

Abstract
Cellular programs coupled to cycles of epithelial–mesenchymal transitions (EMTs) play critical roles during embryogenesis, as well as during tissue development, remodeling, and repair. Research over the last decade has established the importance of an ever-expanding list of master EMT transcription factors, whose activity is regulated by STAT3 and function to stimulate the rapid transition of cells between epithelial and mesenchymal phenotypes. Importantly, inappropriate reactivation of embryonic EMT programs in carcinoma cells underlies their metastasis to distant organ sites, as well as their acquisition of stem cell-like and chemoresistant phenotypes operant in eliciting disease recurrence. Thus, targeted inactivation of master EMT transcription factors may offer new inroads to alleviate metastatic disease. Here we review the molecular, cellular, and microenvironmental factors that contribute to the pathophysiological activities of STAT3 during its regulation of EMT programs in human carcinomas.
Keywords: EGFR, EMP, EMT, STAT3, TGF-β, extracellular matrix, metastasis, signal transduction, tumor microenvironment
Introduction
Signal transducer and activator of transcription 3 (STAT3) was originally discovered nearly two decades ago as a latent transcription factor that was robustly activated by interleukin-6 (IL-6), and by epidermal growth factor (EGF).1 However, shortly thereafter inappropriate STAT3 activity was determined to participate in cellular transformation, resulting in its subsequent designation as a proto-oncogene.2 The activation of STAT3 by IL-6 commences upon its binding to the IL-6 receptor (IL-6R), which belongs to the class I family of cytokine receptors that form heterotetrameric complexes with the common signaling receptor, gp130 (Fig. 1; refs. 3 and 4). Indeed, gp130 dimerization results in the activation of members the Janus family (JAK1, JAK2, or TYK2) of receptor-associated protein tyrosine kinases, which readily phosphorylate gp130 at intracellular SH2-domain docking sites that not only promote the activation of STAT3 (and STAT1), but also that of the MAP kinase and PI3K:AKT pathways.3,4 Following its phosphorylation at a single C-terminal Tyr residue (i.e., Y705) by JAKs, STAT3 undergoes homodimerization and nuclear translocation where it governs the expression of a variety of genes operant in regulating cell transformation, proliferation, survival, and motility,3,4 events essential to the development and metastatic progression of human carcinomas. In stark contrast, immunohistochemical surveys of STAT3 activation in human carcinomas (e.g., breast and prostate) correlated the phosphorylation (Y705) and nuclear localization of STAT3 with better overall patient prognosis and clinical outcome.5-7 This dichotomy in STAT3 function may result from the alternative splicing of STAT3 transcripts at exon 23, resulting in the production of either full-length STAT3α or truncated STAT3β.8,9 Indeed, unlike its full-length STAT3α counterpart, truncated STAT3β lacks Ser727, a residue that is phosphorylated by a variety of Ser/Thr protein kinases (e.g., MAP kinases, mTOR, and PKCδ) and is required for maximal STAT3 transcriptional activity.4 Thus, while initial findings attributed dominant-negative functions to the expression of STAT3β,9 more recent findings demonstrate that the truncated STAT3β variant is indeed biologically and transcriptionally active, and more importantly, is operant in driving the expression of genes that coalesce in eliciting anticancer activities.8 At present, the pathophysiological balance between the tumor promoting and tumor suppressing activities of individual STAT3 isoforms, as well as the splicing events coupled to their production remain to be fully elucidated. Nevertheless, it is abundantly clear that STAT3α is the predominant STAT3 isoform expressed in human carcinomas and regulates multiple aspects of their development and metastatic progression.

Figure 1. The differential modes of STAT3 activation and its genomic and nongenomic functions in responsive cells. (A) The IL-6/IL-6R complex induces dimerization of the common cytokine receptor subunit, gp130 leading the sequential activation of JAK2 and STAT3. (B) OSM binds gp130 and signals through LIF receptor (LIFR)/gp130 heterodimers, or through OSM receptor (OSMR)/gp130 heterodimers to activate JAK2-STAT3. (C) Ligand-activated EGFR forms a molecular complex with STAT3, leading to its activation. (D) FN adhesion induces formation of integrin-EGFR complexes and subsequent EGFR-dependent STAT3 activation. (E) FN-induced cell adhesion is also associated with STAT3 activation via an integrin:Pyk2:JAK2 molecular complex that functions independent of EGFR. (F and G) Tyrosine-phosphorylated STAT3 dimers undergo nuclear translocation (F) where they regulate several “master” EMT transcriptional networks (G). (H) Various miRs discussed in the text either restrict (−) or enhance (+) STAT3 signaling and EMT programs by targeting molecules associated with distinct aspects of the STAT3 pathway. (I) SOCS3 inhibits STAT3 signaling via blockade of upstream signaling through interactions with gp130 and JAK family members. (J) Tyrosine-phosphorylated STAT3 can localize to focal adhesions where it interacts with focal adhesion kinase (FAK) and paxillin, and also regulates the phosphorylation status of the adaptor protein p130Cas. (K) Cytosolic STAT3 also promotes microtubule polymerization by sequestering the microtubule-destabilizing factor stathmin.
An emerging paradigm operant in driving the progression of indolent carcinoma in situ to aggressive metastatic disease is that of epithelial–mesenchymal transition (EMT). At its core, the term EMT relates to the ability of polarized, immotile epithelial cells to undergo a cellular metamorphosis that results in their acquisition of apolar, motile, and mesenchymal-like phenotypes.10-12 EMT programs are essential for normal embryogenesis and tissue development, as well as for the normal remodeling and repair of wounded tissues. Interestingly, the reactivation of embryonic EMT programs in cells that harbor malignant genomes enables carcinoma cells to become metastatic and resistant to chemotherapies, and to exhibit properties reminiscent of cancer stem cells.10,11 Additionally, the ability of disseminated cells to undergo metastatic outgrowth has been linked to mesenchymal-epithelial transition (MET) programs, which function to reverse the phenotypic and morphological features of EMT programs. Furthermore, recent studies have identified carcinoma cells that simultaneously display mixed epithelial and mesenchymal traits, which likely enhances the ability of these cells to rapidly adapt and survive dynamic changes within their microenvironments, ultimately leading to the establishment of secondary tumor lesions. The metastable nature of carcinoma cells to readily transition between epithelial and mesenchymal phenotypes has recently been termed epithelial–mesenchymal plasticity (EMP),13 which greatly enhances the metastatic and chemoresistant properties of developing tumors. At present, the precise manner through which carcinoma cells initiate and resolve EMT programs as they traverse the metastatic cascade remains unclear. However, what is clear is that EMT programs are driven by the activation of an ever-expanding list of master transcription factors, whose activities are governed and fine-tuned by extrinsic cues and microenvironmental signals housed within the primary tumor and subsequent metastatic niche.10,14 Readers desiring additional information related to the pathophysiology of EMT programs are directed to several comprehensive reviews.10,11,15,16 In the succeeding sections, we review recent findings that directly impact our understanding of the role STAT3 plays in regulating the initiation and resolution of EMT programs in normal and malignant cells. In addition, we also discuss how EMT programs influence the function of STAT3 in developing and progressing carcinomas.
Master Regulators of EMT Programs
Numerous studies over the last decade have focused on unraveling the molecular events that drive EMT programs. In doing so, a rapidly expanding list of “master” EMT transcription factors have been established, all of which share the ability of inducing epithelial cells to acquire mesenchymal-like phenotypes and morphologies.10 Most notable among these EMT transcription factors is Twist, which belongs to the family of basic helix-loop-helix transcription factors, and Snail, which belongs to the family of zinc-finger transcription factors.17 Twist and Snail were originally identified as essential players for embryonic dorsoventral patterning, and for mesodermal formation.18,19 Today, Twist and Snail are both well-established drivers of EMT programs and metastatic progression, doing so via direct and indirect mechanisms.10,20,21 Likewise, aberrant expression of either Snail2/Slug,22 Goosecoid,23 FoxC2,24 and Zeb1 and Zeb225,26 can all similarly elicit EMT programs in epithelial cells, as well as induce metastasis in their malignant counterparts. Additionally, the overexpression of individual “master” EMT transcription factors is sufficient to further induce the expression of additional EMT transcription factors,27 indicating the interdependent wiring of EMT transcriptional networks in normal and malignant epithelial cells. Finally, the initiation and resolution of EMT programs is governed by numerous intrinsic and extrinsic factors, including STAT3, whose modes of activation and modulation of EMT programs is discussed below and depicted in Figure 1.
Activators of STAT3
IL-6 and canonical STAT3 activation
The existence of IL-6 was first hypothesized in the late 1960s and ultimately established in the mid-1980s upon the isolation of its cDNA in response to studies of the immune system and its regulation of acute-phase protein responses.28 More recently, aberrant IL-6 expression and its activation of STAT3 have been positively associated with the development and progression of carcinomas in humans,29 particularly with increases in metastatic burden and diminished overall survival.30 Indeed, upregulated IL-6 signaling dramatically induces the expression of Twist and Snail, leading to the initiation of EMT programs and expression of mesenchymal markers in cancers of the breast and head and neck.31,32 Furthermore, enforced expression of Twist upregulates the production of IL-6, leading to its autocrine activation of STAT3.31 Taken together, these findings point to the presence of an autocrine, positive feedback loop involving the activation of an IL-6 → STAT3 → Twist signaling axis coupled to EMT programs in breast cancers. Accordingly, the activation of STAT3 by IL-6 in breast cancers also promotes the selection and expansion of CD44high/CD24low cells that possess stem-like properties,30,33 which is a hallmark of post-EMT carcinoma cells characterized by their enhanced tumorigenicity, pluripotency, and chemoresistance.34 Additionally, cancer stem cells (CSCs) are also responsible for seeding metastatic niches, leading to secondary tumor formation and disease recurrence.34 Interestingly, these IL-6-dependent events are regulated in part by the expression and activity of molecular chaperones. For instance, genetic silencing of the heat shock protein, Hsp27 suppresses the expression of mesenchymal markers and EMT programs driven by the IL-6 → STAT3 → Twist signaling axis, leading to reduced numbers of circulating tumor cells and diminished metastatic burden in mouse models of prostate cancer.35 Collectively, these findings highlight the importance of IL-6 and STAT3 in driving carcinoma progression and “stemness”, and as such, point to the potential clinical benefit that may be gained by the development of effective therapies against IL-6.
Oncostatin M
Oncostatin M (OSM) is an inflammatory cytokine that also belongs to the family of gp130-related cytokines, and as such, OSM stimulates transmembrane signaling by binding and activating either of two receptor complexes: (1) gp130 and LIFR (leukemia inhibitor factor receptor) heterodimers, and (2) gp130 and OSM receptor (OSMR). Similar to IL-6, OSM is a robust stimulator of STAT3, and consequently, is a strong promoter of EMT programs, metastasis, and CSCs.36,37 Through its ability to activate STAT3, OSM also enhances cell migration and upregulates the expression of the extracellular matrix (ECM) protein, fibronectin (FN).38,39 The induction of EMT programs by OSM transpires in part through its ability to downregulate members of the miR-200 family of microRNAs (miRs), which function in silencing the expression of the “master” EMT transcription factors ZEB1 and ZEB2.37,40,41 OSM also downregulates the family of Let-7 miRs, which function as tumor suppressors by silencing a variety of molecules, including the nonhistone chromatin-binding protein HMGA2.37,40,41 Despite the clear associations of OSM with the induction of EMT programs, a recent study has observed the OSM:STAT3 signaling axis to prevent TGF-β from inducing the expression of the “master” EMT transcription factor FoxC2, as well as that of a variety of matricellular proteins (e.g., SPARC, CTGF, tenascin C) known to enhance the functional output of EMT programs.42 At present, it remains uncertain as to whether the anti-TGF-β activities of OSM will be restricted to a few specific tissues, or whether these events will be universally applicable to all tissues. However, a recent study identified c-Myc as a “molecular switch” that enables OSM to collaborate with TGF-β in driving tumorigenicity of breast cancer cells,43 thereby demonstrating the power of oncogenes to usurp signaling pathways to confer a selective advantage for developing carcinomas. Future studies need to identify additional oncogenic pathways and cellular contexts capable of converting OSM:STAT3 function.
EGF receptor
The binding of EGF to its receptor represented one of the first events identified as being capable of inducing robust activation of STAT3.1 Indeed, the activation of STAT3 by EGF receptor (EGFR) requires Src to phosphorylate the catalytic domain of EGFR at Tyr845, as well as at two EGFR autophosphorylation sites located at Tyr1068 and Tyr1086, both of which serve as docking sites for STAT3.44,45 Moreover, EGF simulation of carcinoma cells that express aberrantly high levels of EGFR is sufficient to induce EMT phenotypes.46 Along these lines, Lo and colleagues47 demonstrated that the ability of EGF to induce EMT programs was contingent upon the activation of STAT3 in human tumors that harbored genomic amplifications of EGFR. Interestingly, both of the human carcinoma cell lines employed in this study, namely MDA-MB-468 breast cancer cells and A431 epidermoid carcinoma cells, readily undergo apoptosis when stimulated by EGF, a reaction that involves STAT1-mediated upregulation of caspase-1.48,49 Therefore, it is tempting to speculate that these dichotomous responses to EGF may be driven by the differential activation of STAT1 vs. STAT3. As such, future studies should investigate the molecular mechanisms that underlie the functional balance between growth factors and individual STAT molecules.
Ovarian cancers frequently overexpress EGFR and exhibit constitutive activation of STAT3,50 events that readily enhance the dissemination of ovarian cancer cells into the peritoneum that contains a rich supply of EGF.51 In fact, EGF elicits EMT programs in ovarian cancer cells by inducing their expression of Twist through a JAK2:STAT3-dependent pathway.47,52 Interestingly, a novel role for calcium in coupling EGFR:STAT3 to EMT programs has recently been described in human breast cancers. Indeed, although calcium chelation had no effect on the coupling of EGF to Akt and ERK1/2, this event did specifically prevent EGF from activating STAT3 and, consequently, from inducing EMT programs.53 Furthermore, cellular depletion of the calcium channel TRPM7 was sufficient to uncouple EGF from STAT3 activation and vimentin expression,53 thereby identifying intracellular calcium signaling as a novel facet of EMT programs. Future studies need to determine the extent to which calcium signaling participates in driving EMT programs in other cell types and disease states, as well as investigate the effectiveness of anti-calcium agents in alleviating carcinoma development and metastatic progression.
Sphingosine-1-phosphate and nuclear factor-κB
The activation of STAT3 and nuclear factor-κB (NFκB) in normal cells is typically robust and transient, reflecting the complex integration and coordination of several tightly controlled negative feedback loops.4,54 In stark contrast, carcinomas typically display constitutive activation of both signaling molecules, an event coupled to the development of reactive tumor stroma and inflammatory signaling (i.e., extrinsic signaling), and to the inappropriate initiation of positive feed-forward loops by progressing carcinomas (i.e., intrinsic signaling). Insights into how these events coalesce and conspire to constitutively activate STAT3 and NFκB has recently been elucidated.55 Indeed, Lee et al.56 found that STAT3 was responsible for eliciting constitutive NFκB activity in human melanoma and prostate cancer cells. Mechanistically, phosphorylation of STAT3 at Ser727 facilitates the recruitment of the p300 acetyltransferase into NFκB DNA complexes, where it acetylates active RelA and prevents its inactivation and exportation from the nucleus.56 Importantly, phosphorylated STAT3 and acetylated RelA were observed to colocalize only in the nuclei of malignant human tissues, not their corresponding normal or adjacent tissue counterparts, suggesting the presence of a prominent collaboration between STAT3 and NFκB in driving tumorigenesis. Along these lines, NFκB activation is a major player in promoting the production of IL-6, which stimulates STAT3 activation and completes a STAT3 → NFκB → IL-6 feed-forward signaling loop in developing carcinomas.55 Recently, this model has been further refined to include the aberrant production of sphingosine-1-phosphate (S1P) and its activation of the sphingosine-1-phosphate receptor 1 (S1PR1) in eliciting constitutive STAT3 signaling in human tumors, a reaction that transpires in part via JAK2.57 Interestingly, activated STAT3 reinforces these events by upregulating S1PR1 expression, thereby enhancing carcinoma growth and metastasis,57 as well as driving inflammatory reactions operant in mediating the formation of colitis-associated colorectal cancers.58 Collectively, these findings highlight the importance of feed-forward inflammatory loops in promoting carcinoma development and metastatic progression, as well as point toward novel therapeutic inroads and strategies to alleviate these adverse events in patients with metastatic disease.
Differential Modes of STAT3 Activation during Metastatic Progression
We recently established the central importance of STAT3 activation not only for inducing the transformation of mammary epithelial cells, but also for promoting their metastatic outgrowth.59-61 In doing so, we initially demonstrated that normal mouse mammary gland (NMuMG) cells can be readily transformed by their enforced overexpression of human EGFR, resulting in the generation of NME cells that are nonmetastatic,61 but which readily become metastatic upon completing EMT programs.62 Importantly, this in situ model of breast cancer is absolutely dependent upon the ability of EGFR to activate STAT3, as expression of a mutant EGFR that lacks its dileucine motif (e.g., L679A, L680A) cannot couple to STAT3 and is incompetent to induce cellular transformation.59 Along these lines, breast cancer progression is characterized by the increased production of the ECM protein FN, which derives from both the carcinoma and stromal compartments of developing tumors.38 Interestingly, we identified three distinct modes whereby FN participates in the activation of STAT3 by carcinoma cells. Indeed, two modes function in pre-EMT and nonmetastatic cells, while a final mode is engaged in their post-EMT counterparts (Fig. 2; refs. 59 and 60). With respect to pre-EMT carcinoma cells, STAT3 is predominantly activated by the binding of EGF to EGFR (i.e., EGF-dependent; Fig. 2, left), and to a lesser extent, by the binding of FN to a complex comprised of αvβ3 integrin and EGFR (i.e., EGF-independent; Fig. 2, middle). In both scenarios, the expression and activity of Src are essential to STAT3 activation, leading to cellular transformation and the initiation of EMT programs that support primary tumor growth.59 Interestingly, while post-EMT carcinoma cells clearly remain addicted to STAT3 activity, these metastatic cells were determined to employ a distinct signaling system to elicit STAT3 activity in response to FN. For instance, activation of a β1 integrin:JAK2:Pyk2 signaling axis supports the outgrowth of secondary tumors (Fig. 2, right), which become sensitized to pharmacological inhibition of JAK2 and Pyk2,59 suggesting a potential therapeutic strategy to target disseminated breast cancer cells. Likewise, future studies should also determine the extent to which this alternative route to STAT3 activation is dependent upon carcinoma and/or stromal production of IL-6, thereby offering an additional therapeutic target to alleviate metastatic disease. Collectively, our findings identified several stimulatory mechanisms that enable mammary carcinoma cells to differentially activate STAT3 as they progress from indolent to aggressive disease states, thereby maintaining their addiction to STAT3 and evading traditional chemotherapies directed at disseminated breast cancer cells.
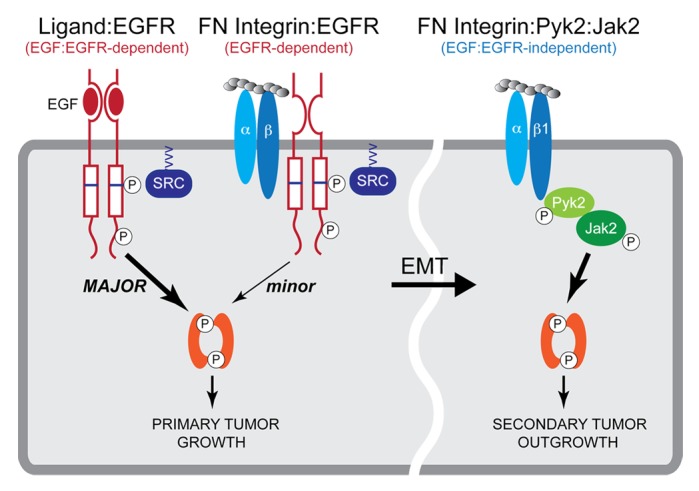
Figure 2. Differential modes of STAT3 activation during the metastatic progression of breast cancers. Early during breast cancer development, pre-EMT and nonmetastatic cells that exhibit high levels EGFR expression engage two distinct mechanisms to activate STAT3 signaling pathways, a major axis involving EGF:EGFR (left) and a minor axis regulated by FN-induced formation of β3 integrin-EGFR complexes (middle). Both of these pathways require Src-dependent phosphorylation of EGFR residue Tyr845 located within the catalytic pocket of the kinase domain. As such, both pathways are sensitive to EGFR and Src inhibition. In contrast, post-EMT and metastatic breast cancer cells engage a third mechanism to activate STAT3 that comprises a FN:β1 integrin:Pyk2:JAK2:STAT3 signaling network (right). The activation of this third mechanism may be clinically relevant at sites of secondary pulmonary tumors and likely contributes to the resistance of metastatic breast cancers to EGFR-targeted therapies. Adapted from reference 59.
Modulators of STAT3 Expression and Activity
microRNAs (miRs)
As mentioned above, miRs have emerged as critical effectors and/or regulators of STAT3 signaling. Indeed, the ability of OSM to induce EMT programs in breast cancer cells proceeds via STAT3-mediated downregulation of both miR-200 and Let-7.37 Likewise, inactivation of STAT3 or re-expression of both miRs proved sufficient to induce MET programs in mesenchymal breast cancers,37 thereby implicating miR-200 and Let-7 as STAT3 effectors during EMT programs. Besides the ability of STAT3 to regulate miR expression, several recent reports have identified miRs that function to enhance the pathophysiology of STAT3.63,64 For instance, the induction of miR-221/222 stimulates EMT programs in part by downregulating the expression of the receptor for adiponectin, adiponectin receptor 1 (ADIPOR1).63 Importantly, the binding of adiponectin to ADIPOR1 typically represses the activation of STAT3. As such, miR-221/222-mediated targeting of ADIPOR1 results in enhanced STAT3 activation in response to IL-6, leading to the initiation of EMT programs. In accordance with their ability to induce EMT programs, miR-221/222 are differentially expressed in basal-like (i.e., mesenchymal-like) vs. luminal (i.e., epithelial-like) breast cancer subtypes,63 suggesting that detecting the expression of these miRs may prove useful as a diagnostic biomarker. Finally, STAT3 potently induces the expression of miR-146b in normal and nontransformed mammary epithelial cells where it functions in preventing the translation of IRAK1 (interleukin-1 receptor-associated kinase 1) and TRAF6 (TNF receptor-associated factor 6),65 thereby inhibiting NFκB activity and its stimulation of IL-6 expression. Importantly, the promoter for miR-146b is hypermethylated and inaccessible to STAT3 in human breast cancers, particularly those lacking estrogen receptor-α (ER-α). Consequently, the uncoupling of STAT3 from miR-146b expression elicits aberrant activation of NFκB and its induction of IL-6 expression, which initiates a positive feed-forward loop that culminates in constitutive STAT3 signaling and its stimulation of migratory and invasive phenotypes in ER-α-negative breast cancers.65
Along these lines, the miR-17-92 cluster member miR-18a is highly expressed in gastric cancers where it targets the expression of PIAS3 (protein inhibitor of activated signal transducer and activator of transcription 3).64 Mechanistically, the loss of PIAS3 results in elevated STAT3 transcriptional activity, thereby driving the expression of genes operant in mediating EMT programs. Collectively, these findings highlight the potential clinical benefit of developing miR-based therapies designed to suppress STAT3 signaling and, consequently, to alleviate metastatic disease in human cancers.
Suppressor of cytokine signaling
Members of the suppressor of cytokine signaling family (SOCS1–7 and CIS) proteins were originally discovered as modulators of immunity, particularly the biology of macrophages and T cells.66 As a group, SOCS proteins function as negative feedback inhibitors of cytokine receptor signaling systems, doing so by (1) preventing STAT:receptor interactions, (2) inhibiting JAK protein tyrosine kinase activity, and (3) targeting STATs and JAKs for ubiquitination and subsequent degradation.66,67 Thus, SOCS proteins have been associated with mediating anti-tumor activity, a designation supported by the finding that SOCS3 expression is frequently silenced via promoter hypermethylation in a variety of human tumors.68-70 Consistent with its ability to inhibit STAT3 activity,66,67 SOCS3 expression has recently been linked to the suppression of EMT programs. Indeed, siRNA-depletion of SOCS3 expression in human hepatocellular carcinoma cells was found to engender increased expression of the “master” EMT transcriptional regulator Snail, as well as enhanced TGF-β1 production and initiation of autocrine signaling.71 These findings highlight the therapeutic potential to suppress STAT3 signaling and its associated EMT by targeting and upregulating endogenous inhibitors of their activation, such as SOCS3. Interestingly, a recent study demonstrated the ability of EGFR to be incorporated into IL-6R/gp130 complexes, an event that elicits a second wave of STAT3 activation that circumvents the inhibitory actions of SOCS3.72 Thus, future attempts to enhance SOCS activity in clinical settings need to remain cognizant for the possibility that cancer cells will rapidly develop innate resistance to such therapies.
Nongenomic STAT3 Signaling
In addition to its role in regulating gene expression, including that of “master” EMT transcription factors, recent evidence has also identified several nongenomic functions for STAT3. Of particular importance is the ability of STAT3 to regulate cell migration and invasion via direct interactions with components and machinery of the cytoskeletal system. For instance, phosphorylated STAT3 colocalizes with focal adhesion kinase (FAK) and paxillin within focal adhesion complexes in migrating ovarian carcinoma cells, events that also required the activity of Src.73 Along these lines, STAT3 activation functions in governing the rate of focal adhesion turnover in keratinocytes, and as such, the strength to which these cells adhere to or move along ECM substrates. Mechanistically, these STAT3-dependent activities derive from its ability to promote the dephosphorylation of the focal adhesion adaptor protein, p130Cas, presumably via recruitment and binding of protein tyrosine phosphatases operant in eliciting focal adhesion turnover.74 Thus, under normal physiological conditions, STAT3 appears to function in balancing the phosphorylation and activation status of p130Cas, while under pathological conditions, aberrant STAT3 activity may enhance metastatic outgrowth by remodeling the phosphorylation status of p130Cas to favor oncogenic signaling by cytokines and growth factors.
In addition to regulating the dynamics of focal adhesion complexes, STAT3 activity has also been associated with alterations in the architecture of the microtubule network, which plays a major role in mediating cell locomotion and focal adhesion turnover.75 Interestingly, more recent models of microtubule function suggest these filamentous proteins are not required for cell movement per se, but instead specify directional cell movements by restraining and remodeling cell morphologies necessary for motility.75,76 Indeed, the formation of stable microtubules at the leading edge of migrating cells inhibits lamellipodial retraction, which results in forward cell protrusion. Moreover, dynamic microtubules located at the trailing edge of the cell are simultaneously remodeled, thereby enabling contractile forces to retract the tail of the cell.75 This balance of microtubule stability between the leading and trailing edges of a cell is essential in directing enhanced cell motility associated with acquisition of a mesenchymal phenotypes. Hence, microtubule remodeling may represent an important step in polarizing post-EMT cells and enabling their escape from the primary tumor. The ability of STAT3 to regulate the microtubule architecture stems from its interaction with the microtubule destabilizing protein stathmin,77 which is a ubiquitously expressed cytosolic phospho-protein that depolymerizes microtubules via binding to microtubule α/β heterodimers. Interestingly, STAT3 interacts directly with stathmin and binds to its C-terminal tubulin-interacting domain, thereby inactivating its ability to destabilize microtubules.77 Whether this nongenomic function of STAT3 is regulated by its phosphorylation status remains controversial.77,78 However, we demonstrated that the pharmacological inhibition of STAT3 phosphorylation and activation in breast cancer cells significantly reduced their formation of filopodia, which are essential actin-rich protrusions that participate in directed cell movement.59,79 Collectively, these findings suggest that STAT3 may play an important role in establishing cell polarity during directed cell migration, processes essential for EMT programs and carcinoma metastasis.
Finally, a metabolic hallmark exhibited by carcinoma cells is reflected in the “Warburg effect”, which refers to their preferential production of ATP through glycolysis as opposed to oxidative phosphorylation.80 Interestingly, hypoxia and its induced expression of hypoxia-inducible factor (HIF)-1α not only promotes tumor angiogenesis, but also functions in (1) linking EMT programs to the acquisition of chemoresistant and stem cell-like phenotypes; and (2) driving the energetic shift away from oxidative phosphorylation to exacerbated glycolysis.81,82 Recently, constitutive STAT3 activity and its stimulation of HIF-1α expression was observed to elicit aerobic glycolysis in human breast cancer cells. Importantly, the growth and glucose uptake of developing mammary tumors was severely compromised in mice treated with the STAT3 inhibitor S3I-201,83 which negated HIF-1α expression and its induction of the noncanonical STAT3 activator, pyruvate kinase M2.84,85 Additionally, STAT3 enhances carcinoma cell survival during metastasis by inhibiting the expression of components of the electron transport chain, thereby decreasing mitochondrial respiration and the production of reactive oxygen species.85 Taken together, these findings highlight novel nongenomic STAT3 activities that figure prominently in driving carcinoma development and metastatic progression.
Other STAT Proteins
Recently, the role of other STAT proteins besides STAT3 have also been investigated to determine their contributions to the progression and prognosis of carcinomas, as well as to assess their function in EMT programs.86 Indeed, STAT5 expression has been linked to favorable prognoses for patients afflicted with breast or nasopharyngeal cancers, suggesting that STAT5 may function to inhibit tumorigenesis.87 Accordingly, breast cancer patients whose tumors exhibit nuclear localization of STAT5 have decreased risks for disease recurrence and mortality as compared with those patients whose tumors lack detectable STAT5 in the nucleus.88 Moreover, constitutive STAT5 activation associates predominantly with less aggressive and highly differentiated ER-α-positive breast cancers as compared with their aggressive and poorly differentiated ER-α-negative counterparts that are addicted to constitutive STAT3 activity and its coupling to the expansion of CSCs.33,89 Interestingly, STAT5 and STAT3 are co-activated in approximately 30% of breast cancers, which tend to be low grade ER-α-positive tumors that exhibit good clinical outcomes. Moreover, breast cancer patients whose tumors house STAT5 gene expression signatures exhibit better overall survival as compared with patients whose tumors only house STAT3 gene expression signatures,89 suggesting that STAT5 activation supersedes and inhibits the oncogenic activities of STAT3 in breast cancers. Indeed, although STAT5 and STAT3 are capable of binding to identical regions within the Bcl6 promoter, only STAT5 occupancy was observed to strongly repress RNA polymerase II binding and Bcl6 transcription, which contrasted sharply with the robust recruitment of RNA polymerase II and induction of Bcl6 expression mediated by STAT3 occupancy. Importantly, simultaneous activation of both STAT proteins in breast cancer cells produced phenotypes reminiscent of STAT5 activation due to its ability to displace STAT3 from the Bcl6 promoter,90 thereby providing a partial mechanistic explanation for how STAT5 activity opposes that of STAT3 in mammary tumors. In stark contrast to breast cancers, STAT5 has been observed to enhance the proliferative and EMT phenotypes of head and neck cancers,91 and of some hematopoietic cancers.92 Along these lines, STAT1 expression is generally associated with favorable outcomes in cancer patients due to its ability to suppress tumor formation93-95 by (1) increasing apoptosis and antitumor immunity,48 and (2) inhibiting cell cycle progression and tumor angiogenesis.96 However, STAT1 fulfills a tumor-promoting role in liquid tumors by enhancing their generation of CSCs as evidenced by the fact that STAT1-deficiency protects against leukemia development by limiting CSC expansion.96-98 Taken together, these findings demonstrate that the biological consequences of aberrant expression and activation of STAT1 and STAT5 are governed in a cell- and context-specific manner, thereby making it difficult to draw more general conclusions about their functions in human malignancies. Moreover, and similar to STAT3, both STAT1 and STAT5 are subject to alternative splicing, events that may also contribute to their dichotomous activities in various human cancers.99,100 As such, future studies need to more thoroughly interrogate the functions of STAT1 and STAT5 variants in developing and progressing carcinomas, as well as in their acquisition of EMT phenotypes.
Concluding Remarks
At present, the reciprocal roles played by STAT3 in regulating EMT programs and, conversely, of EMT programs in regulating STAT3 expression and activity during carcinoma progression and metastasis remain to be fully delineated. Despite these uncertainties, it is nevertheless clear that STAT3 modulates the expression of “master” EMT transcriptional factors operant in integrating signals from multiple extracellular stimuli that influence EMT phenotypes. In addition to its role as a transcription factor, STAT3 also mediates nongenomic effects on the cytoskeleton during the initiation of EMT programs. As our understanding of EMT programs and their role in facilitating distinct aspects of the metastatic cascade increases, it will be necessary to delineate the precise functions played by STAT3 during tumor evolution, particularly with respect to how disseminated carcinoma cells seed and survive within foreign tissues and metastatic niches. Answering these essential questions will enhance the potential effectiveness of STAT3 inhibitors to abrogate carcinoma development and metastasis in cancer patients.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Carlin and Schiemann laboratories for their helpful comments and suggestions. Research support was provided in part by the National Institutes of Health to CRC (GM081498), to W.P.S. (CA129359 and CA177069), to M.K.W. (CA166140), and to N.B. (T32 HL007653).
Glossary
Abbreviations:
- ADIPOR1
adiponectin receptor 1
- CSC
cancer stem cell
- ECM
extracellular matrix
- EGF
epidermal growth factor
- EGFR
EGF receptor
- EMP
epithelial-mesenchymal plasticity
- EMT
epithelial-mesenchymal transition
- FAK
focal adhesion kinase
- FN
fibronectin
- HIF-1α
hypoxia-inducible factor-1α
- IL-6
interleukin-6
- IL-6R
IL-6 receptor
- IRAK1
interleukin-1 receptor-associated kinase 1
- JAK
Janus kinase
- LIFR
LIF receptor
- miR
microRNA
- NFκB
nuclear factor-κB
- NMuMG
normal murine mammary gland
- OSM
Oncostatin M
- OSMR
OSM receptor
- PIAS3
protein inhibitor of activated signal transducer and activator of transcription 3
- S1P
sphingosine-1-phosphate
- S1PR1
S1P receptor 1
- SH2
Src-homology domain 2
- SK1
sphingosine kinase-1
- SOCS
suppressor of cytokine signaling
- STAT3
signal transducer and activator of transcription 3
- STAT5
signal transducer and activator of transcription 5
- TRAF6
TNF receptor-associated factor 6
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/28975
References
- 1.Zhong Z, Wen Z, Darnell JE., Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–8. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 2.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/S0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 3.Darnell JE, Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 4.Stark GR, Darnell JE., Jr. The JAK-STAT pathway at twenty. Immunity. 2012;36:503–14. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torres-Roca JF, DeSilvio M, Mora LB, Khor LY, Hammond E, Ahmad N, Jove R, Forman J, Lee RJ, Sandler H, et al. Activated STAT3 as a correlate of distant metastasis in prostate cancer: a secondary analysis of Radiation Therapy Oncology Group 86-10. Urology. 2007;69:505–9. doi: 10.1016/j.urology.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Sonnenblick A, Shriki A, Galun E, Axelrod JH, Daum H, Rottenberg Y, Hamburger T, Mali B, Peretz T. Tissue microarray-based study of patients with lymph node-positive breast cancer shows tyrosine phosphorylation of signal transducer and activator of transcription 3 (tyrosine705-STAT3) is a marker of good prognosis. Clin Transl Oncol. 2012;14:232–6. doi: 10.1007/s12094-012-0789-z. [DOI] [PubMed] [Google Scholar]
- 7.Dolled-Filhart M, Camp RL, Kowalski DP, Smith BL, Rimm DL. Tissue microarray analysis of signal transducers and activators of transcription 3 (Stat3) and phospho-Stat3 (Tyr705) in node-negative breast cancer shows nuclear localization is associated with a better prognosis. Clin Cancer Res. 2003;9:594–600. [PubMed] [Google Scholar]
- 8.Zammarchi F, de Stanchina E, Bournazou E, Supakorndej T, Martires K, Riedel E, Corben AD, Bromberg JF, Cartegni L. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc Natl Acad Sci U S A. 2011;108:17779–84. doi: 10.1073/pnas.1108482108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caldenhoven E, van Dijk TB, Solari R, Armstrong J, Raaijmakers JA, Lammers JW, Koenderman L, de Groot RP. STAT3β, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J Biol Chem. 1996;271:13221–7. doi: 10.1074/jbc.271.22.13221. [DOI] [PubMed] [Google Scholar]
- 10.Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15:117–34. doi: 10.1007/s10911-010-9178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiery JP, Lim CT. Tumor dissemination: an EMT affair. Cancer Cell. 2013;23:272–3. doi: 10.1016/j.ccr.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinto CA, Widodo E, Waltham M, Thompson EW. Breast cancer stem cells and epithelial mesenchymal plasticity - Implications for chemoresistance. Cancer Lett. 2013;341:56–62. doi: 10.1016/j.canlet.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Gao D, Vahdat LT, Wong S, Chang JC, Mittal V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res. 2012;72:4883–9. doi: 10.1158/0008-5472.CAN-12-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-β in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15:169–90. doi: 10.1007/s10911-010-9181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wendt MK, Allington TM, Schiemann WP. Mechanisms of the epithelial-mesenchymal transition by TGF-β. Future Oncol. 2009;5:1145–68. doi: 10.2217/fon.09.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foubert E, De Craene B, Berx G. Key signalling nodes in mammary gland development and cancer. The Snail1-Twist1 conspiracy in malignant breast cancer progression. Breast Cancer Res. 2010;12:206. doi: 10.1186/bcr2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leptin M. twist and snail as positive and negative regulators during Drosophila mesoderm development. Genes Dev. 1991;5:1568–76. doi: 10.1101/gad.5.9.1568. [DOI] [PubMed] [Google Scholar]
- 19.Simpson P. Maternal-Zygotic Gene interactions during formation of the dorsoventral pattern in Drosophila embryos. Genetics. 1983;105:615–32. doi: 10.1093/genetics/105.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 21.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 22.Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011;71:245–54. doi: 10.1158/0008-5472.CAN-10-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartwell KA, Muir B, Reinhardt F, Carpenter AE, Sgroi DC, Weinberg RA. The Spemann organizer gene, Goosecoid, promotes tumor metastasis. Proc Natl Acad Sci U S A. 2006;103:18969–74. doi: 10.1073/pnas.0608636103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, Kutok JL, Hartwell K, Richardson AL, Weinberg RA. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A. 2007;104:10069–74. doi: 10.1073/pnas.0703900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P, et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene. 2007;26:6979–88. doi: 10.1038/sj.onc.1210508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–78. doi: 10.1016/S1097-2765(01)00260-X. [DOI] [PubMed] [Google Scholar]
- 27.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107:15449–54. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, Kashiwamura S, Nakajima K, Koyama K, Iwamatsu A, et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature. 1986;324:73–6. doi: 10.1038/324073a0. [DOI] [PubMed] [Google Scholar]
- 29.Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012;38:904–10. doi: 10.1016/j.ctrv.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 30.Xie G, Yao Q, Liu Y, Du S, Liu A, Guo Z, Sun A, Ruan J, Chen L, Ye C, et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int J Oncol. 2012;40:1171–9. doi: 10.3892/ijo.2011.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM, Hall BM. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28:2940–7. doi: 10.1038/onc.2009.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res. 2011;9:1658–67. doi: 10.1158/1541-7786.MCR-11-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44⁺CD24⁻ stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723–35. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol. 2013;15:338–44. doi: 10.1038/ncb2717. [DOI] [PubMed] [Google Scholar]
- 35.Shiota M, Bishop JL, Nip KM, Zardan A, Takeuchi A, Cordonnier T, Beraldi E, Bazov J, Fazli L, Chi K, et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013;73:3109–19. doi: 10.1158/0008-5472.CAN-12-3979. [DOI] [PubMed] [Google Scholar]
- 36.West NR, Murray JI, Watson PH. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene. 2014;33:1485–94. doi: 10.1038/onc.2013.105. [DOI] [PubMed] [Google Scholar]
- 37.Guo L, Chen C, Shi M, Wang F, Chen X, Diao D, Hu M, Yu M, Qian L, Guo N. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene. 2013;32:5272–82. doi: 10.1038/onc.2012.573. [DOI] [PubMed] [Google Scholar]
- 38.Park J, Schwarzbauer JE. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene. 2014;33:1649–57. doi: 10.1038/onc.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang F, Li C, Halfter H, Liu J. Delineating an oncostatin M-activated STAT3 signaling pathway that coordinates the expression of genes involved in cell cycle regulation and extracellular matrix deposition of MCF-7 cells. Oncogene. 2003;22:894–905. doi: 10.1038/sj.onc.1206158. [DOI] [PubMed] [Google Scholar]
- 40.Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, Morris M, Wyatt L, Farshid G, Lim YY, et al. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22:1686–98. doi: 10.1091/mbc.E11-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peter ME. Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle. 2009;8:843–52. doi: 10.4161/cc.8.6.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarközi R, Hauser C, Noppert SJ, Kronbichler A, Pirklbauer M, Haller VM, Grillari J, Grillari-Voglauer R, Mayer G, Schramek H. Oncostatin M is a novel inhibitor of TGF-β1-induced matricellular protein expression. Am J Physiol Renal Physiol. 2011;301:F1014–25. doi: 10.1152/ajprenal.00123.2011. [DOI] [PubMed] [Google Scholar]
- 43.Kan CE, Cipriano R, Jackson MW. c-MYC functions as a molecular switch to alter the response of human mammary epithelial cells to oncostatin M. Cancer Res. 2011;71:6930–9. doi: 10.1158/0008-5472.CAN-10-3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shao H, Cheng HY, Cook RG, Tweardy DJ. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 2003;63:3923–30. [PubMed] [Google Scholar]
- 45.Boerner JL, Biscardi JS, Silva CM, Parsons SJ. Transactivating agonists of the EGF receptor require Tyr 845 phosphorylation for induction of DNA synthesis. Mol Carcinog. 2005;44:262–73. doi: 10.1002/mc.20138. [DOI] [PubMed] [Google Scholar]
- 46.Lu Z, Ghosh S, Wang Z, Hunter T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of β-catenin, and enhanced tumor cell invasion. Cancer Cell. 2003;4:499–515. doi: 10.1016/S1535-6108(03)00304-0. [DOI] [PubMed] [Google Scholar]
- 47.Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67:9066–76. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chin YE, Kitagawa M, Kuida K, Flavell RA, Fu XY. Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol Cell Biol. 1997;17:5328–37. doi: 10.1128/mcb.17.9.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Armstrong DK, Kaufmann SH, Ottaviano YL, Furuya Y, Buckley JA, Isaacs JT, Davidson NE. Epidermal growth factor-mediated apoptosis of MDA-MB-468 human breast cancer cells. Cancer Res. 1994;54:5280–3. [PubMed] [Google Scholar]
- 50.Rosen DG, Mercado-Uribe I, Yang G, Bast RC, Jr., Amin HM, Lai R, Liu J. The role of constitutively active signal transducer and activator of transcription 3 in ovarian tumorigenesis and prognosis. Cancer. 2006;107:2730–40. doi: 10.1002/cncr.22293. [DOI] [PubMed] [Google Scholar]
- 51.Puiffe ML, Le Page C, Filali-Mouhim A, Zietarska M, Ouellet V, Tonin PN, Chevrette M, Provencher DM, Mes-Masson AM. Characterization of ovarian cancer ascites on cell invasion, proliferation, spheroid formation, and gene expression in an in vitro model of epithelial ovarian cancer. Neoplasia. 2007;9:820–9. doi: 10.1593/neo.07472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colomiere M, Ward AC, Riley C, Trenerry MK, Cameron-Smith D, Findlay J, Ackland L, Ahmed N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br J Cancer. 2009;100:134–44. doi: 10.1038/sj.bjc.6604794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davis FM, Azimi I, Faville RA, Peters AA, Jalink K, Putney JW, Jr., Goodhill GJ, Thompson EW, Roberts-Thomson SJ, Monteith GR. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene. 2013 doi: 10.1038/onc.2013.187. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perkins ND. The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer. 2012;12:121–32. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 55.Theiss AL. Sphingosine-1-phosphate: Driver of NFκB and STAT3 persistent activation in chronic intestinal inflammation and colitis-associated cancer. JAKSTAT. 2013;2:e24150. doi: 10.4161/jkst.24150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, Forman S, Jove R, Pardoll DM, Yu H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–93. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, Kortylewski M, Horne D, Somlo G, Forman S, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16:1421–8. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23:107–20. doi: 10.1016/j.ccr.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Balanis N, Wendt MK, Schiemann BJ, Wang Z, Schiemann WP, Carlin CR. Epithelial to mesenchymal transition promotes breast cancer progression via a fibronectin-dependent STAT3 signaling pathway. J Biol Chem. 2013;288:17954–67. doi: 10.1074/jbc.M113.475277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Balanis N, Yoshigi M, Wendt MK, Schiemann WP, Carlin CR. β3 integrin-EGF receptor cross-talk activates p190RhoGAP in mouse mammary gland epithelial cells. Mol Biol Cell. 2011;22:4288–301. doi: 10.1091/mbc.E10-08-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wendt MK, Smith JA, Schiemann WP. Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene. 2010;29:6485–98. doi: 10.1038/onc.2010.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wendt MK, Taylor MA, Schiemann BJ, Sossey-Alaoui K, Schiemann WP. Fibroblast growth factor receptor splice variants are stable markers of oncogenic transforming growth factor- β1 signaling in metastatic breast cancers. Breast Cancer Res. 2014;16:R24. doi: 10.1186/bcr3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hwang MS, Yu N, Stinson SY, Yue P, Newman RJ, Allan BB, Dornan D. miR-221/222 targets adiponectin receptor 1 to promote the epithelial-to-mesenchymal transition in breast cancer. PLoS One. 2013;8:e66502. doi: 10.1371/journal.pone.0066502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu W, Takanashi M, Borjigin N, Ohno SI, Fujita K, Hoshino S, Osaka Y, Tsuchida A, Kuroda M. MicroRNA-18a modulates STAT3 activity through negative regulation of PIAS3 during gastric adenocarcinogenesis. Br J Cancer. 2013;108:653–61. doi: 10.1038/bjc.2012.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiang M, Birkbak NJ, Vafaizadeh V, Walker SR, Yeh JE, Liu S, Kroll Y, Boldin M, Taganov K, Groner B, et al. STAT3 induction of miR-146b forms a feedback loop to inhibit the NF-κB to IL-6 signaling axis and STAT3-driven cancer phenotypes. Sci Signal. 2014;7:ra11. doi: 10.1126/scisignal.2004497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–76. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- 67.Inagaki-Ohara K, Kondo T, Ito M, Yoshimura A. SOCS, inflammation, and cancer. JAKSTAT. 2013;2:e24053. doi: 10.4161/jkst.24053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ogata H, Kobayashi T, Chinen T, Takaki H, Sanada T, Minoda Y, Koga K, Takaesu G, Maehara Y, Iida M, et al. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology. 2006;131:179–93. doi: 10.1053/j.gastro.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 69.Tischoff I, Hengge UR, Vieth M, Ell C, Stolte M, Weber A, Schmidt WE, Tannapfel A. Methylation of SOCS-3 and SOCS-1 in the carcinogenesis of Barrett’s adenocarcinoma. Gut. 2007;56:1047–53. doi: 10.1136/gut.2006.111633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, Kitagawa T, Yamamoto J, Kubo T, Yoshikawa H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene. 2005;24:6406–17. doi: 10.1038/sj.onc.1208788. [DOI] [PubMed] [Google Scholar]
- 71.Ji YY, Wang ZD, Li ZF, Li K. Interference of suppressor of cytokine signaling 3 promotes epithelial-mesenchymal transition in MHCC97H cells. World J Gastroenterol. 2013;19:866–73. doi: 10.3748/wjg.v19.i6.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Y, van Boxel-Dezaire AH, Cheon H, Yang J, Stark GR. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc Natl Acad Sci U S A. 2013;110:16975–80. doi: 10.1073/pnas.1315862110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Silver DL, Naora H, Liu J, Cheng W, Montell DJ. Activated signal transducer and activator of transcription (STAT) 3: localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004;64:3550–8. doi: 10.1158/0008-5472.CAN-03-3959. [DOI] [PubMed] [Google Scholar]
- 74.Kira M, Sano S, Takagi S, Yoshikawa K, Takeda J, Itami S. STAT3 deficiency in keratinocytes leads to compromised cell migration through hyperphosphorylation of p130(cas) J Biol Chem. 2002;277:12931–6. doi: 10.1074/jbc.M110795200. [DOI] [PubMed] [Google Scholar]
- 75.Ganguly A, Yang H, Sharma R, Patel KD, Cabral F. The role of microtubules and their dynamics in cell migration. J Biol Chem. 2012;287:43359–69. doi: 10.1074/jbc.M112.423905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaverina I, Straube A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 2011;22:968–74. doi: 10.1016/j.semcdb.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ng DC, Lin BH, Lim CP, Huang G, Zhang T, Poli V, Cao X. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J Cell Biol. 2006;172:245–57. doi: 10.1083/jcb.200503021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Verma NK, Dourlat J, Davies AM, Long A, Liu WQ, Garbay C, Kelleher D, Volkov Y. STAT3-stathmin interactions control microtubule dynamics in migrating T-cells. J Biol Chem. 2009;284:12349–62. doi: 10.1074/jbc.M807761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schober JM, Komarova YA, Chaga OY, Akhmanova A, Borisy GG. Microtubule-targeting-dependent reorganization of filopodia. J Cell Sci. 2007;120:1235–44. doi: 10.1242/jcs.003913. [DOI] [PubMed] [Google Scholar]
- 80.Cairns RA, Harris I, McCracken S, Mak TW. Cancer cell metabolism. Cold Spring Harb Symp Quant Biol. 2011;76:299–311. doi: 10.1101/sqb.2011.76.012856. [DOI] [PubMed] [Google Scholar]
- 81.Jiang J, Tang YL, Liang XH. EMT: a new vision of hypoxia promoting cancer progression. Cancer Biol Ther. 2011;11:714–23. doi: 10.4161/cbt.11.8.15274. [DOI] [PubMed] [Google Scholar]
- 82.Brahimi-Horn MC, Bellot G, Pouysségur J. Hypoxia and energetic tumour metabolism. Curr Opin Genet Dev. 2011;21:67–72. doi: 10.1016/j.gde.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 83.Demaria M, Poli V. PKM2, STAT3 and HIF-1α: The Warburg’s vicious circle. JAKSTAT. 2012;1:194–6. doi: 10.4161/jkst.20662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Demaria M, Camporeale A, Poli V. STAT3 and metabolism: How many ways to use a single molecule? Int J Cancer. 2014 doi: 10.1002/ijc.28767. Forthcoming. [DOI] [PubMed] [Google Scholar]
- 86.Yamashita H, Nishio M, Ando Y, Zhang Z, Hamaguchi M, Mita K, Kobayashi S, Fujii Y, Iwase H. Stat5 expression predicts response to endocrine therapy and improves survival in estrogen receptor-positive breast cancer. Endocr Relat Cancer. 2006;13:885–93. doi: 10.1677/erc.1.01095. [DOI] [PubMed] [Google Scholar]
- 87.Hsiao JR, Jin YT, Tsai ST, Shiau AL, Wu CL, Su WC. Constitutive activation of STAT3 and STAT5 is present in the majority of nasopharyngeal carcinoma and correlates with better prognosis. Br J Cancer. 2003;89:344–9. doi: 10.1038/sj.bjc.6601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peck AR, Witkiewicz AK, Liu C, Stringer GA, Klimowicz AC, Pequignot E, Freydin B, Tran TH, Yang N, Rosenberg AL, et al. Loss of nuclear localized and tyrosine phosphorylated Stat5 in breast cancer predicts poor clinical outcome and increased risk of antiestrogen therapy failure. J Clin Oncol. 2011;29:2448–58. doi: 10.1200/JCO.2010.30.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Walker SR, Xiang M, Frank DA. Distinct roles of STAT3 and STAT5 in the pathogenesis and targeted therapy of breast cancer. Mol Cell Endocrinol. 2014;382:616–21. doi: 10.1016/j.mce.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Walker SR, Nelson EA, Yeh JE, Pinello L, Yuan GC, Frank DA. STAT5 outcompetes STAT3 to regulate the expression of the oncogenic transcriptional modulator BCL6. Mol Cell Biol. 2013;33:2879–90. doi: 10.1128/MCB.01620-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koppikar P, Lui VW, Man D, Xi S, Chai RL, Nelson E, Tobey AB, Grandis JR. Constitutive activation of signal transducer and activator of transcription 5 contributes to tumor growth, epithelial-mesenchymal transition, and resistance to epidermal growth factor receptor targeting. Clin Cancer Res. 2008;14:7682–90. doi: 10.1158/1078-0432.CCR-08-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Moriggl R, Sexl V, Kenner L, Duntsch C, Stangl K, Gingras S, Hoffmeyer A, Bauer A, Piekorz R, Wang D, et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell. 2005;7:87–99. doi: 10.1016/j.ccr.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 93.Simpson JA, Al-Attar A, Watson NF, Scholefield JH, Ilyas M, Durrant LG. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut. 2010;59:926–33. doi: 10.1136/gut.2009.194472. [DOI] [PubMed] [Google Scholar]
- 94.Chen HY, Yu SL, Chen CH, Chang GC, Chen CY, Yuan A, Cheng CL, Wang CH, Terng HJ, Kao SF, et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. N Engl J Med. 2007;356:11–20. doi: 10.1056/NEJMoa060096. [DOI] [PubMed] [Google Scholar]
- 95.Stephanou A, Latchman DS. STAT-1: a novel regulator of apoptosis. Int J Exp Pathol. 2003;84:239–44. doi: 10.1111/j.0959-9673.2003.00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Avalle L, Pensa S, Regis G, Novelli F, Poli V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT. 2012;1:65–72. doi: 10.4161/jkst.20045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kovacic B, Stoiber D, Moriggl R, Weisz E, Ott RG, Kreibich R, Levy DE, Beug H, Freissmuth M, Sexl V. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell. 2006;10:77–87. doi: 10.1016/j.ccr.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 98.Heuser M, Sly LM, Argiropoulos B, Kuchenbauer F, Lai C, Weng A, Leung M, Lin G, Brookes C, Fung S, et al. Modeling the functional heterogeneity of leukemia stem cells: role of STAT5 in leukemia stem cell self-renewal. Blood. 2009;114:3983–93. doi: 10.1182/blood-2009-06-227603. [DOI] [PubMed] [Google Scholar]
- 99.Kazansky AV, Raught B, Lindsey SM, Wang YF, Rosen JM. Regulation of mammary gland factor/Stat5a during mammary gland development. Mol Endocrinol. 1995;9:1598–609. doi: 10.1210/mend.9.11.8584036. [DOI] [PubMed] [Google Scholar]
- 100.Ning Q, Berger L, Luo X, Yan W, Gong F, Dennis J, Levy G. STAT1 and STAT3 α/β splice form activation predicts host responses in mouse hepatitis virus type 3 infection. J Med Virol. 2003;69:306–12. doi: 10.1002/jmv.10290. [DOI] [PubMed] [Google Scholar]