

Abstract
Plasma membrane expression of G protein-coupled receptors (GPCRs) is a dynamic process balancing anterograde and retrograde trafficking. Multiple interrelated cellular processes determine the final level of cell surface expression, including endoplasmic reticulum (ER) export/retention, receptor internalization, recycling, and degradation. These processes are highly regulated to achieve specific localization to subcellular domains (e.g., dendrites or basolateral membranes) and to affect receptor signaling. Analysis of potential ER trafficking motifs within GPCRs requires careful consideration of intracellular dynamics, such as protein folding, ER export and retention, and glycosylation. This chapter presents an approach and methods for qualitative and quantitative assessment of these processes to aid in accurate identification of GPCR trafficking motifs, utilizing the analysis of a hydrophobic extracellular trafficking motif in α2C adrenergic receptors as a model system.
1. INTRODUCTION
G protein-coupled receptors (GPCRs) are synthesized and exported along the secretory pathway from the endoplasmic reticulum (ER) to the plasma membrane by a complex process involving multiple mechanisms to insure proper folding, assembly, quality control, selective retention, and transport (Ellgaard & Helenius, 2003). GPCRs are synthesized in the ER, where they are folded, assembled, and then packaged into ER-derived COPII transport vesicles that traverse the Golgi network for glycolytic processing and eventual transport to their membrane localizations (Duvernay, Filipeanu, & Wu, 2005). The resident time within the ER is dependent upon several factors including folding rates and assembly, which may be modified by pharmacological and/or protein chaperones (Brothers, Janovick, & Conn, 2006; Petaja-Repo et al., 2002), as well as specific sequences within the GPCR that dictate ER or Golgi export or retention. Several such trafficking motifs have been identified in various GPCRs, including variations of a carboxy-terminal F(X)6LL ER export motif (Duvernay et al., 2009; Schulein et al., 1998). Additionally, several arginine-rich ER retention motifs have been identified, including RSRR and RXR motifs in GABAB R1 and vasopressin type 2 receptors, respectively (Hermosilla & Schulein, 2001; Pagano et al., 2001).
Traditional identification of ER trafficking motifs is often done by mutational analysis and expression; however, interpretation can be complex. For example, mutation or removal of a suspected GPCR ER export motif should lead to ER retention; however, a mutant GPCR that is misfolded could also lead to ER retention. Alternatively, mutation of an ER retention motif should lead to ER export, with an increase in the ratio of plasma membrane/total cellular expression. However, a mutant GPCR with an enhanced intracellular half-life will appear to have increased plasma membrane expression due to increased total cellular receptor expression, but the plasma membrane/total cellular expression ratio may not change. Thus, to ensure proper identification of ER trafficking motifs, a complete analysis must be undertaken to ensure proper interpretation of the data. Three concerns should be addressed prior to identification of a GPCR ER trafficking motif:
Removal or mutation of the motif does not affect GPCR function (i.e., it is not misfolded).
Alterations in trafficking lead to changes in intracellular processing (e.g., glycosylation).
Changes in plasma membrane expression are specific and do not simply reflect changes in total cellular expression only.
α2A and α2C adrenergic receptors (ARs) are highly homologous receptors that target to different neuronal sites (extra- vs. presynaptic, respectively), as well as demonstrate cell-type specific trafficking (Angelotti, Daunt, Shcherbakova, Kobilka, & Hurt, 2010; Brum, Hurt, Shcherbakova, Kobilka, & Angelotti, 2006). Utilizing α2A and α2C ARs as model GPCRs, we describe a systematic approach to identifying GPCR trafficking motifs, culminating in a quantitative membrane expression assay. Using this multiassay approach, we have identified a new extracellular hydrophobic trafficking domain within the amino terminus of α2C ARs (ALAAALAAAAA). These methods are optimized for analysis of ER trafficking; however, they can be utilized to study Golgi or other trafficking motifs.
2. MOTIF SCREENING BY IMMUNOFLUORESCENT STAINING
2.1. Epitope tagging of GPCRs
The majority of methods presented in this chapter require a GPCR-specific antibody for immunofluorescence, immunoblot, and flow-activated cell sorting (FACS) analysis. If a specific antiserum against an extracellular epitope does not exist for the native receptor, it can be tagged at the amino terminus with any one of several epitopes (e.g., FLAG, hemagglutinin, or myc) using standard molecular biological techniques. Epitope-tagged GPCRs need to be tested to ensure no alteration in functionality or biochemical processing; a more detailed methodology can be found elsewhere (Wozniak, Saunders, Schramm, Keefer, & Limbird, 2002).
2.2. Identification of trafficking motifs
Potential trafficking motifs can be identified by any one of several methods, including sequence analysis with known consensus trafficking motifs from other proteins, analysis of convergent and divergent protein sequences between homologous receptors, or analysis based on crystal structure. A comparison of α2A&C ARs reveals that the most divergent sequences are found within the third intracellular loop (iC3) between transmembrane domains V and VI and the amino- and carboxy-terminal regions. Using standard molecular biological methods, these regions were swapped between α2A and α2C ARs (Angelotti et al., 2010) to create a series of chimeric α2A/C ARs for rapid screening of trafficking motifs. Once a region/motif of interest is identified, specific mutations can be constructed within this region prior to further characterization. Thus, this process requires iterative steps of rapid immunofluorescent screening and functional assays to hone in on a specific motif followed by a more complete analysis of membrane trafficking.
2.3. Rapid immunofluorescent screening
Immunofluorescent microscopy is a rapid method to screen for regions or motifs involved in GPCR trafficking and to analyze chimeric and/or mutant receptors with respect to the relative distribution of surface and total receptor expression (Kallal & Benovic, 2002). For example, heterologously expressed α2A ARs demonstrate over 90% plasma membrane expression in nonneuronal cell lines, whereas α2C ARs are predominantly localized to an intracellular site (ER), with only 25% plasma membrane expression (Angelotti et al., 2010; Hurt, Feng, & Kobilka, 2000). Therefore, HA-tagged α2A&C AR chimeras were screened to identify regions involved with this trafficking disparity.
Plate HEK293 cells onto 15×15-mm poly-D-lysine (70 kDa, Sigma Chemical)-coated glass coverslips placed into 12-well cell culture plates with DMEM+10% FBS. Plate approximately 100–150,000 cells per well 24 h prior to transfection.
Transfect cells with epitope-tagged GPCR chimeras or mutants using Effectene or other transfection reagent (per manufacturer’s recommendations) and maintain in culture for 48 h prior to staining.
Rinse cells three times with room temperature (RT) PBS and then fix with 4% paraformaldehyde (PFA) in PBS for 5 min at RT. Wash fixed cells three times with PBS at 5-min intervals.
For total cellular immunofluorescent staining, permeabilize and block fixed cells at RT for 30 min with blocking solution (40 mM HEPES, pH 7.4, 5%dry milk, 2%goat serum (Gemini Bio Products), 0.2%Non-idet P-40 in PBS). For surface immunofluorescent staining, use blocking solution without Nonidet P-40.
Label epitope-tagged GPCR chimeras using primary mouse monoclonal antibody (HA epitope=16B12 [Covance]) at a 1:500 dilution in appropriate blocking solution (with or without NP-40) for 1 h at RT. To counterstain ER or Golgi compartments, add primary antibodies against calreticulin or giantin (respectively). Appropriate dilutions for all antibodies should be determined empirically based upon manufacturer’s recommendations.
Rinse cells four to five times at RT with PBS over 15 min and reapply the appropriate blocking solution for 30 min at RT.
Apply fluorescent-conjugated secondary antibodies in appropriate blocking solution for 1 h at RT (e.g., goat antimouse Alexa Fluor 596, Invitrogen). Dilutions should be based upon manufacturer’s recommendations and empiric determination.
Rinse cells four to five times with PBS over 15 min at RT. Mount glass coverslips cell-side down onto glass slides using Vectashield (Vector Labs). Allow to air dry at RT in a dark area.
Examine and photograph stained cells using a fluorescent microscope with appropriate filter sets for the secondary fluorescent antisera utilized. A magnification over 500× should be used.
2.4. Interpretation of total versus surface immunofluorescent staining
Rapid visual screening of total versus surface staining of wild-type (WT) and α2A&C AR chimeras will allow for recognition of potential trafficking motifs, based upon observation of altered surface/total expression. Once regions of interest are identified, sequence analysis within these regions can be utilized to refine possible trafficking motifs for further mutational analysis and screening (Fig. 9.1). However, further analysis is necessary to ensure that the increased or decreased immunofluorescent staining observed represents true alterations in trafficking.
Figure 9.1.
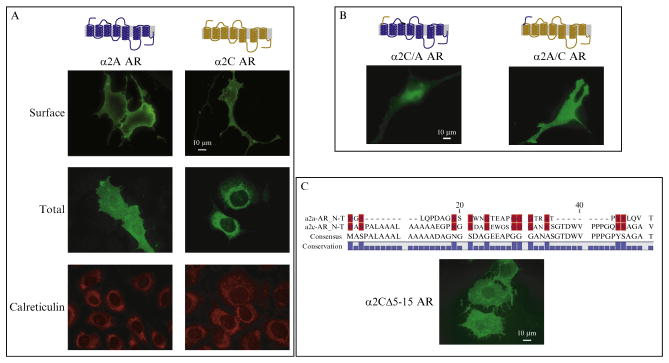
Immunofluorescent screening of α2A&C AR trafficking motifs. (A) Surface and total staining of WT HA-α2A&C ARs, co-stained with the ER marker calreticulin, demonstrate predominant intracellular (ER) localization of α2C, compared to α2A, ARs. α2A ARs are primarily found at the plasma membrane. (B) Total cellular staining of α2A/C AR chimeras demonstrates that the extracellular amino terminus of α2A and α2C ARs is a determinant of their relative expression patterns. (C) Removal of an α2C AR amino-terminal hydrophobic region (ALAAALAAAAA =α2CΔ5-15 AR) enhances cell surface expression, as assessed by total cellular staining, suggesting a possible role for this domain in α2C AR membrane expression. Adapted from Angelotti et al. (2010).
3. ANALYSIS OF RECEPTOR FUNCTIONALITY
3.1. Determination of ligand binding
As described above, a chimeric or mutant GPCR can lead to ER retention or alterations in plasma membrane expression due to misfolding or changes in protein half-life. To ensure that chimeric or mutated GPCRs are operative prior to further analysis of trafficking, their functionality needs to be examined. Ligand binding can aid in determining if a chimeric or mutant GPCR is folded properly. Membrane binding is preferable to whole cell binding to determine a complete pharmacological profile of binding affinities and expression levels (KD and Bmax, respectively).
3.2. Saturation ligand binding assays
Split a 15-cm tissue culture dish of confluent HEK293 cells 1:10 in DMEM with 10% FBS. Allow them to replicate at 37 °C until approximately 90% confluent.
Transfect selected WT, chimeric, or mutant GPCR constructs into a 15-cm dish of HEK293 cells.
After 48 h of growth, lyse cells in 3.0 ml lysis buffer (10 mM Tris, 1 mM EDTA, pH 7.4 with protease inhibitor cocktail [Roche]) and homogenize 15 times on ice using an appropriately sized dounce homogenizer. Centrifuge at 1000×g for 5 min to remove debris/nuclei and centrifuge the supernatant at 15,000×g for 30 min, both at 16 °C.
Resuspend resultant membrane pellet in 1 ml binding buffer (75 mM Tris, 12.5 mM MgCl2, 1 mM EDTA, pH 7.4 with protease inhibitor cocktail) using a 1-ml TB syringe and a 25-g needle on ice. Repeat steps 3 and 4.
Determine protein concentration using RC DC assay (Bio-Rad Laboratories) and freeze membranes at −70 °C at approximately 1 mg/ml, dilute with binding buffer as needed.
Perform saturation binding using an appropriate ligand, as described previously (Daunt et al., 1997); nonspecific binding is determined by adding excess cold ligand. For α2 AR analysis, [3H]RX-821002 (PerkinElmer) is recommended, with 100 μM yohimbine added to determine non-specific binding.
Calculate equilibrium dissociation constants and Bmax values from saturation isotherms and competition curves using GraphPAD software (San Diego, CA).
3.3. Analysis of receptor signaling
In addition to ligand binding, the ability of mutant or chimeric GPCRs to activate second messenger pathways should also be determined to ensure functionality. An immunofluorescent MAP kinase assay can be utilized for rapid screening of GPCRs that activate Gi/Go signaling pathways. For example, following addition of an α2 AR agonist, MAP kinase activation and phosphorylation of extracellular signal-regulated kinase 1 and 2 (ERK1/2) can be detected by specific phospho-ERK1/2 antisera, using an in cell immunofluorescent assay (Angelotti et al., 2010).
Plate, transfect, and grow Rat1 fibroblasts on poly-D-lysine-coated coverslips, as described above under Section 2.3. Other heterologous cell systems can be utilized.
Twenty-four hour after transfection with WT or chimeric GPCRs of interest, serum starve cells in DMEM with 4% bovine serum albumin (BSA) and 20 mM HEPES, pH 7.4 for 24 h at 37 °C.
Incubate cells in DMEM alone (control) or 1 nM dexmedetomidine (agonist) for 10 min at 37 °C in a tissue culture incubator. The appropriate agonist for the GPCR of interest should be chosen.
Immediately fix cells with 4% PFA for 5 min at RT.
Wash three times with PBS at 5-min intervals, followed by permeabilization and blocking with MAP kinase blocking solution (40 mM HEPES, pH 7.4, 3% BSA, 2% goat serum, 0.2% Nonidet P-40 in PBS-CM) for 30 min at RT.
Label epitope-tagged GPCR chimeras using primary mouse monoclonal antibody (HA epitope=16B12 [Covance]) at a 1:500 dilution in MAP kinase blocking solution for 1 h at RT. Detect MAP kinase activity by addition of phospho-ERK1/2 rabbit polyclonal antibody (Millipore) at a 1:1000 dilution. Appropriate dilutions for all antibodies should be determined empirically based upon manufacturer’s recommendations.
Rinse cells four to five times at RT with PBS over 15 min and reapply appropriate blocking solution for 30 min at RT.
Apply fluorescent-conjugated secondary antibodies (e.g., goat antimouse Alexa Fluor 596 and donkey anti-rabbit Alexa Fluor 488 antibodies [Invitrogen]) in appropriate blocking solution for 1 h at RT. Dilutions should be based upon manufacturer’s recommendations and empiric determination.
Rinse cells four to five times with PBS over 15 min at RT. Mount glass coverslips cell-side down onto glass slides using Vectashield (Vector Labs). Allow to air dry at RT in a dark area.
Examine and photograph stained cells using a fluorescent microscope with appropriate filter sets for the secondary fluorescent antisera utilized. A magnification over 500× should be used.
3.4. Interpretation of receptor functionality
Ligand binding and second messenger screening assays can be utilized to ensure that mutant or chimeric GPCRs are functional. The relative expression levels determined by binding may vary; however, ligand affinity should not be altered drastically. If ligand binding is drastically altered, consider using a different ligand (agonist vs. antagonist). In a similar manner, MAP kinase activation may not occur or be relevant to a specific GPCR, thus another second messenger assay should be considered (e.g., cyclase, phosphoinositol turnover). Prior to going forward with trafficking analysis, all mutant or chimeric GPCRs should retain ligand binding and second messenger activation to demonstrate functionality (Fig. 9.2).
Figure 9.2.
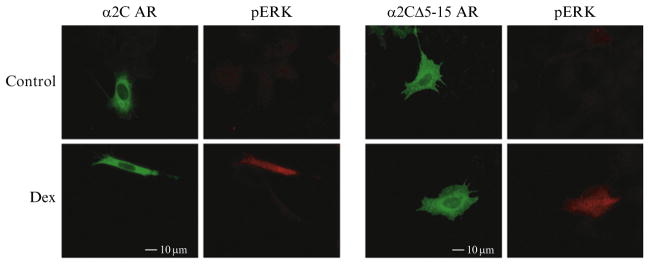
Functional screening of α2C AR mutations by MAP kinase. An immunofluorescent assay for MAP kinase (pERK) in response to agonist (Dex =dexmedetomidine) allows for screening of GPCR chimeras and mutations to ensure pharmacological function prior to further trafficking analysis. Permeabilized cells were stained with monoclonal antibody 16B12 to examine total cellular HA-α2 AR expression. Note minimal pERK staining in the absence of agonist. Both α2CΔ5-15 and α2CA7D ARs can activate MAP kinase and bind RX-821002 (data not shown) suggesting that they are not misfolded and are functional. Adapted from Angelotti et al. (2010).
4. BIOCHEMICAL ANALYSIS OF ER/GOLGI TRAFFICKING
4.1. GPCR glycosylation analysis
As GPCRs move through the secretory pathway from ER to Golgi, glycosidic processing/maturation occurs (Hurt et al., 2000). The glycosylation state can be determined by digestion of GPCR-containing membrane preparations with endoglycosidase H (Endo H) and peptide:N-glycosidase F (PNGase F). Endo H removes asparagine-linked high-mannose-content glycans from glycoproteins that have not trafficked to the cis-medial Golgi apparatus, and thus are residing within the ER as immature proteins. PNGase F removes all asparagine-linked glycans regardless of their level of processing as immature or mature proteins. Therefore, analysis of the relative immature and mature glycoprotein content of a mutant or chimeric GPCR can determine its effect on secretory pathway trafficking.
4.2. Glycolytic analysis of GPCR trafficking
Endo H and PNGase F can be purchased from New England Biolabs. All necessary buffers and accessory reagents are supplied.
Thaw GPCR membrane preparations on ice (see Section 3.2).
Add 50–100 μg of membrane to a 1.5-ml microcentrifuge tube. Three reactions are necessary for each GPCR studied, control (no enzyme), Endo H, and PNGase F.
Manufacturer’s instructions should be followed for the enzymatic digestion; however, due to aggregation of HA-α2A&C ARs at temperatures above 55 °C, detergent and Laemmli loading buffer denaturation reactions were carried out at 55 °C.
Incubate the final glycosidic reaction for 4 h at 37 °C. Stop reaction by adding one-fifth volume of 5× SDS sample buffer (Hurt et al., 2000) and load each sample onto a 10% SDS PAGE gel. Large format gels will enhance detection of reaction products.
Following standard immunoblotting protocols, transfer the electrophoresed proteins onto nitrocellulose (or other membrane) and probe the blot with an appropriate epitope-specific monoclonal antisera and appropriate secondary antibody at dilutions appropriate for your detection methods (e.g., LiCOR, ECL).
4.3. Interpretation of glycosidic processing
Immunoblot analysis of GPCRs demonstrates multiple cell-specific patterns representing mature and immature glycosylation (Hurt et al., 2000). For example, WT-α2A ARs are predominantly found with mature glycosylation (Endo H insensitive), whereas α2C ARs are predominantly found with immature glycosylation (Endo H sensitive) (Fig. 9.3). Removal of the putative extracellular hydrophobic trafficking motif (α2CΔ5-15) enhances mature glycosylation, consistent with the enhanced plasma membrane staining seen by immunofluorescence. In addition, disruption of this domain by a charged point mutation (α2CA7D) has the same effect. The ratio of mature to immature glycosylation (ER retained) correlates with the ratio of surface to intracellular expression seen by immunocytochemistry and FACS analysis. WT, chimeric, and mutant GPCR glycosylation analysis can assist in determining if a putative trafficking motif truly affects movement through the secretory pathway.
Figure 9.3.
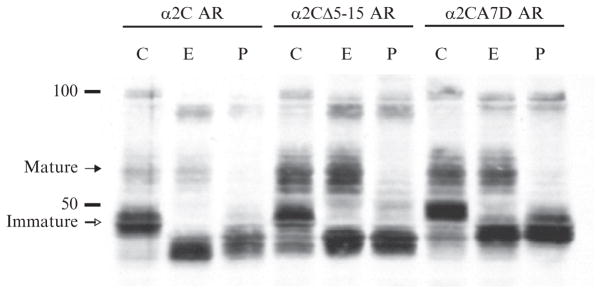
Glycosylation analysis of α2C AR trafficking. Cellular processing and maturation of α2C ARs and various trafficking mutations can be assessed by biochemical analysis of N-linked glycosylation (C=control, E =Endo H, and P=PNGase F). Mature and immature represent Endo H-resistant and -sensitive GPCR forms, respectively. Note that α2CΔ5-15 and α2CA7D ARs both have enhanced mature forms, suggestive of enhanced ER to Golgi trafficking and glycosidic processing. These results are consistent with a role for an extracellular hydrophobic domain in trafficking and plasma membrane expression. Adapted from Angelotti et al. (2010).
5. QUANTITATIVE ANALYSIS OF GPCR TRAFFICKING
If a GPCR trafficking motif is functional, then mutating it should alter the ratio of cell surface to total receptor expression. For example, removal of an ER retention motif should lead to ER export and thus an increase in cell surface/total GPCR expression (Angelotti et al., 2010). Qualitative immunofluorescent microscopy cannot readily discern such changes, especially if transient transfection methods are utilized and/or there is a large intracellular pool of receptor. Therefore, quantification of GPCR expression by FACS can assess the effects of mutating a potential trafficking motif.
5.1. FACS analysis of receptor expression
FACS can be utilized to measure the relative surface and total cell receptor levels for a given WT, chimeric, or mutant GPCR. From these data, the % surface and % intracellular expression can then be calculated to determine if a trafficking motif has affected this ratio. A proper FACS analysis requires measurements of nontransfected cells and determination of cell death to ensure accurate measurements of relative surface and total cell receptor levels. A more complete description of FACS analysis of transfected cells can be found elsewhere (Adams, Lopez, Sellers, & Kaelin, 1997).
Split a 10-cm tissue culture dish of confluent HEK293 cells 1:10 in DMEM with 10%FBS. Allow them to replicate at 37 °C until approximately 50% confluent.
Transfect cells with epitope-tagged GPCR chimeras or mutants using Effectene or other transfection reagent (per manufacturer’s recommendations) and maintain in culture for 72 h prior to analysis. In addition, three separate dishes of nontransfected cells will be required for controls (unstained, stained, and dead cell assessment).
Treat transfected cells with 10 μM cycloheximide (protein synthesis inhibitor) for 90 min at 37 °C to reduce background intracellular retention of GPCRs.
Wash plates two times with Hanks buffered saline solution (HBSS). Lift cells off the dish with 10 ml HBSS+5 mM EDTA.
Centrifuge cells at 200×g for 5 min at 4 °C. Aspirate supernatant and resuspend cell pellet in 10 ml PBS at 4 °C. Repeat centrifugation and resuspend pellet in 1 ml PBS.
Stain one control set of cells with propidium iodide (PI, Sigma) to determine cell death in the preparation. Incubate the sample preparation with PI in PBS (1 mg/ml) at 4 °C for 15 min on ice. Centrifuge and resuspend cell pellet in 250 μl PBS without PI and keep sample on ice in the dark. PI cannot be used on cells that will subsequently undergo fixation, as this will lead to dissociation from DNA of dead cells and binding to DNA of fixed cells. Fluorescent Live/Dead fixable dyes (Becton Dickinson) can be used with fixed cells.
Fix all other cells (transfected and nontransfected controls) by adding 1 ml of 4% PFA (2% final concentration) for 10 min at RT.
Place two 1-ml fractions (nonpermeabilized and permeabilized) into Eppendorf tubes. Centrifuge cells at 2500×g for 5 min at RT to remove PFA and resuspend cell pellet in 1 ml PBS. Repeat three times at 10-min intervals.
For surface expression analysis (nonpermeabilized), centrifuge cells as above and resuspend in 1 ml nonpermeabilized blocking solution (PBS+2% FBS) for 45 min prior to fluorescent labeling.
For total expression analysis (permeabilized), centrifuge cells as above and resuspend in 1 ml permeabilized blocking solution (PBS+2% FBS+0.1% Triton X-100) for 15 min and wash once with 1 ml non-permeabilized blocking solution for 30 min.
Set aside the permeabilized and nonpermeabilized, nontransfected cells for unstained controls.
Centrifuge both cell preparations at 2500×g for 5 min at RT. Resuspend cell pellets in 250 μl blocking solution (in appropriate permeabilized or nonpermeabilized blocking solution) containing fluorescein-conjugated 16B12 antibody (Covance) at 0.75 μg/ml. Incubate for 30 min at RT in the dark.
Similarly, permeabilized and nonpermeabilized, nontransfected cells should be prepared as stained controls in the same manner.
Centrifugecell preparationsat 2500×g for 5 min at RT and wash cell pellets three times at 5-min intervals with 500 μl PBS+2% FBS. Repeat once more and resuspend in 200–400 μl PBS. Cover tubes with foil.
5.2. FACS analysis
FACS analysis can be performed using a LSR-1 (Becton Dickinson Bioscience) or other similar device.
Prior to running cell samples, dissociate cells by passing through a 35-μm pore size, 5-ml round-bottom tube with a cell strainer cap (Becton Dickinson).
Following FACS device recommendations, gate the instrument to remove cell debris and clumps of aggregated cells using the non-permeabilized, unstained control cells.
Dead cells can autofluoresce and have increased nonspecific immunofluorescent staining. Using the PI-stained control cells, the fraction of dead cells in the population being studied should be determined. PI is excited using a blue 488-nm laser and detected using Cy5-PE/PI setting at 665/40 nm for the photomultiplier tube. The fraction of dead cells in these preparations should be low (less than a few percent of the population) so as to not adversely affect data quality.
Depending upon the transfection efficiency, the total number of cells needed to be analyzed can vary between 10,000 to 100,000 cells. Permeabilized and nonpermeabilized, nontransfected, stained control cells are used to set the gates for determination of transfected (stained) versus untransfected (minimally stained) GPCR-expressing cells. Fluorescent intensities should be obtained from 50,000 to 100,000 cells for each WT, mutant, or chimeric construct under permeabilized and non-permeabilized conditions.
For a good representation of the transfected cell population, a minimum of 2000 transfected cells should be used to determine median fluorescent intensity.
A minimum of three independent experiments should be carried out to calculate average median fluorescent intensity for each construct under each condition (permeabilized and nonpermeabilized).
- Calculate the percentage of GPCR surface expression relative to total cellular expression by dividing the average median surface (non-permeabilized) fluorescent intensity by the average median total (permeabilized) fluorescent intensity and report as average±standard error of the mean (SEM):
[9.1] [9.2]
5.3. Interpretation of FACS analysis
FACS analysis allows quantitative measurement of relative surface and total expression for a given WT or mutant GPCR (Fig. 9.4). With this information, the fraction of GPCR at the plasma membrane can be calculated (% surface) and the relative effect of a given trafficking motif on surface expression can be delineated with more certainty over qualitative immunofluorescent microscopic methods. It is hard to discern if an increase in surface expression seen by immunofluorescent microscopy is due to an overall increase in total protein expression or truly represents enhanced trafficking, especially in transiently transfected cells. For example, deletion or mutation of the extracellular hydrophobic domain (α2CΔ5-15 and α2CA7D AR) leads to increased surface expression compared to WT-α2C ARs, demonstrating a change in plasma membrane trafficking.
Figure 9.4.

Quantitative FACS analysis of plasma membrane trafficking. (A) Relative plasma membrane expression can be easily quantified by measuring GPCR levels in permeabilized (total) and nonpermeabilized (surface) cells using a one-step fluorescent labeling procedure (UT=untransfected). Following proper FACS gating to remove dead cells, measurement of single cell fluorescence allows for the determination of median fluorescent intensity for each GPCR under both conditions. Notice the shift in α2C AR median fluorescence intensity following permeabilization, reflecting the larger intracellular pool of receptor B. Calculation of relative surface and intracellular expression levels can be performed using median fluorescent measurements, demonstrating that both α2CΔ5-15 and α2CA7D ARs enhance plasma membrane expression. Adapted from Angelotti et al. (2010).
6. SUMMARY
Appropriate trafficking and localization of GPCRs to specific subdomains within a cell or neuron can be an important aspect of their physiological function (Edwards, Tan, & Limbird, 2000). A more thorough understanding of the biochemical machinery underlying transport of cargo proteins is ongoing (De Matteis & Luini, 2011), and an important aspect of trafficking is understanding protein motifs involved with GPCR transport. However, given the complexities of protein synthesis, folding, assembly, and transport through the ER/Golgi/plasma membrane, determination of trafficking motifs is not as straightforward as it may seem.
We have applied the above series of experimental methods to understand and identify a hydrophobic extracellular trafficking motif within α2C ARs, which is cell-type specific and transferable to α2A ARs (Angelotti et al., 2010). By initially constructing α2A&C AR chimeras and screening them with immunofluorescent microscopy, we were able to determine that a potential ER retention motif resided within the amino terminus of α2C ARs. Sequence analysis of multiple divergent species of α2C ARs suggested that a highly conserved motif may be responsible. Through a reiterative process of immunofluorescent staining, an extracellular hydrophobic domain (ALAAALAAAAA) was identified. Furthermore, alternative explanations such as alterations in receptor folding or half-life were removed from consideration by use of functional assays, including ligand binding and MAP kinase activation. Glycosidic processing clearly demonstrated that the motif regulated trafficking from ER to Golgi, resulting in altered glycosylation. Lastly, quantitative FACS analysis demonstrated that this motif truly regulated cell surface expression, not just total cellular expression. By applying a series of rigorous cell biological and biochemical methods, a novel trafficking motif was identified in α2C ARs. These methods should be applicable to other GPCRs.
Acknowledgments
This work was support in part by research MSTP grant GM07365 from the National Institutes of General Medical Sciences (C. M. H.) and NINDS K08NS050654-01A1 (T. A.). The authors would like to thank Dr. Brian Kobilka for his support during the early stages of this research.
References
- Adams PD, Lopez P, Sellers WR, Kaelin WG., Jr Fluorescence-activated cell sorting of transfected cells. Methods in Enzymology. 1997;283:59–72. doi: 10.1016/s0076-6879(97)83007-8. [DOI] [PubMed] [Google Scholar]
- Angelotti T, Daunt D, Shcherbakova OG, Kobilka B, Hurt CM. Regulation of G-protein coupled receptor traffic by an evolutionary conserved hydrophobic signal. Traffic. 2010;11:560–578. doi: 10.1111/j.1600-0854.2010.01033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brothers SP, Janovick JA, Conn PM. Calnexin regulated gonadotropin-releasing hormone receptor plasma membrane expression. Journal of Molecular Endocrinology. 2006;37:479–488. doi: 10.1677/jme.1.02142. [DOI] [PubMed] [Google Scholar]
- Brum PC, Hurt CM, Shcherbakova OG, Kobilka B, Angelotti T. Differential targeting and function of alpha(2A) and alpha(2C) adrenergic receptor subtypes in cultured sympathetic neurons. Neuropharmacology. 2006;51:397–413. doi: 10.1016/j.neuropharm.2006.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daunt DA, Hurt C, Hein L, Kallio J, Feng F, Kobilka BK. Subtype-specific intracellular trafficking of alpha2-adrenergic receptors. Molecular Pharmacology. 1997;51:711–720. doi: 10.1124/mol.51.5.711. [DOI] [PubMed] [Google Scholar]
- De Matteis MA, Luini A. Mendelian disorders of membrane trafficking. The New England Journal of Medicine. 2011;365:927–938. doi: 10.1056/NEJMra0910494. [DOI] [PubMed] [Google Scholar]
- Duvernay MT, Dong C, Zhang X, Zhou F, Nichols CD, Wu G. Anterograde trafficking of G protein-coupled receptors: Function of the C-terminal F(X)6LL motif in export from the endoplasmic reticulum. Molecular Pharmacology. 2009;75:751–761. doi: 10.1124/mol.108.051623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvernay MT, Filipeanu CM, Wu G. The regulatory mechanisms of export trafficking of G protein-coupled receptors. Cellular Signalling. 2005;17:1457–1465. doi: 10.1016/j.cellsig.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Edwards SW, Tan CM, Limbird LE. Localization of G-protein-coupled receptors in health and disease. Trends in Pharmacological Sciences. 2000;21:304–308. doi: 10.1016/s0165-6147(00)01513-3. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nature Reviews Molecular Cell Biology. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Hermosilla R, Schulein R. Sorting functions of the individual cytoplasmic domains of the G protein-coupled vasopressin V(2) receptor in Madin Darby canine kidney epithelial cells. Molecular Pharmacology. 2001;60:1031–1039. doi: 10.1124/mol.60.5.1031. [DOI] [PubMed] [Google Scholar]
- Hurt CM, Feng FY, Kobilka B. Cell-type specific targeting of the alpha 2c-adrenoceptor. Evidence for the organization of receptor microdomains during neuronal differentiation of PC12 cells. The Journal of Biological Chemistry. 2000;275:35424–35431. doi: 10.1074/jbc.M006241200. [DOI] [PubMed] [Google Scholar]
- Kallal L, Benovic JL. Fluorescence microscopy techniques for the study of G protein-coupled receptor trafficking. Methods in Enzymology. 2002;343:492–506. doi: 10.1016/s0076-6879(02)43154-0. [DOI] [PubMed] [Google Scholar]
- Pagano A, Rovelli G, Mosbacher J, Lohmann T, Duthey B, Stauffer D, et al. C-terminal interaction is essential for surface trafficking but not for heteromeric assembly of GABA(b) receptors. The Journal of Neuroscience. 2001;21:1189–1202. doi: 10.1523/JNEUROSCI.21-04-01189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petaja-Repo UE, Hogue M, Bhalla S, Laperriere A, Morello JP, Bouvier M. Ligands act as pharmacological chaperones and increase the efficiency of delta opioid receptor maturation. The EMBO Journal. 2002;21:1628–1637. doi: 10.1093/emboj/21.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulein R, Hermosilla R, Oksche A, Dehe M, Wiesner B, Krause G, et al. A dileucine sequence and an upstream glutamate residue in the intracellular carboxyl terminus of the vasopressin V2 receptor are essential for cell surface transport in COS. M6 cells. Molecular Pharmacology. 1998;54:525–535. doi: 10.1124/mol.54.3.525. [DOI] [PubMed] [Google Scholar]
- Wozniak M, Saunders C, Schramm N, Keefer JR, Limbird LE. Morphological and biochemical strategies for monitoring trafficking of epitope-tagged G protein-coupled receptors in agonist-naive and agonist-occupied states. Methods in Enzymology. 2002;343:530–544. doi: 10.1016/s0076-6879(02)43156-4. [DOI] [PubMed] [Google Scholar]