

Abstract
Poly(3-hydroxypropionate) (P3HP) is a biodegradable and biocompatible thermoplastic. In our previous study, a pathway for P3HP production was constructed in recombinant Esecherichia coli. Seven exogenous genes in P3HP synthesis pathway were carried by two plasmid vectors. However, the P3HP production was severely suppressed by strain instability due to plasmid loss. In this paper, two strategies, chromosomal gene integration and plasmid addiction system (PAS) based on amino acid anabolism, were applied to construct a genetically stable strain. Finally, a combination of those two methods resulted in the best results. The resultant strain carried a portion of P3HP synthesis genes on chromosome and the others on plasmid, and also brought a tyrosine-auxotrophy based PAS. In aerobic fed-batch fermentation, this strain produced 25.7 g/L P3HP from glycerol, about 2.5-time higher than the previous strain with two plasmids. To the best of our knowledge, this is the highest P3HP production from inexpensive carbon sources.
Introduction
Escherichia coli strains are widely used as hosts for microbial production of valuable compounds, like biofuels, chemicals, polymers, and proteins, and the production processes often depend on expression of heterologous genes carried by plasmid vectors [1]. Plasmids have been regarded as important tools for microbial genetic modifications. However, plasmids are separate genetic elements and autonomously replicated, and the redundant DNA carried by plasmids may cause metabolic burden in host strains, which could result in plasmid loss [2]–[4]. For plasmid maintenance, the cloning and expression vectors harbor antibiotic resistance genes and require the addition of antibiotics to the medium. Though it is feasible at the laboratory scale, the use of antibiotics at industrial scale will increase the production cost and raise the ecological issues. Furthermore, plasmid loss can even occur with presence of antibiotics during cultivation [5].
Plasmid addiction system (PAS) is an efficient strategy to prevent the survival of plasmid-free cells due to selective killing. Up to now, three major groups of PAS have been described: (1) toxin/antitoxin-based systems, (2) metabolism-based systems, and (3) operator repressor titration systems [1]. PASs have been successfully used in metabolically engineered strains to increase the product yield by stabilizing the plasmid in the cells, which carries the genes associated with product synthesis pathway and an addiction system. For instance, an antibiotic-free plasmid selection system based on glycine auxotrophy was constructed and used for overproduction of recombinant protein [6]. To maintenance the plasmid carrying cyanophycin synthesis gene cphA, Kroll et al. [7] established a novel anabolism-based addiction system. This system consisted of two components: an E. coli ispH mutant that cannot synthesize isopentenyl pyrophosphate (IPP), an essential precursor for isoprenoid biosynthesis, and a synthetic plasmid harboring cphA gene and the relevant genes of a foreign IPP-producing mevalonate pathway. The resultant strain revealed a plasmid stability of 100% and improved cyanophycin production.
Chromosomal gene integration (CGI) is another strategy to stabilize foreign genes. For example, pyruvate decarboxylase gene pdc and alcohol dehydrogenase II gene adhB from Zymomonas mobilis were integrated into the E. coli chromosome for ethanol biosynthesis, and the integration improved the stability of the Z. mobilis genes in E. coli and ethanol production [8]. Recently, a method was developed to insert multiple desired genes into target loci on E. coli chromosome and up to six copies of lacZ gene were simultaneously integrated into different loci. The β-galactosidase activity increased corresponding to the copy number of inserted lacZ genes [9]. To a certain extent, multiple insertions have resolved the main problem of CGI, low expression level of recombinant protein due to a low copy number.
Poly(3-hydroxypropionate) (P3HP) is a biodegradable and biocompatible plastic exhibiting high rigidity, ductility, and exceptional tensile strength in drawn films, and was regarded as one of the alternatives to petrochemical-derived plastic [10]. The biosynthesis of P3HP and 3HP-containing copolymers was previously dependent on structurally related precursors, such as 3HP, acrylate and 1,3-propanediol [11]–[14]. However the addition of these expensive precursors increased P3HP production cost. To solve this problem, we constructed a recombinant E. coli strain to synthesize P3HP using inexpensive carbon source glycerol (Figure 1) [15]. The genes involved in P3HP synthesis were cloned into two plasmids: the glycerol dehydratase and its reactivating factor genes, dhaB123 and gdrAB, from Klebsiella pneumoniae were inserted in the expression vector pACYCDuet-1 to generate plasmid pWQ04, and the propionaldehyde dehydrogenase gene pduP from Salmonella typhimurium and polyhydroxyalkanoate synthase gene phaC1 from Cupriavidus necator were carried by pWQ02. Under the optimized culture conditions, the recombinant E. coli strain accumulated 10.1 g/L P3HP (representing 46.4% of the cell dry weight) in a fed-batch fermentation.
Figure 1. P3HP synthesis pathway used in this study.
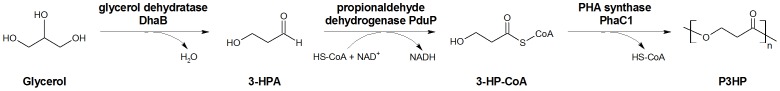
3-HPA, 3-hydroxypropionaldehyde; 3-HP-CoA, 3-hydroxypropionyl coenzyme A.
To optimize the P3HP production strain, 5 strategies (Figure 2) were designed and tested in this study. Strategy I used the previously constructed plasmids pWQ02 and pWQ04. Strategy II was designed to construct a phenylalanine/tyrosine-auxotrophy based PAS and all the genes associated with P3HP synthesis were integrated in E. coli chromosome in Strategy III. Strategy IV strains carried portion of genes involved in P3HP synthesis on chromosome and others on plasmid, which was further developed using a tyrosine-auxotrophy based PAS to improve the plasmid stability in Strategy V. As a result, the Strategy V strain Q1738 produced 25.7 g/L P3HP from glycerol in aerobic fed-batch fermentation, 2.5-time higher than the previous report.
Figure 2. Schematic representation of the strains and strategies for P3HP production used in this study.

Materials and Methods
Bacterial Strains and Growth Conditions
All strains and plasmids used in this study are listed in Table 1. E. coli DH5α was used as the host to construct and store all recombinant plasmids, E. coli χ7213 strain was used for preparation of all suicide vectors, and E. coli BL21(DE3) strain was used for protein expression and P3HP production. Bacteria were grown at 37°C in Luria-Bertani (LB) broth unless specified. Diaminopimelic acid (DAP) (50 µg/ml) was used for the growth of χ7213 strain. When necessary, antibiotics were added at final concentration of 100 µg/ml for ampicillin and 34 µg/ml for chloramphenicol. LB agar containing 10% sucrose was used for sacB gene-based counter selection in allelic exchange experiments.
Table 1. Bacteria strains and plasmids used in this study.
| Strains or plasmid | Description | Source |
| E. coli strains | ||
| DH5α | F– supE44 ΔlacU169 (φ80 lacZ ΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | lab collection |
| BL21(DE3) | F– ompT gal dcm lon hsdSB (rB− mB−) λ(DE3), | lab collection |
| ?7213 | thi-1 thr-1 leuB6 glnV44 fhuA21 lacY1 recA1 RP4-2-Tc::Mu λpir ΔasdA4 Δzhf-2::Tn10 | [29] |
| Q1475 | ΔpheA ΔtyrA | BL21(DE3) |
| Q1463 | ΔprpR::lacI PT7 gdrAB PT7 dhaB123 | BL21(DE3) |
| Q1599 | ΔprpR::lacI PT7 gdrAB PT7 dhaB123 ΔmtlA::PT7 phaC pduP | BL21(DE3) |
| Q1633 | ΔprpR::lacI PT7 gdrAB PT7 dhaB123 ΔascF::PT7 phaC pduP ΔmtlA::PT7 phaC pduP | BL21(DE3) |
| Q1693 | ΔprpR::lacI PT7 gdrAB PT7 dhaB123 ΔascF::PT7 phaC pduP ΔmtlA::PT7 phaC pduP ΔebgR::PT7 phaC pduP | BL21(DE3) |
| Q1736 | ΔprpR::lacI PT7 gdrAB PT7 dhaB123 ΔascF::PT7 phaC pduP ΔmtlA::PT7 phaC pduP ΔebgR::PT7 phaC pduP ΔmelR::PT7 phaC pduP | BL21(DE3) |
| Q1779 | ΔmtlA::PT7 phaC pduP | BL21(DE3) |
| Q1734 | ΔtyrA ΔprpR::lacI PT7 gdrAB PT7 dhaB123 | BL21(DE3) |
| Q1359 | BL21(DE3) carrying pWQ02 and pWQ04 | [15] |
| Q1509 | Q1475 carrying pG01 and pG02 | BL21(DE3) |
| Q1638 | Q1463 carrying pWQ02 | BL21(DE3) |
| Q1802 | Q1779 carrying pWQ04 | BL21(DE3) |
| Q1738 | Q1734 carrying pG01 | BL21(DE3) |
| Recombinant plasmids | ||
| pWQ02 | reppBR322 AmpR lacI PT7 phaC pduP | [15] |
| pWQ04 | repp15A CmR lacI PT7 gdrAB PT7 dhaB123 | [15] |
| pG01 | reppBR322 AmpR lacI PT7 phaC pduP TT Plac1-6 tyrA | pWQ02 |
| pG02 | repp15A CmR lacI PT7 gdrAB TT PpheA pheA PT7 dhaB123 | pWQ04 |
| Suicide plasmids | ||
| pRE112 | oriT oriV sacB cat | [16] |
| pG03 | ΔpheA ΔtyrA | pRE112 |
| pG04 | ΔtyrA | pRE112 |
| pG05 | ΔascF::PT7 phaC pduP | pRE112 |
| pG06 | ΔmtlA::PT7 phaC pduP | pRE112 |
| pG07 | ΔebgR::PT7 phaC pduP | pRE112 |
| pG08 | ΔmelR::PT7 phaC pduP | pRE112 |
| pLC01 | ΔprpR::lacI PT7 gdrAB PT7 dhaB123 | pRE112 |
Construction of Recombinant Plasmids
The primers used are listed in Table S1 in File S1. The plasmid pG01 was constructed using PCR fragments containing the tyrA coding region generated with primers 366 and 367 and BL21(DE3) chromosomal DNA as a template, which were digested with XhoI and then ligated with pWQ02 digested by the same enzyme. The plasmid pG02 was constructed using PCR fragments containing pheA coding region generated with primers 364 and 365 and BL21(DE3) chromosomal DNA as template, which were digested with AflII and HindIII and then ligated with pWQ04 digested by the same enzymes.
Construction of BL21(DE3) Strains with Chromosomal Mutations
The primers used are listed in Table S1in File S1. The mutations were constructed using suicide vector pRE112 as previously described [16]. For the pheA tyrA deletion, two pairs of primers, 369/370 and 371/372, were used to amplify approximately 500-bp fragments upstream and downstream of these genes from BL21(DE3) chromosome, respectively. The two fragments were then joined by PCR using primers 369 and 372. The PCR product was digested with SacI and XbaI and then ligated between the SacI and XbaI sites of vector pRE112 to generate plasmid pG03. The pheA tyrA mutation was introduced into BL21(DE3) by allelic exchange using suicide vectors pG03. A similar strategy was used to construct tyrA mutation using suicide vector pG04 constructed with primers 569/570 and 571/572.
To integrate the dhaB123 and gdrAB genes into prpR locus on BL21(DE3) chromosome, two pairs of primers, 268/269 and 270/271, were used to amplify approximately 500-bp fragments upstream and downstream of prpR gene from BL21(DE3) chromosome, respectively. The two fragments were then joined by PCR using primers 268 and 271. The PCR product was digested with SacI and NheI and then ligated between the SacI and XbaI sites of vector pRE112 to generate plasmid pRE112-ΔprpR. Then the KpnI-XbaI fragment containing dhaB123 gdrAB genes from plasmid pWQ04 was inserted into the corresponding sites of plasmid pRE112-ΔprpR to generate suicide vector pLC01, which was used to mediate the allelic exchange to generate ΔprpR::lacI PT7 gdrAB PT7 dhaB123 strain. A similar strategy was used to construct suicide vectors pG05, pG06, pG07, and pG08 to generate the chromosomal gene integration of ΔascF::PT7 phaC pduP, ΔmtlA::PT7 phaC pduP, ΔebgR::PT7 phaC pduP, and ΔmelR::PT7 phaC pduP, respectively.
Shake Flask Cultivation
The strain was inoculated into 500 ml baffled Erlenmeyer flasks containing 100 mL of minimal medium, which contains 3 g/L glucose, 20 g/L glycerol, 1.5 g/L KH2PO4, 3 g/L (NH4)2SO4, 1 g/L citric acid, 1 g/L citrate sodium, 1.9 g/L KCl, 3 g/L MgSO4, 0.138 g/L FeSO4·7H2O, 4.5 mg/L vitamin B1, and 1 ml of trace element solution. The trace element solution contained (per liter): 0.37 g (NH4)6Mo7O24·4H2O, 2.47 g H3BO4, 1.58 g MnCl2·4H2O, 0.29 g ZnSO4·7H2O, 0.25 g CuSO4·5H2O. The culture broth was inoculated with the overnight culture and incubated in a gyratory shaker incubator at 37°C and 200 rpm. The cells were induced at OD600∼0.6 with 0.05 mM IPTG and further incubated at 30°C. 5 µM of vitamin B12 (VB12) and appropriate antibiotic were added every 12 h. The cell dry weight (CDW) and P3HP yield were determined after 48 h culturing. All shake flask experiments were carried out in triplicates.
Fed-batch Fermentation
Fed-batch cultures were carried out in a Biostat B plus MO5L fermentor (Sartorius Stedim Biotech GmbH, Germany) containing 3 L of minimal medium as described above. During the fermentation process, pH was controlled at 7.0 via automated addition of 5 M KOH and antifoam 204 was used for foam control. The dissolved oxygen (DO) concentration was maintained at 5% saturation by associating with agitation from 400 rmp to 800 rpm and aeration with the airflow rate of 2 liters per min. After the initial carbon sources were nearly exhausted, fed-batch mode was commenced by feeding a solution containing 10 M glycerol at 0.5 mL/min. The expression of exogenous genes was initiated at an OD600 of 12 by adding 0.05 mM IPTG and 5 µM VB12. IPTG, VB12 and appropriate antibiotic were added every 12 h.
Cell Harvest, P3HP Extraction and Characterization
Cells of E. coli were harvested with centrifugation at 5000×g for 15 min, and washed with distilled water twice. To determine CDW, cell pellets were lyophilized, and the CDW was gravimetrically determined. P3HP was extracted from lyophilized cells with hot chloroform in a Soxhlet apparatus, and precipitated by ice-cold ethanol as described [17]. P3HP structure was confirmed by NMR analysis using an Avance III 600 NMR spectrometer (Bruker, Switzerland) as described previously [15].
Analysis of Antibiotic Concentration
Samples were withdrawn from the cultivation flask and centrifuged at 5000×g for 10 min. The supernatant was filtered using 0.22-µm filter, and concentrations of ampicillin and chloramphenicol were determined using Agilent 1200 HPLC System as described previously [18], [19].
Analysis of Plasmid Stability
To determine plasmid stability, samples were withdrawn from the cultivation flask at assigned time, diluted and spread onto LB agar plates with or without antibiotics supplemented. Ampicillin and chloramphenicol were used for plasmids pWQ02/pG01 and pWQ04/pG02, respectively. The plates were incubated for 16 h at 37°C, and the colony-forming units (CFU) were determined and analyzed by comparing the CFU on LB agar plates containing antibiotics and CFU on LB agar plates without antibiotics.
SDS-PAGE
The strain Q1599 and E. coli BL21(DE3) were grown in MM and induced by 0.05 mM IPTG. The cells were harvested by centrifugation and lysed by sonication. The whole-cell lysate was used for SDS-PAGE. Protein concentration was determined using the Bradford Protein Assay Kit (Tiangen, China). Proteins were separated in 12% acrylamide gels and visualized with Coomassie brilliant blue R250.
Results
P3HP Production and Plasmid Stability of Strategy I Strain
In our previous study, the genes involved in P3HP synthesis pathway were carried by two plasmids pWQ02 and pWQ04 [15]. An E. coli BL21(DE3) strain carrying pWQ02 and pWQ04, named as Q1359, was used to test the P3HP production and plasmid stability with the presence and absence of antibiotics. Without the addition of antibiotics, strain Q1359 accumulated 0.34 g/L P3HP representing 15.5% of the CDW under shaking flask condition. When ampicillin and chloramphenicol were added into the culture media, P3HP production and content increased to 0.52 g/L and 17.3%. After 48 h of cultivation, cultures were appropriately diluted and spread onto LB agar plates with and without the antibiotics to calculate the plasmid stability. Most of the cells lost their plasmids even with antibiotic selection (Table 2), and it was assumed that the segregational plasmid instability was caused by two reasons. First, plasmid duplication increased metabolic burden of the strain. Secondly, the antibiotic in medium was degraded during culturing process. To test this speculation, the ampicillin and chloramphenicol concentration in medium was determined by HPLC. Surprisingly, no antibiotic can be detected after 48-h cultivation even with periodic antibiotic addition every 12 h, indicating that the antibiotic degraded very fast under concentrations used in this study.
Table 2. P3HP production and plasmid stability of plasmid-containing strains.
| Strains | antibiotics | CDW (g/L) | P3HP (g/L) | P3HP content | pWQ02 or pG01 stability | pWQ04 or pG02 stability |
| Q1359 | – | 2.27±0.19 | 0.34±0.04 | 15.5±1.2% | 6.7±1.0% | 4.4±1.1% |
| + | 3.07±0.22 | 0.52±0.02 | 17.3±2.1% | 12.8±2.3% | 8.2±1.7% | |
| Q1509 | – | 2.33±0.31 | 0.55±0.03 | 22.5±1.1% | 42.9±3.7% | 27.2±0.9% |
| + | 2.59±0.29 | 0.84±0.07 | 31.2±0.5% | 59.1±2.4% | 36.9±3.1% | |
| Q1638 | – | 4.67±0.23 | 2.01±0.09 | 42.6±1.7% | 76.4±4.0% | – |
| + | 4.54±0.36 | 2.34±0.05 | 52.4±4.2% | 87.3±2.8% | – | |
| Q1802 | – | 2.78±0.06 | 0.56±0.06 | 20.3±1.9% | – | 6.5±0.5% |
| + | 3.32±0.14 | 0.83±0.05 | 25.0±0.9% | – | 13.1±.06% | |
| Q1738 | – | 4.46±0.24 | 1.99±0.06 | 45.0±2.9% | 80.8±1.7% | – |
| + | 4.51±0.18 | 2.40±0.04 | 53.7±3.4% | 90.2±4.0% | – |
The experiment was performed under shake flask condition in triplicate.
Construction and Characterization of Strain with PAS
To improve P3HP production and plasmid stability, a phenylalanine/tyrosine-auxotrophy based PAS was designed and constructed. The biosynthetic pathways of aromatic amino acids phenylalanine and tyrosine share the first step, from chorismate to prephenate catalyzed by bifunctional chorismate mutase/prephenate dehydratase PheA or TyrA. Besides that, PheA also carries out the second step in phenylalanine synthesis, converting prephenate into 2-keto-phenylpyruvate [20], and TyrA is responsible for the formation of 4-hydroxyphenylpyruvate from prephenate in tyrosine synthesis [21]. The pheA and tyrA genes are located next to each other in E. coli chromosome, and transcription of these 2 loci proceeds in opposite direction. In this study, an E. coli ΔpheA ΔtyrA mutant Q1475 was constructed using suicide vector pRE112 [16]. The chromosomal knockout of pheA and tyrA was verified by PCR and DNA sequencing. As shown in Figure 3, strain Q1475 was not able to grow in minimal medium. When phenylalanine and tyrosine were added, growth level and rate was similar to the wild-type E. coli BL21(DE3) strain, confirming the Phe−Tyr− phenotype of strain Q1475.
Figure 3. Growth of E. coli strains with and without the phenylalanine/tyrosine- auxotrophy based PAS, tested using minimal medium.
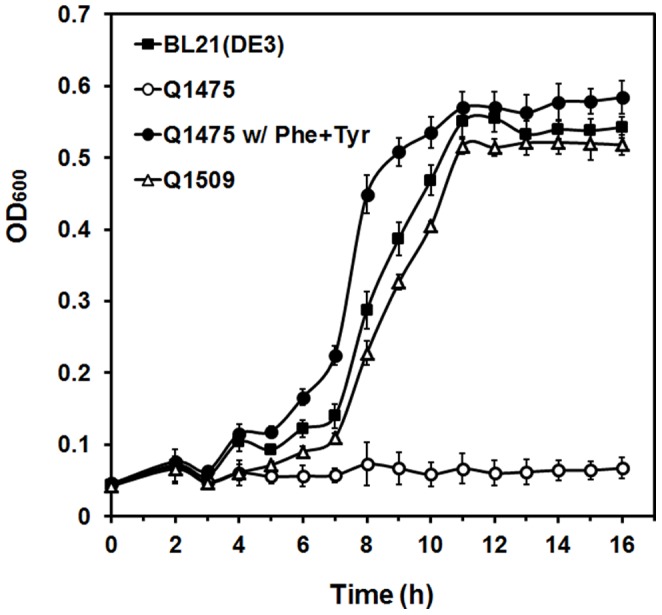
The experiment was performed under shake flask condition in triplicate.
To complement the phenylalanine/tyrosine-auxotrophy phenotype, the tyrA and pheA genes were cloned into pWQ04 and pWQ02, and the resulting plasmids were named as pG01 and pG02, respectively (Fig. S1 in File S1). For pheA gene, the structure gene and its own promoter region were amplified from E. coli chromosomal DNA. As tyrA is the second gene in cluster and does not have its own promoter, a constitutive promoter Plac1–6 [22] was fused to the E. coli tyrA coding region by PCR. In order to rule out the possible interferer between transcription of pheA/tyrA gene and P3HP synthesis associated genes, a bi-directional Rho-independent transcriptional terminator [23] was added behind the pheA and tyrA structural genes. The resulting plasmids pG01 and pG02 were confirmed by DNA sequencing, and transformed into Q1475 to generate Strategy II strain Q1509. In minimal medium without addition of phenylalanine and tyrosine, strain Q1509 revealed a similar growth with the wild-type strain (Fig. 3).
To verify P3HP accumulation and plasmid stability, strain Q1509 was cultivated as described above. The results showed that the phenylalanine/tyrosine-auxotrophy based PAS greatly increased the stabilities of pG01 and pG02 (Table 2). In absence of antibiotics, Q1509 exhibited plasmid stabilities of 42.9% for pG01 and 27.2% for pG02, more than 6-time higher than Strategy I strain Q1359. When antibiotics were added into the medium, the plasmid stabilities of pG01 and pG02 increased to 59.1% and 36.9%, respectively, about 4.5-time higher than strain Q1359. Unfortunately, the P3HP production did not raise in proportion of the plasmid stabilities. Only 0.55 g/L and 0.84 g/L P3HP were harvested under the condition without and with antibiotics addition, about 1.5-time higher compared with Q1359 (Table 2).
Construction and Characterization of CGI Strains
To insert the P3HP synthesis associated genes into E. coli BL21(DE3) chromosome, a set of suicide plasmid was constructed based on the vector pRE112 [16]. For example, the flanking regions of prpR gene were amplified and linked up with each other by overlap extension PCR, and the restriction sites of KpnI and XhoI were introduced at the connection point by primer design. This fragment was cloned into the vector pRE112 to generate pRE112-ΔprpR. Then an XhoI-KpnI fragment from pWQ04, encoding transcriptional regulator LacI and glycerol dehydratase system, was inserted into the corresponding site of pRE112-ΔprpR, and the resulting plasmid was defined as pLC01, which was used to mediate the allelic exchange. After two rounds of selection based on the positive marker chloramphenicol resistance gene cat and negative marker levan-sucrase gene sacB from Bacillus spp. [24], we obtained the strain Q1463 carrying chromosomal copy of dhaB123 and gdrAB genes (Figure S2 in File S1and Table 1). The phaC1 and pduP genes were inserted into the mtlA locus, mediated by suicide vector pG06 similarly, to generate Strategy III strain Q1599 (Figure 2, Figure S3 in File S1 and Table 1). The prpR gene and mtlA gene encode propionate catabolism regulator and mannitol-specific phosphotransferase system (PTS) enzyme IIA component, respectively. Under conditions used in this study, the prpR and mtlA mutations shouldnot affect the cell metabolism.
Cultivated in minimal medium for 48 h, strain Q1599 accumulated 0.26 g/L P3HP, which represented 6.4% of the CDW and was much lower than the P3HP productions of Q1359 and Q1509. To figure out the reason of low P3HP production in strain Q1599, SDS-PAGE analysis of whole-cell lysate was performed. Compared with the control strain E. coli BL21(DE3), DhaB1 and GdrA were observed as distinct bands with the expected molecular weights on SDS-PAGE, however we cannot find the bands for PhaC1 and PduP at appropriate position. It is assumed that the low copy number limited the expression level of phaC1 and pduP genes, so another three copies of these two genes were inserted into the chromosomal loci of ascF, ebgR and melR. These three genes are all involved in degradation of carbon compounds like cellobiose and lactose. As these disaccharide molecules are absent, the inactivation of those genes should not be detrimental to cell metabolism and growth. The result of shake flask cultivation showed that the P3HP production increased with the crescent copy number of phaC1 and pduP genes (Table 3), to the similar level of strains Q1359 and Q1509. Although this strain is stable and antibiotic-free, its P3HP production was still too low.
Table 3. P3HP production of strains with various copy numbers of phaC1 and pduP genes.
| Strains | copy number of phaC1 and pduP genes | CDW (g/L) | P3HP (g/L) | P3HP content |
| Q1599 | 1 | 4.02±0.26 | 0.26±0.04 | 6.4±0.4% |
| Q1633 | 2 | 3.58±0.14 | 0.38±0.03 | 10.7±0.8% |
| Q1693 | 3 | 3.37±0.21 | 0.44±0.01 | 13.2±1.4% |
| Q1736 | 4 | 2.65±0.26 | 0.50±0.01 | 18.9±0.3% |
The experiment was performed under shake flask condition in triplicate.
Combination of CGI Strategy and Plasmid Vector
To further improve the P3HP production, we constructed the Strategy IV strains, in which the CGI strategy and plasmid vector were used simultaneously. The strain Q1638 carries a chromosomal copy of genes encoding glycerol dehydratase and its reactivatase and plasmid-borne phaC1 and pduP genes, while in strain Q1802 the phaC1 and pduP genes were integrated into the mtlA locus and the dhaB and gdrAB genes were carried by plasmid pWQ04 (Figure 2).
The strains Q1638 and Q1802 were inoculated into minimal medium to test the P3HP production and plasmid stability. After 48 h of cultivation in shake flasks, the strain Q1638 accumulated 2.01 g/L and 2.34 g/L P3HP without and with addition of ampicillin, respectively, while the strain Q1802 only produced 0.56 g/L and 0.83 g/L P3HP under the same conditions (Table 2). In respect of plasmid stability, 76.4% of strain Q1638 cells still carried plasmid pWQ02 at the end of cultivation even without addition of ampicillin, however only 13.1% of strain Q1802 cells possessed the expression plasmid pWQ04 with the presence of chloramphenicol (Table 2).
Based on Strategy IV strain Q1638, we developed Strategy V strain Q1738 as following: the chromosomal tyrA gene was knocked out and plasmid pWQ02 was replaced by pG01 (Figure 2). In shake flask cultivation, strain Q1738 revealed similar P3HP production and slightly higher plasmid stability than strain Q1638 (Table 2). Compared with original strain Q1359, strains Q1638 and Q1738 presented about 4.5-time higher P3HP production and only required the addition of ampicillin in the growth process. Even without the usage of antibiotics, the P3HP production of strains Q1638 and Q1738 was still about 4-time higher than that of strain Q1359 with presence of ampicillin and chloramphenicol.
Fed-batch Fermentation
To evaluate the P3HP production in a scalable process, fed-batch fermentation of Q1638 and Q1738 was carried out at 5-L scale under aerobic condition. Cell growth and P3HP accumulation were monitored over the course of fermentation. As shown in Figure 4, CDW and P3HP reached the maximum in 36 h. With presence of ampicillin, the P3HP productions of trains Q1638 and Q1738 were 24.3 g/L (58.1% of CDW) and 25.7 g/L (67.9% of CDW), respectively. Even without antibiotic addition, 15.1 g/L and 16.2 g/L P3HP was accumulated by trains Q1638 and Q1738, respectively, higher than the previously reported P3HP yield from glycerol [15].
Figure 4. Time profiles for CDW and P3HP production during aerobic fed-batch fermentation of Strain Q1638 (A) and Q1738 (B).

The experiment was performed in triplicate.
Discussion
In this study, we are trying to construct a genetically stable strain for P3HP biosynthesis. As shown in Table 2, the stability of plasmid pWQ02 in stain Q1638 and plasmid pWQ04 in strain Q1802 was significantly improved when compared with strain Q1359. In strains Q1738 and Q1509, similar phenotype of plasmid pG01 was also observed. All these three strains with increased plasmid stability contain only one plasmid, whereas two in strains Q1359 and Q 1509. This phenomenon indicated that the plasmid stability would decrease if two or more types of plasmid exist in the same strain. It was reported that the segregative plasmid stability decreased with the size increasing, and the metabolic burden caused by plasmid duplication is a major reason for plasmid loss [25], [26]. Multiple plasmids brought about heavier burden on cell metabolism obviously, and a single plasmid had better stability reasonably.
Another noticeable result is that the CDW and P3HP production of strain Q1638 were much higher than that of Q1359, and the only difference between these strains is the copy number of glycerol dehydratase genes. Besides burden caused by plasmid duplication, the toxic product of glycerol dehydratase was also responsible for the low P3HP content. Glycerol dehydratase converts glycerol into 3-hydroxypropionaldehyde, which is a major component of antimicrobial substance Reuterin and inhibits the growth of some bacteria, yeasts and protozoa [27], [28]. The difference between strains Q1509 and Q1738 can be explained in the same way, and the intermediate toxicity was also the reason that the CDW and P3HP production were significantly lower when ampicillin was not supplemented in strains Q1638 and Q1738 (Figure 4). They carry a chromosomal copy of glycerol dehydratase gene, and the other genes involved in P3HP synthesis were borne by plasmids with ampicillin resistance. When ampicillin was absent, the plasmid instability increased, resulting in intracellular accumulation of 3-hydroxypropionaldehyde and growth depression.
In sum, the microbial P3HP production from glycerol was improved greatly by constructing a genetically stable E. coli recombinant strain. To overcome the strain instability due to plasmid loss, two strategies, amino acid anabolism based PAS and chromosomal integration, were tested. Finally, a combination of those two methods led to the best result. Our recombinant strain Q1738 produced 25.7 g/L P3HP from glycerol in aerobic fed-batch fermentation. To the best of our knowledge, this is the highest P3HP production from inexpensive carbon sources.
Supporting Information
This file contains Figure S1–S3 and Table S1.
(DOCX)
Acknowledgments
We acknowledge Dr. Roy Curtiss III (Arizona State University) for supplying the suicide vector pRE112 and strain χ7213.
Funding Statement
This research was financially supported by the 100-Talent Project of CAS (for GZ), Natural Science Foundation of Shandong Province (ZR2013EMZ002), National Natural Science Foundation of China (21206185, 21376255), and Qingdao Sci-Tech Development Programme (12-1-4-9-(3)-jch). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kroll J, Klinter S, Schneider C, Voss I, Steinbuchel A (2010) Plasmid addiction systems: perspectives and applications in biotechnology. Microbial Biotechnol 3: 634–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peredelchuk MY, Bennett GN (1997) A method for construction of E. coli strains with multiple DNA insertions in the chromosome. Gene 187: 231–238. [DOI] [PubMed] [Google Scholar]
- 3. Jones KL, Keasling JD (1998) Construction and characterization of F plasmid-based expression vectors. Biotechnol Bioeng 59: 659–665. [PubMed] [Google Scholar]
- 4. Wang Z, Xiang L, Shao J, Wegrzyn A, Wegrzyn G (2006) Effects of the presence of ColE1 plasmid DNA in Escherichia coli on the host cell metabolism. Microb Cell Fact 5: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zabriskie DW, Arcuri EJ (1986) Factors influencing productivity of fermentations employing recombinant microorganisms. Enzyme Microb Technol 8: 706–717. [Google Scholar]
- 6. Vidal L, Pinsach J, Striedner G, Caminal G, Ferrer P (2008) Development of an antibiotic-free plasmid selection system based on glycine auxotrophy for recombinant protein overproduction in Escherichia coli . J Biotechnol 134: 127–136. [DOI] [PubMed] [Google Scholar]
- 7. Kroll J, Steinle A, Reichelt R, Ewering C, Steinbuchel A (2009) Establishment of a novel anabolism-based addiction system with an artificially introduced mevalonate pathway: complete stabilization of plasmids as universal application in white biotechnology. Metab Eng 11: 168–177. [DOI] [PubMed] [Google Scholar]
- 8. Ohta K, Beall DS, Mejia JP, Shanmugam KT, Ingram LO (1990) Genetic improvement of Escherichia coli for ethanol production: chromosomal integration of Zymomonas mobilis genes encoding pyruvate decarboxylase and alcohol dehydrogenase Il. J Bacteriol 57: 893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koma D, Yamanaka H, Moriyoshi K, Ohmoto T, Sakai K (2012) A convenient method for multiple insertions of desired genes into target loci on the Escherichia coli chromosome. Appl Microbiol Biot 93: 815–829. [DOI] [PubMed] [Google Scholar]
- 10. Andreeben B, Steinbuchel A (2010) Biosynthesis and biodegradation of 3-hydroxypropionate-containing polyesters. Appl Environ Microbiol 76: 4919–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ichikawa M, Nakamura K, Yoshie N, Asakawa N, Inoue Y, et al. (1996) Morphological study of bacterial poly(3-hydroxybutyrate-co-3-hydroxypropionate). Macromol Chem Phys 197: 2467–2480. [Google Scholar]
- 12. Green PR, Kemper J, Schechtman L, Guo L, Satkowski M, et al. (2002) Formation of short chain length/medium chain length polyhydroxyalkanoate copolymers by fatty acid beta-oxidation inhibited Ralstonia eutropha . Biomacromolecules 3: 208–213. [DOI] [PubMed] [Google Scholar]
- 13. Zhou Q, Shi ZY, Meng DC, Wu Q, Chen JC, et al. (2011) Production of 3-hydroxypropionate homopolymer and poly(3-hydroxypropionate-co-4-hydroxybutyrate) copolymer by recombinant Escherichia coli . Metab Eng 13: 777–785. [DOI] [PubMed] [Google Scholar]
- 14. Meng DC, Shi ZY, Wu LP, Zhou Q, Wu Q, et al. (2012) Production and characterization of poly(3-hydroxypropionate-co-4-hydroxybutyrate) with fully controllable structures by recombinant Escherichia coli containing an engineered pathway. Metab Eng 14: 317–324. [DOI] [PubMed] [Google Scholar]
- 15. Wang Q, Yang P, Liu C, Xue Y, Xian M, et al. (2013) Biosynthesis of poly(3-hydroxypropionate) from glycerol by recombinant Escherichia coli . Bioresour Technol 131: 548–551. [DOI] [PubMed] [Google Scholar]
- 16. Edwards RA, Keller LH, Schifferli DM (1998) Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene 207: 149–157. [DOI] [PubMed] [Google Scholar]
- 17. Brandl H, Gross RA, Lenz RW, Fuller RC (1988) Pseudomonas oleovorans as a source of poly(β-hydroxyalkanoates) for potential applications as biodegradable polyesters. Appl Environ Microbiol 54: 1977–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burns DT, O’Callaghan M, Smyth WF, Ayling CJ (1991) High-performance liquid chromatographic analysis of ampicillin and cloxacillin and its application to an intramammary veterinary preparation. Fresenius’ J Anal Chem 340: 53–56. [Google Scholar]
- 19. Shen HY, Jiang HL (2005) Screening, determination and confirmation of chloramphenicol in seafood, meat and honey using ELISA, HPLC–UVD, GC–ECD, GC–MS–EI–SIM and GCMS–NCI–SIM methods. Anal Chim Acta 535: 33–41. [Google Scholar]
- 20. Dopheide TA, Crewther P, Davidson BE (1972) Chorismate mutase-prephenate dehydratase from Escherichia coli K-12. II. Kinetic properties. J Biol Chem 247: 4447–4452. [PubMed] [Google Scholar]
- 21. Sampathkumar P, Morrison JF (1982) Chorismate mutase-prephenate dehydrogenase from Escherichia coli. Purification and properties of the bifunctional enzyme. Biochim Biophys Acta 702: 204–211. [DOI] [PubMed] [Google Scholar]
- 22. Liu M, Tolstorukov M, Zhurkin V, Garges S, Adhya S (2004) A mutant spacer sequence between -35 and -10 elements makes the Plac promoter hyperactive and cAMP receptor protein-independent. Proc Natl Acad Sci USA 101: 6911–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lesnik EA, Sampath R, Levene HB, Henderson TJ, McNeil JA, et al. (2001) Prediction of Rho-independent transcriptional terminators in Escherichia coli . Nucleic Acids Res 29: 3583–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gay P, Le Coq D, Steinmetz M, Ferrari E, Hoch JA (1983) Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis: expression of the gene in Escherichia coli . J Bacteriol 153: 1424–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wojcik K, Wieckiewicz J, Kuczma M, Porwit-Bobr Z (1993) Instability of hybrid plasmids in Bacillus subtilis . Acta Microbiol Pol 42: 127–136. [PubMed] [Google Scholar]
- 26. Friehs K (2004) Plasmid copy number and plasmid stability. Adv Biochem Engin/Biotechnol 86: 47–82. [DOI] [PubMed] [Google Scholar]
- 27. Vollenweider S, Grassi G, Konig I, Puhan Z (2003) Purification and structural characterization of 3-hydroxypropionaldehyde and its derivatives. J Agric Food Chem 51: 3287–3293. [DOI] [PubMed] [Google Scholar]
- 28. Talarico TL, Dobrogosz WJ (1989) Chemical characterization of an antimicrobial substance produced by Lactobacillus reuteri . Antimicrob Agents Chemother 33: 674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roland K, Curtiss R 3rd, Sizemore D (1999) Construction and evaluation of a Dcya Dcrp Salmonella typhimurium strain expressing avian pathogenic Escherichia coli O78 LPS as a vaccine to prevent airsacculitis in chickens. Avian Dis 43: 429–441. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains Figure S1–S3 and Table S1.
(DOCX)