

Abstract
The drugs available for Chagas disease treatment are toxic and ineffective. We studied the in vivo activity of a new drug, lychnopholide (LYC). LYC was loaded in nanocapsules (NC), and its effects were compared to free LYC and benznidazole against Trypanosoma cruzi. Infected mice were treated in the acute phase at 2.0 mg/kg/day with free LYC, LYC-poly-ε-caprolactone NC (LYC-PCL), and LYC-poly(lactic acid)-co-polyethylene glycol NC (LYC-PLA-PEG) or at 50 mg/kg/day with benznidazole solution by the intravenous route. Animals infected with the CL strain, treated 24 h after infection for 10 days, evaluated by hemoculture, PCR, and enzyme-linked immunosorbent assay exhibited a 50% parasitological cure when treated with LYC-PCL NC and 100% cure when treated with benznidazole, but 100% of the animals treated during the prepatent period for 20 days with these formulations or LYC-PLA-PEG NC were cured. In animals with the Y strain treated 24 h after infection for 10 days, only mice treated by LYC-PCL NC were cured, but animals treated in the prepatent period for 20 days exhibited 100, 75, and 62.5% cure when treated with LYC-PLA-PEG NC, benznidazole, and LYC-PCL NC, respectively. Free LYC reduced the parasitemia and improved mice survival, but no mice were cured. LYC-loaded NC showed higher cure rates, reduced parasitemia, and increased survival when used in doses 2five times lower than those used for benznidazole. This study confirms that LYC is a potential new treatment for Chagas disease. Furthermore, the long-circulating property of PLA-PEG NC and its ability to improve LYC efficacy showed that this formulation is more effective in reaching the parasite in vivo.
INTRODUCTION
Chagas disease (CD) is recognized by the World Health Organization (1) as one among 13 of the world's most neglected tropical diseases. This disease remains a relevant social and economic problem in Latin America, where eight million people are infected with the causative intracellular parasite, Trypanosoma cruzi. More than 25 million people are at risk of infection, and 12,500 deaths are attributed to American trypanosomiasis (Chagas disease). Furthermore, the majority of people affected do not receive effective treatment for several reasons (1). As a consequence of the intense migration of individuals from areas of endemicity in Latin America to North America, Europe, and Asia, CD is now considered a global disease and represents a critical public health problem in several countries of the Northern Hemisphere (2, 3). Similarly to many other neglected diseases, CD chemotherapy research suffers from limited economic potential, because it is not the main focus of interest of the pharmaceutical industry (4). T. cruzi infection involves different morphological forms along its life cycle. This protozoan proliferates alternatively between hematophagous triatomines insect and vertebrate hosts. The disease presents two successive phases: a short acute phase characterized by patent parasitemia, and a long and chronic phase that may progress after several years or decades to cardiomyopathy and/or digestive megasyndromes in 30 to 40% of the infected individuals (1, 5).
Although CD was discovered more than 100 years ago, no efficient chemotherapy is available to treat this disease in either the acute or chronic phases of infection. Nitroheterocyclics, benznidazole (BZ), and nifurtimox (NF) are far from ideal medicines, particularly due to their long periods of treatment, the frequency of serious side effects, and their poor activity in the late chronic phase (6, 7). Moreover, differences in the susceptibility and natural resistance of different T. cruzi isolates to both nitroderivatives have also been reported (8, 9).
The main challenge of CD pharmacotherapy is the lack of ability of anti-T. cruzi agents with the suitable selectivity to reach infected cells and attain the intracellular parasites, because the plasma membrane and complex microenvironment of the host cells prevent the selective and massive delivery of drugs to the intracellular amastigote nests (10). Thus, a drug or a drug delivery system that provides a high volume of distribution, long plasmatic half-lives, and high efficacy in both the acute and the chronic phases of the infection is especially desirable (11).
Lychnopholide (LYC) (Fig. 1) is a sesquiterpene lactone (SL) that was isolated from Lychnophora trichocarpha (12, 13). The anti-T. cruzi activity of LYC in vitro was first reported by Oliveira et al. (14). This substance presents many other activities (15–17), including clastogenic and cytotoxic effects (18). Despite its pharmacological potential, the therapeutic application of LYC is limited due to its physicochemical properties that hamper oral administration, such as poor aqueous solubility, high lipophilicity (log P = 5.03), and potential chemical instability in alkaline media (13). Recently, our group developed a pharmaceutical formulation of LYC loaded in polymeric nanocapsules (NC) (19). These NC can be easily dispersed in water and are therefore suitable for administration by oral and parenteral routes. These LYC-NC also possess controlled release properties (13, 19). Furthermore, the validated methods were also developed to quantify this formulation in this new dosage form, which are necessary for preclinical evaluation in vivo (13).
FIG 1.
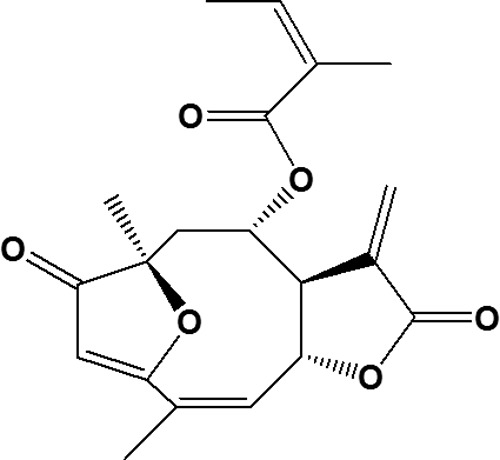
Chemical structure of lychnopholide, a sesquiterpene lactone extracted from Lychonophora trichocarpha.
The advantages of associating a CD drug with a nanocarrier has already been reviewed (20). Polymeric NC are formed by an oil droplet surrounded by a polymeric membrane stabilized by surfactants (21) and have been used successfully for the following purposes: to increase the dispersibility of poorly water-soluble drugs, to protect drugs against inactivation (22), to reduce drug toxicity (23), to control drug release (13), and to prolong blood circulation time after intravenous (i.v.) administration (24).
Thus, the aim of the study was to evaluate the efficacy of LYC-NC in vivo during the acute phase of infection in mice experimentally infected with T. cruzi strains with different susceptibility patterns to BZ. Two types of polymeric NC were used: one conventional form that is rapidly cleared from blood circulation by phagocytes of the mononuclear phagocyte system (PCL NC) and the other (PLA-PEG NC) that circulates longer in blood due to its polymeric steric stabilization which reduces uptake by phagocytes (25). Thus, the probability of LYC to target sites of inflammation induced by the presence of the parasite, particularly during the acute phase of infection, is expected to be increased due to increased plasma circulation time of PEG sterically stabilized NC (20, 24, 25, 26).
MATERIALS AND METHODS
Plant Collection, isolation and identification of lychnopholide.
L. trichocarpha Spreng was collected at Ouro Preto city in Minas Gerais state, Brazil, in August 2006. A voucher specimen (no. 20635) was deposited in the Herbarium of the Instituto de Ciências Exatas e Biológicas, Universidade Federal de Ouro Preto, Ouro Preto, MG, Brazil. All ethanol extract preparation, isolation, and identification of LYC were performed as described by Saúde et al. (12) and Branquinho et al. (13). LYC characterization and isolation from L. trichocarpha were performed as described previously by Saúde et al. (12) and Branquinho et al. (13), and its chemical structure is shown in Fig. 1.
Chemicals, adjuvants, and solvents.
Benznidazole (BZ) (N-benzyl-2-nitro-1-imidazole-acetamide) as Rochagan tablets obtained from LAFEPE (Brazil) was used. The analytical standard BZ (97.0%) was purchased from Sigma-Aldrich (St. Louis, MO). Epikuron 170 (soy lecithin with ∼70% phosphatidylcholine) was purchased from Lucas Meyer (Le Blanc Mesnil, France). Poly-ε-caprolactone (PCL) with average Mn of 42,500 g/mol and Poloxamer 188 (nonionic surfactant) were provided by Sigma-Aldrich. PLA-PEG [poly(d,l-lactide)-co-polyethylenoglycol average Mn of 49,000 g/mol] with a PEG block of Mn of 5,000 g/mol was obtained from Alkermes (Cambridge, MA) and used without further purification. PLA [poly(d,l-lactide)] Resomer 203R 203H (molecular mass, 18,000 g/mol) was purchased from Boehringer Ingelheim (Germany). Miglyol 810N (capric/caprilic triglyceride) was purchased from Hulls (Germany). Polyethylene glycol 300 (PEG 300), dimethylacetamide (DMA), ethyl acetate (AcOEt), methanol (MeOH), and acetone, all analytical grade, were purchased from Vetec (Rio de Janeiro, Brazil). The analytical-grade revelators were N-(1-naphthyl)-ethylenediamine-dihydrochloride and tin(II) chloride and were obtained from Merck (Germany). Silica gel 60 GF254 was purchased from Merck (Darmstadt, Germany) and was used for thin-layer chromatography (TLC). Milli-Q water was purified using a Symplicity System (Millipore, Bedford, MA) and used throughout the experiments.
Preparation of lychnopholide solution and lychnopholide-loaded NC.
To administer LYC i.v. solution (2 mg/ml), LYC was dissolved in a DMA-PEG 300 mixture at 40:60 (vol/vol) as described by Leite et al. (23) and further diluted in 5% (wt/vol) glucose to reach the correct dose of LYC of 2.0 mg/kg/day to be administered i.v. Both formulations were filtered in 0.45-μm-pore-size sterile filter before injection. Conventional NC (PCL NC) were loaded with LYC according to the method of Branquinho et al. (13). These NC were prepared with 0.8% (wt/vol) polymer (PCL) dissolved in acetone solution containing 0.4% (wt/vol) Epikuron 170, 2.5% (vol/vol) Miglyol 810N, and LYC to obtain a 2-mg/ml final concentration. This organic solution was poured into the external aqueous phase containing 0.75% (wt/vol) Poloxamer 188 and mixed. All solvents were evaporated under reduced pressure (Laborota 4000; Heidolph Instruments, Germany) to render a colloidal NC suspension (10 ml). NC sterically stabilized with a corona of PEG were prepared using PLA-PEG diblock polymer as reported by Mosqueira et al. (25). Briefly, a 1:1 mixture of PLA-PEG and Resomer 203 (1.2% [wt/vol]) was dissolved in acetone containing Epikuron 170 (0.4% [wt/vol]), Miglyol 810N (2.5% [vol/vol]), and LYC. This organic phase was poured into an external aqueous phase and mixed for 10 min. The solvents were then eliminated under reduced pressure to render the desired concentration of LYC and polymers in the colloidal suspension (2.0 mg/ml). The unloaded NC were prepared by the same methods described above, but without LYC in the formulation. The mean size and the polydispersity index of the NC were determined as previously described (25, 26).
Extraction, purification, and characterization of benznidazole and preparations of BZ solution.
BZ was not obtained from suppliers in its pure form, so it was extracted from tablets and subjected to purification according to the procedure described below. Rochagan tablets (20) were pulverized, dissolved in MeOH, and stirred in the dark for 20 min. The resulting suspension was filtered through quantitative paper filter (Whatman filter), and the filtrate was concentrated in a rotary evaporator until dried (Heidolph Instruments, Germany). The material was recrystallized twice in MeOH-H2O to render acicular crystals. The crystals were filtered and dried under a vacuum in a desiccant containing anhydrous silica. Melting points were determined, and TLC was used for chemical identification and determination of BZ purity. BZ purity was verified by comparison with a standard BZ in TLC of silica gel eluted with AcOEt-MeOH (85:15 [vol/vol]), with a detection system consisting of UV light (λ = 254 nm), tin(II) chloride, and N-(1-naphthyl)-ethylenediamine-dihydrochloride (27).
The BZ solution for i.v. administration was prepared similarly to a method described by Leite et al. (23). The solution contained DMA-PEG 300 mixture at 40:60 proportions was diluted in isotonic 5% (wt/vol) glucose up to 4.0 mg/ml. Subsequently, the solution was filtered in a sterile 0.80-μm-pore-size filter before i.v. injection in mice at a dose of 50 mg/kg of bodyweight.
Parasites.
The CL and Y strains of T. cruzi weren determined to be susceptible and partially resistant, respectively, to BZ by Filardi and Brener (8) and were therefore used in the present study. The original isolates were maintained as blood trypomastigotes in liquid nitrogen and cultured in liver infusion tryptose (LIT) medium (28). Mice were subsequently inoculated with trypomastigotes from the culture.
Mouse infection.
Groups of eight female Swiss mice aged 28 to 30 days and weighing 20 to 25 g were used and maintained according to the guidelines established by the Colégio Brasileiro de Experimentação Animal. Mice were maintained in a specific-pathogen-free room at 20 to 24°C under a 12/12-h light/dark cycle and were provided with sterilized water and chow ad libitum. Infection was performed via intraperitoneal injection of 104 blood trypomastigotes. Uninfected and age-matched mice were maintained under identical conditions as a control. The experiments were approved by the Ethical Committee on Animal Experimentation of the Universidade Federal de Ouro Preto, Brazil (protocol 2009/13).
Treatment schedules.
The dose of LYC used in all experiments was determined after a pilot experiment performed with T. cruzi-infected Swiss mice at 1, 2, and 4 mg/kg/day for three consecutive days, evaluating only the parasitemia level. Since the parasitemia reduction rate and extent were not improved above 2 mg/kg/day, this dose of LYC was adopted for the treatment of the animals during 10 and 20 days, as shown in Table 1. All animals were treated during the acute phase of the infection. Four independent experiments were carried out to evaluate the in vivo efficacy of free LYC, LYC-PCL NC, LYC-PLA-PEG NC, BZ (as a reference drug), and the following controls: control untreated (infected but not treated), control unloaded NC (UN-NC), and control DMA-PEG 300 (control solution). Four experiments of each treatment were used for each T. cruzi strain: in experiments I and III, the infection was carried out with the CL and Y strains, respectively. Treatment started 24 h after infection according to the method of Filardi and Brener (8). In experiments II and IV, mice were infected with the CL and Y strains, and treatment was started in the prepatent period of the T. cruzi strains used (CL strain, 7 days; Y strain, 4 days after inoculation) as described by Filardi and Brener (8). All formulations were administered i.v. in the tail veins at the doses described in Table 1.
TABLE 1.
Treatment schedules for T. cruzi-infected mice with lychnopholide and control formulations during the acute phase of infection
| Expt no. (treatment scheme) and T. cruzi strain | Drug/formulationa | Treatment period (days) | Dose (mg/kg/day), i.v.b |
|---|---|---|---|
| Ic (24 h after infection), CL strain (sensitive to BZ) | LYC-PCL NC | ||
| Free LYC | 10 | 2.0 | |
| BZ | 10 | 50.0 | |
| Unloaded NC | 10 | 2.0* | |
| DMA-PEG 300 (control i.v. solution) | 10 | 50.0* | |
| Untreated control | |||
| II (prepatent period, 7th day), CL strain (sensitive to BZ) | LYC-PCL NC | 20 | 2.0 |
| LYC-PLA-PEG NC | 20 | 2.0 | |
| Free LYC | 20 | 2.0 | |
| BZ | 20 | 50.0 | |
| Unloaded NC | 20 | 2.0* | |
| DMA-PEG 300 (control i.v. solution) | 20 | 50.0* | |
| Untreated control | |||
| III (24 h after infection), Y strain (partially resistant to BZ) | LYC-PCL NC | 10 | 2.0 |
| Free LYC | 10 | 2.0 | |
| BZ | 10 | 50.0 | |
| Unloaded NC | 10 | 2.0* | |
| DMA-PEG 300 (control i.v. solution) | 10 | 50.0* | |
| Untreated control | 2.0 | ||
| IV (prepatent period, 4th day), Y strain (partially resistant to BZ) | LYC-PCL NC | 20 | 2.0 |
| LYC-PLA-PEG NC | 20 | 2.0 | |
| BZ | 20 | 50.0 | |
| Unloaded NC | 20 | 2.0* | |
| DMA-PEG 300 (control i.v. solution) | 20 | 50.0* | |
| Untreated control |
NC, nanocapsules.
*, the amount of excipients used in DMA-PEG solution and in unloaded NC was the same as that used in BZ and LYC NC formulations.
Infection was confirmed by hemoculture in all animals.
Parasitemia and survival rates.
The level of parasitemia was checked by fresh blood examination using the Brener method (29). Mice were individually examined daily by direct counting of parasites in 5 μl of blood under optical microscopy. Mortality of the animals was checked daily until 6 months postinfection in order to determine the percentage of survival expressed in percentage.
Parasitological cure assessment.
The cure criterion was based on parasitological methods (fresh blood examination, hemoculture, and PCR on peripheral blood) and conventional serology. These methodologies are described below. Animals with negative parasitological and serological outcomes were considered cured.
Parasitological methods. (i) Fresh blood examination.
To determine the reduction, suppression, and/or reactivation of parasitemia in treated animals, fresh blood examination was performed before the prepatent period, throughout experimental period, and for 5 days after complete negative parasitemia. Parasitemia curves were plotted by using the mean parasitemia values of eight mice recorded daily.
(ii) Hemoculture.
Hemoculture was carried out 30 days after the end of treatment as described by Filardi and Brener (8). Blood samples collected from the orbital sinus vein were inoculated into 3 ml of LIT medium and maintained at 28°C. Each tube was examined under microscopy for parasites detection at 30, 60, 90, and 120 days after culture.
(iii) PCR on peripheral blood.
Blood samples were collected from the orbital sinus veins 60 and 120 days after treatment. DNA extraction and PCR protocols were adapted and standardized for rodent samples as previously reported (30). Briefly, 200-ml portions of blood were diluted in a 1:2 volume of guanidine solution (guanidine-HCl, 6 M; EDTA, 0.2 M) and heated for 90 s in boiling water to cleave the parasite kDNA (DNA of the kinetoplast). PCR was performed using the primers 5′-AAATAATGTACGGG[T/G]GAGATGCATGA-3′ and 5′-GGTTCGATTGGGGTTGGTGTAATATA-3′ (Invitrogen, São Paulo, Brazil), which amplify a 330-bp sequence from kDNA as previously described (31). A 2-μl blood DNA sample was added to the reaction mixture, which was then overlaid with 30 ml of mineral oil to avoid evaporation. Following an initial denaturation step of 5 min at 94°C, 35 amplification cycles consisting of 1 min at 95°C for DNA denaturation, 1 min at 65°C for primer annealing, and 1 min at 72°C for primer extension were performed, followed in turn by a final extension performed in a thermal cycler (MJ Research, model PTC-150). The amplified DNA was visualized by electrophoresis on a 6% polyacrylamide gel and revealed by silver staining (32). Positive and negative controls and reagents were included in each test. The presence of inhibitors in negative samples was evaluated by the addition of 100 fg of DNA of T. cruzi in 30 samples randomly selected from different samples with negative results followed by a new PCR.
Serological methods.
Conventional serology using a modified enzyme-linked immunosorbent assay (ELISA) approach based on that described by Voller et al. (33) was performed. Serum samples were collected 3 and 6 months after treatment and stored at −20°C. Samples were tested at 1:80 dilution in phosphate-buffered saline using the antigen of T. cruzi Y strain cultivated in LIT medium and prepared by alkaline extraction in the exponential growth phase. Antibody binding was detected by peroxidase-labeled anti-mouse immunoglobulin G (Sigma Immunochemical Reagents, St. Louis, MO). The absorbance was read in a spectrophotometer with a 490-nm filter (model 3550; Bio-Rad). Positive and negative controls were processed in parallel with each assay. The cutoff value calculated for each plate was the mean absorbance of 10 negative-control serum samples plus two standard deviations.
Statistical analysis.
Statistical analyses of data were carried out using Prism software v5.02 (GraphPad Software, San Diego, CA). The data were initially assessed by one-way analysis of variance (ANOVA). When interactions were significant, a Tukey test was used to determine specific differences between mean values. The Kolmogorov-Smirnov test was used to compare parasitemia between infected groups that were either treated or untreated. One-way ANOVA or Mann-Whitney U tests were used to compare maximum peak values of parasitemia between the different groups. The log-rank (Mantel-Cox) method was used to estimate the media of survival for the different experimental groups. Values were expressed as means ± the standard deviations. Differences in mean values were considered significant at P ≤ 0.05.
RESULTS
LYC was encapsulated in NC, at a high yield, more than 95% in conventional PCL NC (13) and with 100% loading in PLA-PEG NC. The mean diameter of LYC PCL NC was 182.5 ± 3.2 nm and that of PLA-PEG NC was 105.3 ± 2.3 nm, as determined by quasi-elastic light scattering method. The nanoparticle populations were monodispersed with a polydispersion index lower than 0.3.
Since pure BZ was not provided in sufficiently large amounts to perform in vivo experiments, the drug was extracted and purified from Rochagan tablets. This process yielded 99.0% ± 1.2% acicular yellowish crystals. The purity of the BZ crystals was confirmed by its melting point of 188.5 to 190°C (methanol SM Lux Leitz apparatus) determination that was performed in accordance with the literature (27) and also by TLC. TLC showed only one stain correspondent in position (retention factor = 9.48 cm), color, and intensity to that obtained with standard BZ.
Toxicity at the injection site with the parenteral BZ formulation was observed in mice during the first days of treatment. Itch and edema around the site were verified immediately after the injection and disappeared within 15 min. No similar effects were observed in the control formulations of NC excipients or in the parenteral solution, LYC solution, or LYC-NC formulations.
Curve of parasitemia and survival rates. (i) Animals infected with CL strain (sensitive to BZ).
Animals treated as described in the experiment I schedule (24 h after infection for 10 days) (Fig. 2A) exhibited a significant parasitemia reduction (P < 0.05), which became subpatent during and after treatment with BZ (Table 2). Mice treated with LYC-PCL NC showed higher parasitemia (P < 0.05) than animals treated with BZ, but this parasitemia was significantly lower (P < 0.05) than that observed in the control groups (untreated control, unloaded-NC, and control solution). Four animals of eight treated with LYC-PCL NC showed subpatent parasitemia during and after treatment (Table 2). Five and one animal(s) of eight treated with LYC-PCL NC and free LYC, respectively, showed subpatent parasitemia during and after treatment (Table 2). Analyses of the parasitemia peak of the treated groups revealed a reduction (P < 0.05) of MPP (maximum peak of parasitemia) of 98.6% (BZ), 56.3% (free LYC), and 96.3% (LYC-PCL NC) compared to the untreated control group (Table 2).
FIG 2.

Parasitemia curves of T. cruzi-infected mice treated i.v. with lychnopholide at 2.0 mg/kg/day and benznidazole at 50 mg/kg. (A) Mice infected with CL strain treated with 10 doses (started 24 h after infection). (B) Mice treated with 20 doses (started in the prepatent period [day 7 after infection]). (C) Mice infected with Y strain treated with 10 doses (started 24 h after infection). (D) Mice treated with 20 doses (started in the prepatent period [day 4 after infection]). dpi, days postinfection; LYC-PCL NC, lychnopholide loaded in conventional NC; LYC-PLA-PEG NC, lychnopholide loaded in PEG sterically stabilized NC; free LYC, lychnopholide in i.v. solution; BZ, benznidazole i.v. solution; control groups, untreated (infected and not treated) or Un-NC (unloaded NC); control solution, DMA-PEG 300.
TABLE 2.
Parasitemia and maximum peaks of parasitemia in T. cruzi-infected mice treated with lychnopholide i.v. using different treatment schemes during acute infectiona
| Treatment (dose [mg/kg])d | Strain and treatment schemeb |
|||||||
|---|---|---|---|---|---|---|---|---|
| CL strain, expt I |
CL strain, expt II |
Y strain, expt III |
Y strain, expt IV |
|||||
| Subpatent PAR/totalc | MPP ± SE (104) (% reduction)e | Subpatent PAR/total | MPP ± SE (104) (% reduction) | Subpatent PAR/total | MPP ± SE (104) (% reduction) | Subpatent PAR/total | MPP ± SE (104) (% reduction) | |
| Untreated | 0/8 | 223.4 ± 40.8 (ND) | 0/8 | 244.7 ± 24.3 (ND) | 0/8 | 163.9 ± 12.9 (ND) | 0/8 | 123.8 ± 7.8 (ND) |
| Benznidazole (50) | 8/8 | 2.8 ± 0.9 (98.6)A | 8/8 | 1.2 ± 0.6 (99.2)A | 0/8 | 5.3 ± 1.3 (94.35)A | 7/8 | 2.2 ± 0.5 (97.9)A,D |
| Free LYC (2) | 1/8 | 88.6 ± 26.8 (56.3)A,B,C | 1/8 | 86.5 ± 27.4 (57.7)A,B,C,D | 0/8 | 90.6 ± 19.4 (37.85)A,B,C | 0/8 | ND |
| LYC-PCL NC (2) | 5/8 | 7.1 ± 2.7 (96.3)A,C | 8/8 | 3.3 ± 1.3 (98.3)A,C,D | 4/8 | 4.0 ± 1.7 (96.9)A | 6/8 | 5.3 ± 1.4 (94.9)A,D |
| LYC-PLA-PEG NC (2) | ND | ND | 8/8 | 1.5 ± 0.2 (99.3)A | ND | ND | 8/8 | 0.6 ± 0.1 (99.7)A |
Female Swiss albino mice (20 to 25 g) were infected with 104 blood trypomastigotes/ml.
Treatment schemes are shown in more detail in Table 1.
Subpatent PAR/total, number of mice with a negative fresh blood examination during the acute phase of the infection divided by the total number of infected animals; MPP, maximum peak of parasitemia; ND, not determined.
Untreated, in this table the untreated group represents all the control groups, including untreated, unloaded PCL NC, unloaded PLA-PEG, and control solution.
The percent parasitemia reduction was based on untreated controls because there were no significant differences between the control groups (untreated, unloaded NC, and control solution). P values of <0.05 indicated significant differences and are denoted by superscript capital letters as follows: A, different from controls; B, different from LYC-PCL NC; C, different from BZ; and D, different from LYC-PLA-PEG).
The percent animal survival for infected and treated groups is shown in Table 3. The untreated control group showed a mean survival time of 20 days. In groups treated with LYC-PCL NC and BZ, 100% survival was observed until 6 months posttreatment, when mice were necropsied. In animals treated with free LYC, the survival was 75% at the same period, and the other two mice (25%) survived 24 days. No significant difference (P > 0.05) in survival was observed between groups treated with the formulations of LYC-PCL NC, free LYC and BZ. Taking into consideration that the results of all control groups (untreated, unloaded NC, and excipients of i.v. solution) were similar (P > 0.05), Tables 2 and 3 show only the results of the untreated group.
TABLE 3.
Efficacy of lychnopholide in mice experimentally infected with T. cruzi and treated with lychnopholide i.v. at 2.0 mg/kg/day using different schemes and formulations during the acute phase of the infection
| Expt (strain)a | Expt groupb | Parasitological cure (%)c | Survival (%) |
|---|---|---|---|
| I (CL strain) | Controls | 0 | 0 |
| LYC-PCL NC | 50 | 100 | |
| Free LYC | 0 | 75 | |
| BZ (50 mg/kg, i.v.) | 100 | 100 | |
| II (CL strain) | Controls | 0 | 0 |
| LYC-PCL NC | 100 | 100 | |
| LYC-PLA-PEG NC | 100 | 100 | |
| Free LYC | 0 | 50 | |
| BZ (50 mg/kg, i.v.) | 100 | 100 | |
| III (Y strain) | Controls | 0 | 0 |
| LYC-PCL NC | 50 | 100 | |
| Free LYC | 0 | 50 | |
| BZ (50 mg/kg, i.v.) | 0 | 100 | |
| IV (Y strain) | Controls | 0 | 0 |
| LYC-PCL NC | 62.5 | 100 | |
| LYC-PLA-PEG NC | 100 | 100 | |
| BZ (50 mg/kg, i.v.) | 75 | 100 |
For more detail about the treatment schemes, see Table 1.
Controls refers to untreated mice, unloaded NC, and control solution.
Defined as a negative HC, PCR, or ELISA result.
The animal treated as described in the experiment II schedule (started at the patent period [the seventh day] and continued for 20 days) (Fig. 2B) with LYC-PCL NC, LYC-PLA-PEG NC, or BZ showed similar parasitemias (P > 0.05). Parasitemia decreased in the three groups of mice and became subpatent during and after treatment in 100% of these animals (Table 2). Treatment with free LYC reduced parasitemia, but the parasitemia was higher and significantly different compared to animals treated with LYC loaded in NC (PCL and PLA-PEG) or even compared to the BZ-treated group (Table 2). Control groups always showed higher patent parasitemia compared to all treated groups. Significant reductions in MPP (P < 0.05) of 99.2% for BZ, 57.7% for free LYC, 98.2% for LYC-PCL NC, and 99.30% for LYC-PLA-PEG NC were observed compared to the untreated control group (Table 2).
Animal survival is illustrated in Table 3. The untreated control groups showed a mean survival time of 27 days. All animals (100%) treated with the formulations LYC-NC (PCL and PLA-PEG) or BZ survived the acute phase and showed similar survival rates until be necropsied 6 months after treatment. In animals treated with free LYC, 50% (4/8) of the animals survived up to 34 days posttreatment.
(ii) Animals infected with Y strain (partially resistant to BZ).
Animals treated, as described in experiment III schedule (started 24 h postinfection with Y strain for 10 days) with the formulation of LYC-PCL NC and BZ presented more intense and significant (P < 0.05) reduction of the parasitemia than control groups (Fig. 2C). No significant difference in the MPP (maximum peak of parasitemia) of mice treated with LYC-PCL NC and BZ. The parasitemia of animals treated with free LYC was higher than that of mice treated with LYC-PCL NC and BZ (P < 0.05) but was significantly lower than that observed in control groups.
Animal survival is shown in Table 3. The control group showed a mean survival time of 16 days, while 100% of the animals treated with BZ or LYC in NC formulations survived the acute phase up to necropsy at 6 months posttreatment. MPP was reduced in 94.3% by BZ, 37.8% by free LYC, and 96.9% by LYC-PCL NC relative to the untreated control group (Table 2). Animals treated as described in the experiment IV schedule (after patent parasitemia [4 days for 20 days]) with LYC PCL NC or LYC-PLA-PEG NC exhibited a reduction in parasitemia similar to that observed in animals treated with BZ (Fig. 2D).
All animals (100%) treated with LYC-PLA-PEG NC, 75% of the animals treated with LY-PCL NC, and 87.5% of the animals treated with BZ displayed subpatent parasitemia (Table 2). Compared to the untreated control group, MPP was reduced in 99.55, 95.76, and 98.20% in mice treated with LYC-PCL NC, LYC-PLA-PEG NC, or BZ, respectively (Table 2). Free LYC was not tested in this experiment because it did not improve the survival of mice and was not effective against the Y strain compared to BZ in experiment III. The animal survival in experiment IV is shown in Table 3. The control group showed a mean survival time of 16 days, while 100% of the animals treated with NC or BZ formulations survived the acute phase up to necropsy at 6 months after treatment.
Treatment efficacy. (i) Animals infected with CL strain (susceptible to BZ).
In experiment I (Table 3), the parasitological cure assessed by HC, PCR, and ELISA revealed that mice treated with either LYC-PCL NC or BZ formulations displayed cure rates of 50% (4/8) and 100% (8/8), respectively, whereas animals treated with free LYC were not cured. In experiment II (Table 3) mice treated with LYC-PLA-PEG NC, LYC-PCL NC, or BZ displayed 100% cure, whereas no parasitological cure was observed in animals treated with free LYC.
(ii) Animals infected with Y strain (partially resistant to BZ).
In experiment III (Table 3), animals treated with LYC-PCL NC showed 50% (4/8) parasitological cure. No cure was observed in mice treated with BZ or free LYC. In experiment IV (Table 3), mice treated with LYC-PLA-PEG NC or LY-PCL NC showed parasitological cures of 100% (8/8) and 62.5% (5/8), respectively. Animals treated with BZ showed a 75% percent cure, and no cure was observed in any control group for all experiments.
DISCUSSION
There is an urgent need for the development of new compounds or new strategies to increase the effectiveness and reduce the toxicity of chemotherapy against Chagas disease (6). Developing such strategies was the main purpose of the present study, in which we investigated the efficacy of a sesquiterpenic lactone named lychnopholide of natural origin isolated from Lychnophora trichocarpha.
The tests for evaluating the therapeutic efficacy of LYC in mice infected with strains of T. cruzi were motivated by the in vitro results reported by Chiari et al. (34) and Oliveira et al. (14). Oliveira et al. conducted an in vitro study wherein the lactone lychnopholide, isolated from L. trichocarpha, inhibited 50% of the growth of blood trypomastigotes of CL and Y strains at a concentration of 150 μg/ml (0.42 μM) (14). In this first work reporting lychnopholide activity, the MIC of LYC was overestimated, because the 100% inhibitory concentration (IC100) could not be precisely determined due to the LYC low solubility in the test conditions against bloodstream trypomastigotes (14). The IC50 was then approximately determined to be 150 μg/ml for the Y and CL strains, using a parasite density of 2 × 106 parasites/ml, even if LYC precipitation in test media had been detected. Considering the parasitemia curves of untreated animals infected with CL and Y strains (Fig. 2), we determined that LYC was able to reduce parasitemia to very low levels just after the first dose, even at LYC concentrations estimated to be approximately 25 μg/ml (2 mg/kg) in the total blood of mice immediately after i.v. administration, assuming the blood volume of mice to be 78 ml/kg (35). Table 2 shows that in vivo the maximum peaks of parasitemia (untreated animals) are around 2 million and 1.6 million trypomastigotes/ml of blood for the CL and Y strains, respectively, similar to the findings of the parasite density studies of Oliveira et al. (14). In fact, blood parasite exposure to LYC in vivo at these daily concentrations was enough to reduce parasitemia by 56 to 99% and by 37 to 99% in CL and Y strains, respectively, depending on the type of LYC formulation used and dosage schedule. If we take into consideration the complexity of in vivo animal model with high efficacy at 25 μg/ml, the in vitro MIC found in previous studies was probably overestimated. On the other hand, an increasing in dose could provide better efficacy results in CL and Y strains by increasing LYC blood exposure. Blood and tissue exposures were not identified here, as a limitation of the present work. However, pharmacokinetic and biodistribution experiments are being performed by our group to clarify this point. In the present study, highly stringent in vivo protocols were used to perform the LYC efficacy studies of two parasite strains with different sensitivities to BZ and NF according to methods of Filardi and Brener (8).
In the recent years, the use of nanocarriers loaded with substances of vegetal origin has attracted much attention because of their several advantages (36). The association of LYC with the polymeric colloidal carrier NC improved the water dispersion of this poorly soluble substance and allowed its safe i.v. administration. NC provided also a modified LYC release profile (13, 19). LYC NC formulations were prepared by interfacial deposition of preformed polymers, followed by solvent displacement (13, 19), as first reported by Fessi et al. (21). This method is notably simple and provides homogeneous dispersion of colloidal particles containing lipophilic drugs suitable for parenteral administration (37). Compared to the parasitic disease leishmaniasis, only a small number of studies have reported the treatment of T. cruzi experimental infection using nanocarriers (38–41).
Under our experimental conditions, all animals survived the treatment without signs of general toxicity related to LYC administration. Transitory toxicity with the BZ i.v. formulation was observed at the injection site in mice, as observed in humans with the antimalarial drug halofantrine i.v. solution in the same excipients (42). In contrast, no signs of toxicity or abnormal behavior were observed when free LYC or LYC-NC (LYC-PCL NC or LYC-PLA-PEG NC) was injected into mice. Furthermore, these NC preparations were stable for 6 months at 4°C.
Surprisingly, BZ in solution at a dose of 50 mg/kg/day cured 75% of the mice, more than when administered by oral route at a dose of 100 mg/kg/day where the index of cure is 50%. This different result could be attributed to the difference in the administration schedule, which could not be compared in different doses and administration routes. The reason for this difference in efficacy, observed with a nitroheterocyclic compound (BZ) in our experiments, was not explained and has not already been determined in other circumstances (7). However, these results may be related to unfavorable pharmacokinetic properties of BZ, such as a relatively short half-life and limited tissue penetration (7, 11, 20) in the conventional dosage form.
Analysis and comparison of the results of treated animals infected with the CL strain (experiment I) showed a reduction in parasitemia and an increase in animal survival among animals treated with free LYC or LYC-PCL NC compared to control groups. These effects, however, were not better than those obtained with BZ. A parasitological cure of 50% in animals treated with LYC-PCL NC and of 100% in animals treated with BZ were obtained. The better efficacy of BZ in this experiment may be attributed to the high sensitivity (100%) of the CL strain to this drug (8) and to the short 10-day period of treatment compared to 20 days normally used in experimentally infected mice treated with BZ and NF. In this dosage schedule, blood exposure to LYC was probably not enough to eliminate blood and tissue parasites (pseudocysts), even in a prolonged drug release NC formulation.
On the other hand, in experiment II LYC-PCL NC, LYC-PLA-PEG NC, and BZ showed 100% subpatent parasitemia, full survival, and complete parasitological cure, whereas animals treated with free LYC were not cured, when the treatment started at day 7 after infection (prepatent period) and continued for 20 days. It is important to highlight that in this case better therapeutic effects were observed, as measured by the higher index of parasitological cure and survival compared to the treatment 24 h after inoculation. These results are in agreement with the findings of Urbina et al. (43) and may indicate that the etiological treatment is more effective, particularly at the moment that a higher number of blood trypomastigotes are exposed to drug, and immediately after the rupture of higher number of pseudocysts (in the prepatent period), besides a longer period of treatment (20 days). Under these conditions, drug would be more effective than in the beginning of infection (24 h after inoculation) when the number of pseudocysts is lower and the majority of the parasites are still under amastigote forms within host cells (44), as in the case of experiment I. These same effects were observed in experiments conducted with the Y strain, which is partially resistant to BZ. The efficacy of LYC NC compared to free LYC, particularly when loaded in sterically stabilized PLA-PEG NC, may be attributed to the ability of the nanocarriers to maintain the LYC release for longer times in biological media compared to free LYC, as we recently reported (13, 19). LYC diffusion from polymeric NC to biological media was reduced in rate by 20-fold compared to apparent LYC dissolution in the same media. The nanocapsule property of controlling LYC release in vitro could be the main factor responsible for LYC's greater parasitological effects in vivo, probably by improving the time of body exposure to the drug. Consequently, the results of the efficacy of LYC on experimental CD are probably related to the LYC association with these nanometrical carriers (13).
In experiment III, mice were infected with the BZ partially resistant Y strain and treated 24 h after infection for 10 days. The number of animals with subpatent parasitemia was higher when treated with LYC-PCL NC (50%) relative to BZ (0%) and free LYC (0%). Parasitological cure was obtained only with LYC-PCL NC (50%). One of the possible reasons for the therapeutic failure of free LYC in experiments I and III may be due to the highly stringent protocols used, in which treatment periods of 10 days instead of 20 days were used (8, 29).
In experiment IV, mice were infected with the Y strain but underwent treatment beginning at the fourth day after infection for 20 days. All animals (100%) treated with LYC-PLA-PEG NC formulation showed subpatent parasitemia and showed the highest reduction in MPP compared to the CL strain (experiment II). Again, an evident improvement of treatment efficacy (100%) and parasitological cure was observed with LYC-PLA-PEG NC compared to BZ (75%) and LYC-PCL NC (62.5%). Free LYC was not used for treatment in this experiment because previous experiments showed that the therapeutic efficacy of NC formulations was always better.
In all experiments, animals treated with free LYC showed higher peaks of parasitemia, a significant number of animals showed patent parasitemia, and 50% did not survive. None of the animals receiving free LYC exhibited parasitological cure compared to animals treated with LYC loaded in NC formulations.
Taken together, these results clearly demonstrated that LYC-NC showed therapeutic effects leading to the reduction of parasitemia, the improvement of animal survival in experimental T. cruzi infections, and higher rates of cure in mice infected with a T. cruzi strain partially resistant to BZ and NF (8), even when used in very low doses (2.0 mg/kg/day) compared to BZ (50 mg/kg/day) i.v. The mechanism of sesquiterpene lactones action against T. cruzi, including LYC, is still unknown. However, the presence of alkylant groups in the LYC molecule could be responsible for these activities (14, 16, 17, 18).
In general, LYC-PLA-PEG NC (experiments II and IV) showed higher efficacy than LYC-PCL NC, except in animals infected with sensitive T. cruzi strain as CL. Both types of NC were important to maintain LYC efficacy in sensitive and in partially resistant strains. However, PLA-PEG NC showed greater efficacy. This result confirms the previously reported long-circulating properties of PLA-PEG NC (24). PLA-PEG NC have already improved the antiprotozoal efficacy of halofantrine by increasing the blood circulation times of this drug (37). Halofantrine is also a very lipophilic molecule, and PLA-PEG NC was able to modify its efficacy in experimental model of malaria (37). The effect of LYC was probably improved by the NC long circulating property in blood, resulting in pharmacokinetic profile modification. It may facilitate the contact of the active substance with the target parasite. In LYC-PLA-PEG NC formulations, the NC surface is modified by PEG. PEG chains retard the rapid removal of NC from the bloodstream by macrophages, consequently prolonging the drug-associated plasma half-life (24, 25, 37). Thus, these nanoparticles have the chance to extravasate to tissues infected by T. cruzi, particularly at inflammatory sites (26). LYC pharmacokinetics are under investigation by our group.
Our results showed that treatment with LYC-NC was able to produce higher levels of parasitological cure in infected mice. This result was particularly striking in studies with a partially resistant T. cruzi strain (Y) during the acute phase of experimental Chagas infection. The results for the efficacy of LYC in the present study are better than the current investigational treatments with several ergosterol biosynthesis inhibitors, particularly the inhibitors of C14-α-demethylase (CYP51), such as posaconazole and ravuconazole, under similar experimental conditions. The ergosterol biosynthesis inhibitor drugs are currently the most promising alternative drugs studied in mice (43–46) and dogs (47, 48). However, they are extremely expensive for the poor population normally affected by CD in Latin American countries (49). It is also important to note that a potent drug may completely lose its efficacy when inappropriately delivered or even when evaluated in an inadequate treatment scheme. Thus, one of the main contributions of the present study is that LYC represents an important alternative for treatment of CD that should be further evaluated in a BZ-resistant strain. At dose of 2.0 mg/kg/day, LYC associated with NC is a potent, fast-acting drug that is useful in short treatment regimens.
Finally, we demonstrated here for the first time the in vivo efficacy of LYC against T. cruzi infection, as well as the important contribution of nanotechnology to the improvement of LYC therapeutic effects for treating Chagas disease. Our results suggest that the use of an isolated substance of vegetal origin encapsulated in polymeric NC formulations may offer promising possibilities for the treatment of experimental CD in mice.
ACKNOWLEDGMENTS
We thank FAPEMIG (processes APQ00566-11, APQ00455/12 and REDES NANOBIOMG and TOXIFAR), as well as CNPq and CAPES financial support. CNPq researcher fellowships to V.C.F.M. and M.D.L. are also acknowledged.
Footnotes
Published ahead of print 21 January 2014
REFERENCES
- 1.World Health Organization/Special Programme for Research and Training in Tropical Diseases. 2012. Research priorities for Chagas disease, human African trypanosomiasis, and leishmaniasis. World Health Organization, Geneva, Switzerland: [PubMed] [Google Scholar]
- 2.Coura JR, Borges-Pereira J. 2010. Chagas disease: 100 years after its discovery: a systemic review. Acta Trop. 115:5–13. 10.1016/j.actatropica.2010.03.008 [DOI] [PubMed] [Google Scholar]
- 3.Leslie M. 2011. Infectious diseases: a tropical disease hits the road. Science 333:934. 10.1126/science.333.6045.934 [DOI] [PubMed] [Google Scholar]
- 4.Buckner FS. 2011. Experimental chemotherapy and approaches to drug discovery for Trypanosoma cruzi infection. Adv. Parasitol. 75:89–119. 10.1016/B978-0-12-385863-4.00005-8 [DOI] [PubMed] [Google Scholar]
- 5.Rassi A, Jr, Rassi A, Marin-Neto JA. 2010. Chagas disease. Lancet 375:1388–1402. 10.1016/S0140-6736(10)60061-X [DOI] [PubMed] [Google Scholar]
- 6.Coura JR, Castro SL. 2002. A critical review on Chagas disease chemotherapy. Mem. Inst. Oswaldo Cruz 97:3–24. 10.1590/S0074-02762002000900001 [DOI] [PubMed] [Google Scholar]
- 7.Urbina JA. 2010. Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Trop. 115:55–68. 10.1016/j.actatropica.2009.10.023 [DOI] [PubMed] [Google Scholar]
- 8.Filardi LS, Brener Z. 1984. A rapid method for testing in vivo the susceptibility of different strains of Trypanosoma cruzi to active chemotherapeutic agents. Mem. Inst. Oswaldo Cruz 79:221–225. 10.1590/S0074-02761984000200008 [DOI] [PubMed] [Google Scholar]
- 9.Murta SMF, Gazzinelli RT, Brener Z, Romanha AJ. 1998. Molecular characterization of susceptible and naturally resistant strains of Trypanosoma cruzi to benznidazole and nifurtimox. Mol. Biochem. Parasitol. 93:203–214. 10.1016/S0166-6851(98)00037-1 [DOI] [PubMed] [Google Scholar]
- 10.Prokop A, Davidson JM. 2008. Nanovehicular intracellular delivery systems. J. Pharm. Sci. 97:3518–3590. 10.1002/jps.21270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urbina JA, Docampo R. 2003. Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasitol. 19:495–501. 10.1016/j.pt.2003.09.001 [DOI] [PubMed] [Google Scholar]
- 12.Saúde DA, Raslan DS, De Souza Filho JD. 1998. Constituents from the aerial parts of Lychnophora trichocarpha. Fitoterapia 69:90–91 [Google Scholar]
- 13.Branquinho RT, Mosqueira VC, Kano EK, de Souza J, Dorim DD, Saúde-Guimarães DA, Lana M. 2012. HPLC-DAD and UV-spectrophotometry for the determination of lychnopholide in nanocapsule dosage form: validation and application to release kinetic study. J. Chromatogr. Sci. 10.1093/chromsci/bms199 [DOI] [PubMed] [Google Scholar]
- 14.Oliveira AB, Saúde DA, Perry KSP, Duarte DS, Raslan DS, Boaventura MAD, Chiari E. 1996. Trypanocidal sesquiterpenes from Lychnophora species. Phytother. Res. 10:292–295. [DOI] [Google Scholar]
- 15.Saúde DA, Barrero AF, Oltra JE, Jusícia J, Raslan DS, Silva EA. 2002. Atividade antibacteriana de furanoeliangolidos. Rev. Bras. Farmacogn. 12:7–10 [Google Scholar]
- 16.Ferrari FC, Ferreira LC, Souza MR, Grabe-Guimarães A, Paula CA, Rezende SA, Saúde-Guimarães DA. 2012. Anti-inflammatory sesquiterpene lactones from Lychnophora trichocarpha Spreng (Brazilian Arnica). Phytother. Res. 10.1002/ptr.4736 [DOI] [PubMed] [Google Scholar]
- 17.Souza MR, De Paula CA, Pereira de Resende ML, Grabe-Guimarães A, Souza Filho JD, Saúde-Guimarães DA. 2012. Pharmacological basis for use of Lychnophora trichocarpha in gouty arthritis: anti-hyperuricemic and anti-inflammatory effects of its extract, fraction and constituents. J. Ethnopharmacol. 142:845–850. 10.1016/j.jep.2012.06.012 [DOI] [PubMed] [Google Scholar]
- 18.Canalle R, Burim RV, Callegari Lopes JL, Takahashi CS. 2001. Assessment of the cytotoxic and clastogenic activities of the sesquiterpene lactone lychnopholide in mammalian cells in vitro and in vivo. Cancer Detect. Prev. 25:93–101 [PubMed] [Google Scholar]
- 19.Branquinho RT, Mosqueira VC, Saúde-Guimarães DA, Lana M. 2012. Composições farmacêuticas contendo lactonas sesquiterpênicas da classe dos furanoeliangolidos para tratamento de infecções parasitárias e de tumores. Document PCTBR/2012/00039 World Intellectual Property Organization, Geneva, Switzerland [Google Scholar]
- 20.Romero EL, Morilla MJ. 2010. Nanotechnological approaches against Chagas disease. Adv. Drug Deliv. Rev. 62:576–588. 10.1016/j.addr.2009.11.025 [DOI] [PubMed] [Google Scholar]
- 21.Fessi H, Piusieux F, Devissaguet JP, Ammoury N, Benita S. 1989. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 55:R1–R4. 10.1016/0378-5173(89)90281-0 [DOI] [Google Scholar]
- 22.Losa C, Marchal-Heussler L, Orallo F, Vila Jato JL, Alonso MJ. 1993. Design of new formulations for topical ocular administration: polymeric nanocapsules containing metipranolol. Pharm. Res. 10:80–87. 10.1023/A:1018977130559 [DOI] [PubMed] [Google Scholar]
- 23.Leite EA, Grabe-Guimarães A, Guimarães HN, Machado-Coelho GL, Barratt G, Mosqueira VCF. 2007. Cardiotoxicity reduction induced by halofantrine entrapped in nanocapsule devices. Life Sci. 80:1327–1334. 10.1016/j.lfs.2006.12.019 [DOI] [PubMed] [Google Scholar]
- 24.Mosqueira VCF, Legrand P, Morgat JL, Vert M, Mysiakine E, Gref R, Devissaguet JP, Barratt G. 2001. Biodistribution of long-circulating PEG-grafted nanocapsules in mice: effects of PEG chain length and density. Pharm. Res. 18:1411–1419. 10.1023/A:1012248721523 [DOI] [PubMed] [Google Scholar]
- 25.Mosqueira VCF, Legrand P, Gulik A, Bourdon O, Gref R, Labarre D, Barratt G. 2001. Relationship between complement activation, cellular uptake and surface physicochemical aspects of novel PEG-modified nanocapsules. Biomaterials 22:2967–2979. 10.1016/S0142-9612(01)00043-6 [DOI] [PubMed] [Google Scholar]
- 26.Pereira MA, Mosqueira VCF, Carmo VA, Ferrari CS, Reis EC, Ramaldes GA, Cardoso VN. 2009. Biodistribution study and identification of inflammatory sites using nanocapsules labeled with (99m) Tc-HMPAO. Nucleic Med. Commun. 30:749–755. 10.1097/MNM.0b013e32832f2b59 [DOI] [PubMed] [Google Scholar]
- 27.Agência Nacional de Vigilância Sanitária. 2010. Farmacopeia Brasileira, 5th ed. Brasília 2:131–133 [Google Scholar]
- 28.Camargo EP. 1964. Growth and differentiation in Trypanosoma cruzi. I. Origin of metacyclic trypanosomes in liquid media. Rev. Inst. Med. Trop. São Paulo. 12:93–100 [PubMed] [Google Scholar]
- 29.Brener Z. 1962. Therapeutic activity and criterion of cure on mice experimentally infected with Trypanosoma cruzi. Rev. Inst. Med. Trop. São Paulo. 4:389–396 [PubMed] [Google Scholar]
- 30.Gomes ML, Macedo AM, Vago AR, Pena SDJ, Galvão LMC, Chiari E. 1998. Trypanosoma cruzi: optimization of polymerase chain reaction for detection in human blood. Exp. Parasitol. 88:28–33. 10.1006/expr.1998.4191 [DOI] [PubMed] [Google Scholar]
- 31.Avila HA, Sigman DS, Cohen LM, Millikan RC, Simpson L. 1991. Polymerase chain reaction amplification of Trypanosoma cruzi kinetoplast minicircle DNA isolated from whole blood lysates: diagnosis of chronic Chagas' disease. Mol. Biochem. Parasitol. 48:211–221. 10.1016/0166-6851(91)90116-N [DOI] [PubMed] [Google Scholar]
- 32.Santos FR, Pena SDJ, Epplen JT. 1993. Genetic and population study of a Y-linked tetranucleotide repeat DNA polymorphism with a simple non-isotopic technique. Hum. Genet. 90:655–656 [DOI] [PubMed] [Google Scholar]
- 33.Voller A, Bidwell DE, Bartlett A. 1976. Enzyme immunoassays in diagnostic medicine: theory and practice. Bull. World Health Organ. 53:55–65 [PMC free article] [PubMed] [Google Scholar]
- 34.Chiari E, de Oliveira AB, Raslan DS, Mesquita AAL, Tavares K. 1991. Screening in vitro of natural products against blood forms of Trypanosoma cruzi. Trans. R. Soc. Trop. Med. Hyg. 85:372–374. 10.1016/0035-9203(91)90296-B [DOI] [PubMed] [Google Scholar]
- 35.Jacoby RO, Fox JG. 1984. Biology and diseases of mice, p 31–89 In Fox JG, Cohen BJ. (ed), Laboratory animal medicine. Academic Press, Inc, New York, NY [Google Scholar]
- 36.Saraf AS. 2010. Applications of novel drug delivery system for herbal formulations. Fitoterapia 81:680–689. 10.1016/j.fitote.2010.05.001 [DOI] [PubMed] [Google Scholar]
- 37.Krishna S, ter Kuile F, Supanaranond W, Pukrittayakamee S, Teja-Isavadharm P, Kyle D, White NJ. 1993. Pharmacokinetics, efficacy and toxicity of parenteral halofantrine in uncomplicated malaria. Br. J. Clin. Pharmacol. 36:585–591. 10.1111/j.1365-2125.1993.tb00419.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molina J, Brener Z, Romanha AJ, Urbina JA. 2000. In vivo activity of the bis-triazole D0870 against drug-susceptible and drug-resistant strains of the protozoan parasite Trypanosoma cruzi. J. Antimicrob. Chemother. 46:137–140. 10.1093/jac/46.1.137 [DOI] [PubMed] [Google Scholar]
- 39.González-Martín G, Figueroa C, Merino I, Osuna A. 2000. Allopurinol encapsulated in polycyanoacrylate nanoparticles as potential lysosomatropic carrier: preparation and trypanocidal activity. Eur. J. Pharm. Biopharm. 49:137–142. 10.1016/S0939-6411(99)00076-4 [DOI] [PubMed] [Google Scholar]
- 40.González-Martín G, Merino I, Rodriguez-Cabezas MN, Torres M, Nuñez R, Osuna A. 1998. Characterization and trypanocidal activity of nifurtimox-containing and empty nanoparticles of polyethylcyanoacrylates. J. Pharm. Pharmacol. 50:29–35. 10.1111/j.2042-7158.1998.tb02229.x [DOI] [PubMed] [Google Scholar]
- 41.Sãnchez G, Cuellar D, Zulantay I, Gajardo M, González-Martin G. 2002. Cytotoxicity and trypanocidal activity of nifurtimox encapsulated in ethylcyanoacrylate nanoparticles. Biol. Res. 35:39–45 [DOI] [PubMed] [Google Scholar]
- 42.Mosqueira VCF, Loiseau P, Bories C, Legrand P, Devissaguet JP, Barratt G. 2004. Efficacy and pharmacokinetics of intravenous nanocapsule formulation of halofantrine in Plasmodium berghei-infected mice. Antimicrob. Agents Chemother. 48:1222–1228. 10.1128/AAC.48.4.1222-1228.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Urbina JA, Payares G, Sanoja C, Lira R, Romanha AJ. 2003. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Int. J. Antimicrob. Agents 21:27–38. 10.1016/S0924-8579(02)00273-X [DOI] [PubMed] [Google Scholar]
- 44.Espuelas S, Plano D, Nguewa P, Font M, Palop JA, Irache JM, Sanmartín C. 2012. Innovative lead compounds and formulation strategies as newer kinetoplastid therapies. Curr. Med. Chem. 19:4259–4288. 10.2174/092986712802884222 [DOI] [PubMed] [Google Scholar]
- 45.Molina J, Martins-Filho O, Brener Z, Romanha AJ, Loebenberg D, Urbina JA. 2000. Activities of the triazole derivative SCH 56592 (posaconazole) against drug-resistant strains of the protozoan parasite Trypanosoma (Schizotrypanum) cruzi in immunocompetent and immunosuppressed murine hosts. Antimicrob. Agents Chemother. 44:150–155. 10.1128/AAC.44.1.150-155.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bahia MT, Andrade IM, Martins TAF, Nascimento Á, Diniz FSLF, Caldas IS, Talvani A, Trunz BB, Torreele E, Ribeiro I. 2012. Fexinidazole: a potential new drug candidate for Chagas disease. PLoS Negl. Trop. Dis. 6:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guedes PM, Urbina JA, Lana M, Afonso LC, Veloso VM, Tafuri WL, Machado-Coelho GL, Chiari E, Bahia MT. 2004. Activity of the new triazole derivative albaconazole against Trypanosoma (Schizotrypanum) cruzi in dog hosts. Antimicrob. Agents Chemother. 48:4286–4292. 10.1128/AAC.48.11.4286-4292.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diniz LDF, Caldas IS, Guedes PMDM, Crepalde G, Lana M, Carneiro CM, Talvani A, Urbina JA, Bahia MT. 2010. Effects of ravuconazole treatment on parasite load and immune response in dogs experimentally infected with Trypanosoma cruzi. Antimicrob. Agents Chemother. 54:2979–2986. 10.1128/AAC.01742-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clayton J. 2010. Chagas disease: pushing through the pipeline. Nature 465:S12–S15. 10.1038/nature09224 [DOI] [PubMed] [Google Scholar]