

Abstract
Dendritic cells (DCs) capture and process antigens in peripheral tissues, migrate to lymphoid tissues, and present the antigens to T cells. PTPN12, also known as PTP-PEST, is an intracellular protein tyrosine phosphatase (PTP) involved in cell-cell and cell-substratum interactions. Herein, we examined the role of PTPN12 in DCs, using a genetically engineered mouse lacking PTPN12 in DCs. Our data indicated that PTPN12 was not necessary for DC differentiation, DC maturation, or cytokine production in response to inflammatory stimuli. However, it was needed for full induction of T cell-dependent immune responses in vivo. This function largely correlated with the need of PTPN12 for DC migration from peripheral sites to secondary lymphoid tissues. Loss of PTPN12 in DCs resulted in hyperphosphorylation of the protein tyrosine kinase Pyk2 and its substrate, the adaptor paxillin. Pharmacological inhibition of Pyk2 or downregulation of Pyk2 expression also compromised DC migration, suggesting that Pyk2 deregulation played a pivotal role in the migration defect caused by PTPN12 deficiency. Together, these findings identified PTPN12 as a key regulator in the ability of DCs to induce antigen-induced T cell responses. This is due primarily to the role of PTPN12 in DC migration from peripheral sites to secondary lymphoid organs through regulation of Pyk2.
INTRODUCTION
Dendritic cells (DCs) play a critical role in initiation of immune responses against pathogens and cancer cells (1). They also have a key role in inflammation and autoimmune diseases (2). These effects relate in part to the fact that DCs are the most efficient antigen-presenting cells (APCs). DCs are broadly distributed at areas of contact between the body and the environment, including skin. From these sites, DCs circulate in lymph and blood and reach secondary lymphoid tissues, where they can interact with other immune cells, including T cells. The ability of DCs to migrate in and out of these various compartments is critical for their functions.
In peripheral tissues, DCs primarily exist in an “immature” state (1). In response to pathogens or cytokines like interleukin 1 (IL-1) and tumor necrosis factor alpha (TNF-α), DCs undergo “maturation.” The best-studied receptors triggering DC maturation are Toll-like receptors (TLRs), which recognize pathogen-associated constituents, such as lipopolysaccharide (LPS) or viral DNA. DC maturation leads to increased expression of major histocompatibility complex class I (MHC-I) and class II (MHC-II) molecules, costimulatory molecules, such as CD80 and CD86, and cytokines. It also promotes migration of DCs to secondary lymphoid tissues, where DCs present antigens to T cells.
PTPN12, also referred to as PTP-PEST (protein tyrosine phosphatase, proline-, glutamic acid-, serine-, and threonine-rich), is a nonreceptor PTP expressed broadly, particularly in hematopoietic cells (3–5). It belongs to the PEST family, with PTPN22 and PTPN8 (3). PTPN12 can dephosphorylate several proteins, such as Cas, focal adhesion kinase (FAK), Pyk2, paxillin, Shc, and receptor protein tyrosine kinases (PTKs), and can regulate multiple cellular processes, including migration, adhesion, and receptor-triggered proliferation (6–12).
Genetic evidence supports the idea that PTPN12 carries out key functions in several cell types. It is critical for embryonic development and viability (10). Analysis of a conditional PTPN12-deficient mouse (Ptpn12fl/fl) indicated that this requirement relates to a role of PTPN12 in endothelial cells (11). PTPN12 is also critical for secondary T cell activation responses as a result of the capacity to promote adhesion between activated T cells (7). Moreover, it is essential for fusion of macrophages into multinucleated giant cells and osteoclasts (13). Last, there is evidence that PTPN12 is a tumor suppressor (12).
Here we examined the role of PTPN12 in DCs by specifically eliminating expression of PTPN12 in DCs from the mouse. We found that PTPN12 was not critical for DC differentiation, maturation, and cytokine production. It also had little or no role in antigen presentation to T cells in vitro. However, PTPN12 expression in DCs was required for T cell-dependent responses in vivo, including induction of T cell-mediated autoimmune disease. This function correlated with the capacity of PTPN12 to promote DC migration and to dephosphorylate the substrates Pyk2 and paxillin.
MATERIALS AND METHODS
Mice.
Mice with a conditional allele of Ptpn12 (Ptpn12fl/fl) have been described (7). They were backcrossed for >12 generations to a C57BL/6 background. To produce mice lacking PTPN12 in DCs, Ptpn12fl/fl mice were crossed with Cd11c-Cre mice (14). OT-II T cell receptor (TCR) transgenic mice were from The Jackson Laboratory (Bar Harbor, ME) (15). All experimentation was given approval by the IRCM Animal Care Committee and was done according to the rules of the Canadian Council for Animal Care.
Cells.
To generate bone marrow-derived DCs (BMDCs), tibiae and femora were flushed with culture medium and grown for ∼9 days in petri dishes, using culture medium containing 10% (vol/vol) X63Ag8 conditioned medium (which contains granulocyte macrophage colony-stimulating factor [GM-CSF]) or recombinant Flt-3L. To isolate splenic DCs, splenic tissue was digested with DNase I and Liberase (Roche, Mississauga, ON, Canada), and DCs were identified by staining for CD11c and MHC-II. To ascertain the ability of DCs to mature in response to inflammatory stimuli, BMDCs were incubated for 24 h with lipopolysaccharide (LPS) (InvivoGen, Burlington, Ontario, Canada) or CpG DNA (Invivogen). CD4+ T cells, B cells and neutrophils were isolated from spleen or bone marrow, using kits from Stem Cell Technologies (Vancouver, BC, Canada). Cell purity was >90% (data not shown). Bone marrow-derived macrophages were produced as described elsewhere (13).
Flow cytometry and antibodies.
Flow cytometry was performed using anti-CD11c (N418), anti-MHC-II (M5/114.11.2), anti-CD11b (M1/70), anti-CD40 (1C10), anti-CD80 (16-10A1), anti-CD86 (GL1), anti-TLR2 (T2.5), anti-TLR4 (MTS-510), anti-PDCA-1 (eBio927), anti-CXCR4 (2B11), and anti-CCR7 (4B12). These antibodies were bought from BD Biosciences (Mississauga, Ontario, Canada) or eBioscience (San Diego, CA). Data acquisition was done on a BD BioSciences FACSCalibur cytometer using CellQuest software (BD BioSciences), and data were analyzed using the FlowJo software program (Trees Star Inc., Ashland, OH). Rabbit antibodies against PTPN12, Pyk2, Csk, signal regulatory protein alpha (SIRPα), proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP-1), and Syk have been described (7, 13, 16, 17). Antiphosphotyrosine monoclonal antibody (MAb) 4G10 was purchased from Millipore (Billerica, CA). Antibodies recognizing FAK (catalog no. sc-558), Cas (sc-860), paxillin (catalog no. 610569), and activated Src (catalog no. 2010) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA), BD Transduction Laboratories, or Cell Signaling Technology (Danvers, MA).
Cytokine production.
BMDCs (1 × 105 cells per well) were stimulated in 96-well plates for 24 h in the presence of LPS or CpG. Cytokines were quantitated by enzyme-linked immunosorbent assay (ELISA), as specified by the manufacturer (R&D Systems, Burlington, Ontario, Canada). Assays were performed in triplicate.
Antigen presentation.
Mice were immunized in the footpad with ovalbumin protein (OVA) (100 μg in 25 μl of phosphate-buffered saline [PBS]; Sigma-Aldrich, St. Louis, MO) plus an equal volume of complete Freund adjuvant (CFA) (Sigma-Aldrich). After 9 days, CD4+ T cells were purified from popliteal lymph nodes and restimulated in vitro for 4 to 5 days in the presence of OVA and irradiated splenocytes from wild-type C57BL/6 mice or for 2 to 3 days with phorbol myristate acetate (PMA) and ionomycin (Sigma-Aldrich). Proliferation and cytokine production were assayed. For in vitro antigen presentation, BMDCs were preincubated with OVA protein or OVA peptide (amino acids 323 to 339 [OVA323–339]) and used to activate OVA-specific CD4+ T cells from OT-II transgenic mice. After 4 to 5 days, proliferation and cytokine production were examined.
Experimental autoimmune encephalomyelitis.
Mice were immunized subcutaneously with 200 μg of myelin oligodendrocyte glycoprotein (MOG) amino acids 35 to 55 (MOG35–55) in CFA plus 300 μg of Mycobacterium tuberculosis H37Ra (Difco Laboratories, Detroit, MI). Twenty-four and seventy-two hours after immunization, mice were injected intraperitoneally with pertussis toxin (300 ng in 500 μl of PBS). They were scored daily for neurological deficits, as follows: 0, no clinical signs; 1, loss of tail tonicity; 2, flaccid tail; 3, hind leg paralysis; 4, hind leg and hind body paresis; 5, hind and fore leg paralysis; 6, death.
Conjugation assays.
Bone-marrow-derived DCs and purified CD4+ T cells from OT-II mice were labeled with 2.5 μM 5- and 6-carboxyfluorescein diacetate succinimidyl ester (CFSE) and 10 μM 5- and 6-(((4-chloromethyl)benzoyl)amino)tetramethylrhodamine (CMTMR), respectively, as specified by the manufacturer (Invitrogen) (18). CFSE-labeled DCs were first incubated or not with various concentrations of OVA peptide (OVA323–339). Then, DCs (106/well) were incubated with CD4+ T cells (5 × 105/well) for 30 min at 37°C. Conjugates were enumerated by flow cytometry.
Migration assays.
BMDCs (106 cells) were loaded in serum-free RPMI in the upper chamber of a Transwell apparatus (5-μm pore size; Cornings, Lowell, MA), while serum-free RPMI (600 μl) with or without chemokines (stromal cell-derived factor 1α [SDF-1α], 200 ng/ml; macrophage inflammatory protein 3β [MIP-3β], 200 ng/ml; Peprotech, Burlington, ON, Canada) was added in the lower chamber. After 1.5 h at 37°C, migrated cells were harvested from the lower chamber and counted by flow cytometry, using Flow Cytometry Absolute Count Standard (Bangs Laboratories Inc., Fishers, IN). For in vivo migration, LPS-matured BMDCs were labeled with CFSE or CMTMR. Then, equal numbers of CFSE- and CMTMR-labeled BMDCs (106 each) were injected subcutaneously into the footpads of syngeneic mice. On day 2, lymph nodes were isolated and numbers of CFSE+ or CMTMR+ cells were determined by flow cytometry.
Replating assays.
Petri dishes were precoated overnight at 4°C with fibronectin (10 μg/ml; Roche), dissolved in PBS. They were subsequently rinsed with PBS and prewarmed before use. BMDCs at day 9 of culture were harvested, and replated or not for 30 min at 37°C onto fibronectin-coated dishes. Cells were lysed and analyzed by immunoprecipitation.
Pyk2 inhibition.
BMDCs were incubated for 24 h with the specified concentrations of the Pyk2/FAK-specific inhibitor (Synkinase, Melbourne, Australia) (19, 20). Migration was then evaluated.
RNA interference.
Small interfering RNAs (siRNAs) against Pyk2 (catalog no. GS14083) were obtained from Qiagen (Toronto, Ontario, Canada). They were a pool of four target-specific siRNAs. Control siRNAs (catalog no. 1027280) were also obtained from Qiagen. Transfection of BMDCs was performed using a mouse dendritic cell Nucleofector kit (catalog no. VPA-1011; Lonza [Amaxa], Burlington, Ontario, Canada), according to the manufacturer's protocol. In brief, BMDCs were resuspended in Nucleofector solution at a final concentration of 2.5 × 105 cells/100 μl. They were then transfected with a total of 40 pmol of the indicated siRNAs, using the program Y-001. Cells transfected using nucleofection (nucleofected) were grown at 37°C for 24 to 36 h and processed for experimentation.
Immunoprecipitations and immunoblotting.
Immunoprecipitations and immunoblotting were done as outlined previously (21).
Statistical analyses and quantitation.
Unpaired Student's t tests (two-tailed) were executed using the Prism software program. Bands in autoradiograms were quantified with the Gel-Pro Analyzer 6.0 software program (Media Cybernetics, L.P.).
RESULTS
Dendritic cell-targeted PTPN12-deficient mouse.
To address the role of PTPN12 in DCs, Ptpn12fl/fl mice were bred with a transgenic mice expressing Cre under the control of the Cd11c promoter (Cd11c-Cre; also known as Itgax-Cre) (Fig. 1A) (14). This Cd11c-Cre mouse enables efficient Cre-mediated deletion in most conventional DCs (cDCs) and plasmacytoid DCs (pDCs) but not in other cells, including T cells, and has been extensively used to delete conditional alleles in DCs (2).
FIG 1.

Generation of dendritic cell-targeted PTPN12-deficient mouse. (A) A diagram of the wild-type Ptpn12 allele is shown at the top. The targeting construct, including loxP and frt sites, is represented below. The floxed allele (Ptpn12fl/fl) was generated by removal of a neo-tk cassette with Flpe recombinase. The expression of PTPN12 in dendritic cells was eliminated by breeding Ptpn12fl/fl mice with mice expressing the Cre recombinase in dendritic cells (DCs), using the Cd11c (Itgax) promoter. (B) Expression of PTPN12 was analyzed by immunoblotting of lysates from bone marrow-derived DCs (BMDCs) from control (wild-type [WT]) mice or mice lacking PTPN12 in DCs (knockout [KO]). Expression of Csk was analyzed as the control. (C) Expression of PTPN12 in purified splenic DCs was tested as detailed for panel B. (D) Expression of PTPN12 was analyzed in purified naive CD4+ T cells, activated CD4+ T cells, B cells, neutrophils, bone marrow-derived macrophages (BMMϕs), and BMDCs from Ptpn12+/+; Cd11c-Cre+ (WT) mice and Ptpn12fl/fl; Cd11-Cre+ (KO) mice. Quantitation of the levels of PTPN12 expression in KO cells, relative to that in WT cells for several independent experiments is shown at the bottom. Average values with standard deviations are depicted. Results are representative of three (left panels) or two (right panels) independent experiments.
To ascertain the efficiency of PTPN12 deletion in DCs, BMDCs and CD11c+ splenic DCs were obtained from Ptpn12fl/fl × Cd11c-Cre+ mice (henceforth termed PTPN12-deficient mice) and control mice (Ptpn12+/+ × Cd11c-Cre+ or Ptpn12fl/fl × Cd11c-Cre− mice). Immunoblotting with anti-PTPN12 antibodies demonstrated that PTPN12 expression was reduced by ∼70 to 90% in BMDCs from PTPN12-deficient mice (Fig. 1B). Similarly, it was diminished by ∼90% in CD11c+ splenic DCs (Fig. 1C). To verify that Cd11c-Cre did not cause deletion of PTPN12 in other cells, PTPN12 expression was also analyzed in other immune cells (Fig. 1D). Cd11c-Cre had no impact on PTPN12 expression in CD4+ T cells, B cells, or macrophages. Little or no expression of PTPN12 was detected in neutrophils, either from control or from PTPN12-deficient mice. Hence, Cd11c-Cre caused efficient and specific deletion of PTPN12 in DCs.
PTPN12 has no influence on DC differentiation, maturation, and cytokine production.
We next studied the impact of PTPN12 deficiency in DCs on the ability of hematopoietic cell precursors to differentiate into DCs in vitro. This was done by growing bone marrow cells in the presence of GM-CSF or Flt-3 ligand (Flt-3L), which favors differentiation into cDCs or pDCs, respectively (data not shown) (22). Lack of PTPN12 had no influence on the generation of either DC population. We also examined whether loss of PTPN12 had any impact of DC differentiation in vivo (Fig. 2). Lack of PTPN12 had no impact on the abundance of CD11c+ MHC-II+ cells in lymph nodes or spleen (Fig. 2A). These cells represent lymphoid tissue-resident migratory cDCs (22). There was also no difference in the numbers of B220+ CD11c+ PDCA-1+ cells, which represent pDCs (Fig. 2B) (22).
FIG 2.

Effects of PTPN12 deficiency in dendritic cells on development of dendritic cells and T cells. Cells were isolated from lymph nodes (LN) and spleens of control (“wild-type” [WT]) mice or mice lacking PTPN12 in DCs (“knockout” [KO]). They were then analyzed by flow cytometry, using antibodies against the indicated markers. (A) Conventional DCs (cDCs) were identified as CD11chi MHC-IIhi cells. Two major populations of cDCs (circled) were identified in LN. Cells expressing larger amounts of MHC-II correspond to migratory cDCs, whereas cells with smaller amounts of MHC-II are lymphoid tissue-resident cDCs. Only lymphoid tissue-resident cDCs (boxed) are found in the spleen. (B) Plasmacytoid DCs (pDCs) (boxed) were identified as CD11c+ B220+ PDCA-1+ cells. (C) Regulatory T cells (TRegs) were identified as CD3+ CD4+ Foxp3+ cells. (D) Total T cells were identified by staining spleen cells for CD3 and CD4 (first set of panels), CD3 and CD8 (second set of panels), or CD4 and CD8 (third set of panels). CD4+ cells were also gated and stained for CD44 and CD62L (fourth set of panels). Naive CD4+ T cells are CD44lo CD62Lhi, whereas effector memory CD4+ T cells are CD44hi CD62Llo. All data are representative of four experiments.
DCs regulate several facets of T cell biology (1). The latter include not only antigen-induced T cell activation but also tonic TCR triggering required for T cell survival. They are also needed for accumulation and persistence of regulatory T cells (TRegs). Thus, to test the impact of PTPN12 expression in DCs on T cell homeostasis, T cell subsets in spleen, lymph nodes, and thymus were analyzed (Fig. 2C and D; Table 1; also data not shown). Loss of PTPN12 had no impact on the proportions of CD4+ T cells and CD8+ T cells in these tissues. Likewise, it had no effect on the proportions of CD44lo CD62Lhi and CD44hi CD62Llo CD4+ T cells, which represent naive and effector-memory CD4+ T cells, respectively. There was also no influence on the accumulation of CD4+ Foxp3+ T cells, which identify TRegs.
TABLE 1.
Impact of PTPN12 deficiency on T cell populationsa
| Source | Mouse group | % of cell population or P value |
|||
|---|---|---|---|---|---|
| CD4+ T cells | CD8+ T cells | CD4+ T cells |
|||
| Naive | Effector/memory | ||||
| Spleen | WT | 31.90 ± 1.53 | 31.58 ± 2.73 | 71.69 ± 5.36 | 10.05 ± 0.75 |
| KO | 32.00 ± 1.81 | 26.35 ± 5.66 | 73.36 ± 4.45 | 9.36 ± 0.22 | |
| P value | 0.94 | 0.15 | 0.70 | 0.88 | |
| LN | WT | 16.45 ± 2.01 | 12.31 ± 0.74 | 61.80 ± 9.01 | 11.66 ± 3.61 |
| KO | 15.65 ± 1.01 | 9.85 ± 3.58 | 59.90 ± 5.40 | 13.63 ± 7.21 | |
| P value | 0.50 | 0.23 | 0.77 | 0.69 | |
Relative T cell numbers (in percentages) in spleen or lymph nodes (LN) were analyzed in control (wild-type [WT]) mice or mice lacking PTPN12 in DCs (knockout [KO]). Total CD4+ and CD8+ T cells were quantitated relative to all cells. Naive and effector memory CD4+ T cells were quantitated relative to total CD4+ T cells. Naive cells are CD44lo CD62Lhi, whereas effector memory cells are CD44hi CD62Llo. Average values, standard deviations, and P values for three independent mice of each type are shown.
To address the impact of PTPN12 deficiency on DC maturation and cytokine production, BMDCs were stimulated or not with LPS or CpG DNA, two TLR ligands (Fig. 3). Expression of markers of DC maturation was analyzed by flow cytometry (Fig. 3A). Exposure of BMDCs to LPS or CpG DNA resulted in increased expression of CD80, CD86, CD40, and MHC-II. No difference was noted between PTPN12-deficient and control DCs. Secretion of IL-6 and IL-12 was also analyzed by ELISA (Fig. 3B). PTPN12 deficiency had no impact on the ability of DCs to secrete either cytokine in the absence or in the presence of TLR ligands.
FIG 3.

Impact of PTPN12 deficiency on dendritic cell maturation and cytokine production. (A) Bone marrow-derived DCs from control (WT) mice or mice lacking PTPN12 in DCs (KO) were stimulated with lipopolysaccharide (LPS) (1,000 ng/ml) or CpG (100 nM) for 24 h. Then, cells were harvested and the indicated surface markers were analyzed by flow cytometry. Staining with isotype control antibodies is shown by the shaded gray curves. (B) The experiment was performed as for panel A, except that the indicated concentrations of LPS and CpG were used. Also, production of IL-6 and IL-12 by the different ranges of LPS or CpG was measured by ELISA. Standard deviations of triplicate values are shown. Data are representative of three experiments.
Therefore, expression of PTPN12 in DCs was not needed for DC differentiation in vitro and in vivo or for T cell homeostasis in vivo. It was also not needed for the capacity of DCs to mature and produce cytokines in response to TLR ligands.
PTPN12 expression in DCs is required for antigen-induced T cell responses in vivo.
We next tested the influence of PTPN12 deficiency in DCs on the ability to induce CD4+ T cell responses in vivo (Fig. 4). We focused our studies on T cell responses initiated via the subcutaneous route. Therefore, mice were immunized in the footpad with OVA in the presence of CFA. After 9 days, CD4+ T cells were isolated from draining popliteal lymph nodes and restimulated in vitro with OVA in the presence of irradiated splenocytes from wild-type mice. Thymidine incorporation and gamma interferon (IFN-γ) secretion were monitored. In this assay, DCs capture and process OVA in the skin while undergoing maturation in response to CFA. They then migrate to lymph nodes, where they present processed antigen to CD4+ T cells. Restimulation with OVA in vitro enables detection of OVA-specific CD4+ T cells generated in vivo.
FIG 4.

Compromised CD4+ T cell-dependent responses in vivo in mice lacking PTPN12 in dendritic cells. (A and B) Control (WT) mice or mice lacking PTPN12 in DCs (KO) were immunized with ovalbumin (OVA) and complete Freund adjuvant (CFA) in the footpad. After 9 days, popliteal lymph nodes were isolated and CD4+ T cells were purified. CD4+ T cells were then restimulated in vitro in the presence of irradiated splenocytes from nonimmunized normal C57BL/6 mice and the indicated concentration of OVA protein. After 3 to 5 days, cell proliferation was examined by measuring thymidine incorporation (A), and production of gamma interferon (IFN-γ) was measured by ELISA. Standard deviations of triplicate values are shown. (C) Various isotypes of Ig directed against OVA mice were measured by ELISA. The dilutions of serum used in the ELISA are indicated. Standard deviations of triplicate values are shown. Data are representative of four experiments.
CD4+ T cells isolated from mice lacking PTPN12 in DCs exhibited no defect in thymidine incorporation when restimulated with OVA in vitro (Fig. 4A). However, they had a reduction of IFN-γ production (Fig. 4B). This was seen in several independent experiments (data not shown). To confirm that CD4+ T cell functions were compromised in vivo, we also measured production of OVA-specific antibodies. OVA-specific antibodies are produced as a result of the ability of OVA-specific CD4+ T cells to trigger B cell differentiation, isotype switching, and affinity maturation. Whereas mice lacking PTPN12 in DCs had no defect in production of immunoglobulin M (Ig M), IgG1, and IgG2b against OVA, they displayed a decrease in OVA-specific IgG2c (Fig. 4C). In C57BL/6 mice, IFN-γ production by T cells stimulates secretion of IgG2c but not of other Ig isotypes (1).
Thus, expression of PTPN12 in DCs was required for full induction of antigen-specific CD4+ T cell responses in vivo.
Compromised T cell-dependent pathology in mice lacking PTPN12 in DCs.
The finding that Ptpn12fl/fl × Cd11c-Cre+ mice had a compromised CD4+ T cell response to OVA suggested that these mice might be less susceptible to immune pathologies mediated by CD4+ T cells. To address this possibility, mice were tested for susceptibility to experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis (Fig. 5) (23). They were immunized subcutaneously with a MOG-derived peptide in the presence of CFA. Under these conditions, the MOG peptide is taken up by local DCs, which mature and migrate to draining lymph nodes, where they present antigen to CD4+ T cells. Activated MOG-specific CD4+ T cells then migrate to the nervous system, where they react with endogenous MOG and provoke neurological defects. In comparison with control mice, mice lacking PTPN12 in DCs had a lower propensity to develop EAE. Hence, expression of PTPN12 in DCs was needed for mice to develop a CD4+ T cell-dependent immune pathology.
FIG 5.
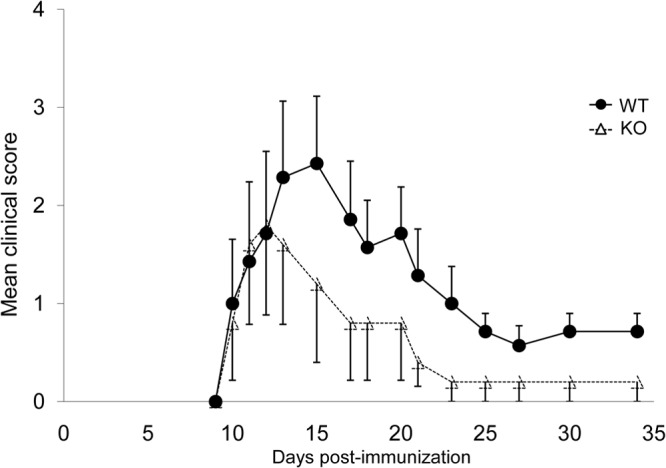
Mice lacking PTPN12 in dendritic cells are less susceptible to experimental autoimmune encephalomyelitis. Control (WT) mice or mice lacking PTPN12 in DCs (KO) (5 to 7 mice per group) were immunized with a myelin oligodendrocyte peptide (MOG35–55) mixed with complete Freund adjuvant and Mycobacterium tuberculosis H37Ra. After 24 and 72 h, additional treatments with pertussis toxin were administered. Mice were then monitored clinically every day for the development of signs of experimental autoimmune encephalomyelitis (EAE). Mean values and standard errors of clinical scores are shown. Clinical scores are as follows: 0, no clinical signs; 1, loss of tail tonicity; 2, flaccid tail; 3, hind leg paralysis; 4, hind leg paralysis with hind body paresis; 5, hind and fore leg paralysis; 6, death. Data are representative of three experiments.
Influence of PTPN12 on antigen presentation in vitro.
To understand how PTPN12 deficiency in DCs led to defects in CD4+ T cell-dependent responses in vivo, we first evaluated the possibility that PTPN12 was critical for the ability of DCs to form stable conjugates with T cells (Fig. 6). This step is critical for initiating antigen-specific T cell responses (6, 7, 11). In a conjugate formation assay using DCs and OVA-specific CD4+ T cells from OT-II TCR transgenic mice (15), loss of PTPN12 had no impact on conjugate formation between DCs and CD4+ T cells. This was seen at different concentrations of the OVA peptide OVA323–339, which is recognized by the OT-II TCR.
FIG 6.
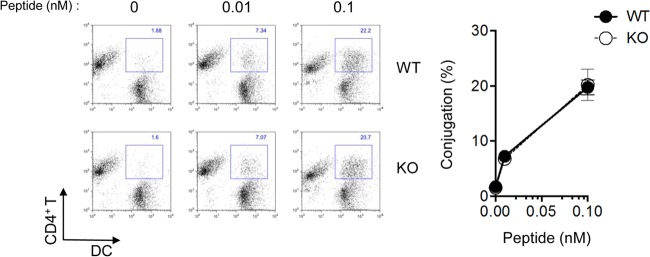
Impact of loss of PTPN12 on the capacity of dendritic cells to form conjugates with T cells. Bone marrow-derived dendritic cells (BMDCs) from control (WT) mice or mice lacking PTPN12 in DCs (KO) were labeled with carboxyfluorescein succinimidyl ester (CFSE), while CD4+ T cells from ovalbumin (OVA)-specific T cell receptor (TCR) transgenic OT-II mice were labeled with 5- (and 6-)(((4-chloromethyl)benzoyl)amino)tetramethylrhodamine (CMTMR). Cells were then mixed and incubated for 30 min at 37°C in the absence or presence of the indicated concentrations of OVA peptide. Conjugate formation was analyzed by flow cytometry (left). Average values with standard deviations are shown on the right and are representative of two independent experiments.
Next, we tested the role of PTPN12 in antigen presentation in vitro (Fig. 7). BMDCs were used to present antigens to CD4+ T cells from OT-II mice (15). Either the OVA peptide OVA323–339 (which does not require processing by DCs) or the OVA protein (which requires processing by DCs) was used for stimulation. When OVA323–339 was utilized for stimulation, there was no defect in the ability of PTPN12-deficient DCs to trigger thymidine incorporation or IFN-γ secretion by OT-II CD4+ T cells (Fig. 7A). When the full OVA protein was used for triggering, there also was no difference in thymidine incorporation (Fig. 7B). However, a moderate decrease (by ∼50%) in IFN-γ secretion was seen (Fig. 7B). This defect was observed in several experiments (data not shown).
FIG 7.

Influence of PTPN12 deficiency on the ability of dendritic cells to activate T cells in vitro. (A and B) Purified CD4+ T cells from ovalbumin (OVA)-specific T cell receptor (TCR) transgenic OT-II mice expressing PTPN12 were activated with bone marrow-derived dendritic cells (BMDCs) from control (WT) mice or mice lacking PTPN12 in DCs (KO) in the absence or presence of OVA peptide (OVA pep.) (A) or OVA protein (OVA pro.) (B). After 3 to 5 days, cell proliferation and IFN-γ production were measured by thymidine incorporation and ELISA, respectively, as described for Fig. 4. Standard deviations of triplicate values are shown. Data are representative of four experiments.
Thus, PTPN12 was not required for the abilities of DCs to form conjugates with CD4+ T cells and to present antigenic peptides to CD4+ T cells in vitro. However, it had a partial role in the aptitude of DCs to present unprocessed antigens.
Loss of PTPN12 results in compromised DC migration.
The finding that loss of PTPN12 in DCs seemed to have a greater impact on CD4+ T cell-dependent responses in vivo than in vitro suggested that PTPN12 was necessary for an additional DC function involved in initiation of T cell responses. One possibility was that PTPN12 was required for the ability of DCs to traffic to secondary lymphoid tissues, where they encounter T cells. To examine this notion, DC migration was first tested in vitro, using a Transwell migration assay (Fig. 8A). BMDCs, lacking or not lacking PTPN12, were loaded in the upper chamber of the Transwell apparatus. The lower chamber contained (or not) the chemokine SDF-1α or MIP-3β. Loss of PTPN12 in DCs resulted in a reduction of the ability of DCs to migrate in response to either chemokine. Importantly, it had no effect on the expression levels of CXCR4 and CCR7, which are the receptors for SDF-1α and MIP-3β, respectively (data not shown).
FIG 8.

Reduced migration by PTPN12-deficient dendritic cells. (A) Migration of bone marrow-derived dendritic cells (BMDCs) was assessed in vitro, using a Transwell migration assay. BMDCs (1 × 106 cells) from control (WT) mice or mice lacking PTPN12 in DCs (KO) were loaded in the upper chamber, while chemoattractants (stromal cell-derived factor 1α [SDF-1α] [200 ng/ml] or macrophage inflammatory protein 3β [MIP-3β] [200 ng/ml]) were placed in the lower chamber. After 1.5 h of incubation at 37°C, migrated cells in the lower chamber were harvested and counted by flow cytometry. Standard deviations of triplicate values and P values are shown. Data are representative of three experiments. (B) For in vivo migration, equal numbers of lipolysaccharide (LPS)-matured BMDCs from WT or KO mice (1 × 106 cells each), labeled either with carboxyfluorescein succinimidyl ester (CFSE) or 5- (and 6-)(((4-chloromethyl)benzoyl)amino)tetramethylrhodamine (CMTMR), were coinjected subcutaneously into the footpad of normal C57BL/6 mice. After 48 h, popliteal lymph nodes were isolated and the total number of CFSE+ or CMTMR+ cells was measured by flow cytometry. A representative experiment is shown at the top. Migrated BMDCs are boxed. Data from multiple independent mice are shown at the bottom. Mean values are represented by horizontal lines, and the P value is shown. Similar results were obtained whether WT and KO BMDCs were labeled with CFSE and CMTMR, respectively, or CMTMR and CFSE, respectively. Data are representative of three experiments.
The effect of PTPN12 deficiency on DC migration was also tested in vivo, using a competitive migration assay (Fig. 8B) (18). In brief, BMDCs from PTPN12-deficient or control mice were labeled with CFSE (green fluorescence) or CMTMR (red fluorescence), respectively. They were subsequently combined in equal numbers and injected into the footpads of normal mice. After 24 h, DC migration to draining lymph nodes was quantitated by flow cytometry. The recovery of PTPN12-deficient DCs in popliteal lymph nodes, which represent the immediate draining site, was significantly reduced (by ∼50%) in comparison to that of control DCs. No labeled DCs were detected in inguinal or axillary lymph nodes, which are not the immediate draining sites for the footpad (data not shown). The latter finding confirmed that DC migration occurred through lymphatic channels and not through blood vessels.
Therefore, expression of PTPN12 was critical for DC migration in vitro and in vivo.
Altered tyrosine phosphorylation of Pyk2 and paxillin in PTPN12-deficient DCs.
To identify the deregulated tyrosine phosphorylation substrates responsible for the defects in PTPN12-deficient DCs, protein tyrosine phosphorylation was examined (Fig. 9A). Immunoblotting of total cell lysates with anti-phosphotyrosine antibodies showed that loss of PTPN12 had minimal effects on global protein tyrosine phosphorylation, with the exception of an ∼110-kDa substrate (pp110). This product was more phosphorylated in PTPN12-deficient DCs and was reminiscent of the PTK Pyk2. Immunoprecipitation of known PTPN12 substrates showed that tyrosine phosphorylation of Pyk2 was indeed increased 3- to 5-fold in PTPN12-deficient DCs. Similar changes were seen for paxillin, an adaptor molecule bound and phosphorylated by Pyk2 (24–27). In contrast, there was no alteration in tyrosine phosphorylation of PSTPIP-1, another adaptor that can be regulated by PTPN12 (28). There was also no impact on tyrosine phosphorylation of SIRPα, Syk, or Src family kinases. Little or no expression of FAK, a PTK closely related to Pyk2, or Cas was noted in these cells. The difference in tyrosine phosphorylation of Pyk2 between PTPN12-deficient and control DCs was not appreciably enhanced by treatment of cells with SDF-1α or MIP-3β (Fig. 9B) or by replating cells on fibronectin (Fig. 9C).
FIG 9.

Increased tyrosine phosphorylation of Pyk2 and paxillin in PTPN12-deficient dendritic cells. (A) Bone marrow-derived dendritic cells (BMDCs) from control (WT) mice or mice lacking PTPN12 in DCs (KO) were lysed. Tyrosine phosphorylation was analyzed by immunoblotting with antiphosphotyrosine (pTyr) antibodies or antibodies against the activating tyrosine of Src family kinases. The loading was verified by reprobing the immunoblot membranes with substrate-specific antibodies. Little or no Cas and FAK was detected in these cells. Data are representative of at least four experiments. IP, immunoprecipitates. (B) BMDCs were stimulated for 5 min with the indicated chemokines, and tyrosine phosphorylation on Pyk2 was assessed as detailed for panel A. Data are representative of three experiments. (C) BMDCs were harvested and replated for 30 min at 37°C on fibronectin (FN)-coated dishes (+). Control cells (-) were kept in suspension. Tyrosine phosphorylation of Pyk2 and paxillin was ascertained as detailed for panel A. Results are representative of three independent experiments.
To assess if Pyk2 deregulation contributed to the migration defect of PTPN12-deficient DCs, BMDCs were first treated with PF-431396, a pharmacological inhibitor of Pyk2 (Fig. 10) (19, 20). Treatment of PTPN12-deficient DCs with PF-431396 resulted in dose-dependent inhibition of tyrosine phosphorylation of Pyk2 and its substrate, paxillin (Fig. 10A). In a Transwell migration assay, it significantly interfered with the ability of control DCs to migrate in response to SDF-1α and MIP-3β (Fig. 10B). However, it caused no further reduction of the compromised ability of PTPN12-deficient DCs to migrate. Moreover, it did not rescue the migration defect observed in PTPN12-deficient DCs (Fig. 10C). These results implied that proper enzymatic activity of Pyk2 was needed for migration of normal DCs in response to chemokines.
FIG 10.

Influence of pharmacological inhibition of Pyk2 on dendritic cell migration. Bone marrow-derived dendritic cells (BMDCs) from control (WT) mice or mice lacking PTPN12 in DCs (KO) were pretreated for 24 h without or with the indicated concentrations of PF-431396, a pharmacological inhibitor of Pyk2. (A) Tyrosine phosphorylation of Pyk2 or paxillin was analyzed as detailed for Fig. 9A. Quantitation of relative tyrosine phosphorylation is shown at the bottom of each lane. (B and C) Migration was analyzed using a Transwell migration assay, as described for Fig. 8A. Standard deviations of triplicate values are shown. Data are representative of three experiments.
To confirm the involvement of Pyk2 in DC migration, expression of Pyk2 was also downregulated in control DCs using small interfering RNAs (siRNAs) (Fig. 11). In comparison to control siRNAs, Pyk2-specific siRNAs caused an ∼50% reduction of Pyk2 expression (Fig. 11A). Importantly, they also triggered a diminution of migration in response to SDF-1α and MIP-3β (Fig. 11B).
FIG 11.
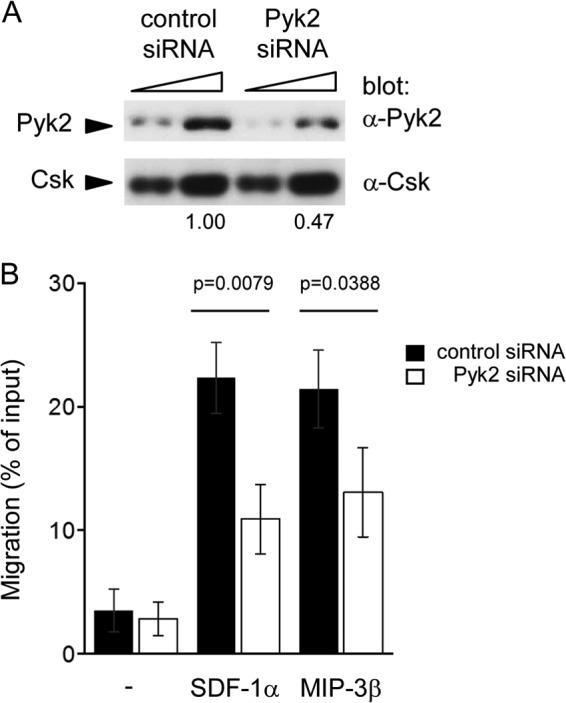
Impact of downregulated expression of Pyk2 on dendritic cell migration. Bone marrow-derived dendritic cells (BMDCs) from wild-type mice were transfected with small interfering RNAs (siRNAs) against Pyk2 or control siRNAs, using nucleofection. (A) After 24 to 36 h, cells were lysed and analyzed for Pyk2 expression. Two different quantities of lysates were used for each cell population. Quantitation of relative abundance of Pyk2 is shown at the bottom. (B) After 24 to 36 h, migration was analyzed as detailed for Fig. 8A. Data are representative of three experiments.
Hence, PTPN12 was necessary for dephosphorylation of Pyk2 and paxillin. Moreover, Pyk2 activity and expression were required for migration of DCs.
DISCUSSION
To determine the role of PTPN12 in DCs, Ptpn12fl/fl mice were crossed with mice expressing Cre under the control of the Cd11c promoter (14). Cd11c-Cre resulted in ∼70 to 90% reduction of PTPN12 expression in splenic DCs and BMDCs. No reduction was seen in other immune cell types, including CD4+ T cells. Hence, Cd11c-Cre enabled efficient deletion of PTPN12 in DCs but not in other immune cell types. These findings were in keeping with data obtained for other conditional alleles deleted by Cd11c-Cre (2).
Loss of PTPN12 in DCs had no appreciable impact on the proportions of cDCs and pDCs in spleen and skin draining lymph nodes. Moreover, it had no influence on expression of differentiation markers on DCs. PTPN12 deficiency also had no effect on the proportions of mature T cells and TRegs in lymphoid tissues. DCs are required to maintain these cell populations under homeostatic conditions (1). There was also no impact on the ability of bone marrow cells to differentiate into cDCs or pDCs in vitro in the presence of GM-CSF or Flt3-L, respectively. Furthermore, there was no effect on the capacity of BMDCs to mature and secrete cytokines in response to TLR ligands in vitro. Thus, PTPN12 was not needed for DC differentiation and maturation in vivo and in vitro.
To examine if PTPN12 expression in DCs was critical for initiating antigen-specific immune responses, mice were immunized with the OVA protein in the presence of CFA. CD4+ T cells were then isolated from draining lymph nodes and restimulated in vitro with OVA in the presence of normal APCs. Expression of PTPN12 in DCs was crucial for the ability to CD4+ T cells to produce IFN-γ upon antigenic restimulation. It was also needed for production of OVA-specific IgG2c, which is controlled by IFN-γ. By opposition, there was no effect on thymidine incorporation. We also examined the impact of PTPN12 deficiency in DCs on the capacity to trigger EAE, a CD4+ T cell-mediated immune pathology (23). PTPN12 deficiency resulted in reduced severity of EAE. Coupled with the findings of the OVA immunization experiments, these data indicated that PTPN12 expression in DCs was necessary for antigen-specific CD4+ T cell responses in vivo.
Subsequent experiments were aimed at elucidating why expression of PTPN12 in DCs was required for antigen-evoked T cell responses. When processed OVA peptides (OVA323–339) were used for stimulation, PTPN12 deficiency had no consequence for the capacity of DCs to form conjugates with OVA-specific CD4+ T cells or induce thymidine incorporation or IFN-γ secretion by OVA-specific CD4+ T cells. However, when unprocessed OVA was utilized, a small (∼50%) albeit reproducible decrease in IFN-γ release was noted, although no effect on thymidine incorporation was seen. These observations suggested that PTPN12 expression in DCs was needed for optimal capture, processing, or presentation of intact protein antigens but not antigenic peptides. Additional studies will be needed to clarify this matter.
The defects in OVA-specific responses in vivo appeared to be more severe than those noted in vitro. Considering this observation, we tested the involvement of PTPN12 in DC migration, which is needed to bring antigen-loaded DCs from peripheral sites to lymphoid tissues (1). Using a Transwell migration assay, we observed that PTPN12-deficient DCs had compromised migration in response to SDF-1α or MIP-3β. Likewise, in a competitive in vivo migration assay, the capacity of PTPN12-deficient DCs to migrate from subcutaneous tissues to draining lymph nodes was reduced. Hence, PTPN12 had a critical role in DC migration.
In order to elucidate the molecular mechanism by which PTPN12 controlled DC migration, the extent of tyrosine phosphorylation of known PTPN12 substrates was ascertained. Loss of PTPN12 resulted in augmented phosphorylation of Pyk2 and paxillin (24, 27). Pyk2 hyperphosphorylation was constitutive and was not significantly altered by exposure to chemokines or replating on fibronectin. Inhibition of Pyk2 by a pharmacological inhibitor or downregulation of Pyk2 expression by siRNAs also resulted in a reduction of DC migration by normal DCs, confirming previous findings that Pyk2 is critical for migration in other cell types (24). It is also likely that deregulation of paxillin contributed to the defect of DC migration caused by PTPN12 deficiency. Paxillin is a substrate of Pyk2, and its tyrosine phosphorylation influences migration through regulation of actin reorganization (27).
It may seem paradoxical that either enhanced tyrosine phosphorylation or inactivation of Pyk2 was accompanied by compromised DC migration. One possibility is that either alteration led to a similar deregulation of the migration machinery. In order to function properly, migration-regulating substrates need to be tyrosine phosphorylated in the right amount at the right time. Alternatively, it is possible that the two alterations of Pyk2 had distinct effects on the migration machinery. For example, one alteration may interfere with membrane protrusion at the leading edge, whereas the other may impede membrane retraction at the uropod. Since membrane protrusion and subsequent membrane retraction are needed for migration, either alteration might yield compromised migration. Future studies will be needed to distinguish these possibilities.
Depending on the cell type involved, loss of PTPN12 augments tyrosine phosphorylation of different substrates. In mouse embryo fibroblasts, it resulted in hyperphosphorylation of Cas, FAK, and PSTPIP-1 (6, 28). In endothelial cells, it yielded enhanced tyrosine phosphorylation of Cas, paxillin, and Pyk2 (11). Hence, in nonimmune cells, it seems that Cas is a preferential target of PTPN12. In contrast, in T cells, absence of PTPN12 caused hyperphosphorylation of Pyk2 (7). In macrophages, it led to increased tyrosine phosphorylation of Pyk2 and paxillin (13). A similar effect is reported herein for DCs. Thus, in immune cells, it appears that Pyk2 is a privileged target of PTPN12. Although the basis for this distinction between nonimmune and immune cells is not known, it may reflect the types of pathways that are preferentially utilized in the functioning of these cell types. This notion may also be consistent with the finding that Cas was not detectably expressed in macrophages and DCs, whereas Pyk2 is primarily expressed in immune cells.
Based on our findings, we propose the following model. Upon exposure to foreign elements, such as pathogens, DCs located in peripheral tissues capture and process foreign antigens. They also undergo maturation, which renders them more competent at presenting antigens to T cells. These processes are largely independent of PTPN12, although a small role for PTPN12 may exist in capture, processing, or presentation of intact antigens but not of antigenic peptides. Then, in order to contact T cells, antigen-loaded DCs migrate to secondary lymphoid tissues where T cells are located. This migration is highly dependent on PTPN12.
In summary, PTPN12 is a key component of the machinery controlling DC migration from peripheral subcutaneous sites to secondary lymphoid tissues. As a result, it is critical for initiation of antigen-induced T cell responses. The machinery regulating DC migration also includes SDF-1α and MIP-3β, which interact with their receptors CXCR4 and CCR7 (29), as well as intracellular effectors, such as Src kinases, Pyk2, and the small G protein Rac-1 (30, 31). It should be pointed out, though, that DCs are located not only in the skin but also in other tissues, including the intestinal tract, thymus, and bone marrow (1). DCs need to migrate in and out of these tissues for their functions, and this process is regulated by chemokines. Although no difference in the numbers of DCs in lymphoid tissues was seen under steady-state conditions in our study, it is tempting to speculate that PTPN12 may also play a key role in trafficking of DCs at and from these sites during inflammation and infections. Studies addressing this possibility are warranted.
ACKNOWLEDGMENTS
We thank the members of our laboratory for discussions.
This work was supported by grants from the Canadian Institutes of Health Research to A.V. and C.C. and from the Canadian Cancer Society Research Institute to A.V. A.V. holds the Canada Research Chair in Signaling in the Immune System; C.C. is a Chercheur-Boursier Junior of Fonds de Recherche du Québec-Santé.
Footnotes
Published ahead of print 23 December 2013
REFERENCES
- 1.Murphy K, Travers P, Walport M, Janeway C. 2008. Immunobiology, 7th Ed. Garland Science, New York, NY [Google Scholar]
- 2.Ganguly D, Haak S, Sisirak V, Reizis B. 2013. The role of dendritic cells in autoimmunity. Nat. Rev. Immunol. 13:566–577. 10.1038/nri3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veillette A, Rhee I, Souza CM, Davidson D. 2009. PEST family phosphatases in immunity, autoimmunity, and autoinflammatory disorders. Immunol. Rev. 228:312–324. 10.1111/j.1600-065X.2008.00747.x [DOI] [PubMed] [Google Scholar]
- 4.Yang Q, Co D, Sommercorn J, Tonks NK. 1993. Cloning and expression of PTP-PEST. A novel, human, nontransmembrane protein tyrosine phosphatase. J. Biol. Chem. 268:6622–6628 (Erratum, 268:17650.) [PubMed] [Google Scholar]
- 5.Rhee I, Veillette A. 2012. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat. Immunol. 13:439–447. 10.1038/ni.2246 [DOI] [PubMed] [Google Scholar]
- 6.Angers-Loustau A, Cote JF, Charest A, Dowbenko D, Spencer S, Lasky LA, Tremblay ML. 1999. Protein tyrosine phosphatase-PEST regulates focal adhesion disassembly, migration, and cytokinesis in fibroblasts. J. Cell Biol. 144:1019–1031. 10.1083/jcb.144.5.1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davidson D, Shi X, Zhong MC, Rhee I, Veillette A. 2010. The phosphatase PTP-PEST promotes secondary T cell responses by dephosphorylating the protein tyrosine kinase Pyk2. Immunity 33:167–180. 10.1016/j.immuni.2010.08.001 [DOI] [PubMed] [Google Scholar]
- 8.Garton AJ, Tonks NK. 1999. Regulation of fibroblast motility by the protein tyrosine phosphatase PTP-PEST. J. Biol. Chem. 274:3811–3818. 10.1074/jbc.274.6.3811 [DOI] [PubMed] [Google Scholar]
- 9.Sastry SK, Lyons PD, Schaller MD, Burridge K. 2002. PTP-PEST controls motility through regulation of Rac1. J. Cell Sci. 115:4305–4316. 10.1242/jcs.00105 [DOI] [PubMed] [Google Scholar]
- 10.Sirois J, Cote JF, Charest A, Uetani N, Bourdeau A, Duncan SA, Daniels E, Tremblay ML. 2006. Essential function of PTP-PEST during mouse embryonic vascularization, mesenchyme formation, neurogenesis and early liver development. Mech. Dev. 123:869–880. 10.1016/j.mod.2006.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Souza CM, Davidson D, Rhee I, Gratton JP, Davis EC, Veillette A. 2012. The phosphatase PTP-PEST/PTPN12 regulates endothelial cell migration and adhesion, but not permeability, and controls vascular development and embryonic viability. J. Biol. Chem. 287:43180–43190. 10.1074/jbc.M112.387456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I, Nguyen DX, Pavlova NN, Botero M, Huang J, Bernardi RJ, Schmitt E, Hu G, Li MZ, Dephoure N, Gygi SP, Rao M, Creighton CJ, Hilsenbeck SG, Shaw CA, Muzny D, Gibbs RA, Wheeler DA, Osborne CK, Schiff R, Bentires-Alj M, Elledge SJ, Westbrook TF. 2011. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 144:703–718. 10.1016/j.cell.2011.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhee I, Davidson D, Souza CM, Vacher J, Veillette A. 2013. Macrophage fusion is controlled by the cytoplasmic protein tyrosine phosphatase PTP-PEST/PTPN12. Mol. Cell. Biol. 33:2458–2469. 10.1128/MCB.00197-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caton ML, Smith-Raska MR, Reizis B. 2007. Notch–RBP-J signaling controls the homeostasis of CD8− dendritic cells in the spleen. J. Exp. Med. 204:1653–1664. 10.1084/jem.20062648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnden MJ, Allison J, Heath WR, Carbone FR. 1998. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76:34–40. 10.1046/j.1440-1711.1998.00709.x [DOI] [PubMed] [Google Scholar]
- 16.Cloutier JF, Veillette A. 1996. Association of inhibitory tyrosine protein kinase p50csk with protein tyrosine phosphatase PEP in T cells and other hemopoietic cells. EMBO J. 15:4909–4918 [PMC free article] [PubMed] [Google Scholar]
- 17.Davidson D, Cloutier JF, Gregorieff A, Veillette A. 1997. Inhibitory tyrosine protein kinase p50csk is associated with protein-tyrosine phosphatase PTP-PEST in hemopoietic and non-hemopoietic cells. J. Biol. Chem. 272:23455–23462. 10.1074/jbc.272.37.23455 [DOI] [PubMed] [Google Scholar]
- 18.Martin-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, Sallusto F. 2003. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J. Exp. Med. 198:615–621. 10.1084/jem.20030448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tse KW, Dang-Lawson M, Lee RL, Vong D, Bulic A, Buckbinder L, Gold MR. 2009. B cell receptor-induced phosphorylation of Pyk2 and focal adhesion kinase involves integrins and the Rap GTPases and is required for B cell spreading. J. Biol. Chem. 284:22865–22877. 10.1074/jbc.M109.013169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han S, Mistry A, Chang JS, Cunningham D, Griffor M, Bonnette PC, Wang H, Chrunyk BA, Aspnes GE, Walker DP, Brosius AD, Buckbinder L. 2009. Structural characterization of proline-rich tyrosine kinase 2 (PYK2) reveals a unique (DFG-out) conformation and enables inhibitor design. J. Biol. Chem. 284:13193–13201. 10.1074/jbc.M809038200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veillette A, Bookman MA, Horak EM, Bolen JB. 1988. The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck. Cell 55:301–308. 10.1016/0092-8674(88)90053-0 [DOI] [PubMed] [Google Scholar]
- 22.Villadangos JA, Schnorrer P. 2007. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat. Rev. Immunol. 7:543–555. 10.1038/nri2103 [DOI] [PubMed] [Google Scholar]
- 23.Zhong MC, Kerlero de Rosbo N, Ben-Nun A. 2002. Multiantigen/multiepitope-directed immune-specific suppression of “complex autoimmune encephalomyelitis” by a novel protein product of a synthetic gene. J. Clin. Invest. 110:81–90. 10.1172/JCI15692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avraham H, Park SY, Schinkmann K, Avraham S. 2000. RAFTK/Pyk2-mediated cellular signalling. Cell Signal. 12:123–133. 10.1016/S0898-6568(99)00076-5 [DOI] [PubMed] [Google Scholar]
- 25.Bonnette PC, Robinson BS, Silva JC, Stokes MP, Brosius AD, Baumann A, Buckbinder L. 2010. Phosphoproteomic characterization of PYK2 signaling pathways involved in osteogenesis. J. Proteomics 73:1306–1320. 10.1016/j.jprot.2010.01.011 [DOI] [PubMed] [Google Scholar]
- 26.Hagel M, George EL, Kim A, Tamimi R, Opitz SL, Turner CE, Imamoto A, Thomas SM. 2002. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol. Cell. Biol. 22:901–915. 10.1128/MCB.22.3.901-915.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turner CE. 2000. Paxillin and focal adhesion signalling. Nat. Cell Biol. 2:E231–E236. 10.1038/35046659 [DOI] [PubMed] [Google Scholar]
- 28.Cote JF, Chung PL, Theberge JF, Halle M, Spencer S, Lasky LA, Tremblay ML. 2002. PSTPIP is a substrate of PTP-PEST and serves as a scaffold guiding PTP-PEST toward a specific dephosphorylation of WASP. J. Biol. Chem. 277:2973–2986. 10.1074/jbc.M106428200 [DOI] [PubMed] [Google Scholar]
- 29.Riol-Blanco L, Sanchez-Sanchez N, Torres A, Tejedor A, Narumiya S, Corbi AL, Sanchez-Mateos P, Rodriguez-Fernandez JL. 2005. The chemokine receptor CCR7 activates in dendritic cells two signaling modules that independently regulate chemotaxis and migratory speed. J. Immunol. 174:4070–4080 [DOI] [PubMed] [Google Scholar]
- 30.Benvenuti F, Hugues S, Walmsley M, Ruf S, Fetler L, Popoff M, Tybulewicz VL, Amigorena S. 2004. Requirement of Rac1 and Rac2 expression by mature dendritic cells for T cell priming. Science 305:1150–1153. 10.1126/science.1099159 [DOI] [PubMed] [Google Scholar]
- 31.Hauck CR, Klingbeil CK, Schlaepfer DD. 2000. Focal adhesion kinase functions as a receptor-proximal signaling component required for directed cell migration. Immunol. Res. 21:293–303. 10.1385/IR:21:2-3:293 [DOI] [PubMed] [Google Scholar]