

Abstract
The majority of patients with BCR-ABL1-negative myeloproliferative neoplasms (MPN) harbor mutations in JAK2 or MPL, which lead to constitutive activation of the JAK/STAT, PI3K, and ERK signaling pathways. JAK inhibitors by themselves are inadequate in producing selective clonal suppression in MPN and are associated with hematopoietic toxicities. MK-2206 is a potent allosteric AKT inhibitor that was well tolerated, including no evidence of myelosuppression, in a phase I study of solid tumors. Herein, we show that inhibition of PI3K/AKT signaling by MK-2206 affected the growth of both JAK2V617F or MPLW515L-expressing cells via reduced phosphorylation of AKT and inhibition of its downstream signaling molecules. Moreover, we demonstrate that MK-2206 synergizes with Ruxolitinib in suppressing the growth of JAK2V617F mutant SET2 cells. Importantly MK-2206 suppressed colony formation from hematopoietic progenitor cells in patients with primary myelofibrosis (PMF) and alleviated hepatosplenomegaly and reduced megakaryocyte burden in the bone marrows, livers and spleens of mice with MPLW515L-induced MPN. Together, these findings establish AKT as a rational therapeutic target in the MPNs.
Keywords: Myelofibrosis, PI3K, MPL, JAK2
Introduction
The BCR-ABL negative myeloproliferative neoplasms (MPNs) are among the most common hematologic malignancies in the US with a prevalence of at least 130,000-150,000(1). MPNs, including polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF), arise in genetically transformed hematopoietic stem cells that retain the capacity for multi-lineage differentiation and effective myelopoiesis. In 2005, a novel activating mutation involving the Janus kinase 2 gene (JAK2), which resulted in expression of the V617F activated mutant, was identified in a substantial fraction of patients with all three subtypes of MPNs (2-6). This discovery led to significant developments in the diagnosis of MPNs and the advent of novel therapies (7, 8).
JAK2 V617F as well as exon 12 mutant alleles seen in JAK2V617F-negative MPN lead to enhanced JAK2 kinase activity and cytokine-independent growth of primary cells and cell lines. Mutations in JAK2 are associated with the vast majority of cases of PV and up to 50% of patients with ET and PMF (9). Sequencing of cytokine receptors in MPN patients lacking a JAK2 mutation led to the discovery of somatic mutations at codon 515 of the thrombopoietin receptor (MPLW515L) in ET (8% of patients) and PMF (10-15% of patients) (10, 11). Similar to the JAK2V617F mutation, expression of MPLW515L leads to cytokine-independent growth of murine and human hematopoietic cells and constitutive activation of the JAK/STAT pathway (10). In a murine retroviral transplant model, MPLW515L resulted in abnormal megakaryocyte expansion and myelofibrosis (10), in contrast to the PV phenotype seen in recipients of JAK2V617F-transformed hematopoietic cells (12-15). It should be noted that no significant differences in overall or leukemia free survival was noted among JAK2 mutated MPL mutated, or JAK2/MPL unmutated patients (16). Apart from mutations in JAK2 and MPL, MPN cells harbor mutations in TET2, ASXL1, SF3B1, EZH2, IDH, DNMT3a, among others, and that the presence of some of these mutations affect outcome (17-20).
Until very recently, management strategies for the MPNs were largely empiric, and depending on the phenotype, consisted of anti-platelet therapy, phlebotomy, hydroxyurea, androgens, anagrelide, immunomodulatory agents, erythropoietin stimulating agents and IFN-α. Recently, the FDA approved the small molecule Ruxolitinib as the first oral JAK inhibitor in patients in myelofibrosis. In clinical trials, Ruxolitinib reduced splenomegaly and improved constitutional symptoms, however, was associated with the development of anemia and thrombocytopenia in a significant subset of MF patients (8, 21). A number of other JAK inhibitors are in varying stages of pre-clinical and clinical development (22, 23). While as a group JAK inhibitors suppress kinase activity in vitro, they show varying effects on JAK2 mutant allele burden in patients and none has been shown to eliminate the malignant clone in an animal model of MPN (15) or in patients. Thus, although JAK inhibitors provide relief of many MPN associated pathologies, they are not curative and should be used in a select group of MF patients whose symptoms justify the need for JAK inhibitor therapy (24).
While much of the research to date has focused on the activation of JAK/STAT signaling in MPN patients, other pathways downstream of the class I cytokine receptors, including PI3K/AKT are also prominently activated in JAK2V617 and MPLW515L induced MPNs (10, 25-29). Of note, dependence of tumor cells on PI3K/AKT signaling has been observed in several oncogenic networks. For example, the PI3K/AKT pathway is required for BCR-ABL induced leukemia in animal models of Ph+ B-ALL (30). Moreover, PI3K/AKT/mTOR inhibitors have been shown to effectively and selectively target MPN cells (31, 32), leukemia cells (33, 34) and solid tumors in pre-clinical and/or clinical studies (35, 36).
Here, using MPN cell lines and patient specimens, we show that inhibition of PI3K/AKT signaling with the selective AKT inhibitor MK-2206 induces proliferative arrest and apoptosis of MPN cells in vitro and reduces MPN tumor burden in vivo. We also demonstrate that MK-2206 and Ruxolitinib cooperate to suppress the growth of SET2 cells that harbor the JAK2V617F mutation, suggesting that combining these two agents represents a rational therapeutic strategy for MPNs with sufficient rationale to support clinical investigation.
Materials and Methods
Reagents
MK-2206, 8-[4-(1-aminocyclobutyl)phenyl]-9-phenyl-1,2,4-triazolo[3,4-f] [1,6]naphthyridin-3(2H)-one hydrochloride [1:1], was generously provided by Merck. For in vitro experiments, 10 μM stock solutions of MK-2206 were formulated in DMSO and subsequently diluted in RPMI-1640 media for HEL and SET2 cells. All other compounds were purchased from either Sigma or Calbiochem. Antibodies used for Western blotting included phosphorylated and total AKT, PRAS-40, and BAD (Cell Signaling).
Cell lines and retroviral transduction
HEL and SET2 cells (37) were grown in RPMI-1640 with 10% fetal bovine serum (FBS). 293T cells were grown in DMEM with 10% FBS. Transient transfection of 293T cells and generation of retroviral supernatant were performed using Fugene (Roche, New Jersey, United States) according to manufacturer's guidelines.
Analysis of growth, cell cycle and apoptosis
Logarithmically growing cells were seeded in a 48-well plate and exposed to the designated concentrations of MK-2206 for 48 hours and viable cells were quantified by Trypan blue staining. Values were transformed to percent inhibition relative to vehicle control (0.1% DMSO) and EC50 curves were fitted according to non-linear regression analysis of the data using PRISM Graphpad. For proliferation assays, cells were labeled with 30 μg/ml bromodeoxyuridine (BrdU) for 30 min, fixed with 2% paraformaldehyde (PFA) for 10 min at room temperature, permeabilized with ethanol (400 μl of 150 mM NaCl, 850 μl of 100% ethanol) for 30 min on ice, and fixed (1% PFA and 0.1% Tween 20 in Hanks balanced salt solution) overnight at 4°C. After permeabilization, cells were treated with 30 μg DNAse for 1 hr at 37° C, stained with Alexa 647-labeled anti-BrdU antibody for 1 hour at room temperature, and DAPI was added before analysis with flow cytometry. For annexin V staining, cells were incubated with an annexin V-Cy5 antibody (BioVision) in staining buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4) for 10 min. The viability dye Sytox-blue was added before the cells were assayed for apoptosis and necrosis by flow cytometry. Flow cytometry was performed on an LSRII (BD), and data were analyzed with FlowJo software (Tree Star, Ashland, OR).
Patient samples
Use of MF samples was approved by the IRBs at Northwestern University and the Mayo Clinic. Peripheral blood was collected from PMF patients in EDTA tubes and mononuclear cells were separated on a ficoll gradient. Mononuclear cells were washed with serum-free IMDM and depleted of red cells before CD34+ cells were purified by immuno-magnetic beads conjugated with anti-CD34 antibody (Miltenyi Biotec). CD34+ cells were cultured in HPGM in the presence of recombinant human SCF (25 ng/ml), TPO (20 ng/ml) and FLT-3L (10 ng/ml) for 48 hrs to allow expansion. 1500 (CFU-M and BFU-E) or 5000 (CFU-MK) cells were then plated in methylcellulose-based colony assays (Methocult H4435, Stem Cell technologies) in the presence of 1-10 μM MK-2206 or DMSO (0.1%) and scored for CFU-GM and BFU-E colonies on days 11-12 respectively. In parallel 5×103 CD34+ cells were plated in CFU-MK colony assays in collagen-based media (Megacult-C #04901) in chamber slides in the presence of 1-10 μM MK-2206 or DMSO (0.1%) and scored after 14 days by staining with an anti-CD41 antibody. The levels of significance for the differential sensitivities of PMF versus normal cell colony assays were determined by ANCOVA.
Murine model of MPN
The MPLW515L bone marrow transplants were performed as previously described (10). Briefly, bone marrow cells were harvested from 5-FU pre-treated female Balb/c donor mice and transduced with viral supernatants containing MSCV-MPLW515L-GFP. 500,000 bone marrow cells were then injected into the tail veins of irradiated recipient mice along with 100,000 support cells from healthy Balb/c mice. Tail bleeds were performed at day 21 to document disease as measured by >50% GFP positivity in the peripheral blood and elevated WBC counts. Mice were then randomized into 3 groups (n=8/group) and treated with vehicle or MK-2206 at 60 mg/kg or 120 mg/kg for 2 weeks and then euthanized. The drug was administered by oral gavage once daily on a Mon-Wed-Fri schedule. All mice were treated for 14 days or until any one of several criteria for sacrifice was met, including severe lethargy or loss of >20% of body weight. After sacrifice, peripheral blood was collected and peripheral counts were measured on a HemaVet 950FS (Drew scientific). Sternum, liver and spleen samples were fixed in formalin and then embedded in paraffin for histopathology. H&E staining was performed by the pathology core. Immunohistochemistry was performed for Von Willebrand Factor using the Dako A0082 antibody. For flow cytometry, bone marrow and spleen cells were washed and stained in PBS+0.1% BSA buffer. Antibodies used included CD41-DyLight 649 (Emfret), CD42-PE (Emfret), Mac1-APC and Gr1-PE (BD Bioscience). A separate cohort of 9 mice was transplanted with malignant cells for pharmacodynamic studies. These mice were randomized into 3 groups (n=3/group) and treated with vehicle or MK-2206 at 60 mg/kg or 120 mg/kg for 1 week and then euthanized 24 hours after the last dose. Whole bone marrow and spleen lysates were used for western blot analysis. Three other cohorts of 4 mice each were treated with vehicle or MK-2206 at 60 mg/kg or 120 mg/kg for 2 weeks and then euthanized 24 hours after the last dose to evaluate the effect on hematopoiesis in healthy animals. Animal studies were approved by the Northwestern University Institutional Animal Care and Use Committee.
Results
MK-2206 induces cell cycle arrest and apoptosis in JAK2V617F cell lines
MK-2206, a highly selective non-ATP competitive allosteric AKT inhibitor (38), is orally bioavailable and has demonstrated excellent tolerability in clinical trials in the solid tumor setting (36). To better understand the consequences of AKT inhibition in MPNs, we cultured human HEL and SET2 cells that harbor the JAK2V617F mutation. We treated these lines with increasing doses of MK-2206 and enumerated live cells at 24 and 48 hours respectively by Trypan blue staining. We found the 50% effective concentration (EC50) to be 4.1 μM for SET2 cells and 1.0 μM for HEL cells and (Fig. 1A and B). Next, to determine how MK-2206 reduced the growth of these cell lines, we assayed the effects of this inhibitor on cell cycle distribution, proliferation and induction of apoptosis. We observed a significant induction of necrosis in SET2 cells at doses above 1 μM, as determined by Annexin V/Sytox staining with the percentage of viable cells to less than 25% at 5 μM (Fig 1C). HEL cells also showed a dramatic induction of apoptosis and necrosis at doses above 1 μM (Fig 1D). In addition to a significant effect on cell death, we observed a dose-dependent cell cycle G0/G1 block in HEL cells treated with MK-2206, as assayed by BrdU staining (Fig 1E). Together, these results suggest that induction of apoptosis and cell cycle arrest are an important basis of the observed cellular effect of MK-2206 in the HEL and SET2 cell lines.
Figure 1. MK-2206 inhibits cell growth by inducing cell cycle arrest and apoptosis in HEL and SET2 cells.
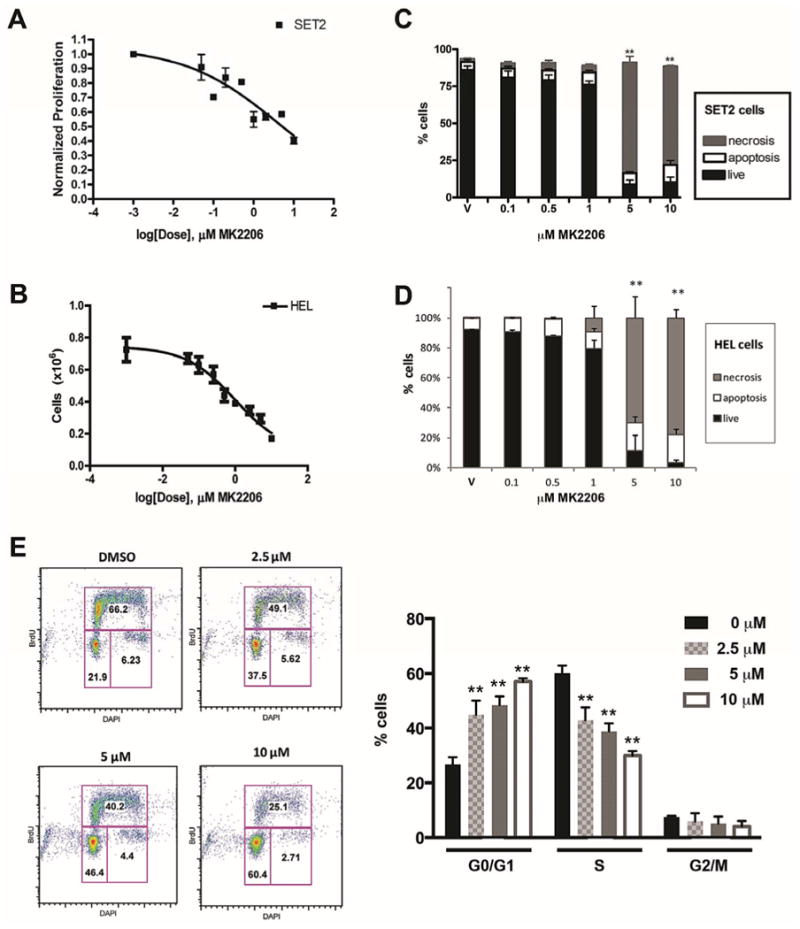
SET2 (A) and HEL (B) cells were cultured with a range of doses of MK-2206 from 0 to 10 μM. Live cells were enumerated at 48 hours using trypan blue staining and the 50% effective concentration (EC50) was calculated using Graph-Prism software. (C,D) Induction of apoptosis and necrosis across a range of concentrations of MK-2206 was assessed in SET2 (C) and HEL (D) cells at 24 hrs using Annexin V surface expression and Sytox-blue permeability dye. Means +/- SD are shown, n=3. (E) BrdU/DAPI staining of viable HEL cells treated with MK-2206 for 24 hours at concentrations from 0 to 10 μM. Representative plots (left) and bar graph of means (n=4) +/- SEM (right), are shown. **p<0.01
MK-2206 inhibits PI3K/AKT signaling in MPN cells
To assess the effects of AKT inhibition on signaling pathways, we extracted protein from HEL cells and primary human CD34+ cells from a PMF patient, treated the cells with MK-2206 and then performed western blot analysis. Treatment of HEL cells with MK-2206 for 6 hours blunted phosphorylation of AKT at concentrations as low as 1 μM (Fig 2A). Concomitant with the striking decrease in pAKT, we also observed inhibition of the downstream signaling molecule pPRAS-40. There was also a decrease in the phosphorylated form of the pro-apoptotic protein BAD, whose phosphorylation at Ser136 is dependent on the PI3K/AKT pathway. Dephosphorylation of BAD is required for its release from sequestration and induction of apoptosis. Of note, we also saw diminished p-AKT levels in peripheral blood CD34+ cells obtained from a PMF patient after exposure to a 1 and 5μM MK2206 for 6 hours. This result confirms that MK-2206 targets AKT in human MPN cells (Fig. 2B).
Figure 2. Treatment with MK-2206 reduced the level of PI3K-AKT signaling in MPN cells.
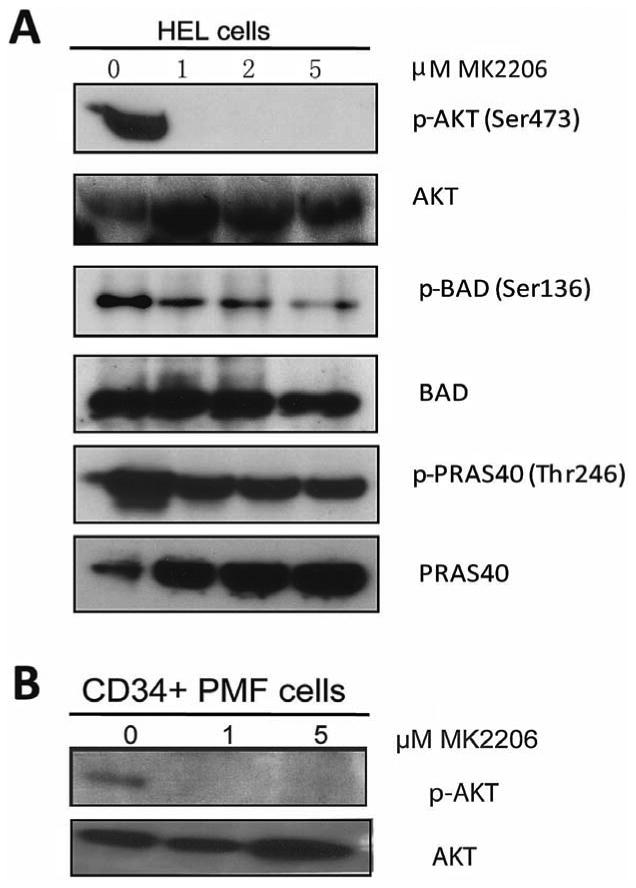
(A) HEL cells were treated with increasing concentrations of MK-2206 for 6 hours and extracts from these cells were subjected to immunoblot analysis for phosphorylated (p) or total forms of proteins associated with the PI3K/AKT signaling pathways. (B) AKT phosphorylation was assessed in PMF CD34+ cells treated with 1 μM and 5 μM of MK-2206 after 6 hours.
Sensitivity of human MPN progenitors to MK-2206
We next cultured peripheral blood CD34+ cells from PMF patients harboring the JAK2V617F mutation or mobilized CD34+ cells from healthy individuals in methylcellulose assays in the presence of a dose titration of MK-2206. We found that exposure of these cells to MK-2206 led to a dose dependent inhibition of colony formation (Fig. 3). Interestingly, while we observed that CFU-M derived from PMF cells were significantly more sensitive than their normal counterparts (p=0.022), and BFU-E from PMF tended to be more sensitive (p=0.068), CFU-MK formation was inhibited in PMF and control cells in a similar fashion. These findings suggest that megakaryocytes are more dependent on AKT signaling than other lineages. This observation is consistent with the existence of crucial crosstalk between AKT and Notch in megakaryocyte specification (39)
Figure 3. AKT inhibition by MK-2206 suppressed the growth of myeloid, erythroid and megakaryocytic colonies.
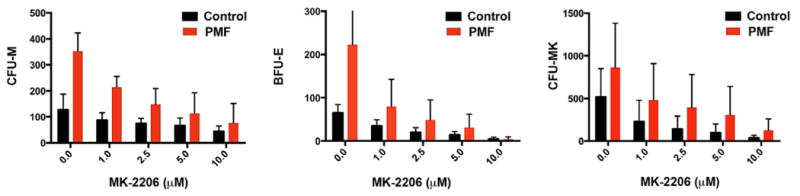
Assays for CFU-M (left), BFU-E (middle) and CFU-MK (right) were performed with a series of PMF and control specimens in the presence of 1-10 μM MK-2206. The experiment shows the average +/- SD for three independent samples for duplicate experiments performed on 3 PMF and 3 control CD34+ cells. P values for the difference in sensitivity between PMF and controls: CFU-M, p=0.022; BFU-E, p=0.069; CFU-MK, p=0.47.
MK-2206 reduces disease burden in a mouse model of myelofibrosis
To assess the in vivo efficacy of MK-2206, we first evaluated the impact of the drug on hematopoiesis in healthy Balb/c mice (n=4) at doses of 60 and 120 mg/kg and compared the phenotype to vehicle-treated controls. After 2 weeks of treatment, the mice were healthy with no changes in body weight and no changes in peripheral blood counts (Supplemental Fig S1). These results are consistent with human phase I/II data that show that MK-2206 is not myelosuppressive (36). This result also indicates that although CFU-MK was inhibited by MK-2206, treatment of healthy mice did not result in thrombocytopenia.
We next tested whether MK-2206 is efficacious in an in vivo model of MPLW515L associated myeloproliferative neoplasm. Transplantation of MPLW515L expressing Balb/c hematopoietic progenitor cells into lethally irradiated recipient mice leads to a phenotype that has several features in common with primary myelofibrosis, including peripheral leukocytosis, hepatosplenomegaly, megakaryocyte expansion and reticulin deposition in the marrow and sites of extramedullary hematopoiesis (10). At day 21 after transplantation, the mean white blood cell count (WBC) for the entire cohort exceeded the normal range for Balb/c mice. Mice were then randomized into 3 groups (n=8/group) and treated with vehicle or MK-2206 at 60 mg/kg or 120 mg/kg for 2 weeks by oral gavage once daily on a Mon-Wed-Fri schedule. After 2 weeks of treatment, mice were euthanized and evaluated for disease. Treatment with MK-2206 led to a significant reduction in liver and spleen size in the higher dose treatment group compared to vehicle-treated mice (Fig. 4A). Treatment also resulted in a reduction in the median WBC count in the peripheral blood from 73.6 ×103 in the vehicle-treated group to 20.4 ×103 in the 60 mg/kg dosed group and 18.9 ×103 in the 120 mg/kg dosed group (Fig 4B). Two of the treated animals displayed WBC counts much higher than other mice in the study for reasons we don't understand. If these outliers were excluded, the differences between the treated and untreated groups would be statistically significant (p=.043, Mann-Whitney test). Staining of peripheral smears confirmed a reduction in circulating immature erythroid cells and granulocytes (Fig 4C). These biologic effects correlated well with the pharmacodynamic effect of the drug assessed by immunoblot, showing inhibited phosphorylation of AKT at Ser473 and Thr308 in the bone marrow of MPLW515L transduced mice treated with MK-2206 at 60 and 120 mg/kg for 7 days (Fig 4D). Platelet and red cell counts, as well as the body weights remained largely constant throughout the experiment (Supplemental Fig S2).
Figure 4. MK-2206 reduces MPN disease burden in vivo.
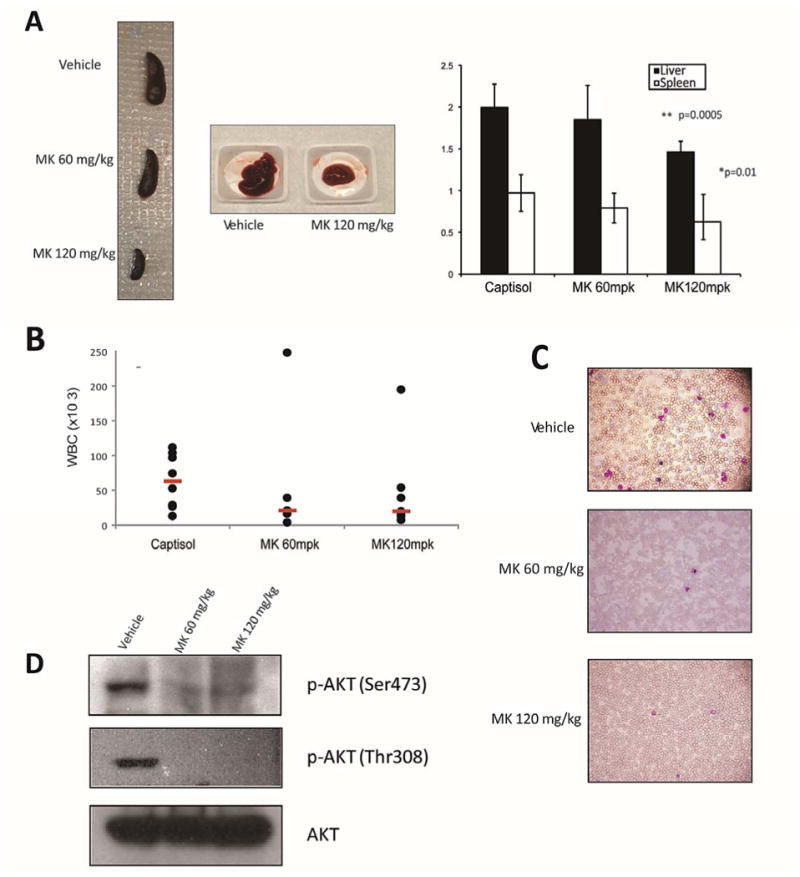
Balb/c recipients of bone marrow donor cells expressing MPLW515L were divided into 3 groups and treated with captisol (vehicle control), 60 mg/kg MK-2206 or 120 mg/kg MK-2206 (n = 8 per group) by oral gavage three times a week on a Mon-Wed-Fri schedule. Mice were sacrificed after 14 days of therapy and disease burden assessed. (A) Liver and spleen weights of treatment groups. Means +/- SD are shown, n=8. P values represent spleen/liver weights in 120 mg/kg group versus weights of vehicle treated mice. (B) White Blood Cell count (WBC) of mice from the 3 groups after 14 days of therapy, median values are highlighted in red. (C) May-Grunwald-Giemsa stained peripheral blood smears of control and MK-2206 treated animals (40X). (D) Western blot analysis of MPLW515L whole bone marrow demonstrated abrogation of AKT phosphorylation after 7 days of MK-2206 treatment (60 mg/kg and 120 mg/kg).
MK-2206 inhibits megakaryocyte expansion in MPLW515L recipient mice
The composition of the bone marrow and spleen of MPLW515L recipients treated with vehicle or MK-2206 were analyzed by flow cytometry after staining for myeloid precursors with Mac-1 and Gr-1, and megakaryocytes with CD41 antibodies. We observed an expansion of CD41+ cells in the bone marrow of transplanted mice that was significantly reduced by MK-2206 treatment (Fig. 5A, B). In contrast, no significant changes were seen in the mature myeloid populations in the bone marrow after treatment for 14 days (Fig 5B). Histologic evaluation of the bone marrow, liver, and spleen revealed extensive extramedullary hematopoiesis with effacement of liver and spleen architecture and hypercellular bone marrow with granulocyte hyperplasia in transplanted mice. Of note, there was a visible reduction in megakaryocytic expansion in the liver, spleen and bone marrow of mice that received the higher dose of 120 mg/kg MK-2206 (Fig 5C-E). This effect was confirmed by immunohistochemical staining with an antibody against von Willebrand Factor (vWF). In addition we performed reticulin staining on bone marrow slides, which were scored on a scale ranging from 0-3 independently by a pathologist who was blinded to the randomization groups (S.G.). We noted a reduction in the severity of fibrosis with vehicle-treated mice exhibiting an average score of 1 while the 120 mg/kg MK-2206 treatment group score reduced to 0.57 (n=7 mice per group). Of note, none of the drug treated mice had a score >1, whereas grade 2 fibrosis was seen in 2/8 vehicle treated mice.
Figure 5. MK-2206 reduces the megakaryocyte burden in a mouse model of PMF.
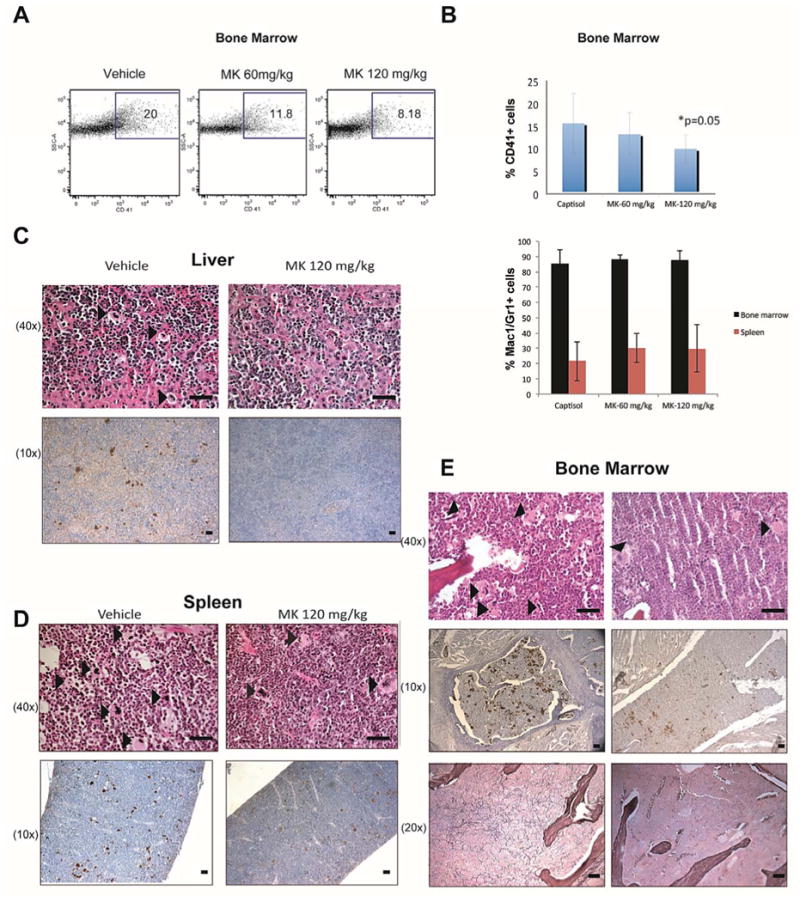
Cells from bone marrows of MPLW515L recipients treated with captisol (vehicle control), 60 mg/kg MK-2206, or 120 mg/kg MK-2206 for 14 days were harvested and stained with fluorescently conjugated antibodies against CD41or Mac1/Gr1. (A) Representative flow cytometry plots. (B) Bar graphs depicting mean +/- SEM of bone marrow megakaryocytes and myeloid cells (n=7 mice per group, p=0.05). 2 mice were excluded from the analysis due to poor engraftment. (C-E) Histologic sections of the liver (C), spleen (D) and bone marrow (E) after 14 days of treatment. Scale bars represent 25 μM. The upper row represents H&E staining and the lower row displays immuno-histochemical staining of von Willebrand Factor (vWF). Arrows highlight the megakaryocytes in the H&E sections. Reticulin staining of bone marrow is shown in the bottom panels.
MK-2206 synergizes with the JAK inhibitor Ruxolitinib in MPN cells
Given the toxicities of Ruxolitinib on erythroid cells and megakaryocytes and the absence of this effect of MK-2206 in our mouse study, use of a lower dose of a JAK inhibitor in combination with MK-2206 may have a more beneficial effect in patients. To investigate the potential for combining these therapies, we cultured SET2 cells with a range of doses of Ruxolitinib and MK-2206 spanning the EC50 for both drugs and then counted live cells by trypan blue exclusion. At all doses tested, the combination was synergistic, based on combination index (CI) calculations (Fig 6A; note CI<1 indicates synergy). Co-treatment with MK-2206 and Ruxolitinib synergistically induced apoptosis and necrosis of the SET 2 cells (Fig. 6B). These data suggest that combining these two agents may provide therapeutic efficacy at lower doses of Ruxolitinib.
Figure 6. MK-2206 and Ruxolitinib synergize to inhibit growth of JAK2V617 mutant SET2 cells.
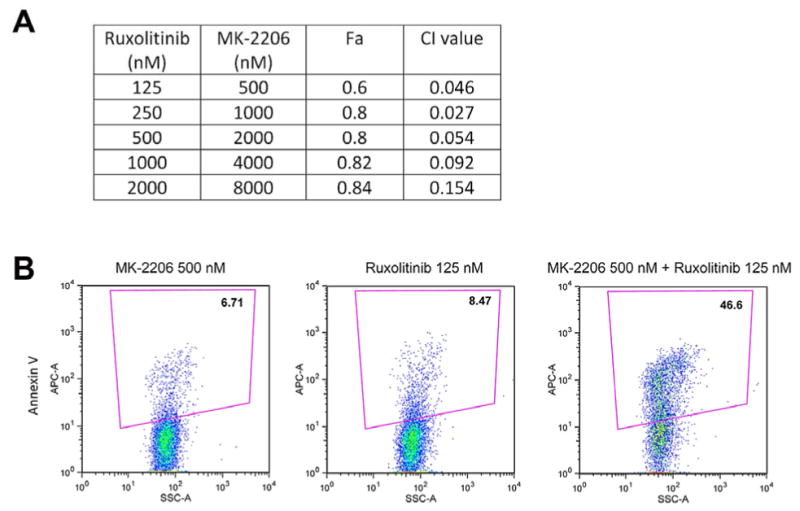
(A) All combinations of MK-2206 and Ruxolitinib tested give rise to strong synergy as evidenced by combination index (CI value). Fa, fraction of cells affected by the drugs. Data are representative of three independent replicates. (B) The combination of 500 nM MK-2206 and 125 nM Ruxolitinib, which alone do not show much of an effect on SET2 cell viability, synergize to give rise to robust cell death as measured by Annexin V staining. Representative plots of three independent biological replicates are shown.
Discussion
In pre-clinical studies, JAK2 inhibitors reduced the proliferation of JAK2V617F and MPLW515L mutant cells and attenuated disease development in murine models of MPN (40-43). Early clinical trials in patients with myelofibrosis resulted in clinical improvement, although the effects on the burden of JAK2 mutant clone were less impressive than anticipated (8, 22, 44). Moreover, given that JAK2 is essential for normal hematopoiesis (45), treatment with JAK2 inhibitors has been limited by hematologic toxicities, including anemia and thrombocytopenia.
With the realization that Ruxolitinib, although effective at relieving many symptoms of myelofibrosis, is not a cure for MPNs, there is a great interest in the development of improved JAK2 inhibitors and combinatorial therapies that target the disease. Compounds that have demonstrated single-agent efficacy in clinical trials include immunomodulators such as pomalidomide (46), which alleviates the anemia associated with myelofibrosis, and drugs that affect remodeling of chromatin such as Givinostat (47, 48). Pre-clinical studies of other HDAC inhibitors, including Panobinostat, for MPN have also shown promising results, but have been associated with myelosuppression, in particular thrombocytopenia (28, 49). Oncoproteins such as JAK2V617F are dependent on the chaperone function of heat shock protein 90 (hsp90) and this has also been validated as a therapeutic target in MPNs (50, 51). Moreover, in a recent phase I/II study of the mTOR inhibitor Everolimus, patients with myelofibrosis showed improvement in splenomegaly, systemic symptoms, and pruritus, reproducing many of the effects seen with JAK inhibitors (52). Myelosuppression was modest, and hematologic toxicity was mainly represented by a grade 2/3 reversible decrease of hemoglobin. Of note, in pre-clinical studies other groups have found that PI3K/mTOR inhibitors show effective against MPN cells alone and in combination with Ruxolitinib (31, 32).
The PI3K/AKT pathway is frequently activated in human cancers and plays a critical role in cell growth, proliferation, survival, apoptosis, and autophagy (53). Here we confirm that the PI3K/AKT pathway is activated in the myeloproliferative neoplasms downstream of both JAK2V617F and MPLW515L, and further, that MPN cells are dependent on this pathway for proliferation, survival and clonogenic expansion. The novel allosteric AKT inhibitor MK-2206 has demonstrated cytotoxic activity against T-ALL cell lines and patient primary cells (54) and synergism with epidermal growth factor receptor inhibitors, such as erlotinib or lapatinib in breast cancer cells (38), with gefitinib in malignant glioma (55) and with MEK inhibitors in non-small cell lung cancers (56). The added benefit of an allosteric inhibitor of AKT rather than an ATP-competitive inhibitor is reduced off-target effect. Indeed, the first phase I trial of this drug in solid tumors showed no hematologic toxicity and was very well tolerated (36). Of note, we observed no overt hematologic toxicity with MK-2206 in healthy mice. Our studies further demonstrate that MK-2206 synergizes with the JAK kinase inhibitor Ruxolitinib in vitro in a JAK2V617F mutant cell line.
MPNs are characterized by extramedullary hematopoiesis with abnormal megakaryocyte morphology and hyperplasia. PMF hematopoietic progenitor cells have demonstrated an increased ability to generate megakaryocytes and a decreased rate of apoptosis (57). In our studies, MK-2206 dramatically suppressed megakaryocyte colony formation from PMF CD34+ cells, although it also showed activity against CFU-MK from healthy progenitors. We surmise that this is due to a strong requirement for AKT in megakaryocyte specification (39). MK-2206 also shows activity against megakaryocytic leukemia cell lines (58). Of note, selectivity for MK-2206 on malignant hematopoiesis has been noted by others, such as one study that found MK-2206 had a minimal effect on the proliferation of peripheral blood CD4+ T cells and clonogenic potential of cord blood CD34+ cells from healthy donors (54). Moreover in our murine model of MPLW515L induced myelofibrosis, treatment with MK-2206 decreased extramedullary hematopoiesis, reduced megakaryocyte expansion in the bone marrow, and reduced the severity of reticulin fibrosis in the marrow without inducing peripheral cytopenias. Moreover, this same treatment course had no overt effect on hematopoiesis in healthy mice.
Together, our findings establish AKT as a rational therapeutic target for the treatment of patients with MPNs. As we become cognizant of the limitations of anti-JAK therapy, inhibition of AKT kinase activity may emerge as an important therapeutic option. Finally, because MK-2206 has already shown excellent tolerability in phase I trials for solid tumors, clinical trials of MK-2206 in combination with Ruxolitinib should be considered in MPN patients.
Supplementary Material
Acknowledgments
The authors thank Jonathan Licht and Lou Dore for helpful advice and critical reading of the manuscript. The authors also thank Merck for supplying MK-2206. This work was supported in part by grants from the NIH (CA101774 to JDC) and the Leukemia and Lymphoma Society, the Samuel Waxman Cancer Research Foundation, National Natural Science Foundation of China (Grant No. 30700412 and 81070406 to Z. Huang). IK was supported by a T32 grant to Northwestern University. IK is a recipient of the American Society of Hematology Translational Research Training in Hematology (TRTH) Award.
Footnotes
Conflict of interest: None of the authors has a competing financial interest.
Author Contributions: IK, ZH, QW, LG, BG performed the experiments, analyzed the data and wrote the manuscript. PK, LD, MJS and RS assisted with experiments and animal studies. SG analyzed the data and assisted with the manuscript. CF, TL, QW, MS, AP, BS, JA and AT assisted with patient specimen experiments. RL, AT, and JC analyzed the data and wrote the manuscript.
Supplementary information is available at Leukemia's website.
References
- 1.Ma X, Vanasse G, Cartmel B, Wang Y, Selinger HA. Prevalence of polycythemia vera and essential thrombocythemia. Am J Hematol. 2008;83(5):359–62. doi: 10.1002/ajh.21129. Epub 2008/01/09. [DOI] [PubMed] [Google Scholar]
- 2.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 3.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 4.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 5.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 6.Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280(24):22788–92. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–51. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 8.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–27. doi: 10.1056/NEJMoa1002028. Epub 2010/09/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tefferi A. Mutations galore in myeloproliferative neoplasms: would the real Spartacus please stand up? Leukemia. 2011;25(7):1059–63. doi: 10.1038/leu.2011.92. Epub 2011/07/14. [DOI] [PubMed] [Google Scholar]
- 10.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006;3(7):e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beer PA, Campbell PJ, Scott LM, Bench AJ, Erber WN, Bareford D, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112(1):141–9. doi: 10.1182/blood-2008-01-131664. [DOI] [PubMed] [Google Scholar]
- 12.Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107(11):4274–81. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zaleskas VM, Krause DS, Lazarides K, Patel N, Hu Y, Li S, et al. Molecular Pathogenesis and Therapy of Polycythemia Induced in Mice by JAK2 V617F. PLoS ONE. 2006;1:e18. doi: 10.1371/journal.pone.0000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108(5):1652–60. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 15.Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17(6):584–96. doi: 10.1016/j.ccr.2010.05.015. Epub 2010/06/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pardanani A, Guglielmelli P, Lasho TL, Pancrazzi A, Finke CM, Vannucchi AM, et al. Primary myelofibrosis with or without mutant MPL: comparison of survival and clinical features involving 603 patients. Leukemia. 2011;25(12):1834–9. doi: 10.1038/leu.2011.161. Epub 2011/06/22. [DOI] [PubMed] [Google Scholar]
- 17.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 18.Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adelaide J, Rey J, et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009;23(11):2183–6. doi: 10.1038/leu.2009.141. Epub 2009/07/18. [DOI] [PubMed] [Google Scholar]
- 19.Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013 Apr 26; doi: 10.1038/leu.2013.119. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 20.Lasho TL, Finke CM, Hanson CA, Jimma T, Knudson RA, Ketterling RP, et al. SF3B1 mutations in primary myelofibrosis: clinical, histopathology and genetic correlates among 155 patients. Leukemia. 2012;26(5):1135–7. doi: 10.1038/leu.2011.320. [DOI] [PubMed] [Google Scholar]
- 21.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. doi: 10.1056/NEJMoa1110557. Epub 2012/03/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM, et al. Safety and Efficacy of TG101348, a Selective JAK2 Inhibitor, in Myelofibrosis. J Clin Oncol. 2011 doi: 10.1200/JCO.2010.32.8021. Epub 2011/01/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pardanani A, Laborde RR, Lasho TL, Finke C, Begna K, Al-Kali A, et al. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia. 2013 Mar 5; doi: 10.1038/leu.2013.71. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tefferi A. JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood. 2012 doi: 10.1182/blood-2011-11-395228. Epub 2012/01/27. [DOI] [PubMed] [Google Scholar]
- 25.Ugo V, Marzac C, Teyssandier I, Larbret F, Lecluse Y, Debili N, et al. Multiple signaling pathways are involved in erythropoietin-independent differentiation of erythroid progenitors in polycythemia vera. Exp Hematol. 2004;32(2):179–87. doi: 10.1016/j.exphem.2003.11.003. Epub 2004/04/23. [DOI] [PubMed] [Google Scholar]
- 26.Laubach JP, Fu P, Jiang X, Salter KH, Potti A, Arcasoy MO. Polycythemia vera erythroid precursors exhibit increased proliferation and apoptosis resistance associated with abnormal RAS and PI3K pathway activation. Exp Hematol. 2009;37(12):1411–22. doi: 10.1016/j.exphem.2009.09.009. Epub 2009/10/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grimwade LF, Happerfield L, Tristram C, McIntosh G, Rees M, Bench AJ, et al. Phospho-STAT5 and phospho-Akt expression in chronic myeloproliferative neoplasms. Br J Haematol. 2009;147(4):495–506. doi: 10.1111/j.1365-2141.2009.07870.x. Epub 2009/09/15. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Fiskus W, Chong DG, Buckley KM, Natarajan K, Rao R, et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood. 2009;114(24):5024–33. doi: 10.1182/blood-2009-05-222133. Epub 2009/10/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood. 2010;115(17):3589–97. doi: 10.1182/blood-2009-04-215848. Epub 2010/03/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kharas MG, Janes MR, Scarfone VM, Lilly MB, Knight ZA, Shokat KM, et al. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J Clin Invest. 2008;118(9):3038–50. doi: 10.1172/JCI33337. Epub 2008/08/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bogani C, Bartalucci N, Martinelli S, Tozzi L, Guglielmelli P, Bosi A, et al. mTOR inhibitors alone and in combination with JAK2 inhibitors effectively inhibit cells of myeloproliferative neoplasms. PLoS One. 2013;8(1):e54826. doi: 10.1371/journal.pone.0054826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fiskus W, Verstovsek S, Manshouri T, Smith JE, Peth K, Abhyankar S, et al. Dual PI3K/AKT/mTOR inhibitor BEZ235 synergistically enhances the activity of JAK2 inhibitor against cultured and primary human myeloproliferative neoplasm cells. Mol Cancer Ther. 2013 Feb 27; doi: 10.1158/1535-7163.MCT-12-0862. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 33.Chapuis N, Tamburini J, Green AS, Vignon C, Bardet V, Neyret A, et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin Cancer Res. 2010;16(22):5424–35. doi: 10.1158/1078-0432.CCR-10-1102. Epub 2010/10/05. [DOI] [PubMed] [Google Scholar]
- 34.Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A, et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res. 2010;70(20):8097–107. doi: 10.1158/0008-5472.CAN-10-1814. Epub 2010/09/30. [DOI] [PubMed] [Google Scholar]
- 35.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, et al. Phase I, Dose-Escalation Study of BKM120, an Oral Pan-Class I PI3K Inhibitor, in Patients With Advanced Solid Tumors. J Clin Oncol. 2012;30(3):282–90. doi: 10.1200/JCO.2011.36.1360. Epub 2011/12/14. [DOI] [PubMed] [Google Scholar]
- 36.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29(35):4688–95. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 37.Uozumi K, Otsuka M, Ohno N, Moriyama T, Suzuki S, Shimotakahara S, et al. Establishment and characterization of a new human megakaryoblastic cell line (SET-2) that spontaneously matures to megakaryocytes and produces platelet-like particles. Leukemia. 2000;14(1):142–52. doi: 10.1038/sj.leu.2401608. [DOI] [PubMed] [Google Scholar]
- 38.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9(7):1956–67. doi: 10.1158/1535-7163.MCT-09-1012. Epub 2010/06/24. [DOI] [PubMed] [Google Scholar]
- 39.Cornejo MG, Mabialah V, Sykes SM, Khandan T, Lo Celso C, Lopez CK, et al. Crosstalk between NOTCH and AKT signaling during murine megakaryocyte lineage specification. Blood. 2011;118(5):1264–73. doi: 10.1182/blood-2011-01-328567. Epub 2011/06/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wernig G, Kharas MG, Mullally A, Leeman DS, Okabe R, George T, et al. EXEL-8232, a small-molecule JAK2 inhibitor, effectively treats thrombocytosis and extramedullary hematopoiesis in a murine model of myeloproliferative neoplasm induced by MPLW515L. Leukemia. 2011 doi: 10.1038/leu.2011.261. Epub 2011/10/19. [DOI] [PubMed] [Google Scholar]
- 41.Koppikar P, Abdel-Wahab O, Hedvat C, Marubayashi S, Patel J, Goel A, et al. Efficacy of the JAK2 inhibitor INCB16562 in a murine model of MPLW515L-induced thrombocytosis and myelofibrosis. Blood. 2010;115(14):2919–27. doi: 10.1182/blood-2009-04-218842. Epub 2010/02/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood. 2010;115(25):5232–40. doi: 10.1182/blood-2009-05-223727. Epub 2010/04/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G, et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia. 2007;21(8):1658–68. doi: 10.1038/sj.leu.2404750. [DOI] [PubMed] [Google Scholar]
- 44.Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G, et al. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood. 2010;115(6):1131–6. doi: 10.1182/blood-2009-10-246363. Epub 2009/12/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93(3):385–95. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 46.Begna KH, Mesa RA, Pardanani A, Hogan WJ, Litzow MR, McClure RF, et al. A phase-2 trial of low-dose pomalidomide in myelofibrosis. Leukemia. 2011;25(2):301–4. doi: 10.1038/leu.2010.254. Epub 2010/11/06. [DOI] [PubMed] [Google Scholar]
- 47.Guerini V, Barbui V, Spinelli O, Salvi A, Dellacasa C, Carobbio A, et al. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F) Leukemia. 2008;22(4):740–7. doi: 10.1038/sj.leu.2405049. Epub 2007/12/15. [DOI] [PubMed] [Google Scholar]
- 48.Rambaldi A, Dellacasa CM, Finazzi G, Carobbio A, Ferrari ML, Guglielmelli P, et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150(4):446–55. doi: 10.1111/j.1365-2141.2010.08266.x. Epub 2010/06/22. [DOI] [PubMed] [Google Scholar]
- 49.Mascarenhas J, Lu M, Li T, Petersen B, Hochman T, Najfeld V, et al. A phase I study of panobinostat (LBH589) in patients with primary myelofibrosis (PMF) and post-polycythaemia vera/essential thrombocythaemia myelofibrosis (post-PV/ET MF) Br J Haematol. 2013;161(1):68–75. doi: 10.1111/bjh.12220. [DOI] [PubMed] [Google Scholar]
- 50.Marubayashi S, Koppikar P, Taldone T, Abdel-Wahab O, West N, Bhagwat N, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010;120(10):3578–93. doi: 10.1172/JCI42442. Epub 2010/09/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fiskus W, Verstovsek S, Manshouri T, Rao R, Balusu R, Venkannagari S, et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin Cancer Res. 2011;17(23):7347–58. doi: 10.1158/1078-0432.CCR-11-1541. Epub 2011/10/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guglielmelli P, Barosi G, Rambaldi A, Marchioli R, Masciulli A, Tozzi L, et al. Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a phase 1/2 study in patients with myelofibrosis. Blood. 2011;118(8):2069–76. doi: 10.1182/blood-2011-01-330563. Epub 2011/07/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–62. doi: 10.1038/nrc2664. Epub 2009/07/25. [DOI] [PubMed] [Google Scholar]
- 54.Simioni C, Neri L, Tabellini G, Ricci F, Bressanin D, Chiarini F, et al. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia. 2012 doi: 10.1038/leu.2012.136. Published on line May 22, 2012. [DOI] [PubMed] [Google Scholar]
- 55.Cheng Y, Zhang Y, Zhang L, Ren X, Huber-Keener KJ, Liu X, et al. MK-2206, a Novel Allosteric Inhibitor of Akt, Synergizes with Gefitinib against Malignant Glioma via Modulating Both Autophagy and Apoptosis. Mol Cancer Ther. 2012;11(1):154–64. doi: 10.1158/1535-7163.MCT-11-0606. Epub 2011/11/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meng J, Dai B, Fang B, Bekele BN, Bornmann WG, Sun D, et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS One. 2010;5(11):e14124. doi: 10.1371/journal.pone.0014124. Epub 2010/12/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ciurea SO, Merchant D, Mahmud N, Ishii T, Zhao Y, Hu W, et al. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood. 2007;110(3):986–93. doi: 10.1182/blood-2006-12-064626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stankiewicz MJ, Crispino JD. AKT collaborates with ERG and Gata1s to dysregulate megakaryopoiesis and promote AMKL. Leukemia. 2013 Feb 5; doi: 10.1038/leu.2013.33. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.