

Summary
Human embryonic stem cells (hESCs) hold great promise for cell therapy as a source of diverse differentiated cell types. One key bottleneck to realizing such potential is allogenic immune rejection of hESC-derived cells by recipients. Here, we optimized humanized mice (Hu-mice) reconstituted with a functional human immune system that mounts a vigorous rejection of hESCs and their derivatives. We established knock-in hESCs that constitutively express CTLA4-Ig and PD-L1 before and after differentiation, denoted CP hESCs. We then demonstrated that allogenic CP hESC-derived teratomas, fibroblasts, and cardiomyocytes are immune protected in Hu-mice, while cells derived from parental hESCs are effectively rejected. Expression of both CTLA4-Ig, which disrupts T-cell co-stimulatory pathways, and PD-L1, which activates T-cell inhibitory pathway, is required to confer immune protection as neither was sufficient on their own. These findings are instrumental for developing a strategy to protect hESC-derived cells from allogenic immune responses without requiring systemic immune suppression.
Introduction
Human embryonic stem cells (hESCs) can undergo unlimited self-renewal and retain the pluripotency to differentiate into all cell types in the body. Therefore, as a renewable source of various cells in human body, hESCs hold great potential for cell replacement therapy. Since the successful establishment of hESCs in 1998 (Thomson et al., 1998), significant progress has been made in establishing the conditions necessary to differentiate hESCs into various lineages of biologically active cells, including cardiomyocytes, oligodendrocytes and pancreatic β cells (Cohen and Melton, 2011; Fu and Xu, 2011). Despite this tremendous progress, several major obstacles must be overcome prior to the successful application of hESC-based cell replacement therapies in the clinic. One such obstacle is the immune-mediated rejection of hESC-derived cells by the recipient because these cells are allogeneic to the recipient patients (Boyd et al., 2012). While persistent systemic immune suppression can delay the allograft rejection, the typical immunosuppressant regimens are especially toxic to patients with chronic disabling diseases (Wekerle and Grinyó 2012). In addition, chronic immunosuppression greatly increases the risk for cancer and infection (Gallagher et al., 2010). Therefore, to achieve the potential of hESC-based therapy, it will be critical to develop new effective strategies to protect hESC-derived cells from alloimmune rejection. While extensive studies on allogeneic immune responses have been performed in mouse models, much less is know about the human immune responses to allografts due to the lack of relevant model system to study such human immune responses (Zhang et al., 2009). Therefore, it is critical to develop new models with a functional human immune system that can mount robust alloimmune responses and mediate allograft rejection.
Extensive effort has been devoted to develop new strategies to induce immune tolerance of allogeneic transplants. Pre-clinical and clinical studies indicate that induction of mixed chimerism by transplantation of bone marrow or hematopoietic stem cells (HSCs) can induce allograft tolerance (Ciancio et al., 2001; Kawai et al., 2008; Tillson et al., 2006). Immature dendritic cells can further facilitate allogeneic hematopoietic stem cell engraftment, ameliorating host responses to allografts and preventing graft-versus-host disease (GVHD) (Fugier-Vivier et al., 2005). Considerable effort has been devoted to the potential benefits of using these cells to induce immune tolerance to allografts (Wood et al., 2012). Therefore, tolerance to allogeneic hESC-derived cells could be achieved by the induction of chimerism using hESC-derived HSCs and/or dendritic cells. If successful, hESC-derived cells could then be transplanted without the adverse effects of long-term immunosuppressive treatments. However, despite a series of publications reporting the differentiation of hESCs into hematopoietic progenitor cells that are multi-potent in vitro (Davis et al., 2008; Ledran et al., 2008; Vodyanik et al., 2005; Woods et al., 2011), none of these hESC-derived HSCs are capable of efficiently repopulating hematopoietic lineages in mouse models. Therefore, the potential for achieving immune tolerance of hESC-derived cells by mixed chimerism depends on the feasibility to derive authentic HSCs from hESCs.
Cytotoxic T lymphocyte antigen 4 (CTLA4) and programmed death ligand-1 (PD-L1) are critical immune inhibitory molecules in maintaining peripheral tolerance by restraining T cell activity. CTLA4 binds CD80 and CD86 with higher affinity and avidity than CD28, which are the primary co-stimulation pathways for T cell activation. Therefore, CTLA4-immunoglobulin fusion protein (CTLA4-Ig) has been developed to inhibit T cell-mediated immune responses,(Walker and Abbas, 2002). PD-L1 binds to PD-1, which is expressed on T cell surface, and inhibits T cell activity (Fife and Bluestone, 2008). In this context, PD-L1 plays a central role in maintaining T cell anergy and preventing autoimmunity (Keir et al., 2008). While data in mouse models have demonstrated the critical roles of CTLA4 and PD-L1 in suppressing allograft and xenograft rejection (Fife and Bluestone, 2008; Francisco et al., 2010; Tian et al., 2007; Walker and Abbas, 2002), the efficacy of these immunosuppressive molecules such as CTLA4-Ig in suppressing human allogeneic immune responses remains unclear as indicated by human clinical trial data. Due to the apparent cellular and physiological differences between mouse and humans, much of the information on immune regulation gathered in the mouse models cannot be completely recapitulated in human clinical trials (Elster et al., 2004; Van Der Touw and Bromberg, 2010). By developing a humanized mouse model with functional human immune system that could mount vigorous allogeneic immune rejection of hESC-derived cells, we investigated the importance of the co-stimulatory pathway and inhibitory pathway on human allogeneic immune responses. Our findings provide a novel strategy to protect the hESC-derived cells from allogeneic immune response without inducing systemic immune suppression.
Results
Developing Hu-mice that can mount vigorous allogeneic immune rejection
To examine the allogeneic immune rejection of transplanted human cells, we established the humanized mice reconstituted with functional human immune system by the combined transplantation of human fetal thymus tissues and isogenic CD34+ fetal liver cells into the immunodeficient NOD/SCID/IL-2γ-/- (NSG) mice as described (Lan et al., 2006; Tonomura et al., 2008). Because human immune cells develop de novo in the recipients, the human immune system is thus tolerant of the recipient mouse and does not mediate graft versus host reaction (Rongvaux et al., 2013; Shultz et al., 2012). The risk to develop GVHD caused by the residue donor mature T cells in the transplanted fetal thymus is further minimized by one round of freeze/thaw of fetal thymus as recently described (Kalscheuer et al., 2012). These humanized mice, denoted Hu-mouse, are well reconstituted with human B and T cells (Figure 1A, B). In addition to previous report of their capability to mount antigen-specific T cell-dependent antibody responses and mediate xenograft rejection (Lan et al., 2006; Tonomura et al., 2008), we showed that Hu-mice could mount effective allogeneic immune rejection of hESC-derived cells (Figure 1C, D). The HLA typing information of human tissues used to repopulate human immune system in mice and human ESCs was shown in table S1. Therefore, the Hu-mouse model is an excellent in vivo model system for studying human immune responses against hESC-derived allografts.
Figure 1.
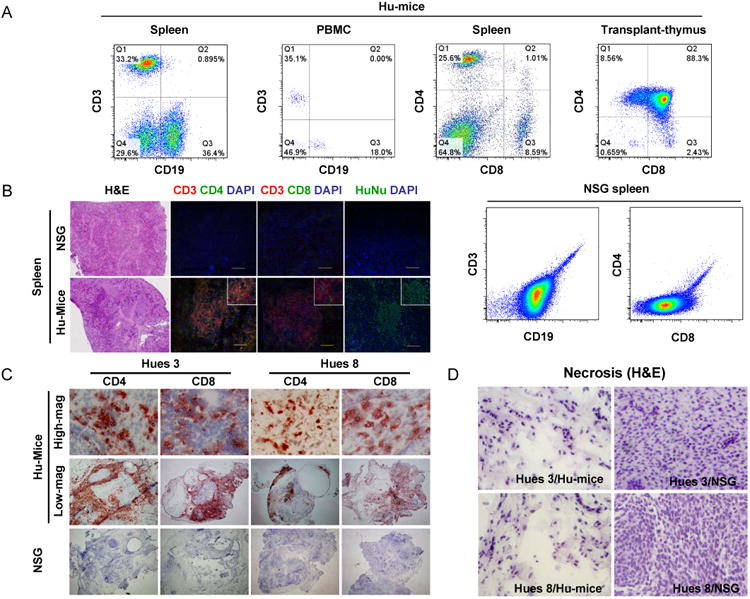
Hu-mice can mount robust immune rejection of hESC-derived allografts. (A) Representative FACS analysis of spleen, peripheral blood mononuclear cells (PBMC) and transplanted human thymus derived from Hu-mice. Single cell suspension was stained for the markers of human T cells (CD3, CD4, and CD8) and B cells (CD19). N>10. Single cell suspension derived from the spleen of NSG mice was used as a negative control. (B) Sections of spleens from NSG and Hu-mice were stained with hematoxylin and eosin, or with antibodies against human CD3, CD4, CD8, and human nuclei to show the repopulation of human T cells in spleen. Nuclei were counterstained with DAPI. Scale bar, 100 μm. Representative images are shown. N=4 for NSG group, N>20 for Hu-mice group. (C) NSG and Hu-mice were subcutaneously implanted with allogeneic Hues 3 and Hues 8 hESCs around the hindlegs. Six-to-eight weeks after implantation, teratomas were harvested, sectioned and stained with anti-human CD4 and CD8 antibodies. (D) Extensive necrosis was detected in the teratomas formed by allogeneic Hues 3 and Hues 8 hESCs in Hu-mice, as revealed by hematoxylin and eosin staining. N=4 for Hues 3/Hu-mice group, N=6 for Hues 8/Hu-mice group, N>10 for Hues 3/NSG group, N>10 for Hues 8/NSG group. See also table S1.
Generation of CTLA4-Ig and PD-L1 knock-in hESCs
To evaluate the strategy to suppress the allogeneic immune response by disrupting the co-stimulatory pathway and activating the T cell inhibitory pathway, we designed a knock-in strategy to express the CTLA4-Ig/PD-L1 in hESC derivatives. To ensure the constitutive expression in hESC-derived cells and prevent random integration that increases cancer risk, we took advantage of a bacterial artificial chromosome (BAC) based targeting strategy, which can achieve high efficiency of homologous recombination at the HPRT locus in hESCs (Song et al., 2010), to insert the CAG-CTLA4-Ig-IRES-PD-L1-PolyA expression cassette around 200bp downstream of the HPRT gene (Figure 2A, B). The LoxP-flanked selection marker was inserted into the 3′ untranslated region of HPRT gene (Figure 2B). Because HPRT is an X-linked gene, the homologous recombination between the targeting vector and the endogenous HPRT locus in male hESCs led to the loss of HPRT expression, which could be easily selected by their insensitivity to 6-TG. Transient expression of Cre enzyme in the targeted hESCs led to the excision of the selection marker from the targeted allele through LoxP/Cre-mediated deletion, leading to the normal HPRT expression, thus the resistance to HAT but sensitivity to 6-TG (Figure 2C, D). The expression and secretion of the CTLA4-Ig dimmer by CP hESCs were confirmed by Western blotting (Figure 2E). In addition, the surface expression of PD-L1 in CP hESCs was confirmed by flow cytometric analysis (Figure 2F). The examination of expression of hESC-specific pluripotency genes and surface markers as well as the capability to form well-differentiated teratomas in SCID mice confirm the pluripotent state of CP hESCs (Figure S1).
Figure 2.
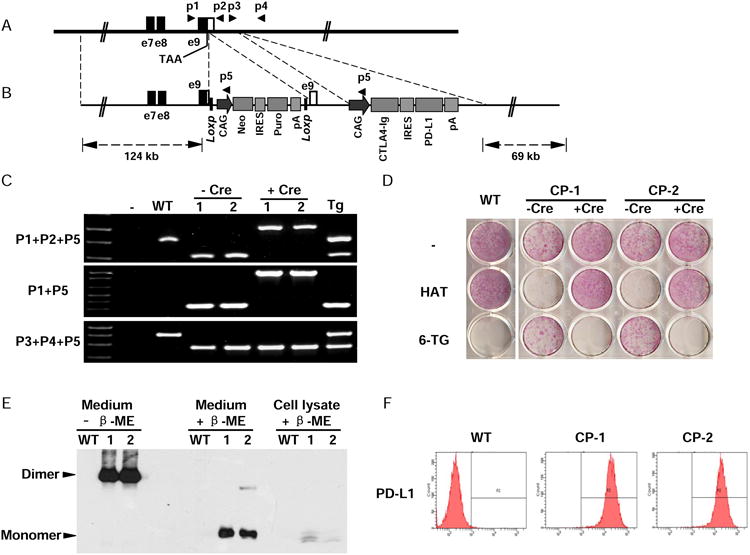
Generation of Hues 3 CP hESCs. (A) The endogenous human hypoxanthine phosphoribosyltransferase 1 (HPRT1) locus. Open box indicates the 3′ UTR of HPRT1. Filled boxes indicate part of HPRT1 coding sequence. The stop codon (TAA) and the binding sites of the primers used for identification of targeting clones are indicated. (B) BAC-based targeting vector. The LoxP flanked selection cassette was inserted between the stop codon and the PolyA signal sequence of HPRT1 to block its expression, introducing both positive and negative selections during targeting process. The CAG promoter-driving expression cassette, CAG/CTLA4-Ig/IRES/PD-L1/pA, was inserted about 600 bps downstream of HPRT1 gene. The sizes of homologous arms are indicated. IRES, internal ribosomal entry site. (C) PCR analysis of the targeted clones in human male ESCs (HUES 3). WT, wide type parental HUES 3; 1 & 2, two targeted clones CP-1 & CP-2; Tg, a random integration clone. The primers used are indicated in A and B. (D) Drug sensitivity assay to confirm the expression and function of HPRT1 in the knock-in clones. Cells were seeded onto 12-well plates. At the following day, the media were changed to that containing hypoxanthine/aminopterin/thymidine (HAT), or 6-thioguanine (6-TG), or without a drug. After being treated for three days, the cells were stained with an alkaline phosphatase detection kit. (E) The expression and secretion of CTLA4-Ig was confirmed. Loading buffer with or without the reducing agent β-mercaptoethanol was used to evaluate the dimerization status of CTLA4-Ig. (F) The surface expression of PD-L1 was confirmed by flow cytometry. See also Figure S1
Teratomas formed by CP hESCs are protected from allogeneic immune responses
To determine whether various lineages of cells derived from CP hESCs were immune rejected by the allogeneic human immune response, we took advantage of the capability of hESCs to form teratomas that contain cells derived from each of the three germ layers. CP hESCs were implanted subcutaneously into the right flank of Hu-mouse with allogeneic immune system. As an internal control, the parental hESCs were implanted into the left flank of the same Hu-mouse. While both CP hESCs and control parental hESCs formed teratomas in Hu-mice, the teratomas formed by parental hESCs regressed rapidly (Figure 3A). When recovered from the Hu-mice six weeks after transplantation, the teratomas formed by parental hESCs were much smaller than that formed by CP hESCs, and was consisted primarily of liquid cyst and few cells (Figure 3A). Because both CP hESCs and parental hESCs could form teratomas of similar size in NSG mice, this ruled out the possibility that the impaired teratoma formation by the parental hESCs in Hu-mice was due to defective pluripotency of these hESCs (Figure 3A).
Figure 3.
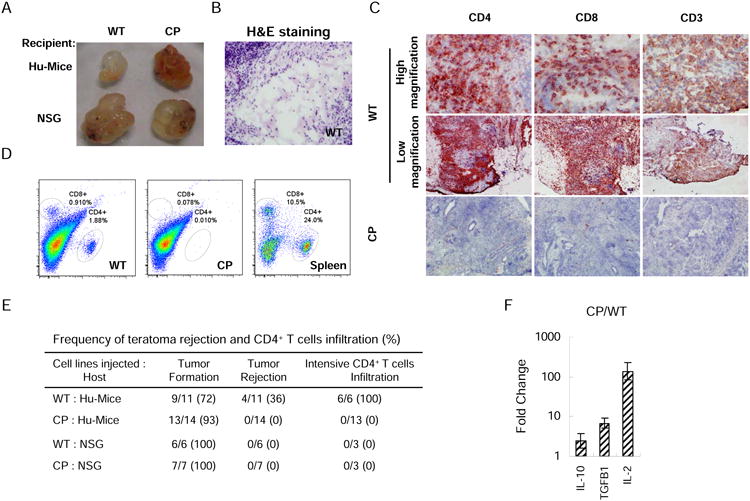
Expression of PD-L1 and CTLA4-Ig protects CP hESC-derived teratomas from allogeneic immune rejection. (A) Images of teratomas derived from WT and CP hESCs formed in Hu-mice and NSG mice. Mice were subcutaneously injected with WT hESCs and CP hESCs around the left and right hindlegs, respectively. Six-to-eight weeks after implantation, the mice were euthanized and teratomas examined. Representative images are shown. (B) Extensive tissue necrosis was detected in the teratomas formed by WT hESCs in Hu-mice. Significant T cell infiltration was detected in the teratomas formed by WT hESCs but not those formed by CP hESCs in Hu-mice as shown by immunohistochemistry (C) and flow cytometry (D). (E) Summary of teratoma formation, immune rejection and CD4+ T cell infiltration. Teratomas with apparent regressing phenotype or containing only liquid-filled cysts without cell mass were classified as rejection. (F) Relative mRNA levels of IL-10, TGFβ1 and IL-2 in T cells isolated from CP hESC- and WT hESC-derived teratomas formed in the same Hu-mouse were determined by real-time PCR. Mean values are presented with SD (N=3). See also Figure S2 and S3.
To test whether the teratomas formed by CP hESCs are protected from the allogeneic immune response, the teratomas formed by parental and CP hESCs in Hu-mice were examined histologically. The teratomas remnant formed by parental hESCs in Hu-mice was consisted mostly of liquid cysts, and was extensively infiltrated with human T cells, indicating robust immune rejection (Figure 3B-D). In contrast, the teratomas formed by CP hESCs in Hu-mice were consisted of well-differentiated tissues with greatly reduced T cell infiltration, indicating that cells derived from CP hESCs were protected from the allogeneic immune system (Figure 3B-E). In this context, the number of human T cells infiltrated into parental hESC-derived teratoma is only a few percent of that in CP hESC-derived teratomas formed in the same Hu-mouse, indicating that CP hESC-derived teratomas can be protected from allogenic immune response without inducing systemic immune suppression (Figure 3D). While the percentage of CD4+CD25+Foxp3+ Treg cells in the T cells infiltrating in the teratomas formed by CP hESCs was similar to that in teratomas formed by WT parental hESCs, the T cells purified from the CP-derived teratomas expressed significantly higher levels of immune suppressive cytokines such as IL-10 and TGF-β than those in parental hESC-derived teratomas (Figure 3F). The majority of teratomas formed by CP hESCs in Hu-mice reached the allowed maximum size by 8 weeks after implantation, some slower growing teratomas formed by CP hESCs were immune protected in Hu-mice for up to three months when they reached the allowed maximum size. Therefore, the greatly reduced T cell infiltration and a local immune suppressive environment contribute to the long-term protection of CP hESC-derived cells from allogeneic immune rejection.
In further support of the conclusion that CP hESC-derived teratomas are protected from allogenic immune response, the teratomas formed by the allogeneic CP hESCs in Hu-mice contained cells derived from each of the three germ layers (Figure 4A, B). In addition, the comparison of the expression of lineage-specific genes in the teratomas formed by CP hESCs in Hu-mice and in NSG mice further suggests that no specific cell lineages differentiated from CP hESCs were rejected (Figure 4C). Using the same strategy, the analysis of another independently generated CP HUES-8 hESC line further supports the conclusion that the expression of CTLA4-Ig and PD-L1 by the cells derived from hESCs can protect them from allogeneic immune response (Figure S2).
Figure 4.
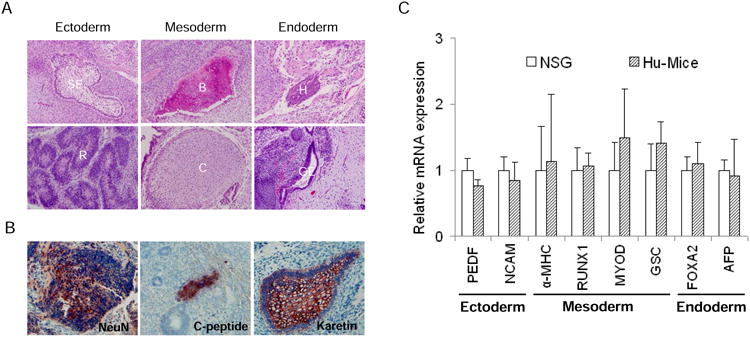
Various lineages of cells derived from CP hESCs are protected from allogeneic immune rejection. (A) Various cell lineages were present in the teratomas formed by CP hESCs in Hu-mice. SE, squamous epithelium; R, rosette; B, bone; C, cartilage; H, hepatocyte-like cells; GE, gut-like epithelium. (B) Established teratomas formed by CP hESCs in Hu-mice contained various cell lineages indicated by immunohistochemical staining. NeuN, neuronal marker; C-peptide, pancreatic β cell marker; Keratin, pan-epidermis marker. (C) The relative expression levels of the three germ layer specific genes in teratomas formed by CP hESCs derived from NSG and Hu-mice. Mean values are presented with SD (for NSG mice, N=7; for Hu-mice, N=12).
To further confirm that the expression of CTLA4-Ig and PD-L1 does not induce systemic immune response, CP hESCs were implanted into Hu-mice, the teratomas were removed six weeks later and the same Hu-mouse implanted with parental hESCs. Six-to-eight weeks after implantation, the teratomas formed by parental hESCs were recovered and analyzed for immune rejection, indicating that the parental hESC-derived teratomas were immune rejected with extensive infiltration of T cells (Figure S3A). In contrast, when parental hESCs mixed with CP hESCs at a ratio of 2:1 were injected into Hu-mice, the resulting teratomas contained cells derived from both WT and CP hESCs without any apparent immune rejection, indicating that the cells derived from CP hESCs can provide local protection of cells derived from parental hESCs from allogeneic rejection (Figure S3B). These findings further support the conclusion that the local expression of CTLA4-Ig and PD-L1 could achieve immune protection of hESC-derived allografts without inducing systemic immune suppression or immune tolerance.
Cells derived from CP hESCs are protected from allogeneic immune response
To further evaluate the immune response to hESC-derived cells, human fibroblasts and cardiomyocytes were derived from CP and control parental hESCs, with their identity confirmed by cell type-specific gene expression (Figure S4C-G). The CP hESC-derived cells were confirmed for the expression of CTLA4-Ig and PD-L1 (Figure S4B). In addition, the increased expression of HLA class I on the differentiated cell types was confirmed by real-time PCR assay (Figure S4A). Consistent with previous findings (Blomer et al., 2004; Xu et al., 2008), the hESC-derived cardiomyocytes could survive for an extended period of time in vivo after being transplanted into the hindleg muscle of NSG mice (Figure S4F). For control WT and CP hESC-derived fibroblasts, they were embedded into gel foam and implanted into left and right side of the same Hu-mice subcutaneously (Figure 5A). For control and CP hESC-derived cardiomyocytes, they were injected directly into the skeletal muscle of the left and right hind legs of Hu-mice (Figure 5B, C). While significant T cell infiltration was detected in the parental hESC-derived cells transplanted in Hu-mice, greatly reduced T cell infiltration was detected in the graft of CP hESC-derived cells in the same Hu-mice, indicating that CP hESC-derived fibroblasts and cardiomyocytes were also protected from the allogeneic immune response (Figure 5). Therefore, these data demonstrate that somatic cells differentiated from CP hESCs are protected from allogeneic human immune system.
Figure 5.
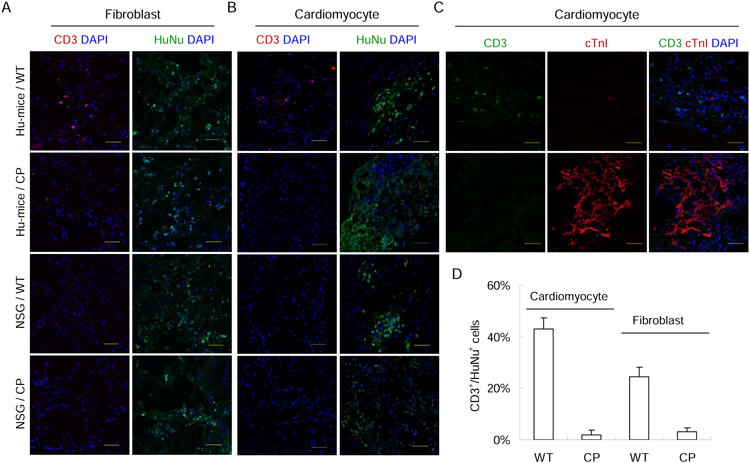
Fibroblasts and cardiomyocytes derived from CP hESCs are protected from allogeneic immune rejection in Hu-mice. Fibroblasts (A) or cardiomyocytes (B) derived from WT and CP hESCs were transplanted into the same Hu-mice or NSG mice. Two weeks later, the grafts were recovered and stained with indicated antibodies. T cells were identified by anti-CD3 antibody, and human cells in the grafts identified by a human nuclei-specific antibody (Hu-Nuclei). Representative images are shown. Parental WT and CP hESC-derived grafts in NSG mice were stained as negative controls. Nuclei were counterstained with DAPI. Scale bar, 50 μm. N=3 per group. (C) hESC-derived cardiomyocyte allografts transplanted in Hu-mice were sectioned and stained for T cells (CD3+) and human cardiomyocytes (cTnl+), indicating the presence of cardiomyocytes but lack of infiltrating T cells in the CP hESC-derived cardiomyocyte allografts. (D) Quantification of T cells infiltrated into fibroblast and cardiomyocyte allografts transplanted into Hu-mice. CD3+ and HuNu+ cells in three randomly selected view fields were counted. The ratio of CD3+ to HuNu+ cells was used to quantify for T cell infiltration. Mean values are presented with SD (N=3). See also Figure S4.
To dissect the contribution of CTLA4-Ig and PD-L1 to immune protection
To determine the contribution of CTLA4-Ig and PD-L1 to the immune protection of hESC-derived allografts, we used the same knock-in strategy to introduce the CTLA4-Ig and PD-L1 expression cassette independently into the HPRT locus of hESCs (Figure S5). Once confirmed for their pluripotency and the expression of CTLA4-Ig or PD-L1 (Figure S5), the parental and knock-in hESCs were transplanted subcutaneously into the left and right flank of Hu-mice. In contrast to the teratomas formed by CP hESCs that are protected from allogeneic immune system in Hu-mice, teratomas formed by knock-in hESCs expressing only CTLA4-Ig or PD-L1 were robustly immune rejected in Hu-mice, similarly to the teratomas formed by parental hESCs (Figure 6A-C). Therefore, CTLA4-Ig and PD-L1 must work together to protect the transplanted cells from the allogeneic immune responses.
Figure 6.
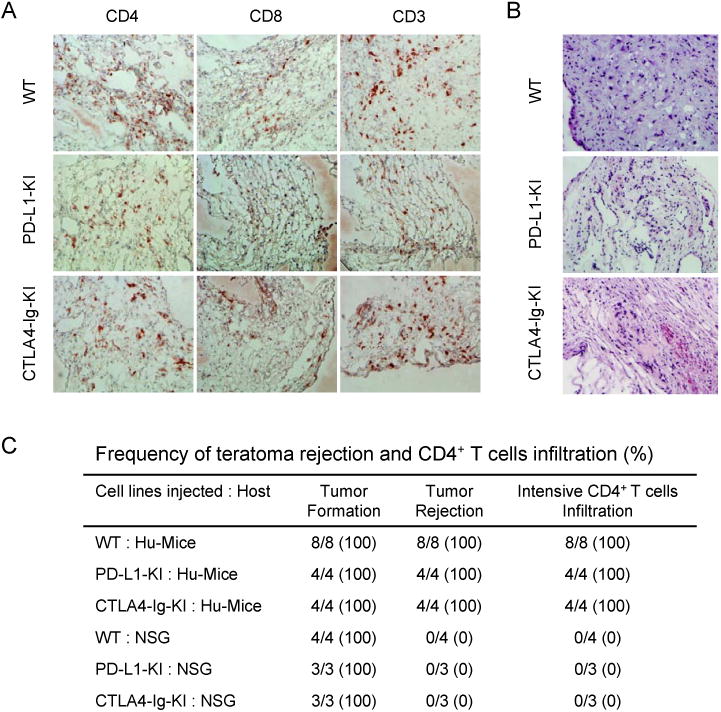
Expression of PD-L1 or CTLA4-Ig alone cannot protect the derivatives of hESCs from allogeneic immune rejection. (A) Extensive T cell infiltration was detected in the teratomas formed by WT hESCs, PD-L1-KI-hESCs and CTLA4-Ig-KI-hESCs in Hu-mice. T cells were identified by anti-CD4, anti-CD8 and anti-CD3 antibodies. (B) Extensive necrosis was detected in the teratomas derived from WT hESCs, PD-L1-KI-hESCs and CTLA4-Ig-KI-hESCs in allogeneic Hu-mice, as revealed by hematoxylin and eosin staining. (C) The frequency of CD4+ T cell infiltration and teratoma rejection in Hu-mice transplanted with parental and various knock-in hESCs. See also Figure S5.
Discussion
Since the successful establishment of hESCs in 1998 (Thomson et al., 1998), tremendous progress has been achieved in generating Good Manufacturing Practice (GMP)-grade hESC lines, in large-scale hESC production, and in the lineage-specific differentiation of hESCs (Fu and Xu, 2011). In addition, the feasibility of hESC-based human cell therapy is further supported by the initiation of two phase I clinic trials of hESC-based cell therapy of spinal cord injury and macular degeneration (Schwartz et al.; Wirth-Iii-etal-2011). However, one major hurdle that hinders the clinic development of hESC-based cell therapy is the allogeneic immune rejection of hESC-derived cells by the recipients, even when the cells are transplanted into immune privileged sites due to the breakdown of blood-brain barrier at the lesion site (Boyd et al., 2012). Therefore, to improve the feasibility of hESC-based cell therapy, it is important to develop effective and scalable approaches to protect hESC-derived allografts from immune rejection.
In this study, we demonstrated that a humanized mouse model reconstituted with functional human immune system could mount allogeneic immune response to hESC-derived allograft. There has been limited effort to study the human allogeneic immune responses in vivo due to the lack of physiologically relevant model systems. While extensive studies of transplantation immunology in mouse models have been instrumental in designing immune tolerance strategies in humans, apparent difference between mouse and human immunity has led to the common dilemma that the findings from mouse models can not be recapitulated by human (Haley, 2003; Mestas and Hughes, 2004). This bottleneck has significantly hindered the effort to develop effective immune tolerance approaches in humans. To address this issue, significant effort has been made to improve humanized mouse models with human immune system during the past three decades (Rongvaux et al., 2013; Shultz et al., 2012). Here, we optimized the Hu-mouse model as a physiologically relevant surrogate to study human allogeneic immune response. In this study, we used Hu-mice to identify a novel combination of immune regulatory molecules to effectively protect hESC-derived grafts from allogenic immune responses. These findings are instrumental for developing effective immune tolerance strategy without inducing systemic immune suppression.
Standard immune suppressant regiments are effective for preventing immune rejection of allogeneic organs, and their use is justified in the case of terminally ill patients (Selzner et al., 2010). However, the high toxicity of the immunosuppressant regiments, and the increased risk of spontaneous cancer and infection associated with systemic immune suppression, significantly raises the risk/benefit ratio of hESC-based cell therapy. Our strategy described here could help to mitigate these problems by inducing local immune protection of hESC-derived cells without using standard immune suppressants or systemic immune suppression. However, one potential risk of this approach is that it allows the grafted cells with tumorigenic potential or viral infection to escape immune surveillance. One feasible way to address this risk is to introduce a suicidal thymidine kinase (TK) gene into CTLA4-Ig/PD-L1 expression cassette so that the transplanted cells can be effectively eliminated by ganciclovir if needed (Springer and Niculescu-Duvaz, 2000). In support of this notion, others and we have reported that TK-expressing tumors derived from hESCs could be effectively eliminated in vivo by administration of ganciclovir (Cheng et al., 2012; Rong et al., 2012; Schuldiner et al., 2003).
In the context of clinic development, the high targeting efficiency to generate CP hESC lines supports the feasibility to genetically modify any clinic-grade hESCs under the GMP conditions. While the genetic modification of hESCs is a known safety concern in human therapy associated with random integration of the exogenous DNA into the human genome, the knock-in approach employed here should minimize such risk because homologous recombination can be achieved without any apparent random integration of the exogenous DNA (Howden et al., 2011; Song et al., 2010). To further address this concern before clinic application of the genetically modified hESCs, the entire genome of CP hESCs will be sequenced to confirm that no other mutations or random insertion of exogenous DNA are introduced during homologous recombination. In summary, the genetic approach described here could be instrumental for developing in the future a safe approach to effectively protect hESC-derived cells from allogeneic immune rejection without inducing systemic immune suppression.
Experimental procedure
Construction of BAC-Based targeting vector
The HPRT1 BAC clone RP11-671P4 was purchased from Invitrogen and the targeting vector was constructed by recombineering as previously described (Rong et al., 2012; Song et al., 2010). Briefly, the pCAG/CTLA4-Ig/IRES/PD-L1/polyA expression cassette was inserted 600 bp downstream of the HPRT1 stop codon. The Loxp flanked selection cassette pCAG/Neo/IRES/Puro/polyA was inserted between the HPRT1 stop codon and its endogenous polyA site. The Cre-mediated deletion of the selection cassette will restore the normal expression of HPRT.
Cell culture
The hESC lines, HUES-3 and HUES-8, were cultured on mouse embryonic fibroblast feeder layer in Knockout DMEM supplemented with 10% KOSR, 10% plasmanate, 0.1 mM nonessential amino acids, 2 mM Glutamax, 1% penicillin/streptomycin, 10 ng/ml bFGF, and 55 μM β-mercaptoethanol. HUES hESCs were dissociated with TryLE and passaged on feeder with 1:4-1:6 dilution. All tissue culture reagents were purchased from Invitrogen unless indicated otherwise.
HAT and 6-TG cytotoxicity assays
hESCs were plated on 12-well plate, and 24 hrs later, media were replaced with that containing 100 μM hypoxanthine/0.4 μM aminopterin/16 μM thymidine (HAT, Sigma), or 1 mM 6-thioguanine (6-TG, Sigma), or mock treated. After 3 days of treatment, the cells were stained with an alkaline phosphatase detection kit according to the manufacturer's instruction (Millipore).
Flow cytometric analysis
The flow cytometric analysis of the surface expression of hESC-specific markers, TRA 1-61, TRA 1-81, SSEA3 and SSEA4, was analyzed as described previously (Rong et al., 2012). Briefly, 5× 105 cells were washed with PBS, stained for 1 hr at room temperature with primary antibody. After being washed twice with PBS, the cells were stained with a FITC/PE-conjugated secondary antibody for 30 min at room temperature and analyzed by a BD LSR-II machine using FACS Diva software (Becton Dickinson). For flow cytometric analysis of spleen and thymus in humanized mouse, single cell suspension was mashed through 40 μM cell strainers and washed with PBS. Red blood cells in spleen samples were removed with ACK lysis buffer. The primary antibodies used: anti-PD-L1 antibody (29E.2A3, Biolegend), PE-anti-hCD3 (eBioScience), FITC-anti-hCD19 (eBioScience), PE-anti-hCD4 (BD Pharmingen), and FITC-anti-hCD8 (BD Pharmingen).
Western blotting assay
To analyze the expression of CTLA4-Ig by CP hESCs and their derivatives, the cells were grown to confluence, the media replaced with DMEM basal media. Twenty-four hrs later, two aliquots of the media were harvested, boiled in 1× loading buffer with or without β-mercaptoethanol, and fractionated by SDS-PAGE and transferred to nitrocellulose membrane. After blocked with 10% milk for 45 min at room temperature, the membrane was probed with a HRP conjugated goat anti-human IgG-Fc antibody (Bethyl) overnight at 4C, and developed with an enhanced chemilluminescent substrate (Pierce).
Humanized mice with functional human immune system
All mouse work was approved by the Institutional Animal Care and Use Committee of UCSD. After conditioned with sublethal (2.25 Gy) total body irradiation, NOD.Cg-PrkdcscidII2rgtm1wjl/SzJ (NOD/SCID/γc-/- or NSG, The Jackson Laboratory) mice of 6-10 weeks of age were transplanted with human fetal thymic tissue piece (previously frozen about 1 mm3) under the kidney capsule, and intravenously transfused 1-5 × 105 human CD34+ fetal liver cells as previously described (Lan et al., 2006). Human fetal tissues of gestational age of 17-20 weeks were obtained from Advanced Bioscience Resource (Alameda, CA). Human CD34+ cells were isolated by a magnetic-activated cell sorter separation system using anti-CD34 microbeads (Miltenyi Biotec).
Immunohistochemical and histologic analysis
Teratoma was fixed in 10% (W/v) buffered formalin, embedded in paraffin and sectioned as previously described (Zhao et al., 2011). Briefly, the sections will be stained with hematoxylin and eosin for histological assessment, and stained with anti-NeuN (Millipore), anti-C-peptide (Abcam) and anti-karetin (Millipore) antibodies for immunohistochemistry analysis. For frozen sections, samples were frozen in optimal cutting temperature (OCT) compound and sectioned. Teratoma sections were fixed in cold acetone for 10 min, sequentially blocked with 0.03% H2O2 for 30 min, 0.1% avidin for 15 min, 0.01% biotin for 15 min and 1% BSA for 30 min. Sections were incubated with primary antibody overnight at 4C, biotinylated secondary antibody for 30 min, HRP streptavidin for 30 min, and developed with AEC substrate solution. After counterstained with hematoxylin, sections were mounted in aqueous gel Vectamount (Vector). Images were captured with an Olympus MVX10 Microscope or an Olympus FluoView 1000 Confocal Microscope. Antibodies used: anti-CD3 antibodies (Epitomics, eBioScience), anti-CD4, anti-CD8 antibodies (BD Pharmingen), anti-human nuclei antibody (Millipore), anti-α-actinin antibody (Sigma), anti-cardiac Troponin I antibody (Epitomics).
Differentiation of hESCs into cardiomyocytes
We used a small molecule-driven cardiomyocyte differentiation protocol (Lian et al., 2012). hESCs were maintained on matrigel-coated plate. When the culture reaches confluence, cells were plated with CH in RPMI/B27-insulin (without insulin) medium for 24 hr (day 0 to day 1), and subsequently changed to RPMI/B27-insulin. On day 3, the cells were exposed to IWP4 (Stemgent) for 2 days. From day 7, the cells were cultured in RPMI/B27 medium with medium change every 3 days.
Transplantation of hESC-derived fibroblasts and cardiomyocytes
Derivation of human fibroblasts from the teratomas formed by hESCs was performed as previously described (Rong et al., 2012). Human fibroblasts were harvested and washed with PBS. About 20 μl of fibroblast suspension (1×107 cells) was loaded onto 6-7 mm diameter × 1.5 mm thick gelatin foam (Gelfoam, Pfizer) discs that were pre-wetted with DMEM basal media. The cells were overlaid with 15-25 μl Matrigel (BD Bioscience) and implanted subcutaneously with cells facing the dermis. hESC-derived cardiomyoctes were harvested, washed with PBS, suspended in PBS with 50% matrigel, and intramuscularly injected into the hind leg gastrocnemius with a 20G needle.
Quantitative real time PCR analysis
The total RNA was purified from hESCs, human T cells isolated from teratoma, hESC-derived fibroblasts and cardiomyocytes as previously described (Rong et al., 2012). The primers used for hESC markers (NANOG, OCT4, SOX2, LIN28, REX1, TDGF-1, GABRB3 and DNMT3B), fibroblast markers (VIMENTIN and FAP) and internal control (GAPDH) were previously described (Rong et al., 2012). The primers used for cardiomyocyte markers (NKX2.5, ISL1, MYL7 and SIRPA) were previously described (Dubois et al., 2011). Other primers used were listed in Supplementary table 2.
Supplementary Material
Highlights.
Optimized Hu-mice for studying human immune responses to hESC-derived allografts
CTLA4-Ig/PD-L1 were knocked into hESCs and provided localized immune protection
Allografts from modified hESCs did not induce systemic immunosuppression
CTLA4-Ig and PD-L1 are both required for immune protection of allografts
Acknowledgments
We thank Dr. Jack Bui for help with HLA genotyping and UCSD pathology core for histologic analysis. This work was supported by the grants from Chinese Ministry of Science and Technology (2013CB966902, 2013CB966903) and NIH (AI-064569 and AI-045897) to Y.G.Y. and H.F.Y, grants from NSFC to X.F. (81172828, 81373166), and grants from California Institute for Regenerative Medicine (RM-0173, TR3-05559, RB4-06244) to A.G and Y.X.
Footnotes
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blomer U, Gruh I, Witschel H, Haverich A, Martin U. Shuttle of lentiviral vectors via transplanted cells in vivo. Gene Ther. 2004;12:67–74. doi: 10.1038/sj.gt.3302384. [DOI] [PubMed] [Google Scholar]
- Boyd AS, Rodrigues NP, Lui KO, Fu X, Xu Y. Concise Review: Immune Recognition of Induced Pluripotent Stem Cells. STEM CELLS. 2012;30:797–803. doi: 10.1002/stem.1066. [DOI] [PubMed] [Google Scholar]
- Cheng F, Ke Q, Chen F, Cai B, Gao Y, Ye C, Wang D, Zhang L, Lahn BT, Li W, et al. Protecting against wayward human induced pluripotent stem cells with a suicide gene. Biomaterials. 2012;33:3195–3204. doi: 10.1016/j.biomaterials.2012.01.023. [DOI] [PubMed] [Google Scholar]
- Ciancio G, Garcia-Morales R, Mathew J, Carreno M, Burke GW, Ricordi C, Kenyon N, Esquenazi V, Cirocco R, Tzakis A, et al. Donor bone marrow infusions are tolerogenic in human renal transplantation. Transplantation Proceedings. 2001;33:1295–1296. doi: 10.1016/s0041-1345(00)02485-4. [DOI] [PubMed] [Google Scholar]
- Cohen DE, Melton D. Turning straw into gold: directing cell fate for regenerative medicine. Nat Rev Genet. 2011;12:243–252. doi: 10.1038/nrg2938. [DOI] [PubMed] [Google Scholar]
- Davis RP, Ng ES, Costa M, Mossman AK, Sourris K, Elefanty AG, Stanley EG. Targeting a GFP reporter gene to the MIXL1 locus of human embryonic stem cells identifies human primitive streak-like cells and enables isolation of primitive hematopoietic precursors. Blood. 2008;111:1876–1884. doi: 10.1182/blood-2007-06-093609. [DOI] [PubMed] [Google Scholar]
- Dubois NC, Craft AM, Sharma P, Elliott DA, Stanley EG, Elefanty AG, Gramolini A, Keller G. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat Biotech. 2011;29:1011–1018. doi: 10.1038/nbt.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elster EA, Hale DA, Mannon RB, Cendales LC, Swanson SJ, Kirk AD. The road to tolerance: renal transplant tolerance induction in nonhuman primate studies and clinical trials. Transplant Immunology. 2004;13:87–99. doi: 10.1016/j.trim.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunological Reviews. 2008;224:166–182. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunological Reviews. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Xu Y. Self-renewal and scalability of human embryonic stem cells for human therapy. Regenerative Medicine. 2011;6:327–334. doi: 10.2217/rme.11.18. [DOI] [PubMed] [Google Scholar]
- Fugier-Vivier IJ, Rezzoug F, Huang Y, Graul-Layman AJ, Schanie CL, Xu H, Chilton PM, Ildstad ST. Plasmacytoid precursor dendritic cells facilitate allogeneic hematopoietic stem cell engraftment. The Journal of Experimental Medicine. 2005;201:373–383. doi: 10.1084/jem.20041399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MP, Kelly PJ, Jardine M, Perkovic V, Cass A, Craig JC, Eris J, Webster AC. Long-Term Cancer Risk of Immunosuppressive Regimens after Kidney Transplantation. Journal of the American Society of Nephrology. 2010;21:852–858. doi: 10.1681/ASN.2009101043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley PJ. Species differences in the structure and function of the immune system. Toxicology. 2003;188:49–71. doi: 10.1016/s0300-483x(03)00043-x. [DOI] [PubMed] [Google Scholar]
- Howden SE, Gore A, Li Z, Fung HL, Nisler BS, Nie J, Chen G, McIntosh BE, Gulbranson DR, Diol NR, et al. Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy. Proceedings of the National Academy of Sciences. 2011;108:6537–6542. doi: 10.1073/pnas.1103388108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalscheuer H, Danzl N, Onoe T, Faust T, Winchester R, Goland R, Greenberg E, Spitzer TR, Savage DG, Tahara H, et al. A Model for Personalized in Vivo Analysis of Human Immune Responsiveness. Science Translational Medicine. 2012;4:125ra130. doi: 10.1126/scitranslmed.3003481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Cosimi AB, Spitzer TR, Tolkoff-Rubin N, Suthanthiran M, Saidman SL, Shaffer J, Preffer FI, Ding R, Sharma V, et al. HLA-Mismatched Renal Transplantation without Maintenance Immunosuppression. New England Journal of Medicine. 2008;358:353–361. doi: 10.1056/NEJMoa071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and Its Ligands in Tolerance and Immunity. Annual Review of Immunology. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. 2006;108:487–492. doi: 10.1182/blood-2005-11-4388. [DOI] [PubMed] [Google Scholar]
- Ledran MH, Krassowska A, Armstrong L, Dimmick I, Renstrˆm J, Lang R, Yung S, Santibanez-Coref M, Dzierzak E, Stojkovic M, et al. Efficient Hematopoietic Differentiation of Human Embryonic Stem Cells on Stromal Cells Derived from Hematopoietic Niches. Cell Stem Cell. 2008;3:85–98. doi: 10.1016/j.stem.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proceedings of the National Academy of Sciences. 2012;109:1848–1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- Rong Z, Fu X, Wang M, Xu Y. A scalable approach to prevent teratoma formation of human embryonic stem cells. Journal of Biological Chemistry. 2012;287:32338–32345. doi: 10.1074/jbc.M112.383810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Takizawa H, Strowig T, Willinger T, Eynon EE, Flavell RA, Manz MG. Human hemato-lymphoid system mice: current use and future potential for medicine. Annu Rev Immunol. 2013;31:635–674. doi: 10.1146/annurev-immunol-032712-095921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, Itskovitz-Eldor J, Benvenisty N. Selective ablation of human embryonic stem cells expressing a “suicide” gene. Stem Cells. 2003;21:257–265. doi: 10.1634/stemcells.21-3-257. [DOI] [PubMed] [Google Scholar]
- Schwartz SD, Hubschman JP, Heilwell G, Franco-Cardenas V, Pan CK, Ostrick RM, Mickunas E, Gay R, Klimanskaya I, Lanza R. Embryonic stem cell trials for macular degeneration: a preliminary report. The Lancet. 379:713–720. doi: 10.1016/S0140-6736(12)60028-2. [DOI] [PubMed] [Google Scholar]
- Selzner N, Grant DR, Shalev I, Levy GA. The immunosuppressive pipeline: Meeting unmet needs in liver transplantation. Liver Transplantation. 2010;16:1359–1372. doi: 10.1002/lt.22193. [DOI] [PubMed] [Google Scholar]
- Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. 2012;12:786–798. doi: 10.1038/nri3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Chung SK, Xu Y. Modeling Disease in Human ESCs Using an Efficient BAC-Based Homologous Recombination System. Cell Stem Cell. 2010;6:80–89. doi: 10.1016/j.stem.2009.11.016. [DOI] [PubMed] [Google Scholar]
- Springer CJ, Niculescu-Duvaz I. Prodrug-activating systems in suicide gene therapy. J Clin Invest. 2000;105:1161–1167. doi: 10.1172/JCI10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Tian C, Ansari MJI, Paez-Cortez J, Bagley J, Godwin J, Donnarumma M, Sayegh MH, Iacomini J. Induction of Robust Diabetes Resistance and Prevention of Recurrent Type 1 Diabetes Following Islet Transplantation by Gene Therapy. The Journal of Immunology. 2007;179:6762–6769. doi: 10.4049/jimmunol.179.10.6762. [DOI] [PubMed] [Google Scholar]
- Tillson M, Niemeyer GP, Welch JA, Brawner W, Swaim SF, Rynders P, Lenz SD, Dean B, Lothrop CD., Jr Hematopoietic chimerism induces renal and skin allograft tolerance in DLA-identical dogs. Experimental Hematology. 2006;34:1759–1770. doi: 10.1016/j.exphem.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Tonomura N, Habiro K, Shimizu A, Sykes M, Yang YG. Antigen-specific human T-cell responses and T cell-dependent production of human antibodies in a humanized mouse model. Blood. 2008;111:4293–4296. doi: 10.1182/blood-2007-11-121319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Touw W, Bromberg JS. Natural Killer Cells and the Immune Response in Solid Organ Transplantation. American Journal of Transplantation. 2010;10:1354–1358. doi: 10.1111/j.1600-6143.2010.03086.x. [DOI] [PubMed] [Google Scholar]
- Vodyanik MA, Bork JA, Thomson JA, Slukvin II. Human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood. 2005;105:617–626. doi: 10.1182/blood-2004-04-1649. [DOI] [PubMed] [Google Scholar]
- Walker LSK, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol. 2002;2:11–19. doi: 10.1038/nri701. [DOI] [PubMed] [Google Scholar]
- Wekerle T, Grinyó JM. Belatacept: from rational design to clinical application. Transplant International. 2012;25:139–150. doi: 10.1111/j.1432-2277.2011.01386.x. [DOI] [PubMed] [Google Scholar]
- Wirth E, Iii, Lebkowski JS, Lebacqz K. Response to Frederic Bretzner et al. “Target Populations for First-In-Human Embryonic Stem Cell Research in Spinal Cord Injury”. Cell Stem Cell. 2011;8:476–478. doi: 10.1016/j.stem.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Wood KJ, Bushell A, Hester J. Regulatory immune cells in transplantation. Nat Rev Immunol. 2012;12:417–430. doi: 10.1038/nri3227. [DOI] [PubMed] [Google Scholar]
- Woods NB, Parker AS, Moraghebi R, Lutz MK, Firth AL, Brennand KJ, Berggren WT, Raya A, Belmonte JCI, Gage FH, et al. Brief Report: Efficient Generation of Hematopoietic Precursors and Progenitors from Human Pluripotent Stem Cell Lines. STEM CELLS. 2011;29:1158–1164. doi: 10.1002/stem.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XQ, Zweigerdt R, Soo SY, Ngoh ZX, Tham SC, Wang ST, Graichen R, Davidson B, Colman A, Sun W. Highly enriched cardiomyocytes from human embryonic stem cells. Cytotherapy. 2008;10:376–389. doi: 10.1080/14653240802105307. [DOI] [PubMed] [Google Scholar]
- Zhang B, Duan Z, Zhao Y. Mouse models with human immunity and their application in biomedical research. Journal of Cellular and Molecular Medicine. 2009;13:1043–1058. doi: 10.1111/j.1582-4934.2008.00347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–215. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.