

Abstract
Lipid-anchored Ras GTPases form transient, spatially segregated nanoclusters on the plasma membrane that are essential for high-fidelity signal transmission. The lipid composition of Ras nanoclusters, however, has not previously been investigated. High-resolution spatial mapping shows that different Ras nanoclusters have distinct lipid compositions, indicating that Ras proteins engage in isoform-selective lipid sorting and accounting for different signal outputs from different Ras isoforms. Phosphatidylserine is a common constituent of all Ras nanoclusters but is only an obligate structural component of K-Ras nanoclusters. Segregation of K-Ras and H-Ras into spatially and compositionally distinct lipid assemblies is exquisitely sensitive to plasma membrane phosphatidylserine levels. Phosphatidylserine spatial organization is also modified by Ras nanocluster formation. In consequence, Ras nanoclusters engage in remote lipid-mediated communication, whereby activated H-Ras disrupts the assembly and operation of spatially segregated K-Ras nanoclusters. Computational modeling and experimentation reveal that complex effects of caveolin and cortical actin on Ras nanoclustering are similarly mediated through regulation of phosphatidylserine spatiotemporal dynamics. We conclude that phosphatidylserine maintains the lateral segregation of diverse lipid-based assemblies on the plasma membrane and that lateral connectivity between spatially remote lipid assemblies offers important previously unexplored opportunities for signal integration and signal processing.
INTRODUCTION
H-, N-, and K-Ras are small GTPases that operate as molecular switches to regulate cell growth, proliferation, and differentiation (1, 2). H-, N-, and K-Ras comprise nearly identical G domains (amino acids 1 to 165), which bind guanine nucleotides and interact with effectors and exchange factors, but they contain highly divergent C-terminal hypervariable regions (HVRs) (3, 4). The HVR undergoes posttranslational processing to attach a membrane anchor, which consists of a C-terminal S-farnesyl cysteine carboxylmethyl ester (common to all Ras proteins) and mono-palmitoylation of N-Ras, di-palmitoylation of H-Ras, and the presence of a polylysine domain in K-Ras (5–8). Ras proteins are distributed heterogeneously over the plasma membrane (PM) in a combination of immobile nanoclusters and freely diffusing monomers (9). A nanocluster comprises ∼7 Ras proteins, has a radius of ∼9 nm, and has an estimated lifetime of 0.5 to 1 s (10, 11). Nanocluster formation is essential for high-fidelity signal transmission (11–16). As a direct consequence of the different lipid anchors and different residues in the flanking HVR and G domain, which directly participate in membrane binding, N-, H-, and K-Ras assemble into spatially nonoverlapping nanoclusters, with further lateral segregation into nonoverlapping GDP and GTP nanoclusters (11, 16–23).
Ras isoforms exhibit different effector activation profiles (1). To account for isoform-specific signal output, Ras proteins must generate compositionally distinct nanoclusters by recruiting specific subsets of lipids. PM lipid organization must also be malleable on time and length scales relevant to nanocluster assembly. In this study, we therefore systematically characterized the molecular association of specific lipids with Ras nanoclusters in intact PMs. The experiments reveal that Ras nanoclusters have different phospholipid compositions, but all share phosphatidylserine (PS) as a constituent. In consequence, spatial interactions occur between laterally segregated nanoclusters, in which H-Ras remotely regulates the structure and operation of K-Ras nanoclusters through induced changes to PS spatiotemporal dynamics. This study illustrates that formation of transient lipid assemblies exerts ripple effects in the PM, with complex outcomes for the assembly and stability of remote lipid assemblies and hitherto-unexplored opportunities for signal integration.
MATERIALS AND METHODS
Materials.
Wild-type BHK cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% bovine calf serum (BCS), while wild-type CHO cells were grown in F-12K medium containing 10% fetal bovine serum (FBS). Mutant Cav-1KD BHK cells were generated as described in our previous studies (24, 29) and grown in DMEM-10% BCS containing 2 μg/ml puromycin. The mutant CHO cell line PSA-3 was a generous gift of Tomohiko Taguchi (University of Tokyo, Japan) and was grown in F-12K medium–10% FBS containing 10 μM ethanolamine. Low PS levels in PSA-3 cells were achieved by growing the cells in F-12K medium containing dialyzed FBS (D-FBS) for at least 72 h. Different PS levels were manipulated by exposing PSA-3 cells suspended in F-12K medium–10% D-FBS to various doses of ethanolamine (2 to 10 μM) and supplementing CHO cells suspended in F-12K medium–10% FBS with 1 to 10 μM ethanolamine. Green fluorescent protein (GFP)-LactC2 was a generous gift of Sergio Grinstein (The Hospital for Sick Children, Toronto, Canada). Guangwei Du (University of Texas Health Science Center, Houston, TX) provided GFP-Spo20, GFP-pleckstrin homology domain (PH)-phospholipase C δ (PLCδ), and GFP-PH-Akt; Tamas Balla (National Institute of Child Health and Human Development, Bethesda, MD) kindly provided GFP-FAPP1. Latrunculin A was purchased from EMD Millipore (Billerica, MA).
Immuno-EM spatial mapping. (i) Univariate K function analysis.
Immuno-electron microscopy (immuno-EM) was performed as described previously. Briefly, BHK cells were transiently transfected with a GFP-tagged protein of interest overnight. Intact-cell PM sheets were attached to EM grids, washed, fixed with 4% paraformaldehyde (PFA) and 0.1% glutaraldehyde, labeled with 4.5-nm gold particles linked to anti-GFP antibody, and embedded in uranyl acetate. Digital images of the PM sheets were obtained using a JEOL JEM-1400 transmission EM at ×100,000 magnification. A 1-μm2 area on a PM sheet was identified, and the x and y coordinates of gold particles were determined using ImageJ. The gold particle distribution and the extent of nanoclustering were calculated by using Ripley's K function, as shown in equations 1 and 2,
| (1) |
| (2) |
where K(r) is the univariate K function for a pattern of n points in an area (A), r is the radius at which K(r) is calculated (we evaluate 1 < r < 240 nm at 1-nm increments), ‖xi − xj‖ is Euclidean distance; 1(‖xi − xj‖ ≤ r) is the indicator function (this takes a value of 1 if ‖xi − xj‖ is ≤r and 0 otherwise), and wij−1 is the proportion of the circumference of the circle with center xi and radius ‖xi − xj‖ contained within A (this term builds in an unbiased edge correction for points at the edge of the study area). L(r) − r is a linear transformation of K(r). Here L(r) − r is standardized on the basis of the 99% confidence interval estimated from Monte Carlo simulations. Under the null hypothesis of complete spatial randomness, L(r) − r has an expected value of 0 for all values of r and thus can be used to quantify the extent of clustering of gold particles. For each condition in this study, 12 to 15 PM sheets were collected, imaged, and analyzed. Bootstrap tests were used to determine statistical differences between replicated point patterns. These bootstrap tests were constructed exactly as described previously (20, 25), and statistical significance was evaluated against 1,000 bootstrap samples.
(ii) Bivariate K function analysis.
The PM sheets of BHK cells coexpressing two different proteins tagged with GFP or red fluorescent protein (RFP) were attached to EM grids, washed, fixed, labeled with 2-nm gold particles coupled to anti-RFP antibody and 6-nm gold particles coupled to anti-GFP antibody, and embedded in uranyl acetate. Gold particle distribution was analyzed using a bivariate K function that characterizes coclustering or colocalization of the two different populations of gold particles (equations 3 to 6),
| (3) |
| (4) |
| (5) |
| (6) |
where the bivariate estimator, Kbiv(r), comprises two bivariate K functions. Kbs(r) maps the distribution of the big gold particles (b) with respect to each small gold particle (s), while Ksb(r) characterizes the distribution of the small gold particles with respect to each big gold particle. Area A contains nb 6-nm gold particles and ns 2-nm small gold particles. Other notations are as shown for equations 1 and 2. Lbiv(r) − r is a linear transformation of Kbiv(r), and we standardize Lbiv(r) − r on the basis of the 95% confidence interval estimated from Monte Carlo simulations. Under the null hypothesis that there is no spatial interaction between the two populations of gold particles, Lbiv(r) − r has an expected value of 0 for all values of r and can therefore be used to quantify the extent of coclustering of the two populations of gold. As a summary statistic, the Lbiv(r) − r function is standardized (Std) on the basis of the 95% confidence interval and the function is integrated over a fixed range to derive the Lbiv(r) − r integrated (LBI), as follows:
| (7) |
For each condition, 12 to 15 PM sheets were collected, imaged, and analyzed. Data are presented as means ± standard errors of the means (SEM). The LBI statistic is, however, not normally distributed; thus, nonparametric Mann-Whitney tests were used to examine the statistical significance of differences between median LBI values of replicated data sets.
FLIM combined with fluorescence resonance energy transfer (FRET).
BHK cells were grown to ∼65% confluence on glass coverslips and transiently transfected with either GFP-tagged protein alone or with both GFP-tagged and RFP-tagged proteins overnight. Cells were then washed and fixed in 4% PFA. The lifetime measurements were performed using a Lambert Instruments (Roden, the Netherlands) fluorescence lifetime imaging microscopy (FLIM) module mounted on a wide-field Nikon Eclipse microscope. GFP was excited using a sinusoidally simulated, modulating 3-watt 497-nm light-emitting diode (LED) at 40 MHz under epi-illumination coupled with a 60× Plan-Apo/1.4-numerical-aperture (NA) oil immersion lens. Three independent experiments were performed under each condition. At least 60 cells were imaged, lifetime values were pooled and averaged, and statistical analysis was performed using one-way analysis of variance (ANOVA).
FRAP.
The TopFluor-PS (TF-PS) labeling protocol and fluorescence recovery after photobleaching (FRAP) protocol followed those in previous studies (26–28), except that cells were allowed to equilibrate for 1 h after being pulse-labeled. Wild-type BHK cells or BHK-Cav-1KD cells were seeded on fibronectin-coated glass-bottom dishes overnight. Approximately 20 μg of TF-PS dissolved chloroform (1 mg/ml) was purged with N2 to evaporate the chloroform and placed under vacuum overnight to completely eliminate residual chloroform. On the day of experiments, 4 ml of ice-cold phosphate-buffered saline (PBS) containing 0.06% bovine serum albumin (BSA) was used to rehydrate the TF-PS film, and the suspension was sonicated in a bath sonicator for 30 min and then cooled to 4°C. Cells were washed with ice-cold PBS-BSA twice, incubated in ice-cold PBS-BSA containing ∼5 μg/ml TF-PS for 10 min at 4°C, and then washed 3 times with ice-cold PBS. BHK cells were placed in a 37°C incubator for ∼1 h to allow equilibration of the fluorescent TF-PS among various cell compartments. All FRAP experiments were performed at room temperature. Wild-type BHK cells and BHK-Cav-1KD cells were directly used for FRAP experiments, while latrunculin (LA) treatment was conducted on the wild-type BHK cells (1 μM LA for 5 min) before FRAP. Cells were imaged using a 60× total internal reflection fluorescence (TIRF)/1.49-NA oil immersion lens mounted on a Nikon A1 confocal microscope. Bleaching was accomplished using a 405-nm diode laser at 38 mW at 100% power, and TF-PS fluorescence intensity was monitored using an argon multiline 458/477/488/514-nm laser. Three regions of interests (ROIs) were used: a bleached region, a reference region, and a background region. In each FRAP experiment, fluorescence intensity in 3 ROIs was monitored simultaneously for 5 s to equilibrate the intensity. The bleached region on the cell PM was then bleached for 5 s using maximal laser power followed by continuous monitoring of fluorescence intensity in all 3 ROIs for 60 s. The raw fluorescence intensity data were then normalized as follows (28),
| (8) |
where F(t)nom is the normalized fluorescence intensity, F(t)ROI is the intensity within the bleached ROI, F(t)Ref is the intensity of the reference area on PM that is not bleached, and F(t)bkgd is the background intensity. Single exponential equations were then fitted to the recovery curves (28) using Prism software and the equation
| (9) |
where F(t)0 is the normalized intensity at time zero, Q is the mobile fraction, K is the rate constant, and t is time. Diffusion coefficients were then estimated (28) as in equation 10,
| (10) |
where D is the diffusion constant and w is the width of the bleached strip. Approximately 20 cells under each condition were imaged. Normalized fluorescence intensities were averaged and used to plot the complete recovery curves, shown as means ± SEM. Mobile fraction and diffusion coefficients were pooled and are shown as means ± SEM. Statistical significance was evaluated by Student's t tests.
Cell signaling assays.
For examining the effects of H-Ras expression on K-Ras signaling, stable cell lines were generated, grown to ∼80% confluence in DMEM with 10% BCS, and harvested. Whole-cell lysates were used to blot against pMEK, pERK, and pAkt. To ensure equal levels of GFP–K-RasG12V, the stable cell lines were sorted by flow cytometry. Cells were incubated in serum-free DMEM with or without 10 μM MEK inhibitor U0126 for 24 h. Cells treated with U0126 were then washed with serum-free DMEM 3 times and incubated in serum-free DMEM for an additional 30 min to allow MEK and mitogen-activated protein kinase (MAPK) activation before being harvested.
For the cancer cell proliferation assay, ∼3,000 cells/well were seeded on day 1 in each 96-well plates, treated with Polybrene (hexadimethrine bromide) (10 μg/ml for 1 h), and infected at a mean multiplicity of infection (MOI) of ∼5:1 with lentiviruses encoding GFP-tH, GFP–H-RasG12V, and GFP–K-RasG12V. Lentivirus (TOPO cloned into the pLenti6.3-V5-TOPO vector) was produced by cotransfecting 293FT cells (Invitrogen) with a ViraPower packaging mix (Invitrogen) using Lipofectamine 2000. After overnight incubation with the virus (titers were estimated with Go-Stix; Clontech), cells were provided with fresh media (100 μl/well) and allowed to grow for 3 days. Cells were then washed with PBS, fixed with 4% PFA for 30 min, and stained with DAPI (4′,6-diamidino-2-phenylindole) at a 1:1,000 dilution in PBS for 10 min at room temperature and then imaged using a 10× Plan-Fluor/0.3-NA lens mounted on a Nikon A1R confocal microscope and excited using a 404-nm-diode, 38-mW laser. Three images were obtained for each well, and three separate experiments were conducted for each condition. At least 36 images were obtained per virus per cell line. DAPI-stained cells were counted using ImageJ and normalized against cell numbers of the corresponding cells expressing GFP-tH.
Basic assumptions and an extended general description of the ODE model.
We constructed a simple multicompartment model that could describe an initial complex spatial data set and predict the consequences of more-extensive perturbations of PM PS organization on K-Ras nanoclustering. This description should be read in conjunction with Tables S1 to S3 in the supplemental material, which define the system parameters, initial conditions, and ordinary differential equations (ODEs). Here, we refer to the various PM compartments in which PS can be distributed as pools. These pools are not necessarily physical entities but can be distributed pools or operational pools with different availabilities for nanocluster assembly. A majority of PS in the PM is immobile (∼60%) on the time and length scales (<30 s) of typical FRAP and fluorescence correlation spectroscopy (FCS) experiments (27); in the model, we assume that this immobile pool is unavailable for partitioning into lipid rafts or various Ras nanoclusters until it is released as a diffusing, mobile pool of PS (DFPS). Regulators of the immobile PS fraction include cortical actin (27) and caveolae (29). We realize these observations by defining actin-PS (APS) and caveolar-PS (CAVPS) pools and allowing exchange of PS between the APS and CAVPS compartments and the DFPS (equations 5, 6, and 7). We further assume that only a fraction of the mobile pool, called the reactive-PS (RPS) pool, is available for the assembly of transient lipid assemblies, including Ras nanoclusters. The implication for cell biology at this stage is somewhat speculative, but the conceptual idea is that not all diffusing PS is likely available on time and length scales required for lipid sorting or domain partitioning or that it is “occupied” with high-affinity counter-ions and therefore unable to interact with K-Ras polybasic-domain lysines, etc. The RPS pool is the sole source of PS for partitioning into lipid rafts and H-Ras nanoclusters and for the formation of K-Ras nanoclusters. Each of these lipid assemblies is realized as a PS pool. Since PS is not required for compartment integrity, the cholesterol-dependent lipid raft compartments (tH) and H-RasG12V (cholesterol-independent) (HGV) operate as passive partitioning compartments, and PS partitions into the compartments as a function of their sizes (equations 3, 4, and 6). In contrast, nanoclusters of polybasic-lipid-anchored proteins, such as K-Ras, are structurally dependent on PM PS (30, 31). In the model, this requirement is realized by having the K-Ras nanocluster (KCLUS) compartment formed by reaction with PS in the RPS pool (equation 1). Once formed, the KCLUS compartment operates like the tH and HGV compartments and exchanges PS with the RPS pool (equations 6 and 10) as a function of size. Thus, PS can appear in any of the three defined types of nanocluster compartments, as revealed by the bivariate EM data in Fig. 1, albeit through different mechanisms that attempt to recapitulate the membrane biochemistry.
FIG 1.
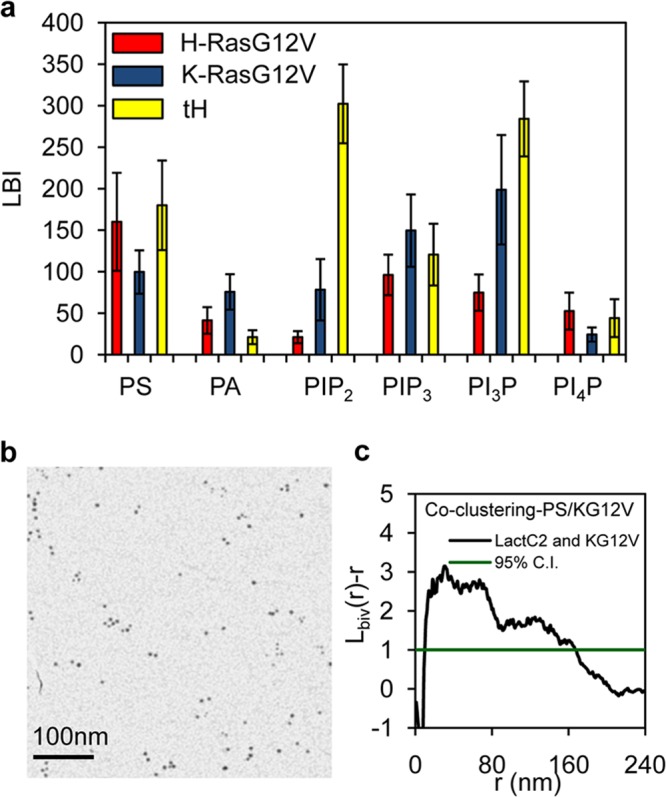
Ras nanoclusters have distinct lipidic compositions. (a) PM sheets prepared from BHK cells coexpressing GFP-LactC2 (to detect PS), GFP-Spo20 (PA), GFP-PH-PLCδ (PIP2), GFP-PH-Akt (PIP3), GFP-PH-FYVE (PI3P), or FAPP1-GFP (PI4P) with RFP–H-RasG12V, RFP–K-RasG12V, or RFP-tH (membrane anchor of H-Ras) were labeled with 2-nm anti-RFP gold particles and 6-nm anti-GFP gold particles and imaged by EM, and the relative distributions of the two gold populations were analyzed using bivariate K functions. The results are summarized as LBI values. The greater the LBI value, the greater the extent of coclustering of the two probes. (b) Sample EM image of 6-nm anti-GFP gold particles and 2-nm anti-RFP gold particles on a PM sheet coexpressing GFP-LactC2 and RFP–K-RasG12V. (c) Bivariate K function analysis of coclustering between the 6-nm and 2-nm gold particles shown in panel b (a 1-μm by 1-μm PM sheet from the experiment whose results are shown in panel b was used). 95% C.I., 95% confidence interval for a random pattern. To obtain the LBI value, the area under the Lbiv(r) − r curve above the confidence interval is calculated as described in the text.
K-Ras proteins present in the system (KMON), in addition to forming nanoclusters directly (equation 1), partition into the HGV compartment in proportion to the total PS content of the HGVPS pool (equation 8); however, this partitioning is constrained over a central range of PS levels to affect the region of effective lateral segregation observed by experimentation. This constraint is implemented as the FC1 variable (see Table S1 in the supplemental material). Certain other well-defined control loops are implemented. Actin positively regulates K-Ras nanoclustering, which is realized using the FV1 variable (see Table S1) as a multiplier of the reaction constants in equations 1 and 2. Actin and caveolin positively regulate lipid raft formation (11, 29), which is implemented by letting the size of the cholesterol-dependent compartment (tH) be regulated by the size of the actin and caveolar compartments (see Table S1). Two other roles of caveolae are implemented to recapitulate biology. First, caveolar depletion enhances the formation of cortical actin, and therefore, in the model, the size of the caveolar compartment negatively regulates the size of the actin compartment (32) (see Table S1). Second, recent work has shown that caveolae maintain the lateral segregation of lipid raft domains from H-RasG12V domains, which is partially realized through the FC2 variable (see Table S1). The model was fully described as a set of ODEs (see Table S3 in the supplemental material), compiled using the Berkeley Madonna v9.0.109 modeling program and simulated with a Runge-Kutta-4 protocol and a time step of 0.0001. The total simulation time used, 200 (arbitrary units), was sufficient for all iterations of the model to reach equilibrium. Initial conditions and rates were determined empirically to reproduce basal K-Ras clustering (see Table S1). Other output parameters are described and listed in Table S2 in the supplemental material. The total amount of K-Ras in the model system was fixed for the simulations in this report, but the sizes of all of the actin, caveolar, and cholesterol-independent compartments and the PS in the system were varied, as described below.
RESULTS
Ras nanoclusters have different lipid compositions, but all contain phosphatidylserine.
To examine whether Ras isoforms differentially sort membrane lipids, we used lipid-binding or PH probes to spatially map the distributions of PS (GFP-LactC2) (27, 31), phosphatidic acid (PA) (GFP-PH-Spo20) (33), phosphatidylinositol 4,5-bisphosphate (PIP2) (GFP-PH-PLCδ) (34), phosphatidylinositol (3,4,5)-trisphosphate (PIP3) (GFP-PH-Akt) (35), phosphatidylinositol 3-phosphate (PI3P) (GFP-PH-FYVE) (36), and phosphatidylinositol 4-phosphate (PI4P) (GFP-FAPP1) (37) with respect to Ras nanoclusters on intact PMs (Fig. 1a). BHK cells were cotransfected with each GFP-tagged lipid probe and RFP–H-RasG12V (a constitutively GTP-bound oncogenic mutant H-Ras), RFP–K-RasG12V (a constitutively GTP-bound oncogenic mutant K-Ras), or RFP-tH. RFP-tH, which is targeted to the PM by the minimal membrane-anchoring domain of H-Ras (tH consists of C-terminal amino acids 180 to 189), comaps extensively with H-Ras–GDP; thus, RFP-tH nanoclusters can be used as surrogates for cholesterol-dependent H-Ras–GDP nanoclusters (21). To quantify coclustering and hence colocalization of each lipid probe with each type of Ras nanocluster, apical PM sheets were attached to EM grids and labeled with anti-RFP 2-nm gold particles and anti-GFP 6-nm gold particles. The grids were imaged using EM, and the congruence of the two gold particle spatial patterns was quantified using bivariate K functions [Lbiv(r) − r] (Fig. 1b and c). To more readily compare the extent of colocalization of each lipid probe with that of each Ras nanocluster, we integrated the Lbiv(r) − r function, to yield the statistic Lbiv integrated, or LBI. Thus, in this set of experiments, LBI is a parameter that provides a quantitative measure of the extent of clustering of a given lipid probe with that of a given Ras nanocluster. The LBI values summarized in Fig. 1a therefore show that PS, as detected by GFP-LactC2, associated extensively with all Ras clusters tested. In contrast, PA associated with K-RasG12V and H-RasG12V but minimally with RFP-tH nanoclusters. PIP2 was extensively enriched in RFP-tH nanoclusters. PIP3 associated with all Ras nanoclusters, while PI3P was most extensively associated with RFP-tH. PI4P comapped with RFP–H-RasG12V and RFP-tH but minimally with RFP–K-RasG12V. These results clearly show that the lipid compositions of H-Ras–GTP (H-RasG12V), H-Ras–GDP (RFP-tH), and K-Ras–GTP (K-RasG12V) nanoclusters are markedly different, indicating that lateral Ras segregation is accompanied by lipid sorting to generate distinct lipid assemblies.
To visualize the overall spatial distribution of PS (Fig. 2a and b), apical PM sheets from BHK cells coexpressing GFP-LactC2 and each RFP-Ras protein were attached to EM grids and labeled with anti-GFP 4.5-nm gold particles. The grids were imaged using EM and the gold particle spatial patterns analyzed using univariate K functions expressed as L(r) − r (Fig. 2c and d). The maximum value of the L(r) − r function (Lmax) quantifies the extent of clustering of the point pattern (11). In this set of experiments, the expression of both RFP-tK (where tK is the minimal membrane anchor of K-Ras, specifically, C-terminal amino acids 175 to 188) and RFP–K-RasG12V increased GFP-LactC2 Lmax values, indicating increased GFP-LactC2 and hence PS clustering, whereas expression of RFP–H-RasG12V significantly decreased GFP-LactC2 Lmax values and expression of RFP-tH had no effect (Fig. 2a and b). Since overall gold labeling of GFP-LactC2 was unchanged (not shown), we conclude that Ras expression can change the lateral distribution but not the PS content of the PM. Specifically, expression of polybasic-lipid-targeted Ras or isolated polybasic anchors increases PS clustering on the PM, likely by sequestering PS at high stoichiometry for nanocluster formation, whereas expression of H-RasG12V reduces PS clustering, presumably by displacing PS from sites of higher to lower stoichiometry. In summary, these experiments show that PS associates extensively with all Ras nanoclusters and that the lateral distribution of PS is differentially sensitive to ubiquitously expressed Ras proteins.
FIG 2.
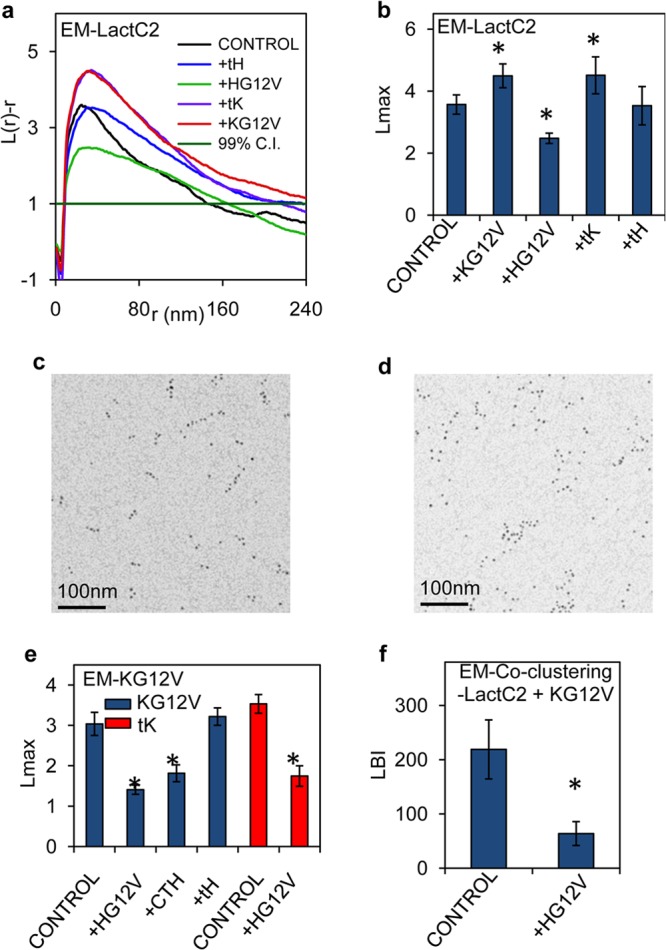
The presence of H-Ras–GTP disrupts nanoclustering of PS and K-Ras–GTP. (a) PM sheets prepared from BHK cells expressing GFP-LactC2 with or without RFP-tH, RFP–H-RasG12V, RFP-tK, or RFP–K-RasG12V were labeled with 4.5-nm anti-GFP gold particles. The spatial distribution of the gold particles visualized by EM was analyzed using univariate K functions. Plots of the weighted mean standardized univariate K functions are shown. (b) A useful summary statistic is the peak value, Lmax, of the univariate K function. Lmax values from the GFP-LactC2 analysis whose results are shown in panel a are means ± SEM. (c and d) Sample EM images of anti-GFP gold particles on a PM sheet from BHK cells expressing GFP-LactC2 alone (c) or GFP-LactC2 with RFP–K-RasG12V (d). (e) Univariate K function analysis as described for panel b, shown as mean Lmax values (±SEM), to evaluate the nanoclustering of GFP–K-RasG12V (KG12V) in BHK cells expressing GFP–K-RasG12V alone (control) or coexpressing RFP–H-RasG12V (+HG12V), RFP-CTH (±CTH), or RFP-tH (±tH). Similar experiments were performed to analyze the nanoclustering of GFP-tK (tK), expressed alone (control) or with RFP–H-RasG12V (±HG12V). Statistical significance of differences between the univariate K functions in panels b to e was evaluated in bootstrap tests (*, P < 0.05). (f) PM sheets prepared from BHK cells expressing GFP-LactC2 and RFP–K-RasG12V alone (control) or with HA-tagged H-RasG12V (+HG12V) were used to evaluate GFP-LactC2 and RFP–K-RasG12V coclustering as in a bivariate EM analysis. Statistical significance of differences between the bivariate K functions, shown as mean LBI values (±SEM), was examined using a Mann-Whitney test (* indicates a P of <0.05).
H-RasG12V expression inhibits K-RasG12V nanoclustering.
Both K-Ras PM binding and nanoclustering are dependent on PM PS content (12, 15). We therefore used EM and univariate K functions to examine whether the changes in PS distribution induced by H-RasG12V expression (shown in Fig. 2b) were sufficient to change K-Ras nanoclustering. Expression of RFP–H-RasG12V caused a significant decrease in the Lmax of GFP–K-RasG12V, indicating reduced GFP–K-RasG12V nanoclustering (Fig. 2e). In contrast, coexpression of RFP-tH, which had no effect on PM PS distribution (Fig. 2b), had no effect on GFP–K-RasG12V clustering (Fig. 2e). Unlike with the minimal H-Ras membrane anchor tH, RFP targeted to the PM by the complete H-Ras HVR (RFP-CTH [C-terminal amino acids 166 to 189]) extensively colocalizes with H-RasG12V (21), suggesting that the lipid-sorting properties of RFP-CTH closely mirror those of activated H-Ras–GTP. Concordantly, coexpression of RFP-CTH also disrupted GFP–K-RasG12V clustering (Fig. 2e). Since expression of signaling-incompetent RFP-CTH had an effect on K-RasG12V nanoclustering similar to that of RFP–H-RasG12V, we conclude that the mechanism is independent of H-Ras signal output and is likely mediated by changes in lateral PM lipid organization, induced by the H-Ras HVR. Expression of RFP–H-RasG12V also significantly inhibited the nanoclustering of the signaling-incompetent GFP-tK (Fig. 2e) that colocalizes extensively with K-RasG12V in PS-dependent nanoclusters (21), further suggesting a membrane lipid-mediated mechanism. Conversely, the nanoclustering of GFP–H-RasG12V was unaffected by expression of RFP–K-RasG12V (not shown). We refer to the regulation of K-Ras lipid assemblies by spatially segregated H-RasG12V as spatial cross talk. Given the importance of PS to K-Ras clustering, the spatial cross talk between H-Ras and K-Ras potentially originates from the ability of H-Ras to competitively bind with PS. Indeed Fig. 2f shows that expression of hemagglutinin-tagged H-RasG12V (HA–H-RasG12V) markedly decreased the LBI value of GFP-LactC2 and RFP–K-RasG12V, indicating that H-Ras–GTP disrupts the interaction between PS and K-Ras. In these experiments, taking into account the known capture ratio of the anti-GFP antibody (11), the GFP-Ras levels expressed on the PM as determined by anti-GFP gold particle labeling are ∼20% of the levels of endogenous H-Ras and K-Ras in BHK cells measured previously (13). Spatial cross talk is therefore not a consequence of high-level ectopic expression.
To validate the EM analysis, we used FLIM-FRET imaging to monitor K-RasG12V nanoclustering in intact cells. FRET between GFP– and RFP–K-RasG12V molecules in the same nanoclusters significantly decreased GFP fluorescent lifetime from that measured in control cells (Fig. 3a and b). Coexpression of HA–H-RasG12V increased GFP fluorescent lifetime, indicating reduced FRET between GFP– and RFP–K-RasG12V molecules, consistent with an inhibitory effect on K-RasG12V nanoclustering (Fig. 3a and b). Similarly, expression of HA–H-RasG12V significantly decreased coclustering between GFP-LactC2 with RFP–K-RasG12V, as reported by an increase in the lifetime of GFP-LactC2 (Fig. 3c and d), indicating reduced FRET. To directly monitor K-Ras and PS interactions, we measured FRET between fluorescent TopFluor-PS (TF-PS) and RFP–K-RasG12V. The fluorescent lifetime of TF-PS in untransfected BHK cells significantly decreased in the presence of RFP–K-RasG12V (Fig. 3e and f), indicating FRET between TF-PS and RFP–K-RasG12V due to K-Ras and PS interaction. The reduced fluorescent lifetime of TF-PS in RFP–K-RasG12V-expressing cells significantly increased when HA–H-RasG12V was coexpressed (Fig. 3e and f), indicating a reduced association between TF-PS and K-Ras in the presence of H-Ras. Concordant data from EM and FLIM imaging therefore confirm that H-Ras nanoclusters sequester PS to attenuate the formation of K-Ras nanoclusters.
FIG 3.

H-Ras expression inhibits K-Ras coclustering with PS on the PM. BHK cells expressing GFP–K-RasG12V alone (GFP-KG12V), GFP–K-RasG12V and RFP–K-RasG12V (GFP/RFP-KG12V), or GFP–K-RasG12V, RFP–K-RasG12V, and HA–H-RasG12V (GFP/RFP+HA-HG12V) were imaged in a FLIM. Representative images (a) and mean GFP lifetimes (calculated per cell) ± SEM (n ≥ 60 cells from 3 independent experiments) (b) are shown. BHK cells expressing GFP-LactC2 alone (GFP-LactC2), GFP-LactC2 and RFP–K-RasG12V (GFP-C2/RFP-Ras), or GFP-LactC2, RFP–K-RasG12V, and HA-tagged H-RasG12V (GFP/RFP+HA-HG12V) were imaged in a FLIM. Representative images (c) and mean GFP lifetimes (calculated per cell) ± SEM (n ≥ 60 cells from 3 independent experiments) (d) are shown. Control BHK cells (TF-PS) and BHK cells expressing RFP–K-RasG12V or coexpressing RFP–K-RasG12V and HA–H-RasG12V were flash-labeled with TF-PS and imaged in a FLIM. Representative images (e) and mean TF-PS lifetimes (calculated per cell) ± SEM (n ≥ 60 cells from 3 independent experiments) (f) are shown. The statistical significance of differences between mean lifetime values was evaluated using one-way ANOVA (*, P < 0.01).
PS mediates lateral segregation of Ras nanoclusters.
To further investigate the role of PS in mediating spatial cross talk between different Ras isoforms, we manipulated PM PS levels using a CHO mutant cell line, PSA-3, which lacks PS synthase 1 (PSS1) and is an ethanolamine auxotroph (38). Interestingly, the level of PS on the PMs of parental CHO cells was also dependent on the ethanolamine content of the culture media (Fig. 4a). Parallel cultures of PSA-3 and CHO cells expressing GFP–K-RasG12V or GFP–H-RasG12V were grown in various ethanolamine concentrations, and the extent of nanoclustering was measured by EM. The results show that GFP–K-RasG12V clustering is linearly dependent on PM PS content (Fig. 4b) but that GFP–H-RasG12V clustering is independent of PM PS content (Fig. 4c).
FIG 4.

K-Ras and H-Ras lateral segregation is dependent on PS level in the PM. (a) PSA-3 and CHO cells expressing GFP-LactC2 were grown in media supplemented with 0 to 10 μM ethanolamine for 72 h. PM sheets prepared from the cells were attached to EM grids, fixed, and labeled with 4.5-nm anti-GFP gold particles. After EM, the number of gold particles per μm2 was counted. PM sheets were prepared from PSA-3 and CHO cells expressing GFP–K-RasG12V (b) or GFP–H-RasG12V (c) and grown under identical conditions as described for panel a. After labeling with 4.5-nm anti-GFP gold particles and EM imaging, univariate K functions were used to quantify the extent of K-Ras and H-Ras clustering. Mean Lmax values ± SEM from these experiments are plotted against the number of GFP-LactC2 gold particles per μm2 (proportional to the inner-leaflet PS content) as measured in panel a. (d) PM sheets prepared from PSA-3 and CHO cells expressing both GFP–H-RasG12V and RFP–K-RasG12V and cultured in different concentrations of ethanolamine exactly as described for panel a were labeled with 2-nm anti-RFP and 6-nm anti-GFP gold particles. Heterotypic coclustering between H-Ras and K-Ras was quantified using bivariate K functions, summarized as mean LBI values ± SEM. The LBI values of H-RasG12V and K-RasG12V for each ethanolamine condition are plotted against the corresponding GFP-LactC2 (PS) gold particle density measured in panel a. (e) Immunogold-labeled PM sheets prepared from CHO or PSA-3 cells expressing GFP–K-RasG12V alone (control) or GFP–K-RasG12V and RFP–H-RasG12V (+HG12V). PSA-3 cells were grown in 10 μM ethanolamine (+Etn) or without it. After EM imaging, the extent of GFP–K-Ras clustering was quantified using univariate K functions, shown as mean Lmax values ± SEM. The statistical significance of differences between values for the univariate K functions was evaluated in bootstrap tests (*, P < 0.05).
We next measured coclustering between GFP–H-RasG12V and RFP–K-RasG12V in PSA-3 and CHO cells using bivariate EM. Figure 4d shows that LBI values change nonlinearly as a function of PS level in the PM. Low LBI values, which correspond to efficient lateral segregation between GFP–H-RasG12V and RFP–K-RasG12V, were observed at PS levels between 350 and 410/μm2 of PM (gold particle labeling) (Fig. 4d). These optimal PS levels were concurrent in wild-type CHO cells and PSA-3 cells supplemented with 10 μM ethanolamine (Fig. 4a). Outside this optimal PS range, at both higher and lower PS levels, LBI values were significantly higher (Fig. 4d), indicating extensive coclustering of H-Ras and K-Ras. Strikingly, these data show that efficient lateral segregation of H-Ras and K-Ras into spatially distinct lipid domains is feasible only over a relatively narrow range of PS concentrations on the PM. If PS levels fall outside this optimal range, the Ras isoforms no longer laterally segregate and form heterotypic clusters. This behavior is analogous with the ability of cholesterol to induce nonlinear lipid demixing in synthetic model bilayers (39–41) and likely correlates with the ability of negatively charged lipids, such as PS and PIP2, to also induce highly efficient domain separation (42, 43).
One prediction from this set of results is that at low PM PS levels, when both K-Ras homotypic nanoclustering and the fidelity of Ras lateral segregation are compromised (Fig. 4b), coexpression of H-RasG12V should paradoxically increase K-Ras nanoclustering by generating PS-independent nanoclusters into which K-Ras proteins can segregate. We found that in wild-type CHO cells, H-Ras disrupted K-Ras clustering (Fig. 4e), as was observed in BHK cells in Fig. 2e. In contrast, in PSA-3 cells with reduced PS levels, H-Ras enhanced K-Ras clustering (Fig. 4e). However, on supplementation with ethanolamine to return PS levels to the optimal level, the ability of H-Ras to perturb K-Ras nanoclustering was recovered (Fig. 4e), clearly illustrating that the PM PS level determines the nature of the spatial cross talk between Ras isoforms.
System modeling identifies complex contributions of PS to Ras nanoclustering.
Combining the results in Fig. 1 to 4 with recent studies on PS PM dynamics, we constructed a computational model to account for our initial observations and predict the consequences on Ras nanoclustering of more-extensive perturbations of PM PS organization (Fig. 5a). The majority of the PS in the PM is immobile (∼60%) on the time and length scales (<30 s) of FRAP and FCS experiments (27). Regulators of this PS immobile fraction include cortical actin (27) and caveolae (29). In the model, we therefore define a mobile pool of PS and allow exchange of PS between the actin- and caveola-regulated PS immobile compartments and the mobile pool (Fig. 5a). A fraction of the mobile pool, called the reactive pool, is deemed available for the assembly of transient lipid assemblies, including Ras nanoclusters. Nanoclusters of polybasic lipid-anchored proteins, such as K-Ras, are structurally dependent on PM PS (30, 31). In the model, this requirement is realized by having polybasic nanoclusters formed by reaction with PS. Conversely, for lipid raft (cholesterol-dependent) and H-RasG12V (cholesterol-independent) nanoclusters, PS passively partitions into the compartments as a function of their size, but PS is not required for compartment integrity. Thus, PS can appear in any of the three defined types of nanocluster compartments, as revealed by the bivariate EM data in Fig. 1, albeit through different mechanisms that recapitulate the membrane biochemistry (Fig. 5a). K-Ras, in addition to forming nanoclusters directly, is allowed to partition into cholesterol-independent nanocluster compartments in proportion to the total PS content of that compartment, but this partitioning is constrained over a central range of PS levels to implement a region of effective lateral segregation. The total amount of K-Ras in the model system is fixed, but the sizes of the actin, caveolar, cholesterol-dependent, and cholesterol-independent compartments and the PS in the system can vary. Certain well-defined control loops are implemented. Actin positively regulates K-Ras nanoclustering and the size of the cholesterol-dependent compartment (11, 44). Caveolae positively regulate the size of the cholesterol-dependent compartment (29) and negatively regulate the size of the actin compartment (32). A fuller description of the model with a more extensive discussion of assumptions and justifications is given in Materials and Methods. The model was compiled as a set of ODEs, and initial conditions and rates were determined empirically to reproduce basal K-Ras clustering (Fig. 5b; see Tables S1 to S3 in the supplemental material).
FIG 5.

An ODE model for K-Ras clustering replicates experimental observations. (a) Flow diagram of a mathematical model of K-Ras clustering and PS partitioning on the PM. A full description is given in the text and in Materials and Methods. (b) Simulation of the model under control conditions, low actin levels (actin molecules = 200), or low caveolar levels (caveolae = 20) in the absence of H-Ras (HGV molecules = 1) as a function of total PS in the system. The graph shows the consequences for K-Ras nanoclustering. These results recapitulate biology. (c) K-Ras homotypic clustering changes approximately linearly with PS levels (blue line). Bivariate coclustering between K-Ras and H-Ras changes nonlinearly as a function of PS (red curve). Homotypic K-Ras clustering in the presence of H-Ras becomes nonlinear (purple curve). Overall total K-Ras univariate clustering becomes nonlinear because of heterotypic mixing with H-Ras (green curve).
In the model, K-Ras homotypic clustering (K-Ras clusters comprised only of K-Ras proteins) increases approximately linearly as a function of the PS level in the system (Fig. 5b and the blue control curve in Fig. 5c), consistent with the result in Fig. 4b. The model also replicates the PS-dependent K-Ras/H-Ras coclustering shown in Fig. 4d (red curve in Fig. 5c). In this context, two aspects of K- and H-Ras interactions are visible in the simulations. The introduction of H-Ras into the system decreases K-Ras homotypic clustering over all PS levels (Fig. 5c, purple curve) as a result of PS sequestration by H-Ras clusters. However, increased K-Ras/H-Ras coclustering outside the optimal range of PS levels exceeds the suppression of K-Ras homotypic clustering. Thus, aggregated K-Ras univariate clustering (which is the combination of K-Ras molecules found in homotypic K-Ras clusters and heterotypic K-Ras/H-Ras clusters) in the presence of H-Ras is a nonlinear function of PS (Fig. 5c, green curve); within the optimal PS range, H-Ras suppresses, and outside the optimal PS range, H-Ras enhances K-Ras clustering. Note that the aggregated K-Ras clustering is what is experimentally measured by a univariate mapping protocol. These simulations replicate all of the observations in Fig. 1 to 4. Thus, a simple PS distribution-driven model system can potentially describe a complex biology.
PS lateral distribution between mobile and immobile pools is dynamically regulated in the PM (27, 45). To predict how changing PS mobile pool size may affect K-Ras nanoclustering and K-Ras/H-Ras segregation, we next simulated the model under conditions of reduced actin or reduced caveolae. These manipulations de facto increase and decrease, respectively, the fraction of PS in the mobile pools available for sorting into nanoclusters (Fig. 6a). With only K-Ras present in the system, actin disruption markedly decreased K-Ras homotypic clustering, whereas eliminating caveolae significantly enhanced K-Ras homotypic clustering (5a and Fig. 6b), concordant with previous experiments (11, 29). The presence of H-Ras significantly decreased the overall K-Ras clustering under control conditions but enhanced overall K-Ras clustering when the actin compartment or caveolar compartment was reduced (Fig. 6b). Under both conditions, this enhanced clustering is due to extensive colocalization of K-Ras in H-Ras clusters, resulting in the formation of heterotypic K-Ras and H-Ras clusters (Fig. 6b, red bars).
FIG 6.

Actin effects on K-Ras clustering are mediated through PS dynamics. (a) Equilibrium distributions of PS in the various model compartments under the conditions of low levels of actin or low numbers of caveolae (as in Fig. 5b) with (HGV = 150) and without (HGV = 1) H-Ras in the simulation. The PS compartments are defined in Tables S1 and S2 in the supplemental material. (b) Simulated K-Ras clustered fractions under conditions of low levels of actin or low numbers of caveolae (i.e., high actin) with or without H-Ras (as described for panel a). Diffusion of TopFluor-PS was measured using FRAP in untreated wild-type BHK cells (CTL), BHK cells treated with 1 μM latrunculin (LA), or BHK-Cav-1KD cells (Cav-1KD). Complete recovery curves (c) and the calculated mobile fraction (d) of TopFluor-PS for each condition are shown as means ± SEM. Statistical significance was evaluated using one-way ANOVA (*, P < 0.05).
Changing the level of mobile PS disrupts lateral segregation between H-Ras and K-Ras.
To test the predictions of the model in Fig. 6a and b, we performed equivalent experiments in vivo. BHK cells were briefly treated with latrunculin to disrupt cortical actin and to release actin-immobilized PS into a mobile pool (27, 45). An increase in mobile PS was confirmed by FRAP experiments on BHK cells, where the TF-PS mobile fraction increased by ∼31% after a 5-min treatment with latrunculin (Fig. 6c and d). As predicted from the modeling, the bivariate coclustering parameter LBI between GFP–H-RasG12V and RFP–K-RasG12V was markedly higher in latrunculin-treated cells than in untreated cells (Fig. 7a), reflecting significant coclustering of K-Ras and H-Ras in the presence of the increased mobile PS pool. Knocking down Cav-1 in BHK-Cav-1KD cells yielded similarly elevated LBI values, illustrating that limiting the PS mobile pool also enhanced coclustering between K-Ras and H-Ras (Fig. 7a) and validating a second model prediction. A decrease in the PS mobile pool in Cav-1KD cells was confirmed in FRAP experiments; the mobile fraction of TF-PS was ∼28% lower in BHK-Cav-1KD than in wild-type BHK cells (Fig. 6c and d).
FIG 7.

Spatial cross talk between K-Ras and H-Ras depends on PS availability in the PM. (a) PM sheets prepared from wild-type BHK cells (WT), wild-type BHK cells treated with 1 μM latrunculin for 5 min (LA), and Cav-1KD BHK cells (Cav-1KD) expressing both GFP–H-RasG12V and RFP–K-RasG12V were labeled with 2-nm anti-RFP and 6-nm anti-GFP gold particles. Heterotypic coclustering between H-Ras and K-Ras was quantified using bivariate K functions, summarized as mean LBI values ± SEM. Similar experiments were conducted on cells coexpressing GFP-LactC2 and RFP–H-RasG12V, and the extent of coclustering of PS and H-RasG12V was quantified using bivariate K functions summarized as mean LBI values ± SEM. Statistical analysis was performed using Mann-Whitney tests (*, P < 0.05). (b) After treatment with 1 μM latrunculin for 5 min (LA), PM sheets were prepared from BHK cells expressing GFP–K-RasG12V alone (control), GFP–K-RasG12V and RFP–H-RasG12V (+HG12V), or GFP–K-RasG12V and RFP-CTH (+CTH). The sheets were immunogold labeled, and after EM imaging, the extent of GFP–K-Ras clustering was quantified using univariate K functions, shown as mean Lmax values ± SEM. Similar experiments were conducted in Cav1-KD cells. (c) BHK cells expressing GFP–K-RasG12V alone (white bar) and BHK cells or Cav-1KD cells coexpressing GFP–K-RasG12V and RFP–H-RasG12V (blue bars) were analyzed in FLIM experiments. Where indicated, measurements were also made on BHK cells treated with 1 μM latrunculin for 5 min (+LA). For a control, reference homotypic K-Ras clustering was examined in BHK cells coexpressing GFP–K-RasG12V and RFP–K-RasG12V (yellow bar). Each GFP fluorescence lifetime value is averaged for a single cell. Data were collected from multiple cells and are shown as means ± SEM (n ≥ 60 cells from 3 independent experiments). Statistical significance was evaluated using one-way ANOVA (*, P < 0.001). (d) Representative FLIM images of the BHK cells analyzed in panel c.
A key expectation is that the PS content of H-Ras nanoclusters should increase under both of these experimental conditions as part of the mechanism that drives increased coclustering with K-Ras. This was directly observed as a significant increase in the bivariate coclustering parameter, LBI, between LactC2 (to detect PS) and H-RasG12V in latrunculin-treated and Cav-1KD cells (Fig. 7a) and was indirectly observed as an increase in overall K-Ras univariate clustering, reflecting the increase in the number of K-Ras proteins missorting to H-RasG12V nanoclusters (Fig. 7b). Enhanced coclustering between K-Ras and H-Ras was also observed in FLIM experiments; Fig. 7c and d show that the fluorescent lifetime of GFP–K-RasG12V in the presence of RFP–H-RasG12V was significantly decreased in latrunculin-treated cells or when expressed in BHK-Cav-1KD cells. These results therefore fully recapitulate the results with H-Ras and K-Ras lateral segregation shown in Fig. 4, but by changing the mobile fraction of PM PS rather than total PM PS.
Signal integration as a consequence of spatial cross talk.
CRAF, KSR-scaffolded MEK, and KSR-scaffolded extracellular signal-regulated kinase (ERK) must be recruited to Ras–GTP nanoclusters on the PM for activation. EM, FRET, and single-molecule video tracking (SFVT) experiments further suggest that Ras–GTP nanoclusters are the sole sites for CRAF-dependent MAPK activation on the PM (10, 13, 20, 46). Thus, total phosphorylated ERK (ERKpp) output from the PM is a function of both the amount of Ras–GTP on the PM and the fraction of that Ras–GTP which is assembled into nanoclusters. For a fixed amount of Ras–GTP, the greater the fraction that assembles into nanoclusters, the greater the MAPK output. Conversely, if Ras–GTP nanoclustering is inhibited, then activation of the MAPK cascade is also inhibited (11, 13, 46). In this context, PS-mediated spatial cross talk between H-Ras and K-Ras should modulate K-Ras signal output.
To evaluate the potential effects of spatial cross talk on downstream MAPK signaling activities, we measured the phosphorylation of CRAF, MEK, ERK, and Akt. BHK cells stably expressing either GFP–K-RasG12V, a combination of GFP–K-RasG12V and RFP–H-RasG12V, or both GFP–K-RasG12V and RFP-CTH were treated with the MEK inhibitor U0126 for 24 h, washed with PBS, and incubated in serum-free DMEM; phosphoS338-CRAF, pMEK, pERK, and pAkt activation was then measured. This use of a MEK inhibitor is useful in stable cell lines expressing oncogenic mutant Ras where negative-feedback mechanisms downstream of activated ERK suppress Ras-dependent Raf recruitment and activation (46, 47). Twenty-four hours of growth in the presence of a MEK inhibitor followed by a 30-min washout effectively resets the negative-feedback mechanisms and leads to robust reactivation of the CRAF/MEK/ERK cascade. In this experimental format, changes or perturbations of oncogenic-Ras-driven MAPK activation are amplified and readily visible. We observed that CRAF, MEK, and ERK activation in BHK cells expressing both GFP–K-RasG12V and RFP–H-RasG12V was markedly lower than in cells expressing GFP–K-RasG12V alone (Fig. 8a). Strikingly, coexpression of GFP–K-RasG12V with signaling-incompetent RFP-CTH also significantly inhibited CRAF, MEK, and ERK activation (Fig. 8a).
FIG 8.
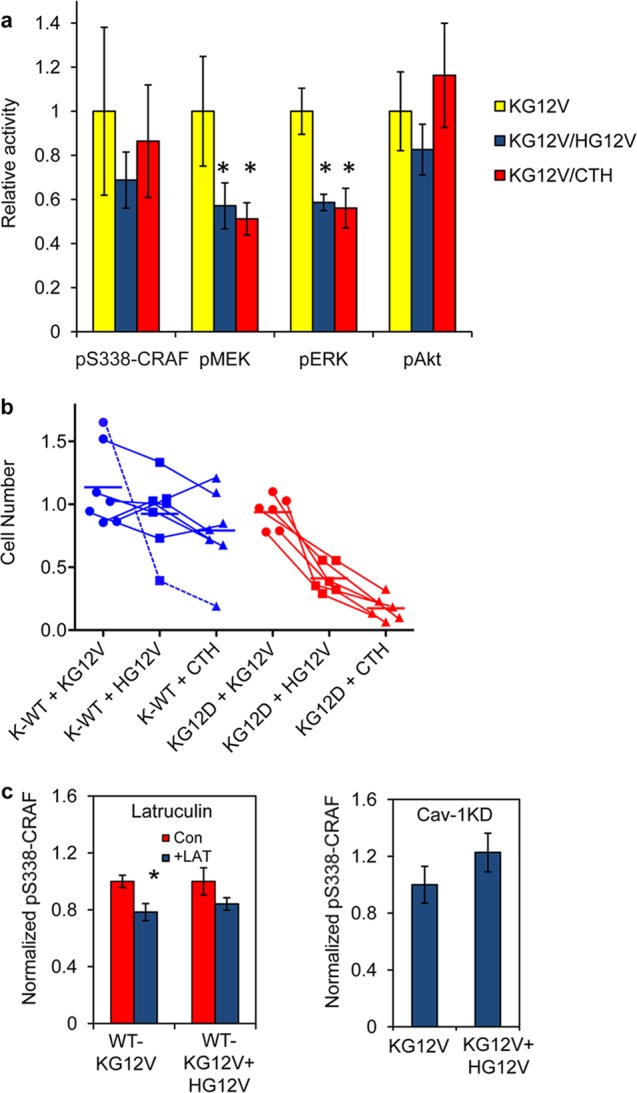
Signal integration as a consequence of spatial cross talk. (a) BHK cells stably expressing GFP–K-RasG12V alone, GFP–K-RasG12V with RFP–H-RasG12V, or GFP–K-RasG12V with RFP-CTH were grown in serum-free media in the presence of the MEK inhibitor U0126 for 24 h, followed by growth for 30 min in the absence of U0126, and then analyzed by quantitative immunoblotting for pS338-CRAF, pMEK, pERK, and pAkt. The results were normalized against those for control cells expressing K-RasG12V alone. The graph shows means ± SEM from 3 independent experiments. The statistical significance of differences was evaluated using one-way ANOVA (*, P < 0.05). (b) Survival of cancer cell lines 3 days after lentiviral transduction of GFP-tH, GFP–K-RasG12V, GFP–H-RasG12V, or GFP-CTH. Cell counts of cells expressing GFP–K-RasG12V (+KG12V), GFP–H-RasG12V (+HG12V), or GFP-CTH (+CTH) are shown relative to cell counts for a cognate GFP-tH control. Cell lines are connected across each lentiviral expression column. Cancer cell lines expressing wild-type K-Ras (K-WT) were HES, ESS1, EFE184, AN3CA, KLE, and RL-95-2 (endometrial) and Bx-PC3 (pancreatic). Cancer cell lines expressing oncogenic K-RasG12D were HEC1a, HEC1b, and HEC50 (endometrial) and MoH, mPanc96, and HPAC (pancreatic). AN3CA cells (dotted blue line) behave like K-RasG12D-expressing cells in this assay. (c) Wild-type BHK cells transiently expressing GFP–K-RasG12V or a combination of GFP–K-RasG12V and RFP–H-RasG12V were serum starved for 1 h and then treated with 1 μM latrunculin for 5 min; pS338-CRAF levels were measured by quantitative immunoblotting (left panel). BHK-Cav-1KD cells transiently expressing GFP–K-RasG12V or a combination of GFP–K-RasG12V and RFP–H-RasG12V were serum starved for 1 h; pS338-CRAF levels were then measured by quantitative immunoblotting (right panel). The graphs show means ± SEM from 3 independent experiments. The statistical significance of differences was evaluated using one-way ANOVA (*, P < 0.05).
To globally assess the effect of H-RasG12V cross talk on K-RasG12V function, we next examined the ability of K-Ras-transformed tumor cell lines to tolerate H-RasG12V expression. A cohort of endometrial and pancreatic cancer cell lines, including HES, EFE184, An3Ca, ESS1, Bx-PC3, RL-95-2, HPNE, HEC1a, HEC1b, HEC50, mPanc96, HPAC, and MoH, that express oncogenic mutant K-Ras or wild-type K-Ras were infected with lentiviruses expressing GFP-tH, GFP–K-RasG12V, GFP–H-RasG12V, or GFP-CTH, and cell survival was quantified. All cell lines tolerated the expression of GFP–K-RasG12V (Fig. 8b). Similarly, expression of GFP–H-RasG12V or GFP-CTH had minimal consequences for the survival of cancer cell lines that express wild-type K-Ras. In contrast, expression of GFP–H-RasG12V markedly decreased the survival of cancer cell lines expressing K-RasG12D, consistent with a suppression of K-Ras signaling to which these cell lines are oncogene addicted (Fig. 8b). Strikingly, expression of signaling-incompetent GFP-CTH also markedly decreased cell survival in cancer cell lines expressing K-RasG12D, in many cases to a greater extent than expression of GFP–H-RasG12V (Fig. 8b). This is consistent with the EM data demonstrating that CTH is as effective at disrupting K-Ras nanoclustering as full-length H-RasG12V (Fig. 2e) and the concomitant signaling data in Fig. 8a. The one exception was exhibited by ANC3A cells, whose growth was also suppressed by GFP–H-RasG12V and GFP-CTH; these cells, while wild type for K-Ras, do not express caveolin-1 and, thus, as shown recently, are exclusively dependent on K-Ras signaling (29). Together, these data are consistent with the EM and FLIM data showing that H-RasG12V remotely disrupts K-RasG12V nanoclustering and hence K-Ras signal transmission from the PM.
Finally, we conducted preliminary experiments to explore the operation of heterotypic, mixed clusters of K-Ras and H-Ras that assemble under conditions of actin disruption by latrunculin treatment or actin stabilization by caveolin-1 knockdown. Although under both of these conditions net K-Ras clustering increases due to K-Ras missorting with H-Ras, Fig. 8c shows that Raf activation is not increased. This result suggests that the signaling capacity of K-Ras incorporated in mixed heterotypic K-Ras/H-Ras clusters is significantly compromised. These observations are consistent with our previous findings that K-Ras-dependent MAPK signaling is compromised in latrunculin-treated cells (11) or when mixed K-Ras and H-Ras clusters are generated in nonsteroidal anti-inflammatory drug (NSAID)-treated cells (16).
DISCUSSION
In this study, we examined the role of lipids in Ras nanocluster operation. We found that Ras proteins engage in lipid sorting to generate lateral assemblies with distinct compositions. We identified a critical role for PS as a global organizer of proteolipid assemblies on the PM, akin to the role of cholesterol in driving liquid-ordered/liquid-disordered (Lo/Ld) domain segregation, and uncovered a novel mechanism for signal integration between spatially remote lipid assemblies on the PM mediated by PS spatiotemporal dynamics. The enrichment of H-Ras clusters for PIP2 and PIP3 concurs with recent studies showing enrichment of these PIs in cholesterol-dependent lipid rafts (48–50) and also with the known effector activation profile of H-Ras as a more potent activator of PI3K than K-Ras. The increased potency may simply reflect a higher concentration of PI3K substrate in H-Ras nanoclusters. Similarly, the enrichment of K-Ras–GTP clusters with PA, an important cofactor and provider of membrane affinity for CRAF (51, 52), can readily account for the increased potency of Raf-MAPK activation by K-Ras compared to that by H-Ras (53–55). Although PS was equivalently present in K-Ras and H-Ras nanoclusters, depletion experiments showed that PS was absolutely required for K-Ras but not H-Ras nanoclustering, suggesting that PS is a key structural component of K-Ras but not H-Ras nanoclusters. The lipid profile of K-Ras nanoclusters, with enrichment of PS and PA but not PIP2, also suggests selective recruitment of specific anionic lipids to engage in electrostatic interactions with the C-terminal polybasic domain. Such specificity implies that in addition to electrostatics, determinants such as lipid packing geometry and lipid cross-sectional area may constrain the interactions between K-Ras and different acidic lipids. Similar levels of selectivity occur in model bilayers and molecular dynamic (MD) simulations, where negatively charged lipids, such as PS and PA, display different capabilities to mediate actin-induced changes in membrane organization (43, 45).
Lateral Ras segregation into compositionally distinct lipid assemblies on the PM is likely related to the phase separation of lipids observed in model membranes (40, 41, 56, 57). Phase separation is generally a nonlinear function of lipid composition; for instance, in 3-component synthetic bilayers composed of cholesterol, saturated 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), and monounsaturated 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), phase separation occurs only at cholesterol levels between 25% and 35% (40, 41). At concentrations below and above this optimal range, cholesterol mixes with other lipids homogeneously and phase separation fails (40, 41). We now show here that lateral segregation of Ras proteins is also a nonlinear function of PM PS levels. Efficient lateral segregation of K-Ras from H-Ras nanoclusters, reflecting formation of specific lipid domains, occurs only over a narrow range of PM PS levels. At levels of PS both above and below this range, mixed clusters of K-Ras and H-Ras form. This behavior is remarkably similar to that described by one-dimensional phase diagrams of cholesterol and other lipids in model membranes and likely reflects a role for PS in driving mixing or demixing of lipid assemblies. In support of this interpretation, biophysical studies also suggest a complex role for PS in membrane lateral heterogeneity (45, 58), where inclusion of actin significantly alters the moles percent of PS required to induce phase separation and highlights a specific role for actin-PS interactions in driving membrane heterogeneity (45). Similarly, our observations on cell PM, coupled with mathematical simulations, now strongly implicate actin-PS interactions in regulating membrane lateral heterogeneity in intact cells (44, 59).
Collecting together the experimental and computational data, we propose a basic model framework to account for the regulatory effects of actin and caveolin on PS and K-Ras spatial distributions (Fig. 9a). The spatial cross talk that we observed between K-Ras and H-Ras nanoclusters follows logically from the phase diagrams in Fig. 9a. At optimal PS levels where there is efficient lateral segregation between the isoforms, H-Ras competes with K-Ras for available PS, effectively depressing the K-Ras clustering/PS isotherm (Fig. 9b). The effect is negative regulation of K-Ras clustering and signaling activity. However, outside the optimal PS range, significant coclustering between K-Ras and H-Ras also occurs for a net increase in overall K-Ras clustering (Fig. 9b). Collecting together these results, we conclude that only a narrow range of PM lipid compositions can support Ras lateral segregation, defined as where optimal PS and cholesterol ranges overlap (Fig. 9c). Since this overlap range may be small, it seems probable that Ras lateral segregation occurs dynamically and heterogeneously. Moreover, since cortical actin controls a major reservoir of immobile PS, the fraction of mobile PS available for lipid assemblies will fluctuate in response to changes in the actin cytoskeleton, with attendant consequences for Ras lateral segregation and spatial cross talk.
FIG 9.

Models of K-Ras clustering as a function of PS levels in the PM. (a) K-Ras homotypic clustering as indicated by Lmax or the clustered fraction (Φ) is plotted against the level of available free PS on the PM (PM mobile PS). A change in PS level without changing actin will increase or decrease K-Ras clustering accordingly (red and gray points). Changing actin organization shifts the K-Ras/PS dependence (green lines). Depleting cortical actin increases the mobile fraction of PS but simultaneously shifts the K-Ras clustering/PS isotherm down, for a net reduction in K-Ras clustering (yellow point). Conversely, caveolin depletion increases cortical actin, decreasing the effective concentration of PS, but simultaneously shifts the K-Ras/PS isotherm up for a net increase in K-Ras clustering (purple point). (b) The formation of H-Ras nanoclusters competes with K-Ras for available PS to suppress K-Ras clustering (green line). At optimal PS levels (pink zone) with efficient lateral segregation, the consequence is negative regulation of K-Ras clustering and signaling activity, as shown by the red circle moving down to the gray circle. However, outside the optimal PS range, significant coclustering between K-Ras and H-Ras occurs, yielding a net increase in overall K-Ras clustering (blue line). At optimal PS levels where there is efficient lateral segregation between the isoforms (pink zone), H-Ras competes with K-Ras for available PS, effectively depressing the K-Ras clustering/PS isotherm. The effect is negative regulation of K-Ras clustering and signaling activity (red and gray points). Note that at optimal PS levels, heterotypic clustering (red curve) does not occur. However, outside the optimal PS range, significant coclustering between K-Ras and H-Ras also occurs (red curve), for a net increase in overall K-Ras clustering (blue curve). (c) The optimal PS range for the most efficient Ras lateral segregation (red lines) overlaps the optimal cholesterol (CHOL) range. The slashed square indicates the two-dimensional phase diagram zone for optimal Ras lateral segregation.
We observed striking suppression of K-Ras signaling in cells where H-RasG12V was coexpressed, to the extent that K-Ras-transformed cancer cell lines were unable to tolerate H-RasG12V expression. This phenotype presumably occurs because spatial cross talk suppresses K-Ras nanoclustering and quenches K-Ras signal output. Oncogenic mutations in H-Ras are extremely rare in human tumors (60); it is tempting to speculate that the quenching of K-Ras signaling that results from H-Ras–GTP expression may be of relevance. Our focus in this study is on spatial cross talk between Ras proteins. Given that the underlying mechanism is mediated by PS spatiotemporal dynamics, it is probable that other polybasic-domain and lipid-anchored membrane proteins will exhibit similar interactions. This is strikingly illustrated by the ability of GFP-CTH, which lacks the H-Ras G-domain, to suppress K-Ras nanoclustering, even though there is no direct signal output from the GFP-CTH lipid assemblies. Furthermore, functionally unrelated membrane proteins, including ion channels and G protein-coupled receptors (GPCRs), all associate with the same continuum of lipids. To the extent that these membrane proteins spatially sequester PS, they will remotely interact with and influence the operation of proteolipid complexes that are structurally or operationally dependent on PS. The network connectivity of signaling complexes in the PM linked by lipid dynamics may therefore be considerably more complex than previously envisaged and may provide mechanisms for many unexplained and seemingly unlinked functional correlations, as, for example, between ion channels and mitogenic signaling.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant GM066717 from the National Institutes of Health, General Medical Sciences (J.F.H.), and the National Health and Medical Research Council of Australia through grants and research fellowships to R.G.P. (511055, 569542).
We thank Sergio Grinstein, Tamas Balla, and Guangwei Du for providing key reagents.
Footnotes
Published ahead of print 23 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01227-13.
REFERENCES
- 1.Hancock JF. 2003. Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 4:373–384. 10.1038/nrm1105 [DOI] [PubMed] [Google Scholar]
- 2.Mor A, Philips MR. 2006. Compartmentalized Ras/MAPK signaling. Annu. Rev. Immunol. 24:771–800. 10.1146/annurev.immunol.24.021605.090723 [DOI] [PubMed] [Google Scholar]
- 3.Prior IA, Hancock JF. 2001. Compartmentalization of Ras proteins. J. Cell Sci. 114:1603–1608 [DOI] [PubMed] [Google Scholar]
- 4.Prior IA, Hancock JF. 2012. Ras trafficking, localization and compartmentalized signalling. Semin. Cell Dev. Biol. 23:145–153. 10.1016/j.semcdb.2011.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casey PJ, Solski PA, Der CJ, Buss JE. 1989. p21ras is modified by a farnesyl isoprenoid. Proc. Natl. Acad. Sci. U. S. A. 86:8323–8327. 10.1073/pnas.86.21.8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutierrez L, Magee AI, Marshall CJ, Hancock JF. 1989. Post-translational processing of p21ras is two-step and involves carboxyl-methylation and carboxy-terminal proteolysis. EMBO J. 8:1093–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hancock JF, Magee AI, Childs JE, Marshall CJ. 1989. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57:1167–1177. 10.1016/0092-8674(89)90054-8 [DOI] [PubMed] [Google Scholar]
- 8.Hancock JF, Paterson H, Marshall CJ. 1990. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 63:133–139. 10.1016/0092-8674(90)90294-O [DOI] [PubMed] [Google Scholar]
- 9.Hancock JF, Parton RG. 2005. Ras plasma membrane signalling platforms. Biochem. J. 389:1–11. 10.1042/BJ20050231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murakoshi H, Iino R, Kobayashi T, Fujiwara T, Ohshima C, Yoshimura A, Kusumi A. 2004. Single-molecule imaging analysis of Ras activation in living cells. Proc. Natl. Acad. Sci. U. S. A. 101:7317–7322. 10.1073/pnas.0401354101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plowman SJ, Muncke C, Parton RG, Hancock JF. 2005. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. U. S. A. 102:15500–15505. 10.1073/pnas.0504114102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho KJ, Park JH, Piggott AM, Salim AA, Gorfe AA, Parton RG, Capon RJ, Lacey E, Hancock JF. 2012. Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. J. Biol. Chem. 287:43573–43584. 10.1074/jbc.M112.424457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. 2007. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 9:905–914. 10.1038/ncb1615 [DOI] [PubMed] [Google Scholar]
- 14.Tian T, Plowman SJ, Parton RG, Kloog Y, Hancock JF. 2010. Mathematical modeling of K-Ras nanocluster formation on the plasma membrane. Biophys. J. 99:534–543. 10.1016/j.bpj.2010.04.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Hoeven D, Cho KJ, Ma X, Chigurupati S, Parton RG, Hancock JF. 2013. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol. Cell. Biol. 33:237–251. 10.1128/MCB.00884-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Y, Cho KJ, Plowman SJ, Hancock JF. 2012. Nonsteroidal anti-inflammatory drugs alter the spatiotemporal organization of Ras proteins on the plasma membrane. J. Biol. Chem. 287:16586–16595. 10.1074/jbc.M112.348490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abankwa D, Gorfe AA, Hancock JF. 2008. Mechanisms of Ras membrane organization and signalling: Ras on a rocker. Cell Cycle 7:2667–2673. 10.4161/cc.7.17.6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abankwa D, Gorfe AA, Hancock JF. 2007. Ras nanoclusters: molecular structure and assembly. Semin. Cell Dev. Biol. 18:599–607. 10.1016/j.semcdb.2007.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorfe AA, Hanzal-Bayer M, Abankwa D, Hancock JF, McCammon JA. 2007. Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-dimyristoylglycero-3-phosphocholine bilayer. J. Med. Chem. 50:674–684. 10.1021/jm061053f [DOI] [PubMed] [Google Scholar]
- 20.Plowman SJ, Ariotti N, Goodall A, Parton RG, Hancock JF. 2008. Electrostatic interactions positively regulate K-Ras nanocluster formation and function. Mol. Cell. Biol. 28:4377–4385. 10.1128/MCB.00050-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prior IA, Muncke C, Parton RG, Hancock JF. 2003. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 160:165–170. 10.1083/jcb.200209091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rotblat B, Prior IA, Muncke C, Parton RG, Kloog Y, Henis YI, Hancock JF. 2004. Three separable domains regulate GTP-dependent association of H-ras with the plasma membrane. Mol. Cell. Biol. 24:6799–6810. 10.1128/MCB.24.15.6799-6810.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy S, Plowman S, Rotblat B, Prior IA, Muncke C, Grainger S, Parton RG, Henis YI, Kloog Y, Hancock JF. 2005. Individual palmitoyl residues serve distinct roles in H-ras trafficking, microlocalization, and signaling. Mol. Cell. Biol. 25:6722–6733. 10.1128/MCB.25.15.6722-6733.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hill MM, Daud NH, Aung CS, Loo D, Martin S, Murphy S, Black DM, Barry R, Simpson F, Liu L, Pilch PF, Hancock JF, Parat MO, Parton RG. 2012. Co-regulation of cell polarization and migration by caveolar proteins PTRF/Cavin-1 and caveolin-1. PLoS One 7:e43041. 10.1371/journal.pone.0043041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diggle PJ, Mateu J, Clough HE. 2000. A comparison between parametric and non-parametric approaches to the analysis of replicated spatial point patterns. Adv. Appl. Probab. 32:331–343. 10.1239/aap/1013540166 [DOI] [Google Scholar]
- 26.Deschout H, Hagman J, Fransson S, Jonasson J, Rudemo M, Loren N, Braeckmans K. 2010. Straightforward FRAP for quantitative diffusion measurements with a laser scanning microscope. Opt. Express 18:22886–22905. 10.1364/OE.18.022886 [DOI] [PubMed] [Google Scholar]
- 27.Kay JG, Koivusalo M, Ma X, Wohland T, Grinstein S. 2012. Phosphatidylserine dynamics in cellular membranes. Mol. Biol. Cell 23:2198–2212. 10.1091/mbc.E11-11-0936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Treeck M, Zacherl S, Herrmann S, Cabrera A, Kono M, Struck NS, Engelberg K, Haase S, Frischknecht F, Miura K, Spielmann T, Gilberger TW. 2009. Functional analysis of the leading malaria vaccine candidate AMA-1 reveals an essential role for the cytoplasmic domain in the invasion process. PLoS Pathog. 5:e1000322. 10.1371/journal.ppat.1000322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ariotti N, Fernández-Rojo MA, Zhou Y, Hill MM, Rodkey T, Inder K, Hancock JF, Parton RG. Caveolae regulate the nanoscale organization of the plasma membrane to remotely control Ras signaling. J. Cell Biol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldenberg NM, Steinberg BE. 2010. Surface charge: a key determinant of protein localization and function. Cancer Res. 70:1277–1280. 10.1158/0008-5472.CAN-09-2905 [DOI] [PubMed] [Google Scholar]
- 31.Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. 2008. Membrane phosphatidylserine regulates surface charge and protein localization. Science 319:210–213. 10.1126/science.1152066 [DOI] [PubMed] [Google Scholar]
- 32.Grande-Garcia A, Echarri A, de Rooij J, Alderson NB, Waterman-Storer CM, Valdivielso JM, del Pozo MA. 2007. Caveolin-1 regulates cell polarization and directional migration through Src kinase and Rho GTPases. J. Cell Biol. 177:683–694. 10.1083/jcb.200701006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeniou-Meyer M, Zabari N, Ashery U, Chasserot-Golaz S, Haeberle AM, Demais V, Bailly Y, Gottfried I, Nakanishi H, Neiman AM, Du G, Frohman MA, Bader MF, Vitale N. 2007. Phospholipase D1 production of phosphatidic acid at the plasma membrane promotes exocytosis of large dense-core granules at a late stage. J. Biol. Chem. 282:21746–21757. 10.1074/jbc.M702968200 [DOI] [PubMed] [Google Scholar]
- 34.Garcia P, Gupta R, Shah S, Morris AJ, Rudge SA, Scarlata S, Petrova V, McLaughlin S, Rebecchi MJ. 1995. The pleckstrin homology domain of phospholipase C-delta 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry 34:16228–16234. 10.1021/bi00049a039 [DOI] [PubMed] [Google Scholar]
- 35.Miao B, Skidan I, Yang J, Lugovskoy A, Reibarkh M, Long K, Brazell T, Durugkar KA, Maki J, Ramana CV, Schaffhausen B, Wagner G, Torchilin V, Yuan J, Degterev A. 2010. Small molecule inhibition of phosphatidylinositol-3,4,5-triphosphate (PIP3) binding to pleckstrin homology domains. Proc. Natl. Acad. Sci. U. S. A. 107:20126–20131. 10.1073/pnas.1004522107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kale SD, Gu B, Capelluto DG, Dou D, Feldman E, Rumore A, Arredondo FD, Hanlon R, Fudal I, Rouxel T, Lawrence CB, Shan W, Tyler BM. 2010. External lipid PI3P mediates entry of eukaryotic pathogen effectors into plant and animal host cells. Cell 142:284–295. 10.1016/j.cell.2010.06.008 [DOI] [PubMed] [Google Scholar]
- 37.Hammond GR, Fischer MJ, Anderson KE, Holdich J, Koteci A, Balla T, Irvine RF. 2012. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 337:727–730. 10.1126/science.1222483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee S, Uchida Y, Emoto K, Umeda M, Kuge O, Taguchi T, Arai H. 2012. Impaired retrograde membrane traffic through endosomes in a mutant CHO cell defective in phosphatidylserine synthesis. Genes Cells 17:728–736. 10.1111/j.1365-2443.2012.01622.x [DOI] [PubMed] [Google Scholar]
- 39.Baumgart T, Hess ST, Webb WW. 2003. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature 425:821–824. 10.1038/nature02013 [DOI] [PubMed] [Google Scholar]
- 40.Veatch SL, Keller SL. 2002. Organization in lipid membranes containing cholesterol. Phys. Rev. Lett. 89:268101. 10.1103/PhysRevLett.89.268101 [DOI] [PubMed] [Google Scholar]
- 41.Veatch SL, Keller SL. 2003. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophys. J. 85:3074–3083. 10.1016/S0006-3495(03)74726-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiselev VY, Leda M, Lobanov AI, Marenduzzo D, Goryachev AB. 2011. Lateral dynamics of charged lipids and peripheral proteins in spatially heterogeneous membranes: comparison of continuous and Monte Carlo approaches. J. Chem. Phys. 135:155103. 10.1063/1.3652958 [DOI] [PubMed] [Google Scholar]
- 43.Kiselev VY, Marenduzzo D, Goryachev AB. 2011. Lateral dynamics of proteins with polybasic domain on anionic membranes: a dynamic Monte-Carlo study. Biophys. J. 100:1261–1270. 10.1016/j.bpj.2011.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma P, Varma R, Sarasij RC, Ira Gousset K, Krishnamoorthy G, Rao M, Mayor S. 2004. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell 116:577–589. 10.1016/S0092-8674(04)00167-9 [DOI] [PubMed] [Google Scholar]
- 45.Garg S, Tang JX, Ruhe J, Naumann CA. 2009. Actin-induced perturbation of PS lipid-cholesterol interaction: a possible mechanism of cytoskeleton-based regulation of membrane organization. J. Struct. Biol. 168:11–20. 10.1016/j.jsb.2009.04.001 [DOI] [PubMed] [Google Scholar]
- 46.Kholodenko BN, Hancock JF, Kolch W. 2010. Signalling ballet in space and time. Nat. Rev. Mol. Cell Biol. 11:414–426. 10.1038/nrm2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. 2005. Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell 17:215–224. 10.1016/j.molcel.2004.11.055 [DOI] [PubMed] [Google Scholar]
- 48.Kalwa H, Michel T. 2011. The MARCKS protein plays a critical role in phosphatidylinositol 4,5-bisphosphate metabolism and directed cell movement in vascular endothelial cells. J. Biol. Chem. 286:2320–2330. 10.1074/jbc.M110.196022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schroeder N, Chung CS, Chen CH, Liao CL, Chang W. 2012. The lipid raft-associated protein CD98 is required for vaccinia virus endocytosis. J. Virol. 86:4868–4882. 10.1128/JVI.06610-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Rheenen J, Achame EM, Janssen H, Calafat J, Jalink K. 2005. PIP2 signaling in lipid domains: a critical re-evaluation. EMBO J. 24:1664–1673. 10.1038/sj.emboj.7600655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andresen BT, Rizzo MA, Shome K, Romero G. 2002. The role of phosphatidic acid in the regulation of the Ras/MEK/Erk signaling cascade. FEBS Lett. 531:65–68. 10.1016/S0014-5793(02)03483-X [DOI] [PubMed] [Google Scholar]
- 52.Ghosh S, Strum JC, Sciorra VA, Daniel L, Bell RM. 1996. Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J. Biol. Chem. 271:8472–8480 [DOI] [PubMed] [Google Scholar]
- 53.Jaumot M, Yan J, Clyde-Smith J, Sluimer J, Hancock JF. 2002. The linker domain of the Ha-Ras hypervariable region regulates interactions with exchange factors, Raf-1 and phosphoinositide 3-kinase. J. Biol. Chem. 277:272–278. 10.1074/jbc.M108423200 [DOI] [PubMed] [Google Scholar]
- 54.Voice J, Klemke R, Le A, Jackson J. 1999. Four human Ras homologs differ in their ability to activate Raf-1, induce transformation and stimulate cell motility. J. Biol. Chem. 274:17164–17170. 10.1074/jbc.274.24.17164 [DOI] [PubMed] [Google Scholar]
- 55.Yan J, Roy S, Apolloni A, Lane A, Hancock JF. 1998. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J. Biol. Chem. 273:24052–24056. 10.1074/jbc.273.37.24052 [DOI] [PubMed] [Google Scholar]
- 56.Brown DA, London E. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136. 10.1146/annurev.cellbio.14.1.111 [DOI] [PubMed] [Google Scholar]
- 57.Brown DA, London E. 1998. Structure and origin of ordered lipid domains in biological membranes. J. Membr. Biol. 164:103–114. 10.1007/s002329900397 [DOI] [PubMed] [Google Scholar]
- 58.Epand RM, Bain AD, Sayer BG, Bach D, Wachtel E. 2002. Properties of mixtures of cholesterol with phosphatidylcholine or with phosphatidylserine studied by (13)C magic angle spinning nuclear magnetic resonance. Biophys. J. 83:2053–2063. 10.1016/S0006-3495(02)73966-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gowrishankar K, Ghosh S, Saha SRC, Mayor S, Rao M. 2012. Active remodeling of cortical actin regulates spatiotemporal organization of cell surface molecules. Cell 149:1353–1367. 10.1016/j.cell.2012.05.008 [DOI] [PubMed] [Google Scholar]
- 60.Prior IA, Lewis PD, Mattos C. 2012. A comprehensive survey of ras mutations in cancer. Cancer Res. 72:2457–2467. 10.1158/0008-5472.CAN-11-2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.