. Antimicrob. Agents Chemother. 43:1584–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Irwin JJ, Raushel FM, Shoichet BK. 2005. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry 44:12316–12328. 10.1021/bi050801k [DOI] [PubMed] [Google Scholar]
- 83.Widmann M, Pleiss J, Oelschlaeger P. 2012. Systematic analysis of metallo-β-lactamases using an automated database. Antimicrob. Agents Chemother. 56:3481–3491. 10.1128/AAC.00255-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oelschlaeger P, Ai N, Duprez KT, Welsh WJ, Toney JH. 2010. Evolving carbapenemases: can medicinal chemists advance one step ahead of the coming storm? J. Med. Chem. 53:3013–3027. 10.1021/jm9012938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vella P, Hussein WM, Leung EW, Clayton D, Ollis DL, Mitić N, Schenk G, McGeary RP. 2011. The identification of new metallo-β-lactamase inhibitor leads from fragment-based screening. Bioorg. Med. Chem. Lett. 21:3282–3285. 10.1016/j.bmcl.2011.04.027 [DOI] [PubMed] [Google Scholar]
- 86.Mohamed MS, Hussein WM, McGeary RP, Vella P, Schenk G, Abd El-Hameed RH. 2011. Synthesis and kinetic testing of new inhibitors for a metallo-β-lactamase from Klebsiella pneumoniae and Pseudomonas aeruginosa. Eur. J. Med. Chem. 46:6075–6082. 10.1016/j.ejmech.2011.10.030 [DOI] [PubMed] [Google Scholar]
- 87.Faridoon Hussein WM, Vella P, Islam NU, Ollis DL, Schenk G, McGeary RP. 2012. 3-Mercapto-1,2,4-triazoles and N-acylated thiosemicarbazides as metallo-β-lactamase inhibitors. Bioorg. Med. Chem. Lett. 22:380–386. 10.1016/j.bmcl.2011.10.116 [DOI] [PubMed] [Google Scholar]
- 88.Aoki N, Ishii Y, Tateda K, Saga T, Kimura S, Kikuchi Y, Kobayashi T, Tanabe Y, Tsukada H, Gejyo F, Yamaguchi K. 2010. Efficacy of calcium-EDTA as an inhibitor for metallo-β-lactamase in a mouse model of Pseudomonas aeruginosa pneumonia. Antimicrob. Agents Chemother. 54:4582–4588. 10.1128/AAC.00511-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yoshizumi A, Ishii Y, Livermore DM, Woodford N, Kimura S, Saga T, Harada S, Yamaguchi K, Tateda K. 2013. Efficacies of calcium-EDTA in combination with imipenem in a murine model of sepsis caused by Escherichia coli with NDM-1 β-lactamase. J. Infect. Chemother. 19:992–995. 10.1007/s10156-012-0528-y [DOI] [PubMed] [Google Scholar]
- 90.Laraki N, Franceschini N, Rossolini GM, Santucci P, Meunier C, de Pauw E, Amicosante G, Frère JM, Galleni M. 1999. Biochemical characterization of the Pseudomonas aeruginosa 101/1477 metallo-β-lactamase IMP-1 produced by Escherichia coli. Antimicrob. Agents Chemother. 43:902–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bush K, Jacoby GA, Medeiros AA. 1995. A functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 39:1211–1233. 10.1128/AAC.39.6.1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee K, Lim YS, Yong D, Yum JH, Chong Y. 2003. Evaluation of the Hodge test and the imipenem-EDTA double-disk synergy test for differentiating metallo-β-lactamase-producing isolates of Pseudomonas spp. and Acinetobacter spp. J. Clin. Microbiol. 41:4623–4629. 10.1128/JCM.41.10.4623-4629.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pitout JD, Gregson DB, Poirel L, McClure JA, Le P, Church DL. 2005. Detection of Pseudomonas aeruginosa producing metallo-β-lactamases in a large centralized laboratory. J. Clin. Microbiol. 43:3129–3135. 10.1128/JCM.43.7.3129-3135.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vaara M. 1992. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 56:395–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Banin E, Brady KM, Greenberg EP. 2006. Chelator-induced dispersal and killing of Pseudomonas aeruginosa cells in a biofilm. Appl. Environ. Microbiol. 72:2064–2069. 10.1128/AEM.72.3.2064-2069.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, Lindblad L, Lewis EF, Drisko J, Lee KL, TACT Investigators 2013. Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: the TACT randomized trial. JAMA 309:1241–1250. 10.1001/jama.2013.2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Falagas ME, Karageorgopoulos DE, Nordmann P. 2011. Therapeutic options for infections with Enterobacteriaceae producing carbapenem-hydrolyzing enzymes. Future Microbiol. 6:653–666. 10.2217/fmb.11.49 [DOI] [PubMed] [Google Scholar]
- 98.Centers for Disease Control and Prevention. 2006. Deaths associated with hypocalcemia from chelation therapy–Texas, Pennsylvania, and Oregon, 2003-2005. JAMA 295:2131–2134. 10.1001/jama.295.18.2131 [DOI] [Google Scholar]
- 99.Franz KJ. 2013. Clawing back: broadening the notion of metal chelators in medicine. Curr. Opin. Chem. Biol. 17:143–149. 10.1016/j.cbpa.2012.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Agrawal A, DeSoto J, Fullagar JL, Maddali K, Rostami S, Richman DD, Pommier Y, Cohen SM. 2012. Probing chelation motifs in HIV integrase inhibitors. Proc. Natl. Acad. Sci. U. S. A. 109:2251–2256. 10.1073/pnas.1112389109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Naas T, Nordmann P. 1999. OXA-type β-lactamases. Curr. Pharm. Des. 5:865–879 [PubMed] [Google Scholar]
- 102.Jacoby GA, Munoz-Price LS. 2005. The new β-lactamases. N. Engl. J. Med. 352:380–391. 10.1056/NEJMra041359 [DOI] [PubMed] [Google Scholar]
- 103.Héritier C, Poirel L, Aubert D, Nordmann P. 2003. Genetic and functional analysis of the chromosome-encoded carbapenem-hydrolyzing oxacillinase OXA-40 of Acinetobacter baumannii. Antimicrob. Agents Chemother. 47:268–273. 10.1128/AAC.47.1.268-273.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bush K, Jacoby GA. 2010. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 54:969–976. 10.1128/AAC.01009-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Poirel L, Naas T, Nordmann P. 2010. Diversity, epidemiology, and genetics of class D β-lactamases. Antimicrob. Agents Chemother. 54:24–38. 10.1128/AAC.01512-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Patel G, Bonomo RA. 2013. “Stormy waters ahead”: global emergence of carbapenemases. Front. Microbiol. 4:48. 10.3389/fmicb.2013.00048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Drawz SM, Bethel CR, Doppalapudi VR, Sheri A, Pagadala SR, Hujer AM, Skalweit MJ, Anderson VE, Chen SG, Buynak JD, Bonomo RA. 2010. Penicillin sulfone inhibitors of class D β-lactamases. Antimicrob. Agents Chemother. 54:1414–1424. 10.1128/AAC.00743-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bou G, Santillana E, Sheri A, Beceiro A, Sampson JM, Kalp M, Bethel CR, Distler AM, Drawz SM, Pagadala SR, van den Akker F, Bonomo RA, Romero A, Buynak JD. 2010. Design, synthesis, and crystal structures of 6-alkylidene-2′-substituted penicillanic acid sulfones as potent inhibitors of Acinetobacter baumannii OXA-24 carbapenemase. J. Am. Chem. Soc. 132:13320–13331. 10.1021/ja104092z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tan Q, Ogawa AM, Painter RE, Park YW, Young K, DiNinno FP. 2010. 4,7-Dichloro benzothien-2-yl sulfonylaminomethyl boronic acid: first boronic acid-derived β-lactamase inhibitor with class A, C, and D activity. Bioorg. Med. Chem. Lett. 20:2622–2624. 10.1016/j.bmcl.2010.02.065 [DOI] [PubMed] [Google Scholar]
- 110.Tan Q, Ogawa AM, Raghoobar SL, Wisniewski D, Colwell L, Park YW, Young K, Hermes JD, Dininno FP, Hammond ML. 2011. Thiophenyl oxime-derived phosphonates as nano-molar class C β-lactamase inhibitors reducing MIC of imipenem against Pseudomonas aeruginosa and Acinetobacter baumannii. Bioorg. Med. Chem. Lett. 21:4363–4365. 10.1016/j.bmcl.2011.04.122 [DOI] [PubMed] [Google Scholar]
- 111.Poirel L, Potron A, Nordmann P. 2012. OXA-48-like carbapenemases: the phantom menace. J. Antimicrob. Chemother. 67:1597–1606. 10.1093/jac/dks121 [DOI] [PubMed] [Google Scholar]
- 112.Héritier C, Poirel L, Lambert T, Nordmann P. 2005. Contribution of acquired carbapenem-hydrolyzing oxacillinases to carbapenem resistance in Acinetobacter baumannii. Antimicrob. Agents Chemother. 49:3198–3202. 10.1128/AAC.49.8.3198-3202.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Queenan AM, Bush K. 2007. Carbapenemases: the versatile β-lactamases. Clin. Microbiol. Rev. 20:440–458. 10.1128/CMR.00001-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bradford PA. 2001. Extended-spectrum β-lactamases in the 21st century: characterization, epidemiology, and detection of this important resistance threat. Clin. Microbiol. Rev. 14:933–951. 10.1128/CMR.14.4.933-951.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin. Microbiol. Rev. 21:538–582. 10.1128/CMR.00058-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shlaes DM. 2013. New β-lactam-β-lactamase inhibitor combinations in clinical development. Ann. N. Y. Acad. Sci. 1277:105–114. 10.1111/nyas.12010 [DOI] [PubMed] [Google Scholar]
- 117.Higgins PG, Stefanik D, Page MG, Hackel M, Seifert H. 2012. In vitro activity of the siderophore monosulfactam BAL30072 against meropenem-non-susceptible Acinetobacter baumannii. J. Antimicrob. Chemother. 67:1167–1169. 10.1093/jac/dks009 [DOI] [PubMed] [Google Scholar]
- 118.Buynak JD. 2013. β-Lactamase inhibitors: a review of the patent literature (2010-2013). Expert. Opin. Ther. Pat. 23:1469–1481. 10.1517/13543776.2013.831071 [DOI] [PubMed] [Google Scholar]
- 119.Boucher HW, Talbot GH, Benjamin DK, Bradley J, Guidos RJ, Jones RN, Murray BE, Bonomo RA, Gilbert D, Infectious Diseases Society of America 2013. 10 x '20 progress–development of new drugs active against gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin. Infect. Dis. 56:1685–1694. 10.1093/cid/cit152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Infectious Diseases Society of America. 2012. White paper: recommendations on the conduct of superiority and organism-specific clinical trials of antibacterial agents for the treatment of infections caused by drug-resistant bacterial pathogens. Clin. Infect. Dis. 55:1031–1046. 10.1093/cid/cis688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Spellberg B, Bartlett JG, Gilbert DN. 2013. The future of antibiotics and resistance. N. Engl. J. Med. 368:299–302. 10.1056/NEJMp1215093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shlaes DM, Sahm D, Opiela C, Spellberg B. 2013. The FDA reboot of antibiotic development. Antimicrob. Agents Chemother. 57:4605–4607. 10.1128/AAC.01277-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dulhunty JM, Roberts JA, Davis JS, Webb SA, Bellomo R, Gomersall C, Shirwadkar C, Eastwood GM, Myburgh J, Paterson DL, Lipman J. 2013. Continuous infusion of β-lactam antibiotics in severe sepsis: a multicenter double-blind, randomized controlled trial. Clin. Infect. Dis. 56:236–244. 10.1093/cid/cis856 [DOI] [PubMed] [Google Scholar]
- 124.Drusano GL, Lodise TP. 2013. Saving lives with optimal antimicrobial chemotherapy. Clin. Infect. Dis. 56:245–247. 10.1093/cid/cis863 [DOI] [PubMed] [Google Scholar]