

Abstract
Mitochondrial morphology is regulated by the balance between two counteracting mitochondrial processes of fusion and fission. There is significant evidence suggesting a stringent association between morphology and bioenergetics of mitochondria. Morphological alterations in mitochondria are linked to several pathological disorders, including cardiovascular diseases. The consequences of stress-induced acetylation of mitochondrial proteins on the organelle morphology remain largely unexplored. Here we report that OPA1, a mitochondrial fusion protein, was hyperacetylated in hearts under pathological stress and this posttranslational modification reduced the GTPase activity of the protein. The mitochondrial deacetylase SIRT3 was capable of deacetylating OPA1 and elevating its GTPase activity. Mass spectrometry and mutagenesis analyses indicated that in SIRT3-deficient cells OPA1 was acetylated at lysine 926 and 931 residues. Overexpression of a deacetylation-mimetic version of OPA1 recovered the mitochondrial functions of OPA1-null cells, thus demonstrating the functional significance of K926/931 acetylation in regulating OPA1 activity. Moreover, SIRT3-dependent activation of OPA1 contributed to the preservation of mitochondrial networking and protection of cardiomyocytes from doxorubicin-mediated cell death. In summary, these data indicated that SIRT3 promotes mitochondrial function not only by regulating activity of metabolic enzymes, as previously reported, but also by regulating mitochondrial dynamics by targeting OPA1.
INTRODUCTION
Sirtuins are class III histone deacetylases (HDACs), which need NAD+ for their catalytic activity. They are emerging as key regulators of diverse biological processes ranging from cellular growth to longevity. Because of their dependency on NAD+, they are highly sensitive to the metabolic state of the cell. The mammalian genome encodes seven sirtuin isoforms (SIRT1 through SIRT7), which are localized to different subcellular compartments and regulate a multitude of cellular functions through posttranslational modification (PTM) of target proteins. Three mammalian sirtuins, SIRT3, 4, and 5, are localized to mitochondria (1, 2). Of these, SIRT3 plays a major role in deacetylating and modifying the enzymatic activities of several mitochondrial proteins (1, 3). Biochemical and genetic studies have demonstrated that SIRT3 protects the organism from metabolic stress, tumorigenesis, development of cardiac hypertrophy, and aging-associated hearing loss by reducing the synthesis of mitochondrial reactive oxygen species (ROS) (4–6). Earlier studies have also shown that SIRT3 expression is reduced in diabetic patients (7) and high SIRT3 expression levels are associated with longevity in humans (8, 9).
Though a role for SIRT3 in regulating mitochondrial metabolic functions is well documented, whether it can also regulate the morphology of mitochondria is not explored. Mitochondria are highly dynamic organelles that continually undergo fusion and fission to maintain the quality of the mitochondrial population (10). The equilibrium between these two antagonistic processes determines mitochondrial morphology. In cardiac cells, mitochondria form a highly interconnected tubular network. The optimal balance between mitochondrial fusion and fission regulates key mitochondrial functions. These include maintaining the electrochemical gradient necessary for oxidative phosphorylation, preserving the integrity of mitochondrial DNA (mtDNA), and exhibiting an appropriate cellular response to apoptotic stimuli. Alterations in fusion-fission processes are observed under both physiologic and pathological conditions (11). The process of fusion rejuvenates mitochondria by mixing their contents and helps the cell to overcome metabolic and/or environmental stress. Fission helps to increase mitochondrial numbers and also alleviates the cellular load of damaged mitochondria (11, 12).
The mitochondrial fusion and fission process is mediated by an evolutionarily conserved family of dynamin-related GTPases (10). In mammals, mitochondrial outer membrane (OM) and inner membrane (IM) fusions are mediated through mitofusins (Mfn1 and 2) and optic atrophy 1 (OPA1) proteins, respectively (10, 13–15). Fission is facilitated via dynamin-related protein 1 (DRP1) and fission protein 1 (Fis1) (16, 17). Although the functional importance of mitochondrial dynamics is evident from numerous human diseases associated with defects in mitochondrial fusion-fission processes (11), the molecular mechanisms involved in the regulation of mitochondrial dynamics are not yet fully understood. Many posttranslational modifications such as phosphorylation, oxidation, sumoylation, and ubiquitination of fusion and fission proteins are reported to play an important role in regulating their activities and the mitochondrial dynamics (18). A major contributor to mitochondrial protein modifications is reversible lysine acetylation within a protein. A proteomic survey revealed acetylation of nearly 20% of mitochondrial proteins (19), and under stress, hyperacetylation of key metabolic enzymes of mitochondria was observed. This indicates that acetylation could be a major regulatory adaptation governing the function of this organelle. The mitochondrial deacetylase SIRT3, which regulates the activity of numerous mitochondrial enzymes, is portrayed as a mitochondrial fidelity protein (1). A role of SIRT3 in regulating mitochondrial biogenesis via activation of the PGC1α/ERRα complex has been demonstrated (20–22). However, its role in regulating mitochondrial dynamics has never been explored.
OPA1, the GTPase anchored to the IM of mitochondria, exists in multiple long and short isoforms and performs multiple functions. Besides being an obligatory protein for the IM fusion, OPA1 is also involved in maintaining crista structure and protecting cells from death stimuli (23, 24). Optimum function of OPA1 depends on its protein expression level and a coordinated balance between its long and short isoforms (25, 26). OPA heterozygous (OPA+/−) mice develop mitochondrial defects in the heart and late-onset cardiomyopathy (27, 28). Although OPA1 was identified as an acetylated protein by proteomic survey, a functional role of this modification was never investigated (29). In the present study, we report OPA1 as a direct target of SIRT3. OPA1 is hyperacetylated under stress conditions, and this modification reduces GTPase activity of OPA1. SIRT3 deacetylates and activates OPA1 and thereby modulates mitochondrial dynamics.
MATERIALS AND METHODS
Antibodies.
The antibodies and conjugates used in this study were anti-OPA1 (BD Biosciences, Santa Cruz), anti-SIRT3 (Cell Signaling, Santa Cruz), anti-Flag (Abcam, Stratagene), antiacetyllysine (Cell Signaling, Santa Cruz), anti-SIRT5 (Orbigen), anti-FlagM2-, antihemagglutinin (anti-HA)-agarose beads (Sigma), and anti-cMyc affinity gel (Clontech). Antibodies for Bax, Bim, FasL, cMyc, GAPDH (glyceraldehyde-3-phosphate dehydrogenase), P300/CBP-associated factor (PCAF), tubulin, and actin were purchased from Santa Cruz. All horseradish peroxidase [HRP]-conjugated secondary antibodies for Western blots were from Santa Cruz. Alexa Fluor 594-conjugated secondary antibodies were bought from Invitrogen, Inc.
Plasmids and adenovirus constructs.
Plasmid constructs Flag-SIRT3 and Flag-SIRT3-HY (SIRT3 catalytic mutant [MUT]) were kindly provided by Eric Verdin, University of San Francisco, CA. Flag-SIRT5 plasmid (plasmid 13816, deposited by Eric Verdin) and human His-SIRT3 plasmid (plasmid 13736, deposited by John Denu) were from Addgene. Human OPA1-myc plasmid was a gift from Daniel Linseman, University of Denver, Denver, CO. Adenovirus for mitochondrial-matrix-targeted green fluorescent protein (mtGFP) was kindly provided by Paul Schumacker from Northwestern University, Evanston, IL. Empty adenovirus (Ad.Vector) and human SIRT3 adenovirus were purchased from Vector BioLabs, Philadelphia, PA.
Animal studies.
All animal protocols were reviewed and approved by the University of Chicago Institutional Animal Care and Use Committee. SIRT3 knockout (SIRT3KO) mice were provided by F. W. Alt, Harvard Medical School, Boston, MA. Transgenic mice with cardiomyocyte-specific (α-myosin heavy chain [MHC] promoter) expression of C-terminal-HA-tagged mouse full-length SIRT3 were essentially generated as described earlier (6). Chronic infusion of angiotensin II (Ang) was done by implanting osmotic minipumps (model 2002; Alzet) in the peritoneal cavity of mice as described earlier (6). To induce pressure overload hypertrophy, transverse aortic constriction (TAC) of ascending aorta was carried out in mice as described elsewhere (30). Hearts from obese, diabetic (db/db, leptin receptor-deficient), and their wild-type (WT) control mice were procured from the laboratory of Christopher Rhodes, University of Chicago, Chicago, IL.
Cell culture, transfection, and adenovirus infection.
Immortalized embryonic fibroblasts (MEFs) from OPA1-WT and OPA1-null mice were generated as described elsewhere (31). Primary cultures of cardiomyocytes were made from ventricles of 2-day-old neonatal rat hearts as described earlier (32). Primary cultures for cardiac fibroblasts from adult (12-month-old) ventricular heart tissue and for embryonic fibroblasts from embryonic day 18 (E18) embryos of WT and SIRT3KO mice were generated using a standard protocol. Experiments were carried out between passages 2 and 4 when using primary fibroblast cultures. SIRT3 WT and KO immortalized MEFs were provided by Marcia Haigis, Harvard Medical School, Boston, MA (33). HeLa stable cells overexpressing either WT or HY mutant SIRT3 are described elsewhere (34). All cells were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 1% penicillin-streptomycin and 10% fetal bovine serum (complete growth medium). Cells were transfected with appropriate plasmids using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's protocol. For all the adenovirus experiments, viruses were used at a multiplicity of infection (MOI) of 100. For doxorubicin (Sigma) experiments, 24 h after plating, cells were treated for 24 h either with vehicle (0.9% saline) or with doxorubicin in serum-free medium. For the ROS inhibitor N-acetyl-l-cysteine (NAC) experiment, WT and SIRT3KO primary MEFs were serum starved for 24 h and then treated with either vehicle or 5 mM NAC for 16 h in serum-free medium, and before harvesting to immunoprecipitate OPA1, cells were treated for 1 h with 2 μM trichostatin A (TSA) (Sigma).
Electron microscopy for heart samples.
Freshly harvested heart tissues from mice were processed in the University of Chicago's electron microscopy core facility for transmission electron microscopy as per the standard protocol. Images were taken at 300 kV with an FEI Tecnai F30 electron microscope equipped with a high-performance Gatan charge-coupled-device (CCD) camera.
Site-directed mutagenesis and retroviral transduction.
Using the QuikChange lightning site-directed mutagenesis kit (Stratagene) and pMSCV-OPA1 retroviral plasmid (35) or human OPA1-myc plasmid as a template, all the K-to-R or -Q OPA1 mutant plasmids were generated. Retrovirus production from 293T cells and infection of immortalized OPA-null MEFs to generate retro-OPA (rOPA) stable MEF cell lines were done using standard protocols. Double mutant (dm) OPA1-myc plasmids were used for transient transfections.
Immunoblotting and immunoprecipitation analyses.
Cell or heart ventricular tissue lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.5], 0.1% Nonidet P-40, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 100 mM phenylmethylsulfonyl fluoride [PMSF], 5 mM sodium orthovanadate, 10 mM β-glycerol phosphate, and 20 mM NaF and Sigma protease inhibitors). Typically, 20 to 50 μg of protein lysates was used for immunoblots (IB). Immunoprecipitation (IP) experiments using 500 to 1,000 μg of total protein lysates were carried out using standard protocols.
Immunostaining.
Cells (10,000 to 20,000) plated on glass coverslips were used for immunostaining. Cells were infected with mito-GFP (mtGFP), SIRT3, or control (Ad.Vector) adenovirus (MOI, 100) as indicated for 24 to 48 h and then immunostained as described earlier (34). Confocal microscopy and imaging analyses were done in the digital light microscopy core facility of the University of Chicago.
Live-cell imaging to assess ΔΨm.
Confocal microscopy was performed with live cells plated on glass bottom dishes (MatTek, Ashland, MA). To monitor mitochondrial membrane potential (ΔΨm), tetramethyl rhodamine methyl ester (TMRM; Invitrogen), a ΔΨm-dependent cationic dye, was used for the adult WT and SIRT3KO cardiac fibroblasts isolated from heart ventricular tissue of 12-month-old mice. These cells were infected with mtGFP adenovirus at an MOI of 100 for 24 to 48 h prior to imaging, to visualize the total mitochondrial population. TMRM was used at a final concentration of 100 nM in complete growth medium (DMEM–10% fetal bovine serum [FBS]) without phenol red. TMRM-loaded live cells were maintained at 37°C, and images were acquired for 5 to 10 min with a Leica SP5 tandem scanner, a two-photon spectral confocal system with a 63× oil objective and 4× digital zoom. Emission windows were 501 to 542 nm and 544 to 683 nm for GFP and TMRM, respectively. Illumination was 3% for a 561-nm laser, using the resonant scanner and HyD detector, to avoid laser toxicity to cells. Cells from six or seven independent fields were imaged from four separate plates per genotype. To evaluate ΔΨm, the ratio of fluorescence intensity for TMRM to mtGFP (red/green) was estimated using Image J software for every time point. The staining ratio was used to assess ΔΨm stability during observations.
Recombinant OPA1-S1 protein purification and in vitro acetylation assay.
A short isoform of human OPA1 (splice form 1; termed here OPA1-S1) with an N-terminal 6-histidine (His) tag was expressed in Escherichia coli BL21(DE3)/pLysS bacteria (Novagen). OPA1-S1 expression and protein purification were essentially done as explained elsewhere (36). His.SIRT3 was also purified similarly. Purified His-OPA1-S1 on Ni-nitrilotriacetic acid (NTA) (His) beads (Qiagen) was acetylated by incubating with either 200 ng of active p300/CBP-associated factor (PCAF) (Millipore) or 2 μg PCAF (Cayman) enzyme, 1 mM acetyl coenzyme A (acetyl-CoA), 50 mM nicotinamide (NAM), and 50 μM TSA in 1× HAT buffer (50 mM Tris-HCl [pH 8.0], 10% glycerol, 0.1 mM EDTA, 1 mM dithiothreitol [DTT]) for 1 h at 30°C on a rotator. In the control reaction mixture, acetyl-CoA was omitted and 63 μM anacardic acid was included to inhibit residual PCAF activity. In vitro-acetylated His-OPA1-S1 was used for downstream applications.
In vitro deacetylation assay.
In vitro-acetylated His-OPA1-S1 bound to Ni-NTA beads was washed three times with 1× HAT buffer, twice with 1× HDAC buffer (50 mM Tris-HCl [pH 8.8], 50 mM NaCl, 1 mM MgCl2, 0.5 mM DTT, and 0.1 mg/ml of bovine serum albumin). Protein-bound beads in the HDAC buffer were incubated with Flag-SIRT3 or His-SIRT3 protein in the absence or presence of 1.5 mM NAD+ for the deacetylation assay. The reaction mixture was incubated for 2 h at 37°C on a rotator. Flag-SIRT3 protein used in these experiments was immunoprecipitated using Flag-tagged beads (Sigma) from overexpressing HEK293T cells.
In vitro OPA1 GTPase assay.
Ni-NTA beads with bound acetylated or deacetylated His-OPA1-S1 protein and their appropriate controls were washed multiple times with 1× OPA1 activity assay buffer (50 mM HEPES [pH 7.0], 1 mM DTT). GTPase activity of bead-bound OPA1 was estimated as described earlier (36) without addition of NaCl or liposomes. GTP was added to a final concentration of 100 μM. Concentration of free phosphate released in the GTPase reaction at different time points was determined using the Malachite green phosphate assay kit (Bioassay Systems) according to the manufacturer's instructions. GTPase activity of OPA1 immunoprecipitated from WT and SIRT3KO immortalized MEFs was similarly estimated.
OPA1-SIRT3 direct binding assay.
For the direct-interaction experiment, in vitro-translated [35S]methionine-labeled Flag-SIRT3 or Flag-SIRT5 (TNT coupled reticulocyte lysate kit; Promega) was incubated at 4°C for 2 h on a rotator with 5 μg of His-OPA1-S1 protein bound to Ni-NTA beads. As a control, labeled Flag proteins were incubated with Ni-NTA (His) beads alone. After five washes of NETN buffer (20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40), bound proteins were resolved on 10% SDS-PAGE and analyzed by autoradiography as well as immunoblotting.
ROS measurement by flow cytometry.
ROS detection was done using the CM-H2DCFDA dye (Invitrogen) according to the manufacturer's instructions. For ROS induction, cells were treated for 15 min with 200 μM H2O2 where indicated. Cells were acquired by FACScalibur, and results were analyzed using FlowJo software. Values for generated ROS were calculated from the mean fluorescence intensity (MFI) of cells positive for CM-H2DCFDA staining.
MS analysis for acetylated lysine residues.
Tryptic digests of the purified OPA1 protein immunoprecipitated either from SIRT3 knockout heart lysates or from other sources were analyzed by mass spectrometry (MS) as described elsewhere (37) at the proteomics core facility of the University of Chicago.
TUNEL assay.
Twenty-four hours after neonatal rat ventricular cardiomyocytes (NRVM) were plated onto laminin-coated glass coverslips, they were infected with either Ad.SIRT3 or vector alone at a MOI of 100. Twenty-four hours after infection, cells were treated either with 100 nM doxorubicin or with vehicle for the next 24 h in a serum-free medium. The terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) reaction was performed as per the manufacturer's instructions using the in situ cell death (TUNEL) detection kit from Roche Diagnostics. TUNEL-positive cells were identified by confocal microscopy at an excitation wavelength of 488 nm. ProLong gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen) was used for staining the nuclei (blue) and mounting the coverslips. Blue nuclei with a well-defined condensed morphology and TUNEL-positive green staining were defined as apoptotic cells. For quantitation, five independent fields with 50 to 100 nuclei each were counted for every image.
Copy number estimation for mitochondrial DNA.
Total (nuclear + mitochondrial) DNA was extracted from different OPA1 cell lines using the QIAamp DNA minikit (Qiagen). By real-time quantitative PCR (Absolute QPCR SYBR green mix; Thermo Scientific), the mtDNA copy number was determined relative to nuclear DNA. Primers used for the mitochondrial gene ND1 and the nuclear gene H19 were as follows: ND1 forward primer, 5′-AATCGCCATAGCCTTCCTAACAT-3′, and reverse primer, 5′-GGCGTCTGCAAATGGTTGTAA-3′; H19 forward primer, 5′-GTACCCACCTGTCGTCC-3′, and reverse primer, 5′-GTCCACGAGACCAATGA CTG-3′. For data analysis, the ΔΔCT (threshold cycle) method was employed. Data are presented as normalized values to mtDNA copy number from WT-OPA1 MEFs. Statistical significance was determined by comparing the normalized values to the mtDNA copy number from OPA1-null MEFs.
OCR measurement.
Baseline oxygen consumption rate (OCR) measurements were done as per the manufacturer's instruction for cells plated at 5 × 104 per well of XF24 Seahorse plates (Seahorse Bioscience, MA). OCR was normalized to the total protein amount per well.
MNF.
To quantify functional networking of the mitochondria, i.e., determine the mitochondrial networking factor (MNF), cells were doubly infected with mitochondrially targeted photoactivatable GFP (mito-PA-GFP) adenovirus (MOI, 100) (38) and mito-Ds-Red2 baculovirus (2 μl/10,000 cells) (39). Imaging experiments were performed 48 to 72 h following the infections as described elsewhere (40, 41).
Statistical analysis.
To compare statistical significance between two groups, a two-tailed paired t test was employed.
RESULTS
OPA1 is hyperacetylated during cardiac stress.
Defects in mitochondrial dynamics have been documented during development of cardiac hypertrophy (28). We found OPA1 to be hyperacetylated in mouse hearts that were subjected to develop cardiac hypertrophy by either angiotensin-II (Ang) infusion or transverse aortic constriction (TAC) (Fig. 1A and B). To determine whether OPA1 was also acetylated under pathological conditions in which cardiac dysfunction is secondary to metabolic stress, we analyzed OPA1 acetylation in hearts of diabetic (db/db) mice. As shown in Fig. 1C, OPA1 was hyperacetylated in hearts of db/db mice compared to their nondiabetic controls. Based on this correlation between pathological stress and OPA1 acetylation, we posited that this PTM could be regulating the enzymatic activity of OPA1. We therefore measured GTPase activity of OPA1 that was subjected to acetylation by the mammalian histone acetylase (HAT) PCAF. The acetylation status of OPA1 was confirmed by Western blotting and mass spectrometry (see Fig. 3B; see also Fig. S4 in the supplemental material). Acetylation consistently reduced GTPase activity of OPA1 by 30 to 40% (Fig. 1D). These results thus indicated that pathological stress induces acetylation of OPA1 and this modification reduces its GTPase activity.
FIG 1.

Pathological stress induces OPA1 acetylation, which reduces GTPase activity of the enzyme. Immunoblots (IB) showing hyperacetylation of immunoprecipitated (IP) OPA1 during hypertrophy of the heart, induced by angiotensin II (Ang) infusion for 30 days (A) or by transthoracic aortic constriction (TAC) for 6 months (B), and from hearts of 2-month-old db/db mice (C), compared to respective wild-type (WT) controls. Ac-K, antiacetyllysine antibody; WCL, whole-cell lysates. (D) Reduced GTPase activity of His-OPA1.S1 protein subjected to acetylation (Ac OPA) compared to that of nonacetylated control (CN OPA). GTP hydrolysis (100 μM) was monitored up to 3 h. Values are means ± standard errors of the means (SEM); n = 4; *, P < 0.001. (E) IB showing increased acetylation of OPA1 in SIRT3 knockout (SIRT3KO) hearts compared to WT controls. (F) Bar graph representing GTPase activity for OPA1 immunoprecipitated from immortalized SIRT3-WT and KO MEF lysates. Values are means ± SEM; n = 4; *, P < 0.05. (G) IB showing the amount of OPA1 protein used in the assay illustrated in panel F. IP:IgG is a negative control for IP:OPA1.
FIG 3.

SIRT3 regulates GTPase activity of OPA1 and mitochondrial fusion. (A) Bar diagram showing GTPase activity of acetylated OPA1, compared to SIRT3-mediated deacetylated OPA1, as assessed by an in vitro enzymatic assay. GTP hydrolysis by His-OPA1.S1 protein was compared for indicated pretreatments: nonacetylated (OPA1+PCAF), acetylated (OPA1+PCAF+AcCoA), deacetylated (Ac.OPA1+SIRT3+NAD+), and subjected to deacetylation in the absence of NAD (Ac.OPA1+SIRT3 − NAD+). Bars represent means ± SEM, n = 4. *, P < 0.001. (B) Immunoblot shows acetylation status of OPA1 and an equal amount of protein used for the activity assay. (C) Reduced mitochondrial fusion in adult SIRT3KO cardiac fibroblasts as measured by diffusion of mtPA-GFP to neighboring mitochondria, compared to WT controls. Total mitochondrial population was visualized by mtDSRed2 expression. Scale bar, 10 μm. (D) Quantitation for mitochondrial networking factor (MNF) from the data acquired in the experiment shown in panel C. Means ± SEM are shown, n = 20 events each. AU, arbitrary units; *, P < 0.001.
SIRT3 directly binds to and deacetylates OPA1.
Since SIRT3 is the major mitochondrial deacetylase whose levels are also reduced in failing hearts (6), we postulated that this may be the HDAC deacetylating OPA1. We therefore examined the acetylation status of OPA1 in SIRT3 knockout (SIRT3KO) hearts. These hearts exhibit progressive aging-associated cardiac hypertrophy and increased ROS synthesis from mitochondria (6, 42). We found robust acetylation of OPA1 in SIRT3KO hearts compared to wild-type controls (Fig. 1E). To test whether SIRT3 deficiency causes a change in OPA1 activity, we immunoprecipitated OPA1 from WT and SIRT3KO MEFs and measured its GTPase activity. As shown in Fig. 1F and G, OPA1 obtained from SIRT3KO cells exhibited significantly reduced GTPase activity compared to OPA1 from WT cells. Since a moderate increase in ROS levels is known to activate HATs, it is possible that OPA1 hyperacetylation results from increased ROS synthesis in SIRT3KO cells. To exclude this possibility, we examined OPA1 acetylation in SIRT3KO MEFs treated with N-acetyl cysteine (NAC), an antioxidant. NAC treatment failed to block hyperacetylation of OPA1 in SIRT3KO cells. However, total acetylation of proteins in the whole-cell lysate (WCL) was suppressed by NAC treatment, which served as a positive control (see Fig. S1A in the supplemental material). These data thus suggested that SIRT3 is likely the enzyme deacetylating OPA1.
To test whether SIRT3 can target OPA1 for deacetylation, we first examined binding of proteins in a cell-free system. 35S-labeled SIRT3 was incubated with His-tagged OPA1 (His.OPA1.S1), and the bound proteins were pulled down with Ni-NTA (His) beads. Another mitochondrial sirtuin, SIRT5, was used as the negative control. We found that His-OPA1 robustly bound to radiolabeled SIRT3 but not to SIRT5 (Fig. 2A, top panel). The blot was also probed with antibodies specific to SIRT3, SIRT5, and OPA1 to confirm protein specificity (Fig. 2A). This experiment demonstrated that SIRT3 and OPA1 are capable of binding to each other directly.
FIG 2.

SIRT3 binds to and deacetylates OPA1. (A) Autoradiogram (uppermost panel) showing direct binding of His.OPA1 to 35S-labeled SIRT3 but not to SIRT5 in an in vitro binding assay. IBs confirm specificity of indicated proteins. (B) SIRT3 and OPA1 bind to each other when overexpressed in 293T cells. Proteins were immunoprecipitated (IP) using Flag or Myc antibodies and immunoblotted (IB) with indicated antibodies. (C) Corresponding controls for panel B, showing that myc-OPA1 does not cross-react with Flag beads (left panels) or that Flag-SIRT3 does not immunoprecipitate with c-myc (right panels). (D) Endogenous SIRT3 was coprecipitated with OPA1 from WT and not from SIRT3KO hearts. (E) Reduced acetylation of OPA1 immunoprecipitated from HeLa stable cells expressing SIRT3-WT, compared to SIRT3 HY mutant-expressing cells. (F) Reexpression of SIRT3 in SIRT3KO MEFs reduced OPA1 acetylation.
We next tested in vivo binding of SIRT3 and OPA1. Cells (293T) were coexpressed with myc-OPA1 and Flag-SIRT3, and immunoprecipitated proteins were analyzed by Western blotting. As shown in Fig. 2B, myc-OPA1 was coprecipitated with Flag.SIRT3 and vice versa. The validity of in vivo binding between OPA1 and SIRT3 was verified by use of appropriate positive and negative controls (Fig. 2C). Binding of endogenous OPA1 to SIRT3 was examined by coimmunoprecipitating proteins from heart lysates of WT and SIRT3KO mice. As shown in Fig. 2D, SIRT3 was pulled down with OPA1 from WT but not from SIRT3KO hearts. We also confirmed binding of endogenous OPA1 to SIRT3 by use of heart lysates from transgenic mice having cardiomyocyte-specific expression of HA-SIRT3 (see Fig. S1B in the supplemental material). These findings demonstrated that SIRT3 binds to OPA1 in vivo. We next tested the ability of SIRT3 to deacetylate OPA1 in vivo. OPA1 was deacetylated in stable HeLa cells expressing WT-SIRT3 but not in the SIRT3-HY catalytic mutant (Fig. 2E) (34). We also confirmed SIRT3-mediated deacetylation of OPA1 in a reconstitution experiment in which SIRT3 was overexpressed in SIRT3KO cells. The results showed reduced acetylation of OPA1 in SIRT3-expressing cells compared to SIRT3-deficient cells (Fig. 2F). Collectively, these results demonstrated that SIRT3 binds to and deacetylates OPA1 in vivo.
SIRT3-mediated deacetylation augments GTPase activity of OPA1 and mitochondrial fusion.
To elucidate the effect of deacetylation on enzymatic activity of OPA1, we performed an in vitro OPA1 activity assay. His.OPA1.S1 was acetylated in vitro with PCAF and then subjected to deacetylation with SIRT3 in the presence or absence of NAD+. Acetylation inhibited OPA1 activity, whereas incubation of OPA1 with SIRT3 significantly recovered its GTPase activity in a NAD-dependent manner (Fig. 3A and B). This confirmed that while acetylation reduces it, SIRT3-mediated deacetylation enhances GTPase activity of OPA1.
Since acetylation reduced the enzymatic activity of OPA1, we investigated whether this might lead to defective mitochondrial dynamics in SIRT3KO cells. To assess mitochondrial fusion capability, we quantified mitochondrial networking in live adult cardiac fibroblasts isolated from 12-month-old WT and SIRT3KO mice. Cardiac fibroblasts were coexpressed with mito-DsRed2 and mito-PAGFP. Mitochondrial fusion was monitored by photoactivating mito-PAGFP in the selected region of cells (40, 41). Spreading of activated GFP through the mitochondrial network was significantly restricted to a limited area in SIRT3KO cells compared to WT controls (Fig. 3C and D), thus suggesting a role for SIRT3 in mitochondrial fusion.
Reduced OPA1 levels are known to cause decline in mitochondrial membrane potential (ΔΨm), which can contribute to defects in mitochondrial networking (43). Since our results showed that acetylation reduced OPA1 activity, we posited that in SIRT3KO cells OPA1 function may be defective in maintaining mitochondrial membrane potential. To test this possibility, TMRM (red) uptake by mitochondria was monitored to measure ΔΨm of adult WT and SIRT3KO cells by live-cell videography. Cells overexpressed with mito-GFP (green) were utilized as a reference control. In the given time frame of the experiment, significantly large numbers of depolarized mitochondria (green) were observed in SIRT3KO cells, unlike the homogenous population seen in the WT cells (Fig. 4A and B), thus suggesting that SIRT3 deficiency leads to heterogeneous population of mitochondria with defective membrane potential. This finding further illustrated the role of SIRT3 in regulating mitochondrial networking. These data are consistent with an earlier study in which knockdown of SIRT3 in hepatocytes caused loss of mitochondrial inner membrane potential with parallel elevation in ROS levels (44).
FIG 4.
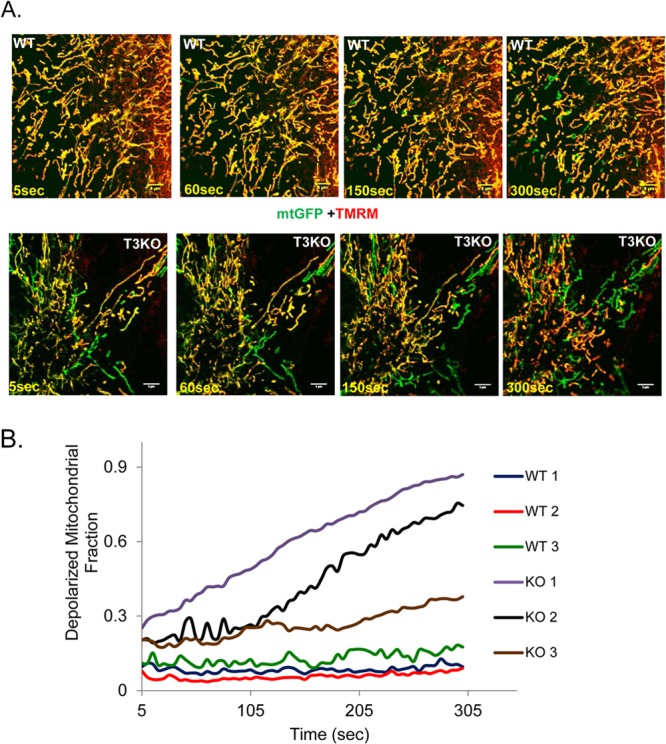
SIRT3KO cardiac fibroblasts exhibit loss of mitochondrial membrane potential (ΔΨm). (A) Still images over a time period of 5 min of WT and SIRT3KO cardiac fibroblasts expressing mtGFP (green) and colabeled with TMRM (red, appearing yellow when merged with green). In live videography, lack and delay in the appearance of yellow mitochondria indicate a defect in ΔΨm. (B) Quantitation of depolarized mitochondria (ratio of TMRM-positive to total mitochondria) for the images shown in panel A. The data shown are representative of four separate experiments. Scale bar, 5 μm.
To test whether OPA1 is needed for SIRT3-mediated networking of mitochondrial population, we monitored the mitochondrial morphology of WT or OPA1-null MEFs overexpressed with Ad.mito-GFP or Ad.SIRT3. As expected, a filamentous mitochondrial network was observed in WT MEFs, whereas OPA1-null MEFs had a highly fragmented mitochondrial population. SIRT3 overexpression in WT cells resulted in “intermediate” mitochondrial morphology (inset) or showed a perinuclear distribution of fused mitochondria (arrow); however, it failed to exhibit a similar effect in OPA1-null MEFs (see Fig. S2A in the supplemental material). These data suggested that the ability of SIRT3 to modulate mitochondrial morphology is dependent on the presence of the fusion protein OPA1. It also indicated that in these cells other contributory factors of mitochondrial dynamics, such as fission machinery, may not be altered by SIRT3. To further examine whether SIRT3 needed OPA1 to manipulate mitochondrial function, we measured ROS synthesis from WT and OPA1-null cells. Increased ROS production is linked to OPA1 mutations in Drosophila (45). We overexpressed SIRT3 in OPA1-WT and OPA1−/− MEFs and subjected them to oxidative stress by treatment with a sublethal dose of H2O2. With H2O2 treatment, ROS levels increased both in OPA1-plus (WT) and -minus (null) cells nearly 4-fold. SIRT3 overexpression reduced ROS to nearly control levels in WT cells but not as much in OPA1-null cells. These data indicated that SIRT3 needs OPA1 as a downstream target to maintain mitochondrial morphology and function (see Fig. S2B in the supplemental material).
OPA1 is acetylated at Lys926 and Lys931 residues.
To identify SIRT3-targeted lysine (K) residues of OPA1, we subjected OPA1 prepared from SIRT3KO hearts to mass spectrometry (MS). The results showed that OPA1 was acetylated at K926 and K931 in the C-terminal GTPase effector domain (GED) (Fig. 5A; see Fig. S3A, B, and C in the supplemental material). These lysine residues were also acetylated when His.OPA-S1 was subjected to acetylation in vitro using PCAF (see Fig. S4A and B in the supplemental material). The OPA1-GED region, where these acetylated lysine residues are present, contains one of the coiled-coil domains necessary for the formation of homotypic complex between different OPA1 isoforms and is required for its role in mitochondrial fusion (15, 46).
FIG 5.

Oxygen consumption rate (OCR) for stable cells expressing OPA1 K mutants in OPA1-null MEFs. (A) Schematic representation of the OPA1 showing locations of acetylated K 926 and K931 residues in the C-terminal GED region. (B) IB showing expression levels of OPA1 mutants in OPA1-null MEFs. (C) Bar diagram depicting basal OCR for immortalized OPA1 WT and OPA1-null MEFs. Values are means ± SEM; n = 3; *, P < 0.001. (D) OPA1-null MEFs were expressed with retroviral vectors (r) synthesizing WT or OPA1 K-to-R mutants. Bar diagram shows basal OCR for different cells. Values are means ± SEM; n = 3; *, P < 0.001. (E and F) Basal OCR of cells expressing K926 and K931 mutated to R or Q. Both K926Q and K931Q (acetylated form) have significantly lower OCR than their respective R mutants. Bars represent means ± SEM; n = 4; *, P < 0.005.
Loss of OPA1 is known to reduce the mitochondrial respiration rate (13). Interestingly, reduced mitochondrial basal oxygen consumption rate (OCR) is also reported for SIRT3KO skeletal muscles (47) and hepatocytes (44). We therefore measured the mitochondrial respiration rate to assess the functional significance of OPA1 lysine acetylation. Acetylation neutralizes the positive charge on lysine (K). Replacement of K with R (arginine) conserves the net positive charge and prevents charge neutralization by acetylation and thus imitates the deacetylated state of the protein. Conversely, a K-to-Q (glutamine) substitution resembles constitutively acetylated lysine in terms of the charge. By site-directed mutagenesis, we made R and Q retroviral mutants for OPA1-K926 and K931 residues. These retroviruses (r) were used to generate stable cell lines from immortalized OPA1-null MEFs. Use of OPA1-null cells gave us the opportunity to assess the effect of only the expressed isoform on mitochondrial function, without interference from the endogenous OPA1. As a negative control, we generated a stable line for K924 (r924R), which was not acetylated in SIRT3KO cells. The expression level of different OPA1 mutants is shown in Fig. 5B. We estimated the basal OCR for these stable cells to evaluate the effect of OPA1-K926- and K931-specific acetylation on mitochondrial respiration. Mitochondrial OCR for wild-type and OPA1-null MEFs served as positive and negative controls, respectively. Consistent with earlier reports (13), OPA1-null MEFs had reduced basal OCR, compared to their WT counterpart (Fig. 5C). OPA1-null MEFs stably expressing WT-OPA1 (rWT-OPA1) showed significantly improved basal OCR for null cells (Fig. 5D). Stable cells expressing r926R and r931R OPA1 also showed similar increase in basal OCR. On the other hand, the r924R OPA1 mutant was unable to elevate the OCR of OPA1-null cells significantly compared to rWT-OPA1, which served as a negative control (Fig. 5D). These results highlighted the significance of K926 and K931 residues in preserving the OPA1 function. The effect of acetylation in these residues was determined by comparing the ability of their respective R and Q mutants to improve upon basal OCR of OPA1-null cells. The deacetylation-mimetic R mutation had significantly higher OCR than the Q mutation, thus implying that K926 and K931 residues play a role in acetylation-mediated regulation of OPA1 function (Fig. 5E and F).
As an additional parameter to assess mitochondrial recovery, we measured mitochondrial DNA copy number for these OPA1-stable MEFs. Earlier studies have reported reduced SIRT3 levels and reduced mtDNA content in diabetic patients (7, 48, 49). Similarly, OPA1 mutations are linked to instability of mtDNA (50, 51). We observed significantly increased mtDNA copy numbers in rWT, r926R, and r931R OPA1-expressing cells, compared to OPA-null MEFs. However, acetylation-mimetic r926Q and r931Q mutants failed to restore mtDNA copy number (see Fig. S5A in the supplemental material). We also checked the effect of the double R and Q mutation on 926/931K residues (dmR- or dmQ-OPA) for their ability to alter mitochondrial copy number. Overexpression of the OPA1-dmR mutant significantly improved the mtDNA content of OPA1-null cells, while the dmQ mutant failed to do so (see Fig. S5B and C in the supplemental material). OPA1-null cells overexpressed with WT-OPA1 (926/931K) served as a positive control for mtDNA recovery. These data thus provided strong evidence that reversible acetylation of 926K and 931K residues regulate functions of OPA1.
One of the major functions ascribed to SIRT3 is its ability to regulate mitochondrial ROS synthesis, thereby protecting cells from oxidative damage. To know whether OPA1 can rescue mitochondrial dysfunction of SIRT3KO cells, we measured ROS production from cells as a readout of mitochondrial recovery. SIRT3KO MEFs were overexpressed with OPA1-dmR or dmQ mutants, and cellular ROS levels were measured. SIRT3KO cells subjected to oxidative stress by H2O2 treatment showed a 4-fold increase in ROS levels compared to those of untreated cells. Overexpression of dmR-OPA1 but not the dmQ mutant significantly reduced ROS levels of these cells (see Fig. S6A and B in the supplemental material). In this experiment, partial rescue from excessive ROS generation in SIRT3KO cells by OPA1-dmR is likely due to defective function of other enzymes involved in mitochondrial ROS synthesis and detoxification (1). Nonetheless, these data further suggested that SIRT3 regulates mitochondrial functions in part by targeting OPA1.
SIRT3 expression modulates morphology of cardiac mitochondria.
Knowing that OPA1 is one of the major regulators of mitochondrial dynamics, we investigated how SIRT3-mediated modulation of OPA1 could impact the morphology of cardiac mitochondria. We performed ultrastructural studies for WT and SIRT3KO adult hearts. Unlike the highly regular roadlike mitochondrial network seen for the adult WT heart (Fig. 6A), SIRT3KO hearts displayed defective organization of mitochondria. In SIRT3KO hearts, mitochondria were mostly clumped in multiple islands (Fig. 6B). Moreover, unlike in WT hearts, mitochondria in SIRT3KO hearts showed a continuous outer membrane but loosely tethered inner membranes (Fig. 6C and D). Similar ultrastructural changes have been reported for OPA+/− hearts (27), in which OPA1 protein expression is significantly reduced. Based on these findings and previous reports, it is reasonable to believe that the mitochondrial defects observed in SIRT3KO hearts result from reduced function of hyperacetylated OPA1.
FIG 6.
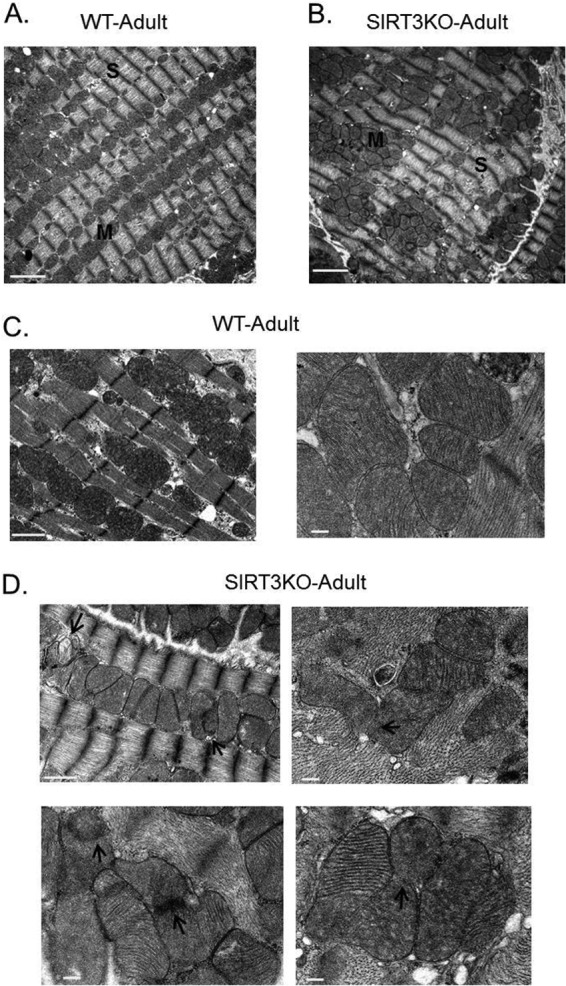
Transmission electron micrographs showing mitochondrial morphology in SIRT3KO hearts. Electron micrographs showing mitochondrial morphology and arrangement in adult wild-type (WT) (A and C) and SIRT3KO hearts (B and D). SIRT3KO micrographs show mitochondria clustered in islands (B). Arrows in SIRT3KO micrographs (D) indicate defective mitochondrial inner membrane fusion. M, mitochondria; S, sarcomeres. Scale bars: 2 μm (A and B); 1 μm (C [left panel] and D [left top panel]); and 200 nm (C [right panel] and D [bottom and right top panels]).
Another model that we examined to study the impact of SIRT3 on mitochondrial morphology was doxorubicin (Dox)-mediated cardiomyocyte cell death. Doxorubicin is a widely used anticancer drug that causes cardiac toxicity due to mitochondrial fragmentation and excessive ROS production (52, 53). Approaches aimed to block mitochondrial fragmentation alleviate the cardiac toxicity of this drug (54). Neonatal rat cardiomyocytes were overexpressed with either Ad.mito-GFP or Ad.SIRT3 followed by treatment with Dox for 24 h. Mitochondrial morphology was monitored by confocal microscopy and cell death by TUNEL staining. Fragmented and swollen mitochondria were seen in Dox-treated, Ad.mito-GFP-expressing cardiomyocytes, whereas in Ad.SIRT3-expressing cells, even following Dox treatment, the normal tubular shape of mitochondria was preserved (Fig. 7A). To examine whether enzymatic activity of SIRT3 is needed for preservation of mitochondrial morphology after Dox treatment, we used stable HeLa cells expressing SIRT3 WT or the HY mutant. As seen in Fig. 7B, cells expressing SIRT3 WT (middle right panel) showed preserved mitochondrial morphology after Dox treatment, but cells expressing catalytic mutant did not (bottom right panel). In control and SIRT3 mutant-expressing cells, mitochondria were largely fragmented and swollen after Dox treatment, whereas the filamentous morphology of mitochondria was preserved in SIRT3 WT-expressing cells. These data thus indicated that SIRT3 needs enzymatic activity to regulate mitochondrial morphology.
FIG 7.
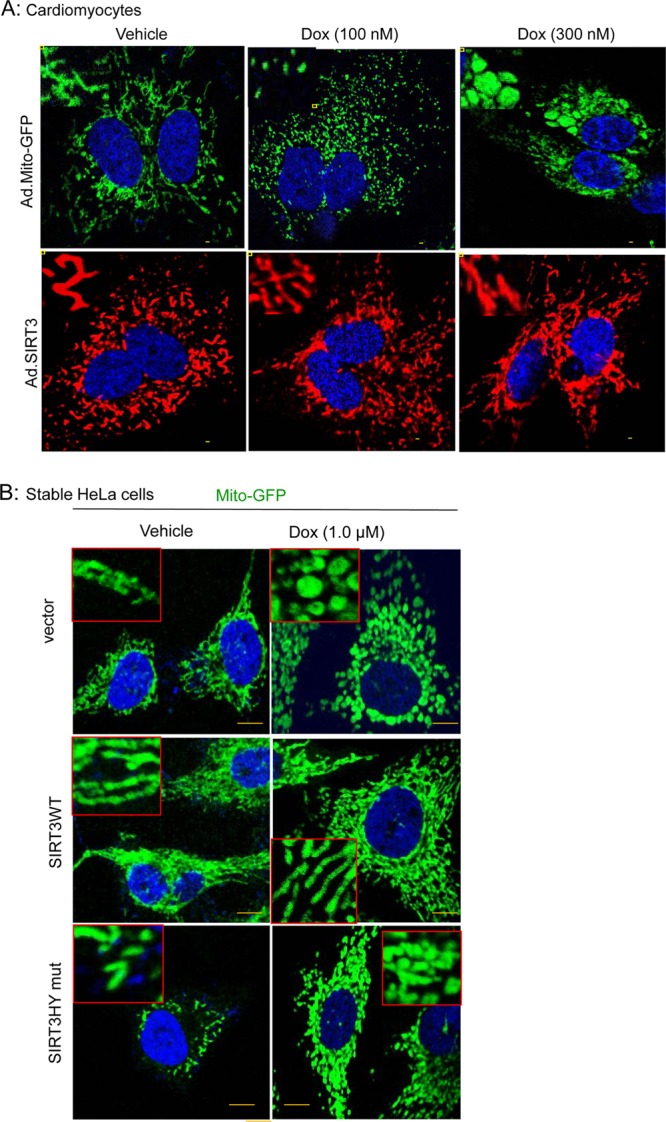
SIRT3 overexpression protects cells from doxorubicin-induced mitochondrial destruction. (A) Representative confocal images of cardiomyocytes showing progressive deterioration of mitochondria when treated with increasing doses of doxorubicin (Dox). Mitochondria are visualized by overexpressing cells with mito-GFP (green, upper panel). SIRT3 overexpression protected the mitochondrial morphology of Dox-treated cardiomyocytes (red, lower panel). Scale bar, 5 μm. Insets display the mitochondrial morphology in an enlarged image in each panel. (B) Unlike stable HeLa cells expressing SIRT3 catalytic mutant (bottom panel) or the vector (top panel), mitochondrial morphology was preserved in SIRT3 WT-expressing cells when treated with Dox (middle panel). Insets show mitochondrial morphology in enlarged images. Scale bar, 1 μm.
Experiments carried out to measure cell death in Dox-treated cardiomyocytes demonstrated a marked reduction in TUNEL-positive nuclei when overexpressed with SIRT3, compared to cells expressed with a mock vector (Fig. 8A and B). The protective effect of SIRT3 on Dox-treated cardiomyocytes was further supported by results of Western analysis. Ad.SIRT3-expressing cardiomyocytes showed reduced expression of cell death markers such as Bax, Bim, and FasL compared to control virus-expressing cells after Dox treatment (see Fig. S7 in the supplemental material). Furthermore, Dox treatment of cardiomyocytes resulted in hyperacetylation of endogenous OPA1, which was reverted back by overexpressing cells with SIRT3 (Fig. 8C). Taken together, these data suggested that SIRT3 overexpression blocks Dox-mediated mitochondrial fragmentation and subsequent cardiomyocyte cell death by targeting OPA1.
FIG 8.
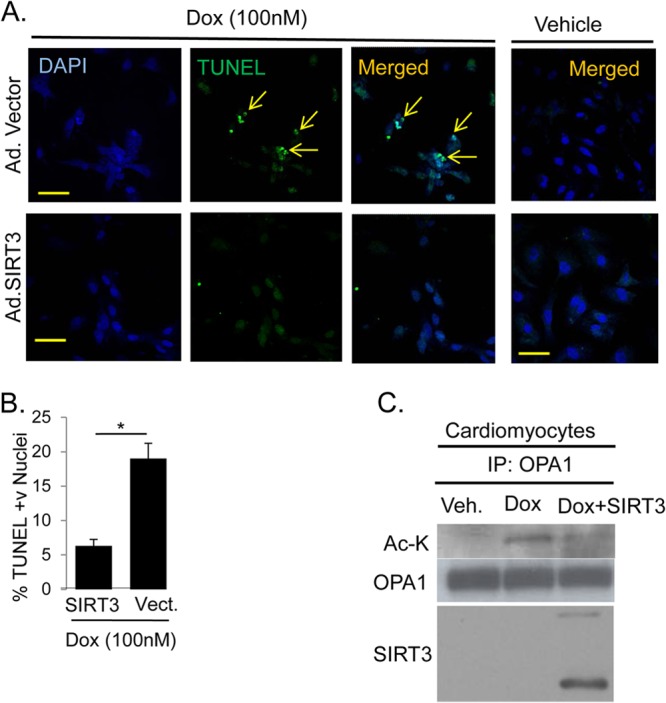
SIRT3 expression protects cells from doxorubicin-mediated cell death. (A) Cell death monitored by TUNEL (green) staining for cardiomyocytes overexpressing either adenoviral vector (Ad.Vector) or SIRT3-adenovirus (Ad.SIRT3) and treated with doxorubicin or vehicle for 24 h. Nuclei are marked with DAPI staining (blue). Scale bar, 10 μm. (B) Quantification of TUNEL-positive cells shown in panel A. Values are means ± SEM, n = 4. *, P < 0.001. (C) IB showing acetylated OPA1 immunoprecipitated from neonatal rat cardiomyocytes treated with 300 nM doxorubicin. The right lane shows that overexpressed SIRT3 binds to and deacetylates endogenous OPA1. Veh., vehicle; vect., vector.
DISCUSSION
In this study, we demonstrated a new role for SIRT3 as a regulator of mitochondrial dynamics. SIRT3 targets the IM fusion protein OPA1 to modulate its enzymatic activity via lysine deacetylation. Our data demonstrated that mitochondrial respiration is differentially affected depending upon the acetylation status of the two lysine residues K926 and K931 residing in the C-terminal GED region of OPA1. We found that these residues were acetylated during cardiac hypertrophy and SIRT3 deficiency. We also showed that SIRT3 deficiency impedes mitochondrial networking, and overexpression of SIRT3 protects cells from doxorubicin-mediated mitochondrial fragmentation and cell death. These changes were associated with change in the acetylation status of OPA1. Thus, in this study we demonstrated for the first time that SIRT3 regulates mitochondrial dynamics by targeting OPA1 to maintain fitness of mitochondrial population.
OPA1 is a functional target of SIRT3.
In addition to matrix, SIRT3 is also reported to be present at mitochondrial inner membrane and crista structures (55, 56). In this study, we found that SIRT3 binds directly to the IM protein OPA1 and activates it by deacetylation. Both OPA1 and SIRT3 are reported to regulate mitochondrial respiration, ROS synthesis, and apoptosis (1, 24, 45, 57). SIRT3 is shown to regulate the oxidative phosphorylation pathway by binding to and deacetylating the Ndufa9 subunit of electron transport chain complex I (ETC-I) and succinate dehydrogenase subunit A (SdhA) of ETC-II (58, 59). Likewise, OPA1 is reported to bind to ETC-I and -II (60). A correlation between downregulation of OPA1, perturbation of crista structures, and reduction in mitochondrial membrane potential is also reported, indicating a potential role for OPA1 in the regulation of mitochondrial oxidative phosphorylation (24, 43). Same as for OPA1-null cells, reduced basal OCR is also reported for SIRT3KO skeletal muscles (47) and hepatocytes (44). Consistent with these reports, our data suggest that under conditions of stress, SIRT3 activates OPA1 to maintain and/or to enhance activity of mitochondrial respiratory complexes.
Mitochondria generate more than 90% of cellular ROS as a by-product of mitochondrial respiration (61). Increased cellular ROS levels are believed to contribute to many aging-associated degenerative diseases (62). SIRT3 has been shown to block cardiac hypertrophy and tumorigenesis by reducing ROS synthesis from mitochondria (4, 6). Reduced activity of SIRT3 and increased mitochondrial ROS synthesis are also reported in aging-associated hearing loss (5). Likewise, OPA1 mutations are also linked to hearing loss (63). Heterozygous mutations of Drosophila OPA1 (dOPA1) result in impairment of the ETC complexes' activity and increased ROS production (45). These dOPA1 mutant flies show a reduced life span, suggesting a similar role for OPA1 in regulating longevity, as it is suggested for SIRT3 (8, 9, 45). Our data demonstrated that SIRT3 can downregulate H2O2-induced ROS levels more efficiently in the presence of OPA1 than in its absence. Similarly, we could significantly reduce ROS generated by SIRT3KO MEFs subjected to oxidative stress by overexpressing the deacetylation-mimetic OPA1 926/931R mutant, thus highlighting the importance of OPA1 as a potential downstream target of SIRT3.
A protective role of SIRT3 to curtail mitochondrially mediated apoptosis has been demonstrated in cardiomyocytes (34, 64). In mitochondrion-dependent apoptosis, cytochrome c release from the inner mitochondrial space to cytosol requires remodeling of crista junctions (65, 66). There are many reports showing that OPA1 oligomers are the building blocks of the crista structures (23). Reduced levels of OPA1 lead to disruption of crista structures and consequently apoptosis (24). OPA1 mutants lacking the functional GTPase domain or the C-terminal GED region sensitize cells to death stimuli (23, 67, 68). In our study, the acetylated lysine residues targeted by SIRT3 are uniquely positioned in the GED region of OPA1. Our findings obtained by using Dox-mediated cell death as a model system indicated that deacetylation of OPA1 via SIRT3 could be one of the mechanisms through which SIRT3 blocks mitochondrion-mediated apoptosis.
Role of OPA1 acetylation in cardiac hypertrophy.
All the mouse models for cardiac hypertrophy used in this study are linked to mitochondrial dysfunction (47, 69–71), and each showed hyperacetylated OPA1. Our study also demonstrated that hyperacetylation reduces OPA's GTPase activity, suggesting that defective OPA1 could be a cause of mitochondrial dysfunction in hypertrophied hearts. Consistent with our results, the adult haplo-insufficient OPA1 heterozygous mice display normal cardiac functions at rest; however, they are highly sensitive to hypertrophic stimuli, same as for SIRT3KO mice (28). OPA1+/− hearts also had clustered mitochondria with delayed opening of mitochondrial permeability transition pores (mPTP) after Ca2+ stimulation, compared to wild-type controls. OPA1 heterozygous mice also developed late-onset cardiomyopathy with characteristics largely resembling those of cardiomyopathy seen in SIRT3KO mice under stress or with aging (28, 42).
Additionally, there is evidence showing a role for both SIRT3 and OPA1 in the development of metabolic stress and diabetes (47, 72, 73). In the ob/ob mouse model, before the onset of type 2 diabetes, a reduced level of OPA1 is seen in pancreatic islets, which contributes to mitochondrial dysfunction (72). Conditional OPA1 knockout in the pancreatic beta cells also showed reduced expression and activity of ETC-IV (73). Similarly, treatment of cardiomyocytes with high glucose levels is reported to cause reduction in OPA1 level, enhanced mitochondrial fragmentation, and diminished activity of ETC-IV (74). In accordance with this, both in type 1 and in type 2 models of diabetes, SIRT3 expression is reduced, which is linked to dysfunctional mitochondria (47). These previous reports support our data presented here, which unravel a new mechanism of action for SIRT3 whereby SIRT3 seems to preserve mitochondrial fitness by deacetylation and activation of OPA1.
In humans and mouse, OPA1 protein exists as a complex mixture of isoforms derived from its eight splice variants (46, 75–77). In our study, retroviral overexpression of OPA1 deacetylation mimic (K-to-R) isoforms could restore the basal OCR and mtDNA copy number of OPA-null cells but not the acetylation mimic (K-to-Q) mutants. Consistent with our results, a marked reduction in mtDNA copy number and defects in mitochondrial respiration concomitant with reduction in OPA1 activity were reported earlier (27). Because the retroviral stable cells used in our study were generated from OPA1-null cells, which lacked OPA1 isoforms normally synthesized from the genomic copy as splice variants, we could not determine the effects of OPA1 K926R and K931R mutants on the fusion ability of mitochondria in OPA1-stable cells. Previous studies have reported that expression of various long and short isoforms of OPA1 in optimum combinations are essential to reinstate all the functional capabilities of this enzyme (35, 36). Such kinds of combinations of OPA isoforms cannot be expected in our cells, which were overexpressed with just one isoform. This is one of the obvious difficulties in studying the effect of PTM on the function of OPA to regulate mitochondrial fusion.
The GTPase effector domain (GED) at the C terminus is predicted to regulate self-assembly of OPA1 and activation of GTP hydrolysis (68). Mitochondrial morphology is reported to be dependent on the integrity of this C-terminal coiled-coil domain of OPA1 (15). The two acetylated lysine residues (926 and 931) of OPA1 identified in our study reside in this GED region and are likely to enhance GTPase activity of OPA1 upon deacetylation by SIRT3. Deletion of the OPA1-GED region (OPA1Δ58) in type I autosomal dominant optic atrophy (ADOA) patients results in 50% percent reduction in the OPA1 protein level, with increased sensitivity to apoptosis (68). In hereditary optic atrophy patients, 9% of the OPA1 mutations, including the infamous R932C mutation, are in the GED region (68). The R932C missense mutation in OPA1 is associated with a large population of shorter mitochondria displaying defects in coupling of oxidative phosphorylation in skin fibroblasts of patients (78). By the same token, our data suggest that under stress conditions GTPase activity of OPA1 may diminish with the addition of a bulky acetyl group on lysine residues (931K and 926K) neighboring R932. Hence, it will be beneficial for the cell to keep these lysine residues deacetylated. Previously, it was reported that a lysine-to-alanine mutation in the Drp1-GED region reduced GTPase activity of Drp1 (79). It will be of future interest to study whether acetylation can affect the interaction between mitochondrial membrane lipids and OPA1 to regulate its catalytic activity. Basal GTPase activity of OPA1 is reported to increase dramatically by its interaction with the mitochondrial IM lipid cardiolipin (36). In summary, our data in this study provide evidence that OPA1 activity is regulated by reversible lysine acetylation and that SIRT3 promotes OPA1 activity to fine-tune mitochondrial dynamics during cellular stress.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the NIH-RO1 grants HL83423, HL117041, and HL111455 to M.P.G.
We thank F. W. Alt and Christopher Rhodes for providing SIRT3KO and db/db mice, respectively. We are thankful to Marcia Haigis for providing SIRT3 WT and KO immortalized MEFs. We express gratitude to Paul Schumacker, Daniel Linseman, and Eric Verdin for providing the DNA vectors mentioned in Materials and Methods. We also thank Vytas Bindokas, Christine Labno, Shirley Bond, and Yimei Chen for the technical help in microscopy work.
Footnotes
Published ahead of print 16 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01483-13.
REFERENCES
- 1.Bell EL, Guarente L. 2011. The SirT3 divining rod points to oxidative stress. Mol. Cell 42:561–568. 10.1016/j.molcel.2011.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He W, Newman JC, Wang MZ, Ho L, Verdin E. 2012. Mitochondrial sirtuins: regulators of protein acylation and metabolism. Trends Endocrinol. Metab. 23:467–476. 10.1016/j.tem.2012.07.004 [DOI] [PubMed] [Google Scholar]
- 3.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV, Jr, Weissman S, Verdin E, Schwer B. 2007. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27:8807–8814. 10.1128/MCB.01636-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, Singh KK, Abdulkadir SA, Spitz DR, Deng CX, Gius D. 2010. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17:41–52. 10.1016/j.ccr.2009.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. 2010. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143:802–812. 10.1016/j.cell.2010.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. 2009. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 119:2758–2771. 10.1172/JCI39162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caton PW, Richardson SJ, Kieswich J, Bugliani M, Holland ML, Marchetti P, Morgan NG, Yaqoob MM, Holness MJ, Sugden MC. 2013. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia 56:1068–1077. 10.1007/s00125-013-2851-y [DOI] [PubMed] [Google Scholar]
- 8.Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, Greco V, Maggiolini M, Feraco E, Mari V, Franceschi C, Passarino G, De Benedictis G. 2005. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics 85:258–263. 10.1016/j.ygeno.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 9.Rose G, Dato S, Altomare K, Bellizzi D, Garasto S, Greco V, Passarino G, Feraco E, Mari V, Barbi C, BonaFe M, Franceschi C, Tan Q, Boiko S, Yashin AI, De Benedictis G. 2003. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp. Gerontol. 38:1065–1070. 10.1016/S0531-5565(03)00209-2 [DOI] [PubMed] [Google Scholar]
- 10.Chan DC. 2006. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 22:79–99. 10.1146/annurev.cellbio.22.010305.104638 [DOI] [PubMed] [Google Scholar]
- 11.Liesa M, Palacin M, Zorzano A. 2009. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 89:799–845. 10.1152/physrev.00030.2008 [DOI] [PubMed] [Google Scholar]
- 12.Westermann B. 2010. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11:872–884. 10.1038/nrm3013 [DOI] [PubMed] [Google Scholar]
- 13.Chen H, Chomyn A, Chan DC. 2005. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 280:26185–26192. 10.1074/jbc.M503062200 [DOI] [PubMed] [Google Scholar]
- 14.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. 2003. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160:189–200. 10.1083/jcb.200211046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. 2004. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. U. S. A. 101:15927–15932. 10.1073/pnas.0407043101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. 2001. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12:2245–2256. 10.1091/mbc.12.8.2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. 2003. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 23:5409–5420. 10.1128/MCB.23.15.5409-5420.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao J, Lendahl U, Nister M. 2013. Regulation of mitochondrial dynamics: convergences and divergences between yeast and vertebrates. Cell. Mol. Life Sci. 70:951–976. 10.1007/s00018-012-1066-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y. 2006. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23:607–618. 10.1016/j.molcel.2006.06.026 [DOI] [PubMed] [Google Scholar]
- 20.Giralt A, Hondares E, Villena JA, Ribas F, Diaz-Delfin J, Giralt M, Iglesias R, Villarroya F. 2011. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J. Biol. Chem. 286:16958–16966. 10.1074/jbc.M110.202390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, Stevens RD, Muehlbauer MJ, Kakar S, Bass NM, Kuusisto J, Laakso M, Alt FW, Newgard CB, Farese RV, Jr, Kahn CR, Verdin E. 2011. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 44:177–190. 10.1016/j.molcel.2011.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, Fang F, Chang Y. 2010. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 5:e11707. 10.1371/journal.pone.0011707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. 2006. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126:177–189. 10.1016/j.cell.2006.06.025 [DOI] [PubMed] [Google Scholar]
- 24.Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G. 2003. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 278:7743–7746. 10.1074/jbc.C200677200 [DOI] [PubMed] [Google Scholar]
- 25.Belenguer P, Pellegrini L. 2013. The dynamin GTPase OPA1: more than mitochondria? Biochim. Biophys. Acta 1833:176–183. 10.1016/j.bbamcr.2012.08.004 [DOI] [PubMed] [Google Scholar]
- 26.Chen L, Gong Q, Stice JP, Knowlton AA. 2009. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 84:91–99. 10.1093/cvr/cvp181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Liu T, Tran A, Lu X, Tomilov AA, Davies V, Cortopassi G, Chiamvimonvat N, Bers DM, Votruba M, Knowlton AA. 2012. OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 1:e003012. 10.1161/JAHA.112.003012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, Fortin D, Huynh le H, Nicolas V, Alavi MV, Brenner C, Ventura-Clapier R, Veksler V, Joubert F. 2012. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc. Res. 94:408–417. 10.1093/cvr/cvs117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. 2009. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840. 10.1126/science.1175371 [DOI] [PubMed] [Google Scholar]
- 30.Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, Cunningham JM, Deng CX, Lombard DB, Mostoslavsky R, Gupta MP. 2012. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat. Med. 18:1643–1650. 10.1038/nm.2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. 2009. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 20:3525–3532. 10.1091/mbc.E09-03-0252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis FJ, Gupta M, Pogwizd SM, Bacha E, Jeevanandam V, Gupta MP. 2002. Increased expression of alternatively spliced dominant-negative isoform of SRF in human failing hearts. Am. J. Physiol. Heart Circ. Physiol. 282:H1521–H1533. 10.1152/ajpheart.00844.2001 [DOI] [PubMed] [Google Scholar]
- 33.Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, Pandolfi PP, Haigis MC. 2011. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 19:416–428. 10.1016/j.ccr.2011.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. 2008. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 28:6384–6401. 10.1128/MCB.00426-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song Z, Chen H, Fiket M, Alexander C, Chan DC. 2007. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 178:749–755. 10.1083/jcb.200704110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ban T, Heymann JA, Song Z, Hinshaw JE, Chan DC. 2010. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 19:2113–2122. 10.1093/hmg/ddq088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samant SA, Courson DS, Sundaresan NR, Pillai VB, Tan M, Zhao Y, Shroff SG, Rock RS, Gupta MP. 2011. HDAC3-dependent reversible lysine acetylation of cardiac myosin heavy chain isoforms modulates their enzymatic and motor activity. J. Biol. Chem. 286:5567–5577. 10.1074/jbc.M110.163865 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. 2002. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 159:931–938. 10.1083/jcb.200209124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. 2006. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 8:1255–1262. 10.1038/ncb1487 [DOI] [PubMed] [Google Scholar]
- 40.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL. 2012. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 110:1484–1497. 10.1161/CIRCRESAHA.111.263848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL. 2012. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 26:2175–2186. 10.1096/fj.11-196543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. 2010. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2:914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. 2008. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27:433–446. 10.1038/sj.emboj.7601963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bao J, Scott I, Lu Z, Pang L, Dimond CC, Gius D, Sack MN. 2010. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic. Biol. Med. 49:1230–1237. 10.1016/j.freeradbiomed.2010.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang S, Le PK, Tse S, Wallace DC, Huang T. 2009. Heterozygous mutation of Opa1 in Drosophila shortens lifespan mediated through increased reactive oxygen species production. PLoS One 4:e4492. 10.1371/journal.pone.0004492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akepati VR, Muller EC, Otto A, Strauss HM, Portwich M, Alexander C. 2008. Characterization of OPA1 isoforms isolated from mouse tissues. J. Neurochem. 106:372–383. 10.1111/j.1471-4159.2008.05401.x [DOI] [PubMed] [Google Scholar]
- 47.Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, Kahn CR. 2011. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. U. S. A. 108:14608–14613. 10.1073/pnas.1111308108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antonetti DA, Reynet C, Kahn CR. 1995. Increased expression of mitochondrial-encoded genes in skeletal muscle of humans with diabetes mellitus. J. Clin. Invest. 95:1383–1388. 10.1172/JCI117790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsieh CJ, Weng SW, Liou CW, Lin TK, Chen JB, Tiao MM, Hung YT, Chen IY, Huang WT, Wang PW. 2011. Tissue-specific differences in mitochondrial DNA content in type 2 diabetes. Diabetes Res. Clin. Pract. 92:106–110. 10.1016/j.diabres.2011.01.010 [DOI] [PubMed] [Google Scholar]
- 50.Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, Campos Y, Rivera H, de la Aleja JG, Carroccia R, Iommarini L, Labauge P, Figarella-Branger D, Marcorelles P, Furby A, Beauvais K, Letournel F, Liguori R, La Morgia C, Montagna P, Liguori M, Zanna C, Rugolo M, Cossarizza A, Wissinger B, Verny C, Schwarzenbacher R, Martin MA, Arenas J, Ayuso C, Garesse R, Lenaers G, Bonneau D, Carelli V. 2008. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus' phenotypes. Brain 131:338–351. 10.1093/brain/awm298 [DOI] [PubMed] [Google Scholar]
- 51.Hudson G, Amati-Bonneau P, Blakely EL, Stewart JD, He L, Schaefer AM, Griffiths PG, Ahlqvist K, Suomalainen A, Reynier P, McFarland R, Turnbull DM, Chinnery PF, Taylor RW. 2008. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain 131:329–337. 10.1093/brain/awm272 [DOI] [PubMed] [Google Scholar]
- 52.Kang YJ, Chen Y, Epstein PN. 1996. Suppression of doxorubicin cardiotoxicity by overexpression of catalase in the heart of transgenic mice. J. Biol. Chem. 271:12610–12616. 10.1074/jbc.271.21.12610 [DOI] [PubMed] [Google Scholar]
- 53.Singal PK, Iliskovic N. 1998. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 339:900–905. 10.1056/NEJM199809243391307 [DOI] [PubMed] [Google Scholar]
- 54.Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, Piantadosi CA. 2007. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J. Clin. Invest. 117:3730–3741. 10.1172/JCI32967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. 2002. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. U. S. A. 99:13653–13658. 10.1073/pnas.222538099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi T, Wang F, Stieren E, Tong Q. 2005. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 280:13560–13567. 10.1074/jbc.M414670200 [DOI] [PubMed] [Google Scholar]
- 57.Park SH, Ozden O, Jiang H, Cha YI, Pennington JD, Aykin-Burns N, Spitz DR, Gius D, Kim HS. 2011. Sirt3, mitochondrial ROS, ageing, and carcinogenesis. Int. J. Mol. Sci. 12:6226–6239. 10.3390/ijms12096226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. 2008. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. U. S. A. 105:14447–14452. 10.1073/pnas.0803790105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. 2010. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 49:304–311. 10.1021/bi901627u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, Wissinger B, Pinti M, Cossarizza A, Vidoni S, Valentino ML, Rugolo M, Carelli V. 2008. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 131:352–367. 10.1093/brain/awm335 [DOI] [PubMed] [Google Scholar]
- 61.Balaban RS, Nemoto S, Finkel T. 2005. Mitochondria, oxidants, and aging. Cell 120:483–495. 10.1016/j.cell.2005.02.001 [DOI] [PubMed] [Google Scholar]
- 62.Brand MD, Buckingham JA, Esteves TC, Green K, Lambert AJ, Miwa S, Murphy MP, Pakay JL, Talbot DA, Echtay KS. 2004. Mitochondrial superoxide and aging: uncoupling-protein activity and superoxide production. Biochem. Soc. Symp. 71:203–213 http://symposia.biochemistry.org/bssymp/071/bss0710203.htm [DOI] [PubMed] [Google Scholar]
- 63.Leruez S, Milea D, Defoort-Dhellemmes S, Colin E, Crochet M, Procaccio V, Ferre M, Lamblin J, Drouin V, Vincent-Delorme C, Lenaers G, Hamel C, Blanchet C, Juul G, Larsen M, Verny C, Reynier P, Amati-Bonneau P, Bonneau D. 2013. Sensorineural hearing loss in OPA1-linked disorders. Brain 136:e236. 10.1093/brain/aws340 [DOI] [PubMed] [Google Scholar]
- 64.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, Sinclair DA. 2007. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130:1095–1107. 10.1016/j.cell.2007.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. 2002. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2:55–67. 10.1016/S1534-5807(01)00116-2 [DOI] [PubMed] [Google Scholar]
- 66.Suen DF, Norris KL, Youle RJ. 2008. Mitochondrial dynamics and apoptosis. Genes Dev. 22:1577–1590. 10.1101/gad.1658508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arnoult D, Grodet A, Lee YJ, Estaquier J, Blackstone C. 2005. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J. Biol. Chem. 280:35742–35750. 10.1074/jbc.M505970200 [DOI] [PubMed] [Google Scholar]
- 68.Olichon A, Landes T, Arnaune-Pelloquin L, Emorine LJ, Mils V, Guichet A, Delettre C, Hamel C, Amati-Bonneau P, Bonneau D, Reynier P, Lenaers G, Belenguer P. 2007. Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis. J. Cell Physiol. 211:423–430. 10.1002/jcp.20950 [DOI] [PubMed] [Google Scholar]
- 69.Abel ED, Doenst T. 2011. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc. Res. 90:234–242. 10.1093/cvr/cvr015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW, II, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. 2011. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 108:837–846. 10.1161/CIRCRESAHA.110.232306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sack MN. 2009. Type 2 diabetes, mitochondrial biology and the heart. J. Mol. Cell Cardiol. 46:842–849. 10.1016/j.yjmcc.2009.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, Stapleton DS, Argmann C, Schueler KL, Edwards S, Steinberg HA, Chaibub Neto E, Kleinhanz R, Turner S, Hellerstein MK, Schadt EE, Yandell BS, Kendziorski C, Attie AD. 2008. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 18:706–716. 10.1101/gr.074914.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Z, Wakabayashi N, Wakabayashi J, Tamura Y, Song WJ, Sereda S, Clerc P, Polster BM, Aja SM, Pletnikov MV, Kensler TW, Shirihai OS, Iijima M, Hussain MA, Sesaki H. 2011. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol. Biol. Cell 22:2235–2245. 10.1091/mbc.E10-12-0933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Makino A, Suarez J, Gawlowski T, Han W, Wang H, Scott BT, Dillmann WH. 2011. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300:R1296–R1302. 10.1152/ajpregu.00437.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D'Adamio L, Derks C, Dejaegere T, Pellegrini L, D'Hooge R, Scorrano L, De Strooper B. 2006. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 126:163–175. 10.1016/j.cell.2006.06.021 [DOI] [PubMed] [Google Scholar]
- 76.Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. 2000. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 26:207–210. 10.1038/79936 [DOI] [PubMed] [Google Scholar]
- 77.Ishihara N, Fujita Y, Oka T, Mihara K. 2006. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 25:2966–2977. 10.1038/sj.emboj.7601184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nochez Y, Arsene S, Gueguen N, Chevrollier A, Ferre M, Guillet V, Desquiret V, Toutain A, Bonneau D, Procaccio V, Amati-Bonneau P, Pisella PJ, Reynier P. 2009. Acute and late-onset optic atrophy due to a novel OPA1 mutation leading to a mitochondrial coupling defect. Mol. Vis. 15:598–608 [PMC free article] [PubMed] [Google Scholar]
- 79.Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C. 2004. Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J. Biol. Chem. 279:35967–35974. 10.1074/jbc.M404105200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.