

Abstract
Trisomy 21 (Down syndrome, DS) is the most common human genetic anomaly associated with heart defects. Based on evolutionary conservation, DS-associated heart defects have been modeled in mice. By generating and analyzing mouse mutants carrying different genomic rearrangements in human chromosome 21 (Hsa21) syntenic regions, we found the triplication of the Tiam1-Kcnj6 region on mouse chromosome 16 (Mmu16) resulted in DS-related cardiovascular abnormalities. In this study, we developed two tandem duplications spanning the Tiam1-Kcnj6 genomic region on Mmu16 using recombinase-mediated genome engineering, Dp(16)3Yey and Dp(16)4Yey, spanning the 2.1Mb Tiam1-Il10rb and 3.7Mb Ifnar1-Kcnj6 regions, respectively. We found that Dp(16)4Yey/+, but not Dp(16)3Yey/+, led to heart defects, suggesting the triplication of the Ifnar1-Kcnj6 region is sufficient to cause DS-associated heart defects. Our transcriptional analysis of Dp(16)4Yey/+ embryos showed that the Hsa21 gene orthologs located within the duplicated interval were expressed at the elevated levels, reflecting the consequences of the gene dosage alterations. Therefore, we have identified a 3.7Mb genomic region, the smallest critical genomic region, for DS-associated heart defects, and our results should set the stage for the final step to establish the identities of the causal gene(s), whose elevated expression(s) directly underlie this major DS phenotype.
Keywords: Down syndrome, trisomy 21, heart defects - congenital, chromosome engineering, mouse models for human genetic disease, genetic mapping
Introduction
Trisomy 21 (Down syndrome, DS) is the most frequent live-born aneuploidy in humans (Epstein 1986; Hassold and Hunt 2001; Roizen and Patterson 2003). In the U.S., it occurs in 1 in every 691 live births, affecting approximately 6,000 newborns per year (Parker et al. 2010). A recent review has shown that, after prenatal diagnosis, termination rates stood at 67% and 85% in the U.S. from population-based studies with 2,593 pregnancies and hospital-based studies with 779 pregnancies, respectively. Evidence suggests that termination rates have decreased in recent years, partly due to improved medical care and social support for DS individuals (Natoli et al. 2012). Trisomy 21 is the most common genetic anomaly associated with congenital heart defects. Although individuals with DS have multiple medical problems, the single greatest risk factor for death during infancy is heart defects (Brookes and Alberman 1996). Heart defects are detected in 40-60% of newborns with DS (Abbag 2006; Freeman et al. 2008; Goodship et al. 1998; Roizen and Patterson 2003; Rowe and Uchida 1961; Torfs and Christianson 1998; Vis et al. 2009). The most frequent heart defects associated with DS are atrioventricular septal defect (AVSD) (23-45%), ventricular septal defect (VSD) (33-43%) and atrial septal defect (17-42%). Other important heart defects include tetralogy of Fallot (TOF), defects associated with valves, aorta, and pulmonary artery (Torfs and Christianson 1998). AVSD is the most frequent cardiac defect in DS in some studies, while VSD is more prevalent in other studies (Abbag 2006; Kava et al. 2004; Paladini et al. 2000). The mechanism underlying DS-associated heart defects is unknown. Although rare genetic variants located outside Hsa21 may play a role in enhancing the frequency, no variants can explain why such a high percentage of newborns with DS have heart defects. Thus, for a majority of DS individuals with heart defects, an extra copy of Hsa21 is necessary and sufficient to cause this phenotype. This is supported by heart defects observed in mouse models. The prevailing hypothesis is that heart defects, like other DS phenotypes, are caused by the dosage increase of a critical gene(s) on Hsa21 (Epstein 1990). It is possible that altered expression level of the critical gene(s) affects one or more key pathways, which in turn results in abnormal heart development as observed in other genetic disorders associated with the cardiovascular system (Jiang et al. 2013; Moskowitz et al. 2011; Terada et al. 2011; Zhang et al. 2006). Although we could seek to identify the disturbance of potentially relevant pathways, it will be strategically most desirable if we could identify the critical gene(s) first. This is because the ensuing analysis of the biological consequences of the triplication of the critical gene(s) would be considerably more focused than the analysis of abnormalities of pathways before the identities of the critical gene(s) is established. Many pathways may be affected by other genes present in three copies that are not causal gene(s) for heart defects. For this reason, human geneticists have, for the last several decades, pursued this critical gene(s) by identifying and analyzing patients carrying segmental trisomy 21. Due principally to the small number of patients with segmental trisomy 21 and to the resulting lack of a complete and informative set of human segmental trisomies, these research efforts have not yet led to identifying the causal gene(s) underlying heart defects in DS (Korbel et al. 2009; Korenberg et al. 1994; Lyle et al. 2009; Sinet et al. 1994).
The genomic regions on Hsa21 are syntenically conserved in three regions in the mouse genome, which are located on mouse chromosome 10 (Mmu10), Mmu16 and Mmu17 (www.ensembl.org) (Fig. 1). Because of this evolutionary conservation, the mouse has become indispensable model organism for DS. To complement the human genetics approach, we are seeking to identify the critical genomic region associated with congenital heart defects in DS based on the syntenic conservation and the shared mutant phenotype of cardiovascular systems between human patients with DS and the mouse models in order to unravel the critical gene(s). In this study, we developed two duplication mouse mutants using recombinase-mediated chromosome engineering and phenotypic characterization of these mutants resulted in the identification of a 3.7-Mb Hsa21 orthologous region underlying congenital heart defects in DS, the smallest critical genomic region associated with this major DS phenotype (Fig. 1).
Fig. 1.
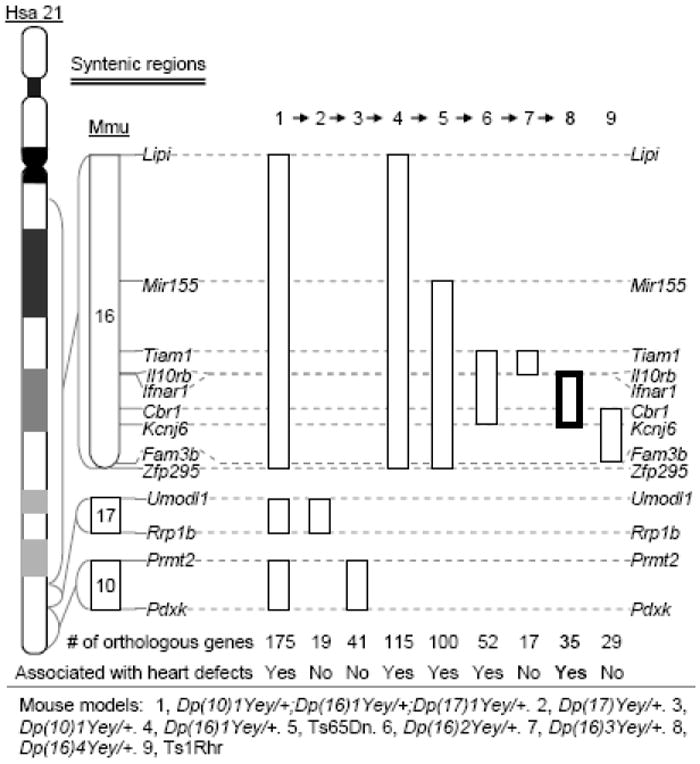
Genetic mapping of DS-associated heart defects in mice.
Materials and methods
Generation of Dp(16Tiam1-Il10rb)Yey/+ mice
To generate Dp(16Tiam1-Il10rb)Yey, MICER clones MHPN374h24 and MHPP321i13 (Adams et al. 2004) were used as the pTVTiam1- and pTVIl10rb-targeting vectors for inserting loxP to the endpoint 1 (EP1) and EP2. AB2.2 embryonic stem (ES) cell line (Bradley et al. 1998), which carries an Hprt-null allele, was used for targeting. The pTVTiam1 and pTVIl10rb vectors were linearized before gene targeting with restriction enzymes EcoNI and KpnI, respectively, at the mouse genome homologous regions in the vectors. After targeting, 8 double targeted ES cell clones were isolated. A Cre-expression vector, pOG231 (O’Gorman et al. 1997), was transfected into these double-targeted ES cells to induce recombination between the two targeted loxP sites. This led to a duplication [Dp(16Tiam1-Il10rb)Yey] and a reciprocal deletion [Df(16Tiam1-Il10rb)Yey] in the mouse genome (Fig. 2a). We used Southern blot analysis to confirm the gene targeting as well as the chromosomal rearrangements (Fig. 2b-2c). The presence of Dp(16)3Yey and Df(16)3Yey was also confirmed by fluorescent in situ hybridization (FISH) (see below) (Fig. 2c). The ES cells carrying the desired genomic rearrangements were microinjected into blastocysts that were isolated from C57BL/6J females to generate germ-line transmitting chimeras. The procedural details of ES cell culture, gene-targeting and induction of Cre/loxP-mediated recombination, Southern blot analysis and injection of ES cells into blastocysts were described previously (Bradley 1987; Bradley et al. 1998; Ramirez-Solis et al. 1993; Ramirez-Solis et al. 1995).
Fig. 2.
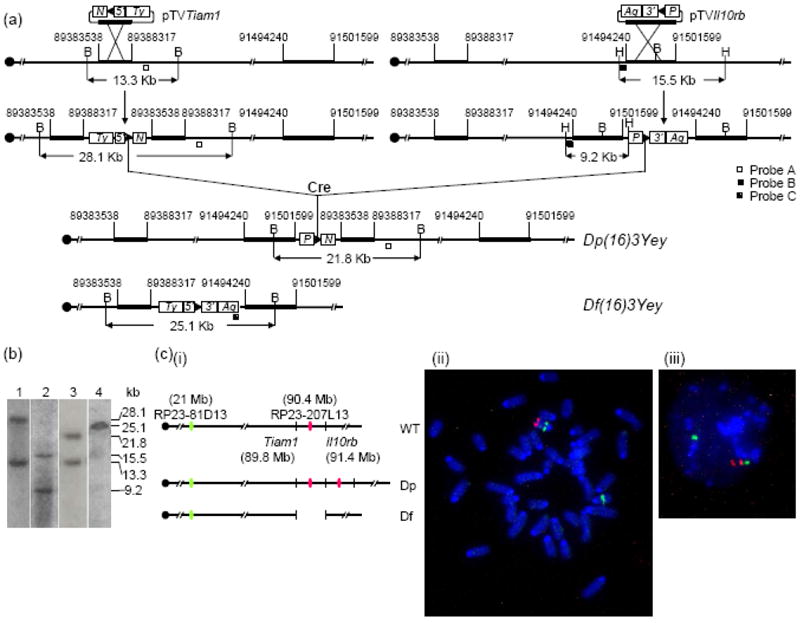
Development of Dp(16)3Yey/+ mice based on recombinase-mediated chromosome engineering. (a) Schematic representation of the strategy used to generate Dp(16)3Yey and Df(16)3Yey. B, BamHI; H, HindIII; 5’, 5’HPRT fragment; 3’, 3’HPRT fragment; N, Neomycin-resistance gene; P, puromycin-resistance gene; Ty, Tyrosinase transgene; Ag, Agouti transgene; arrowhead, loxP site. (b) Southern blot anaylsis of ES cell DNA digested with BamHI (lanes 1, 3 and 4) or HindIII (lane 2) using Probe A (lanes 1 and 3), Probe B (lane 2) or Probe C (lane 4), respectively. (c) FISH analysis of chromosomes of the engineered ES cells. (i) Schematic representation of the genomic locations of BAC probes for FISH analysis. (ii) FISH analysis of metaphase nuclei prepared from the ES cells carrying Dp(16)3Yey/Df(16)3Yey. (iii) FISH analysis of interphase chromosomes prepared from the ES cells carrying Dp(16)3Yey/Df(16)3Yey.
Generation of Dp(16Ifnar1-Kcnj6)Yey/+ mice
To generate Dp(16Il10rb-Kcnj6)Yey/+ mice, MICER clones MHPN247l16 and MHPP54c08 were used as the pTVIfnar1- and pTVKcnj6-targeting vectors for inserting loxP to EP3 and EP4. The pTVIfnar1 and pTVKcnj6 vectors were linearized with restriction enzymes NheI and BaeI, respectively, before gene targeting. Afterwards, 8 double targeted clones were isolated. With Cre/loxP-mediated recombination between the two targeted loxP sites, duplication [Dp(16Ifnar1-Kcnj6)Yey] and the reciprocal deletion [Df(16Ifnar1-Kcnj6)Yey] between EP3 and EP4 were generated (Fig. 3a). Southern blot analysis and FISH were used to confirm the gene targeting as well as the presence of the chromosome rearrangements (Fig. 3b-3c). The procedures of ES cell culture, gene-targeting and induction of Cre/loxP-mediated recombination, Southern blot analysis and injection of ES cells into blastocysts were similar to those used for generating Dp(16)3Yey/+ mice (see above).
Fig. 3.
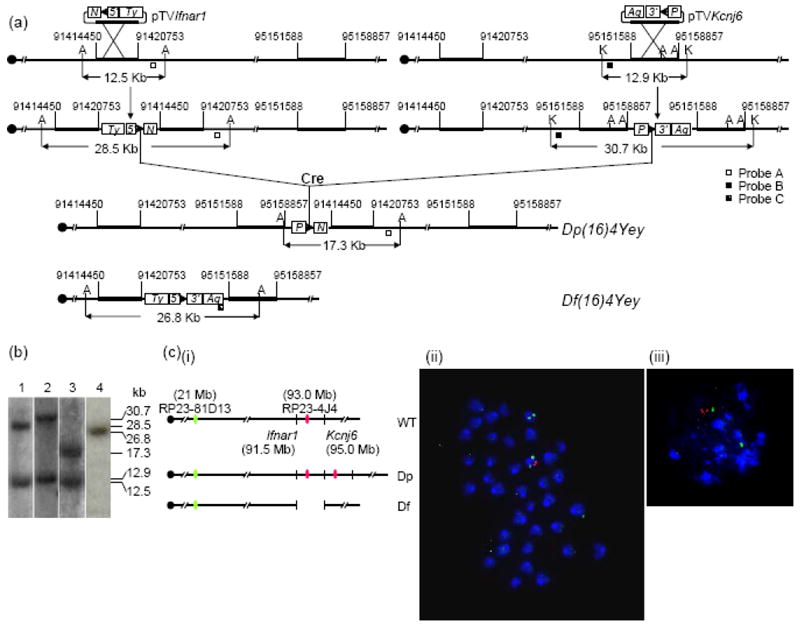
Development of Dp(16)4Yey/+ mice based on recombinase-mediated chromosome engineering. (a) Schematic representation of the strategy to generate Dp(16)4Yey and Df(16)4Yey. A, AflII; K, KpnI. (b) Southern blot anaylsis of ES cell DNA digested with AflII (lanes 1, 3 and 4) or KpnI (lane 2) using Probe A (lanes 1 and 3), Probe B (lane 2) or Probe C (lane 4), respectively. (c) FISH analysis of chromosomes of the engineered ES cells. (i) Schematic representation of the genomic locations of BAC probes for FISH analysis. (ii) FISH analysis of metaphase nuclei prepared from the ES cells carrying Dp(16)4Yey/Df(16)4Yey. (iii) FISH analysis of interphase chromosomes prepared from the ES cells carrying Dp(16)4Yey/Df(16)4Yey.
Fluorescent in situ hybridization
FISH analysis was performed as described previously (Yu et al. 2006). The metaphase chromosome spreads and interphase nuclei of ES cells were prepared as described previously (Robertson 1987). To detect the chromosomal deletion and duplication between Tiam1 and Il10rb or Ifnar1 and Kcnj6, BAC clone RP23-207L13 or RP23-4J4 were labeled with digoxigenin, respectively, and detected with anti-digoxigenin-rhodamine antibody (Figs. 2c and 3c). BAC clone RP23-81D13 was used to identify Mmu16 and labeled with biotin and detected with fluorescin isothiocyanate-avidin (Figs. 2c and 3c). Chromosomes were counter-stained with DAPI (4’,6’-diamidino-2-2phenylindole).
Mice
The mutant mice and their wild-type littermates were maintained at a temperature- and humidity-controlled animal facility. The experimental procedures were approved by the Institutional Animal Care and Use Committee.
RNA extraction
RNA was extracted from the pharyngeal arch region and heart of E10.5 embryos with 129Sv background using PureLink RNA Micro kit (Invitrogen Corp., Carlsbad, CA). The boundaries of the pharyngeal arch region were defined as previously described (Prescott et al. 2005). Prior to the RNA extraction, the embryos were genotyped using yolk sac DNA. After the elution step, RNA samples were concentrated by precipitation and resuspended in DEPC-treated nuclease-free water. The quality of the RNA samples was assessed by a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA).
RNA sequencing and data analysis
For RNA-seq, the pharyngeal arch regions and hearts from 4 E10.5 embryos with the genotype of Dp(16)4Yey/+ or +/+ were pooled before RNA extraction. RNA-Seq libraries were prepared using the Illumina TruSeq Stranded Total RNA Sample Prep Kit. Each cDNA library was sequenced for 50 cycles (single end reads), which generated 50 base pair reads, in a single flow-cell lane on the Illumina HiSeq 2000 (Illumina) at the University at Buffalo Next-Generation Sequencing and Expression Analysis Core Facility. Thus, each read consists of a 50-base-pair sequence of a DNA fragment. The total number of reads was obtained from sequencing all the DNA fragments of each individual cDNA library [116 million reads for Dp(16)4Yey/+ embryos and 136 million reads for the wild-type control embryos]. The sequence reads were aligned to the annotated mouse genome (NCBI build37.1) using TopHat2 version 2.0.8 (Kim et al. 2013). The resulting alignments were further assembled using Cufflinks v2.1.1 (Trapnell et al. 2010). Cuffdiff 2 (Trapnell et al. 2013) was used to calculate the expression at the gene level and detect differentially expressed gene transcripts between Dp(16)4Yey/+ embryos and the wild-type control embryos. Briefly, library sizes were normalized using the geometric mean method. Transcript abundances were estimated from the counts of individual isoforms transcribed from genes and reported as the expected number of fragments per kilobase per million fragments mapped. Fold changes of expression at the gene-level were calculated.
Real-time quantitative reverse transcriptase PCR
Real-time quantitative PCR was used to analyze RNA levels of the selected genes. Gapdh is located on Mmu6 and served as a reference gene of the disomic state for all the mice examined. Total RNAs were isolated from the pharyngeal arch regions and hearts of E10.5 embryos, as described above. 1 μg of RNA from each embryo was used to generate cDNA by using Superscript version III reverse transcriptase (Invitrogen Corp., Carlsbad, CA). The specific primers and probes for the genes were obtained from the TagMan® Gene Expression Assays System of Applied Biosystems, Inc. A 0.5 μg of cDNA from each embryo was analyzed by ABI 7900HT Real-Time Thermocycler (Applied Biosystems, Foster City, CA) with the following amplification conditions: an initial activation and denaturation at 95° C for 10 min, followed by 40 cycles of denaturation at 95° C for 15 sec and primer annealing and extension at 60° C for 1 min.
Results
Development of a mouse model carrying Dp(16Tiam1-Il10rb)Yey using chromosome engineering
We previously established that the triplication of the 5.43-Mb Tiam1-Kcnj6 region on Mmu16, which contains 52 Hsa21 gene orthologs, is necessary and sufficient to cause heart defects (Fig. 1) (Liu et al. 2011b). To dissect the Tiam1-Kcnj6 region, we, therefore in this study, first generated a new mouse model carrying a 2.11-Mb duplication between Tiam1 and Il10rb within the Tiam1-Kcnj6 region, which contain 17 Hsa21 gene orthologs. We developed this model using Cre/loxP-mediated chromosome engineering (Yu and Bradley 2001). MICER vectors (Adams et al. 2004) were used as pTVTiam1 and pTVKcnj6 for targeting loxP to the regions 403.6-Kb proximal and 75.8-Kb distal to the coding regions of Tiam1 and Il10rb, respectively, in AB2.2 ES cells (Fig. 2a) (Bradley et al. 1998). A duplication and the reciprocal deletion were induced in ES cells by the transfection with a Cre expression vector as described (Liu et al. 1998; Ramirez-Solis et al. 1995) and were confirmed by Southern blot analysis and FISH (Fig. 2b-2c). We used these ES cell clones to generate chimeras. The germ-line transmission of the duplication after crossing C57BL/6J and 129Sv females with chimeric males was confirmed by Southern blot analysis. The duplication was designated as Dup(16Tiam1-Il10rb)Yey, abbreviated as Dp(16)3Yey or Ts5Yey. The deletion was designated as Del(16Tiam1-Il10rb)Yey, abbreviated as Df(16)3/Yey or Ms4Yey.
Development of a mouse model carrying Dp(16Ifnar1-Kcnj6)Yey using chromosome engineering
The genomic region surrounding a gene contains cis-regulatory elements, and the size of such a region affecting gene transcription depends on the individual gene. In order to maximize the probability of including all the cis-regulatory elements for the genes near endpoints of the duplications, the proximal endpoint of Dp(16Ifnar1-Kcnj6)Yey is about 87 Kb proximal to the distal endpoint of Dp(16)3Yey (Figs. 2-3; Supplementary Fig. 1). Therefore, Dp(16)3Yey and Dp(16Ifnar1-Kcnj6)Yey share a ~87-Kb overlapping region. We generated Dp(16Ifnar1-Kcnj6)Yey/+ mice using a similar strategy for generating Dp(16)3Yey/+ mice as described above. MICER vectors (Adams et al. 2004) were used as pTVIfnar1 and pTVKcnj6 for targeting loxP to the regions 70.8-Kb proximal and 152.3-Kb distal to the coding regions of Ifnar1 and Kcnj6, respectively, in ES cells (Fig. 3a) (Bradley et al. 1998). A duplication and the reciprocal deletion were induced in ES cells by the transfection with a Cre expression vector and were confirmed by Southern blot analysis and FISH (Fig. 3b-3c). After generating chimeras using these ES cell clones, the germ-line transmission of the duplication after crossing C57BL/6J and 129Sv females with chimeric males was confirmed by Southern blot analysis. However, we could not obtain any Df(16)4Yey/+ mice from these crosses. One possibility is that the genotype of Df(16)4Yey/+ may have led to embryonic lethality. To test this possibility, we crossed the chimeric males with the Dp(16)1Yey/+ females (Li et al. 2007). As predicted, we obtained Dp(16)1Yey/Df(16)4Yey progeny from this cross, providing evidence that the Ifnar1-Kcnj6 region contains a gene(s) associated with haploinsufficient lethality. This gene(s) may underlie the embryonic lethality associated with human monosomy 21 (Chang et al. 2001; Joosten et al. 1997). The duplication was designated as Dup(16Ifnar1-Kcnj6)Yey, abbreviated as Dp(16)4Yey or Ts6Yey. The deletion was designated as Del(16Ifnar1-Kcnj6)Yey, abbreviated as Df(16)4/Yey or Ms5Yey.
Identification of a 3.7-Mb minimal critical genomic region for DS-associated heart defects by genetic mapping in mice
In the process of genetic analysis of heart defects in DS, we have recently identified 5.4-Mb Tiam1-Kcnj6 region on Mmu16 as a critical genomic region for this syndromic phenotype (Liu et al. 2011b). In the current study, we engineered Dp(16)3Yey/+ mice which carries a 2.11-Mb duplication between Tiam1 and Il10rb within the Tiam1-Kcnj6 region (Fig. 2). We examined the cardiovascular phenotypes of embryos carrying Dp(16)3Yey/+ (n=25) at E18.5 and found no heart defects. This result indicates that the presence of three copies of the Tiam1-Il10rb region is not sufficient to cause DS-associated heart defects. On the other hand, examination of Dp(16)4Yey/+ embryos at E18.5 showed that these embryos exhibit heart defects similar to Dp(16)1Yey/+ and Dp(16)2Yey/+ embryos with a similar frequency in either the 129Sv background or after crossing to C57BL/6J mice (Fig. 4; Table 1) (Li et al. 2007; Liu et al. 2011b). Therefore, the causal gene(s) for this phenotype is located in the Ifnar1-Kcnj6 region (Fig. 1).
Fig. 4.
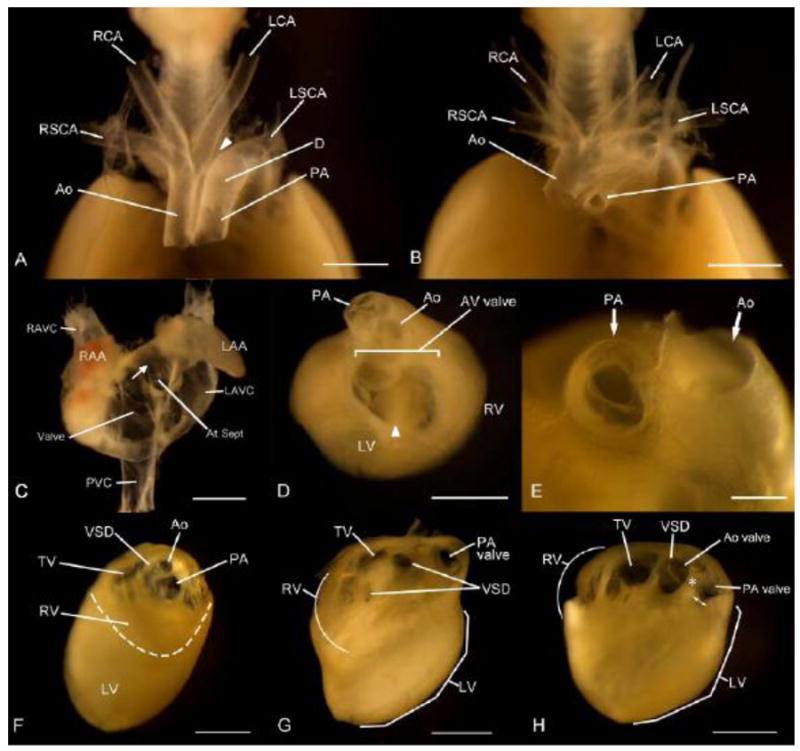
Cardiovascular malformations in Dp(16)4Yey/+ embryos at E18.5. (a-b) Ventral views of the great arteries after the hearts were removed. Interrupted aortic arch is indicated by an arrowhead in (a) and a stenosis of the pulmonary artery (PA) is shown in (b). (c) A ventral view of the atria after the ventricles were removed. The arrow indicates atrial septal defect (ASD). (d-e) Superior views of the hearts after removal of the atria. Atrioventricular (AV) septal defect is shown in (d) with “common AV valve”. The arrowhead points to the location of the ventricular septum. A unicommissural unicuspid PA valve is shown in (e). (f-h) Intracardic views of the right ventricle (RV) after the free wall was removed. A double outlet right ventricle (DORV) and subaortic stenosis is shown in (f), and both an inlet and a perimembranous ventricular septal defects (VSD) are shown in (g). A tetralogy of Fallot (TOF) is shown in (h), which includes a narrowed outflow tract (double arrows) of the right ventricle due to obstruction by the deviation of the parietal band (asterisk), a VSD, and the overriding of the aorta, where the aortic valve is visible through the VSD. Ao, aorta; Ao valve, aortic valve; At Sept, Atrial septum; AV valve, atrioventricular valv; D, ductus arteriosus; LAA, left atrial appendage; LAVC, left anterior vena cava; LCA, left carotid artery; LSCA, left subclavian artery; LV, left ventricle; PA, pulmonary artery; PVC, posterior vena cava; RAA, right atrial appendage; RAVC, right anterior vena cava; RCA, right carotid artery; RSCA, right subclavian artery; RV, right ventricle; TV, tricuspid valve; VSD, ventricular septal defect. Scale bars, 1 mm.
Table 1.
Cardiovascular malformations of Dp(16)4Yey/+ embryos at E18.5
| Genotype | n2/n1 | Type of heart defects | Number of embryos |
|---|---|---|---|
| Dp(16)4Yey/+ | 7/29 | ASD | 3 |
| VSD | 3 | ||
| AVSD | 1 | ||
| TOF | 1 | ||
| Valve defect | 1 | ||
| DORV | 1 | ||
| Interrupted aortic arch | 1 | ||
| Pulmonary stenosis | 1 |
n1, number of embryos examined; n2, number of embryos with heart defects. ASD, atrial septal defect; VSD, ventricular septal defect; AVSD, atrioventricular septal defects; TOF, Tetralogy of Fallot; DORV, double outlet right ventricle.
To examine if the genes located in the duplicated region in Dp(16)4Yey/+ mice are expressed, we performed RNA-seq using RNA isolated from the pharyngeal arch region and heart of the E10.5 Dp(16)4Yey/+ and wild-type embryos. The RNA-seq result is consistent with the elevated expression levels for the duplicated genes (Table 2). In addition, we used TaqMan real-time quantitative PCR to compare the mRNA levels for 4 genes located within the duplicated interval in Dp(16)4Yey/+ mice. Gapdh is located on mouse chromosome 6 and served as a reference gene of the disomic state in the Dp(16)4Yey/+ and +/+ mice. This analysis showed that the duplication altered the transcript levels of the genes in the pharyngeal arch region and heart of the Dp(16)4Yey/+ model at E10.5 (Table 3), reflecting the dosage imbalance for the duplicated region. This result further supports the conclusion that the duplicated genes within the Ifnar1-Kcnj6 region are expressed with the exception for transcriptionally inactive genes and suggests that the cardiovascular abnormalities are a consequence of elevated expression of the duplicated gene(s).
Table 2.
RNA-seq-based relative values of expression*
| Gene name | Dp(16)4Yey/+ over +/+ | Gene name | Dp(16)4Yey/+ over +/+ | Gene name | Dp(16)4Yey/+ over +/+ | Gene name | Dp(16)4Yey/+ over +/+ |
|---|---|---|---|---|---|---|---|
| Ifnar1 | 1.77 | Donson | 1.92 | Smim11 | 1.28 | Chaf1b | 2.26 |
| Ifngr2 | 1.58 | Atp5o | 1.38 | Rcan1 | 1.33 | Pigp | 1.38 |
| Tmem50b | 1.47 | Cryzl1 | 1.68 | Cbr1 | 1.46 | Ttc3 | 1.55 |
| Gart | 1.80 | Itsn1 | 1.49 | Cbr3 | 1.40 | Dscr3 | 1.39 |
| Son | 1.36 | Mrps6 | 1.72 | Morc3 | 1.58 | Dyrk1a | 1.41 |
RNA was isolated from the pharyngeal arch region and the heart of E10.5 Dp(16)4Yey/+ or +/+ embryos. The samples from 4 mutant embryos or the wild-type control embryos were pooled before RNA extraction.
Table 3.
Real-time PCR-based relative values (RQ) of expression*
| Gene name | Dp(16)4Yey/+ over +/+ (RQ ± S.E.M.) |
|---|---|
| Gart | 1.51 ± 0.10 |
| Mrps6 | 1.69 ± 0.10 |
| Pigp | 1.51 ± 0.13 |
| Dyrk1a | 1.52 ± 0.07 |
The values are calculated based on the means of the samples with different genotypes. Gapdh was used as an internal control and is disomic in all strains. RNA was isolated from the pharyngeal arch region and the heart of each E10.5 Dp(16)4Yey/+ or +/+ embryo. Three mutant embryos or the wild-type control embryos were used.
Discussion
The combined Hsa21 orthologous regions on Mmu10, Mmu16 and Mmu17 are about 26.3Mb and contain about 175 Hsa21 gene orthologs (Fig. 1) (www.ensembl.org). We developed Dp(10)1Yey/+, Dp(16)1Yey/+, and Dp(17)1Yey/+ mouse mutants carrying the duplications spanning the entire Hsa21 orthologous regions on three mouse chromosomes, and the model carrying all three duplications simultaneously is considered as a complete genetic model for DS (Lana-Elola et al. 2011; Roubertoux and Carlier 2010; Yu et al. 2010). To genetically dissect DS-associated heart defects, we first examined the cardiovascular system of the embryos at E18.5 with the following genotypes: Dp(10)1Yey/+, Dp(16)1Yey/+, Dp(17)1Yey/+ or Dp(10)1Yey/+;Dp(16)1Yey/+;Dp(17)1Yey/+ carrying the triple duplications. Heart defects were observed only in Dp(16)1Yey/+ and Dp(10)1Yey/+;Dp(16)1Yey/+;Dp(17)1Yey/+ models. We and others (Moore 2006; Williams et al. 2008) detected heart defects in Ts65Dn model (Fig. 1), so we next engineered a pair of duplication and deletion associated with the Tiam1-Kcnj6 region, which spans 5.4Mb and contains 52 Hsa21 gene orthologs, because of the following reasons: (a) the region is located within the triplicated region in Ts65Dn, (b) TIAM1 affects the functions of endothelial cells (Birukova et al. 2007a; Birukova et al. 2007b; Singleton et al. 2005), and (c) KCNJ6 has been implicated in affecting heart rate (Lignon et al. 2008). The result from this effort was surprisingly revealing: Dp(16Tiam1-Kcnj6)Yey/+ (i.e., Dp(16)2Yey/+) embryos exhibited heart defects similar to those observed in Dp(16)1Yey/+ embryos with similar frequency (Liu et al. 2011b). On the other hand, we did not detect any heart defects in the mutants carrying Dp(16)3Yey/+ nor Ts1Rhr which carries a Dp in the Cbr1-Fam3b region (Fig. 1). Our genetic mapping culminated in the discovery that Dp(16)4Yey is sufficient to cause heart defects (Figs. 1-3), so we have reduced the critical regions for DS-associated heart defects from 26.3Mb to 3.7Mb and the number of the candidate genes from 175 to 35 (Figs. 1-3) (Liu et al. 2011a; Liu et al. 2011b; Yu et al. 2010; Zhang et al. 2012).
The data from our previous and current studies have clearly shown that there are different critical Hsa21 orthologous regions for different DS-related phenotypes (Zhang et al., 2013). The Ifnar1-Kcnj6 region on Mmu16 is associated with heart defects, while the Cbr1-Fam3b region on Mmu16 as well as the interaction of the Hsa21 orthologous region on Mmu17 and the Hsa21 orthologous region proximal to Cbr1 on Mmu16 are associated with developmental cognitive deficits (Fig. 1) (Zhang et al., 2013).
Our analysis of gene expression of the Dp(16)4Yey/+ model at E10.5 showed that 20 genes of the 35 genes in the region are expressed at elevated levels in the heart and the pharyngeal arch which contributes to arteries during subsequent developmental processes (Tables 2-3). It is possible that other genes in this region are expressed at an earlier developmental stage or in specific cell types of the cardiovascular system, which may not be detected at a high level in our analysis. The contributions of many genes in the Ifnar1-Kcnj6 region to cardiovascular development are unknown, and our results indicate that one or more of the genes in this region underlies the mutant phenotype. Our hypothesis is supported by the studies showing a mutation or a copy number variation of a single gene, such as Cx40, Cited2, or Nt3, can cause a spectrum of heart defects, including heart septal defects, abnormalities associated with aortic/pulmonary arteries and TOF in mouse mutants (Donovan et al. 1996; Gu et al. 2003; Yin et al. 2002). However, no orthologs of these genes are located on Hsa21.
To identify the causal gene(s), we could generate single-copy transgenic mice for every Hsa21 gene orthologs located in the Ifnar1-Kcnj6 region one by one. However, this approach has some drawbacks. First, to accurately mimic the trisomic state of a gene in human trisomy 21, a transgene construct will need to include all the endogenous regulatory elements. However, we do not know every endogenous regulatory element for most of the Hsa 21 gene orthologs triplicated in Dp(16)4Yey, and it will need a very large amount of effort to identify them. Second, for several genes, transgenic approach is not feasible because the sizes of the structural genes plus the regulatory elements are larger than a BAC clone (www.ensembl.org). Third, if a mutant phenotype is a consequence of a joint effect of two or more triplicated genes, an individual gene transgenic approach may fail to identify the causal genes. Therefore a far more desirable approach is to further reduce the number of the candidate genes by continuing to narrow down the critical genomic region(s) using additional chromosomal duplications and deletions without premature or bias assumptions before initiating the efforts to screen for individual causal gene(s).
The Ifnar1-Kcnj6 region can be divided into the Ifnar1-Cbr1 and Cbr1-Kcnj6 regions. We and the others have shown that the triplication of the Cbr1-Fam3b does not cause heart defects in the Ts1Rhr embryos (Fig. 1) (Dunlevy et al. 2010; Liu et al. 2011b). Because of the presence of a 1.6-Mb overlapping duplicated region between Dp(16)4Yey and Ts1Rhr, which carries 14 Hsa21 gene orthologs (Fig. 1; Supplementary Table 1), there are two possibilities for the genomic locations of the causal gene(s) for heart defects. First, the causal gene(s) may be located in the Ifnar1-Cbr1 region (Fig. 1). Second, a causal gene(s) may be located in the Ifnar1-Cbr1 region and another causal gene(s) may be located in the Cbr1-Kcnj6 region (Fig. 1). Under the latter circumstance, the mutant phenotype is caused by the combined effect of the triplications of the causal genes in both regions, and triplications of the causal gene(s) in either region alone are not sufficient to cause the phenotype. The aforementioned scenarios could be dissected by engineering and characterization of mutants carrying a duplication and a deletion between Ifnar1 and Cbr1. If the causal gene(s) is located in the Ifnar1-Cbr1 region, Dp(16Ifnar1-Cbr1)/+, but not Dp(16)4Yey/Df(16Ifnar1-Cbr1), should lead to heart defects. If the causal genes are located in both the Ifnar1-Cbr1 and Cbr1-Kcnj6 regions and the simultaneous triplications of these genes are required to cause heart defects, neither Dp(16Ifnar1-Cbr1)/+ nor Dp(16)4Yey/Df(16Ifnar1-Cbr1) should lead to heart defects. If we can confirm that the causal gene(s) are located in the Ifnar1-Cbr1 region, this genomic region can be further dissected independently, which contains 21 protein- and 1 miRNA-coding Hsa21 gene orthologs. Because the Slc5a3 gene is located in the middle of the region, the Ifnar1-Slc5a3 and Slc5a3-Cbr1 segments will divide the genomic region into two with a similar number of the Hsa21 gene orthologs in each region. Generation and characterization of Dp(16)4Yey/Df(16Ifnar1-Slc5a3) mice can inform us if the Ifnar1-Slc5a3 region contains a causal gene(s) whose triplication is necessary to cause heart defects. On the other hand, generation and characterization of Dp(16Ifnar1-Slc5a3)/+ mice can inform us if the same genomic region contains the causal gene(s) whose triplication is sufficient to cause the mutant phenotype. This strategy using a duplication alone and in combination with a smaller deletion could also be employed to dissect the Slc5a3-Cbr1 region as well as the sub-regions in order to narrow down the critical genomic regions. After a minimal critical genomic region(s) is established through genetic dissection, the identity of a causal gene can be revealed by generation and characterization of a compound mouse mutant which harbors a duplication of the minimal critical genomic region and a knockout allele of a candidate gene located within the region. This genetic analysis approach will be significantly facilitated by the availability of knockout mice generated by the International Knockout Mouse Consortium (www.knockoutmouse.org).
Through our current effort, we have mapped heart defects-associated critical Hsa21 gene ortholog(s) to the 3.7-Mb Ifnar1-Kcnj6 region, the smallest critical genomic region that has been identified. This success lays the groundwork for the final effort to identify the dosage-sensitive gene(s) underlying heart defects in DS. Because human trisomy 21 is the most common genetic anomaly associated with heart defects, the gene(s) identified will be an entry point to better understand normal and abnormal cardiovascular development. Since human trisomy 21 can be detected by sequencing fetal DNA in maternal blood (Benn et al. 2012; Ehrich et al. 2011; Palomaki et al. 2011) and fetal DNA in maternal blood can be detected at 4-5 weeks of gestation (Norbury and Norbury 2008; Rijnders et al. 2003; Scheffer et al. 2010; Sikora et al. 2010), there is a genuine possibility that the causal gene(s) and/or the corresponding protein(s) could serve as the targets for preventing heart malformations in embryos carrying human trisomy 21.
Supplementary Material
Acknowledgments
The authors would like to thank Dawei Fu for his technical assistance. This study is supported in part by grants from the Children’s Guild Foundation and the NIH (R01HL91519, R01NS66072 and P30CA16056).
Contributor Information
Chunhong Liu, The Children’s Guild Foundation Down Syndrome Research Program and Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263, USA.
Masae Morishima, Department of Anatomy and Developmental Biology, Tokyo Women’s Medical University, Tokyo, Japan.
Xiaoling Jiang, The Children’s Guild Foundation Down Syndrome Research Program and Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263, USA.
Tao Yu, The Children’s Guild Foundation Down Syndrome Research Program and Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263, USA.
Kai Meng, The Children’s Guild Foundation Down Syndrome Research Program and Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263, USA, Department of Physiology and Pathophysiology, Medical School of Xi’an Jiaotong University, Xi’an, Shaanxi, China.
Debjit Ray, School of Molecular Biosciences, Washington State University, Pullman, WA 99164, USA.
Annie Pao, The Children’s Guild Foundation Down Syndrome Research Program and Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263, USA.
Ping Ye, School of Molecular Biosciences, Washington State University, Pullman, WA 99164, USA.
Michael S. Parmacek, Departments of Cell and Developmental Biology and Medicine, University of Pennsylvania Cardiovascular Institute, Philadelphia, PA 19104, USA
Y. Eugene Yu, Email: yuejin.yu@roswellpark.org, The Children’s Guild Foundation Down Syndrome Research Program and Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263, USA, New York State Center of Excellence in Bioinformatics and Life Sciences, Buffalo, NY 14263, USA; Department of Cellular and Molecular Biology, Roswell Park Division of Graduate School, State University of New York at Buffalo, Buffalo, NY 14263, USA.
References
- Abbag FI. Congenital heart diseases and other major anomalies in patients with Down syndrome. Saudi Med J. 2006;27:219–222. [PubMed] [Google Scholar]
- Adams DJ, Biggs PJ, Cox T, Davies R, van der Weyden L, Jonkers J, Smith J, Plumb B, Taylor R, Nishijima I, Yu Y, Rogers J, Bradley A. Mutagenic insertion and chromosome engineering resource (MICER) Nat Genet. 2004;36:867–871. doi: 10.1038/ng1388. [DOI] [PubMed] [Google Scholar]
- Benn P, Borrell A, Cuckle H, Dugoff L, Gross S, Johnson JA, Maymon R, Odibo A, Schielen P, Spencer K, Wright D, Yaron Y. Prenatal Detection of Down Syndrome using Massively Parallel Sequencing (MPS): a rapid response statement from a committee on behalf of the Board of the International Society for Prenatal Diagnosis, 24 October 2011. Prenat Diagn. 2012;32:1–2. doi: 10.1002/pd.2919. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Alekseeva E, Mikaelyan A, Birukov KG. HGF attenuates thrombin-induced endothelial permeability by Tiam1-mediated activation of the Rac pathway and by Tiam1/Rac-dependent inhibition of the Rho pathway. FASEB J. 2007a;21:2776–2786. doi: 10.1096/fj.06-7660com. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Malyukova I, Mikaelyan A, Fu P, Birukov KG. Tiam1 and betaPIX mediate Rac-dependent endothelial barrier protective response to oxidized phospholipids. J Cell Physiol. 2007b;211:608–617. doi: 10.1002/jcp.20966. [DOI] [PubMed] [Google Scholar]
- Bradley A. Production and analysis of chimaeric mice. In: Robertson E, editor. Teratocarcinomas and Embryonic Stem Cells - A Practical Approach. IRL Press; 1987. pp. 113–151. [Google Scholar]
- Bradley A, Zheng B, Liu P. Thirteen years of manipulating the mouse genome: a personal history. Int J Dev Biol. 1998;42:943–950. [PubMed] [Google Scholar]
- Brookes ME, Alberman E. Early mortality and morbidity in children with Down’s syndrome diagnosed in two regional health authorities in 1989. J Med Screen. 1996;3:7–11. doi: 10.1177/096914139600300104. [DOI] [PubMed] [Google Scholar]
- Chang LW, Chen PY, Kuo PL, Chang FM. Prenatal diagnosis of a fetus with megacystis and monosomy 21. Prenat Diagn. 2001;21:512–513. doi: 10.1002/pd.86. [DOI] [PubMed] [Google Scholar]
- Donovan MJ, Hahn R, Tessarollo L, Hempstead BL. Identification of an essential nonneuronal function of neurotrophin 3 in mammalian cardiac development. Nat Genet. 1996;14:210–213. doi: 10.1038/ng1096-210. [DOI] [PubMed] [Google Scholar]
- Dunlevy L, Bennett M, Slender A, Lana-Elola E, Tybulewicz VL, Fisher EM, Mohun T. Down syndrome-like cardiac developmental defects in embryos of the transchromosomic Tc1 mouse. Cardiovasc Res. 2010;88:287–295. doi: 10.1093/cvr/cvq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrich M, Deciu C, Zwiefelhofer T, Tynan JA, Cagasan L, Tim R, Lu V, McCullough R, McCarthy E, Nygren AO, Dean J, Tang L, Hutchison D, Lu T, Wang H, Angkachatchai V, Oeth P, Cantor CR, Bombard A, van den Boom D. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: a study in a clinical setting. Am J Obstet Gynecol. 2011;204:205 e1–11. doi: 10.1016/j.ajog.2010.12.060. [DOI] [PubMed] [Google Scholar]
- Epstein CJ. The consequences of chromosome imbalance: principles, mechanism and models. Cambridge University Press; New York: 1986. [Google Scholar]
- Epstein CJ. The consequences of chromosome imbalance. Am J Med Genet. 1990;(Suppl 7):31–37. doi: 10.1002/ajmg.1320370706. [DOI] [PubMed] [Google Scholar]
- Freeman SB, Bean LH, Allen EG, Tinker SW, Locke AE, Druschel C, Hobbs CA, Romitti PA, Royle MH, Torfs CP, Dooley KJ, Sherman SL. Ethnicity, sex, and the incidence of congenital heart defects: a report from the National Down Syndrome Project. Genet Med. 2008;10:173–180. doi: 10.1097/GIM.0b013e3181634867. [DOI] [PubMed] [Google Scholar]
- Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in infancy. Arch Dis Child. 1998;79:348–351. doi: 10.1136/adc.79.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Smith FC, Taffet SM, Delmar M. High incidence of cardiac malformations in connexin40-deficient mice. Circ Res. 2003;93:201–206. doi: 10.1161/01.RES.0000084852.65396.70. [DOI] [PubMed] [Google Scholar]
- Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet. 2001;2:280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Zhu L, Hu L, Slesnick TC, Pautler RG, Justice MJ, Belmont JW. Zic3 is required in the extra-cardiac perinodal region of the lateral plate mesoderm for left-right patterning and heart development. Hum Mol Genet. 2013;22:879–889. doi: 10.1093/hmg/dds494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten AM, De Vos S, Van Opstal D, Brandenburg H, Gaillard JL, Vermeij-Keers C. Full monosomy 21, prenatally diagnosed by fluorescent in situ hybridization. Prenat Diagn. 1997;17:271–275. doi: 10.1002/(sici)1097-0223(199703)17:3<271::aid-pd51>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Kava MP, Tullu MS, Muranjan MN, Girisha KM. Down syndrome: clinical profile from India. Arch Med Res. 2004;35:31–35. doi: 10.1016/j.arcmed.2003.06.005. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Tirosh-Wagner T, Urban AE, Chen XN, Kasowski M, Dai L, Grubert F, Erdman C, Gao MC, Lange K, Sobel EM, Barlow GM, Aylsworth AS, Carpenter NJ, Clark RD, Cohen MY, Doran E, Falik-Zaccai T, Lewin SO, Lott IT, McGillivray BC, Moeschler JB, Pettenati MJ, Pueschel SM, Rao KW, Shaffer LG, Shohat M, Van Riper AJ, Warburton D, Weissman S, Gerstein MB, Snyder M, Korenberg JR. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci U S A. 2009;106:12031–12036. doi: 10.1073/pnas.0813248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenberg JR, Chen XN, Schipper R, Sun Z, Gonsky R, Gerwehr S, Carpenter N, Daumer C, Dignan P, Disteche C. Down syndrome phenotypes: the consequences of chromosomal imbalance. Proc Natl Acad Sci U S A. 1994;91:4997–5001. doi: 10.1073/pnas.91.11.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana-Elola E, Watson-Scales SD, Fisher EM, Tybulewicz VL. Down syndrome: searching for the genetic culprits. Dis Model Mech. 2011;4:586–595. doi: 10.1242/dmm.008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Yu T, Morishima M, Pao A, LaDuca J, Conroy J, Nowak N, Matsui S, Shiraishi I, Yu Y. Duplication of the entire 22.9-Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum Mol Genet. 2007;16:1359–1366. doi: 10.1093/hmg/ddm086. [DOI] [PubMed] [Google Scholar]
- Lignon JM, Bichler Z, Hivert B, Gannier FE, Cosnay P, del Rio JA, Migliore-Samour D, Malecot CO. Altered heart rate control in transgenic mice carrying the KCNJ6 gene of the human chromosome 21. Physiol Genomics. 2008;33:230–239. doi: 10.1152/physiolgenomics.00143.2007. [DOI] [PubMed] [Google Scholar]
- Liu C, Belichenko PV, Zhang L, Fu D, Kleschevnikov AM, Baldini A, Antonarakis SE, Mobley WC, Yu YE. Mouse models for Down syndrome-associated developmental cognitive disabilities. Dev Neurosci. 2011a;33:404–413. doi: 10.1159/000329422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Morishima M, Yu T, Matsui SI, Zhang L, Fu D, Pao A, Costa AC, Gardiner KJ, Cowell JK, Nowak NJ, Parmacek MS, Liang P, Baldini A, Yu YE. Genetic analysis of Down syndrome-associated heart defects in mice. Hum Genet. 2011b;130:623–632. doi: 10.1007/s00439-011-0980-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Zhang H, McLellan A, Vogel H, Bradley A. Embryonic lethality and tumorigenesis caused by segmental aneuploidy on mouse chromosome 11. Genetics. 1998;150:1155–1168. doi: 10.1093/genetics/150.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle R, Bena F, Gagos S, Gehrig C, Lopez G, Schinzel A, Lespinasse J, Bottani A, Dahoun S, Taine L, Doco-Fenzy M, Cornillet-Lefebvre P, Pelet A, Lyonnet S, Toutain A, Colleaux L, Horst J, Kennerknecht I, Wakamatsu N, Descartes M, Franklin JC, Florentin-Arar L, Kitsiou S, Ait Yahya-Graison E, Costantine M, Sinet PM, Delabar JM, Antonarakis SE. Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur J Hum Genet. 2009;17:454–466. doi: 10.1038/ejhg.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS. Postnatal lethality and cardiac anomalies in the Ts65Dn Down syndrome mouse model. Mamm Genome. 2006;17:1005–1012. doi: 10.1007/s00335-006-0032-8. [DOI] [PubMed] [Google Scholar]
- Moskowitz IP, Wang J, Peterson MA, Pu WT, Mackinnon AC, Oxburgh L, Chu GC, Sarkar M, Berul C, Smoot L, Robertson EJ, Schwartz R, Seidman JG, Seidman CE. Transcription factor genes Smad4 and Gata4 cooperatively regulate cardiac valve development. Proc Natl Acad Sci U S A. 2011;108:4006–4011. doi: 10.1073/pnas.1019025108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli JL, Ackerman DL, McDermott S, Edwards JG. Prenatal diagnosis of Down syndrome: a systematic review of termination rates (1995-2011) Prenat Diagn. 2012;32:142–153. doi: 10.1002/pd.2910. [DOI] [PubMed] [Google Scholar]
- Norbury G, Norbury CJ. Non-invasive prenatal diagnosis of single gene disorders: how close are we? Semin Fetal Neonatal Med. 2008;13:76–83. doi: 10.1016/j.siny.2007.12.008. [DOI] [PubMed] [Google Scholar]
- O’Gorman S, Dagenais NA, Qian M, Marchuk Y. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci U S A. 1997;94:14602–14607. doi: 10.1073/pnas.94.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini D, Tartaglione A, Agangi A, Teodoro A, Forleo F, Borghese A, Martinelli P. The association between congenital heart disease and Down syndrome in prenatal life. Ultrasound Obstet Gynecol. 2000;15:104–108. doi: 10.1046/j.1469-0705.2000.00027.x. [DOI] [PubMed] [Google Scholar]
- Palomaki GE, Kloza EM, Lambert-Messerlian GM, Haddow JE, Neveux LM, Ehrich M, van den Boom D, Bombard AT, Deciu C, Grody WW, Nelson SF, Canick JA. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med. 2011;13:913–920. doi: 10.1097/GIM.0b013e3182368a0e. [DOI] [PubMed] [Google Scholar]
- Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004-2006. Birth Defects Res A Clin Mol Teratol. 2010;88:1008–1016. doi: 10.1002/bdra.20735. [DOI] [PubMed] [Google Scholar]
- Prescott K, Ivins S, Hubank M, Lindsay E, Baldini A, Scambler P. Microarray analysis of the Df1 mouse model of the 22q11 deletion syndrome. Hum Genet. 2005;116:486–496. doi: 10.1007/s00439-005-1274-3. [DOI] [PubMed] [Google Scholar]
- Ramirez-Solis R, Davis AC, Bradley A. Gene targeting in embryonic stem cells. Methods Enzymol. 1993;225:855–878. doi: 10.1016/0076-6879(93)25054-6. [DOI] [PubMed] [Google Scholar]
- Ramirez-Solis R, Liu P, Bradley A. Chromosome engineering in mice. Nature. 1995;378:720–724. doi: 10.1038/378720a0. [DOI] [PubMed] [Google Scholar]
- Rijnders RJ, Van Der Luijt RB, Peters ED, Goeree JK, Van Der Schoot CE, Ploos Van Amstel JK, Christiaens GC. Earliest gestational age for fetal sexing in cell-free maternal plasma. Prenat Diagn. 2003;23:1042–1044. doi: 10.1002/pd.750. [DOI] [PubMed] [Google Scholar]
- Robertson E. Embryo-derived stem cell lines. In: Robertson E, editor. Teratocarcinomas and Embryonic Stem Cells - A Practical Approach. IRL Press; 1987. pp. 77–112. [Google Scholar]
- Roizen NJ, Patterson D. Down’s syndrome. Lancet. 2003;361:1281–1289. doi: 10.1016/S0140-6736(03)12987-X. [DOI] [PubMed] [Google Scholar]
- Roubertoux PL, Carlier M. Mouse models of cognitive disabilities in trisomy 21 (Down syndrome) Am J Med Genet C Semin Med Genet. 2010;154C:400–416. doi: 10.1002/ajmg.c.30280. [DOI] [PubMed] [Google Scholar]
- Rowe RD, Uchida IA. Cardiac malformation in mongolism: a prospective study of 184 mongoloid children. Am J Med. 1961;31:726–735. doi: 10.1016/0002-9343(61)90157-7. [DOI] [PubMed] [Google Scholar]
- Scheffer PG, van der Schoot CE, Page-Christiaens GC, Bossers B, van Erp F, de Haas M. Reliability of fetal sex determination using maternal plasma. Obstet Gynecol. 2010;115:117–126. doi: 10.1097/AOG.0b013e3181c3c938. [DOI] [PubMed] [Google Scholar]
- Sikora A, Zimmermann BG, Rusterholz C, Birri D, Kolla V, Lapaire O, Hoesli I, Kiefer V, Jackson L, Hahn S. Detection of increased amounts of cell-free fetal DNA with short PCR amplicons. Clin Chem. 2010;56:136–138. doi: 10.1373/clinchem.2009.132951. [DOI] [PubMed] [Google Scholar]
- Sinet PM, Theophile D, Rahmani Z, Chettouh Z, Blouin JL, Prieur M, Noel B, Delabar JM. Mapping of the Down syndrome phenotype on chromosome 21 at the molecular level. Biomed Pharmacother. 1994;48:247–252. doi: 10.1016/0753-3322(94)90140-6. [DOI] [PubMed] [Google Scholar]
- Singleton PA, Dudek SM, Chiang ET, Garcia JG. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J. 2005;19:1646–1656. doi: 10.1096/fj.05-3928com. [DOI] [PubMed] [Google Scholar]
- Terada R, Warren S, Lu JT, Chien KR, Wessels A, Kasahara H. Ablation of Nkx2-5 at mid-embryonic stage results in premature lethality and cardiac malformation. Cardiovasc Res. 2011;91:289–299. doi: 10.1093/cvr/cvr037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torfs CP, Christianson RE. Anomalies in Down syndrome individuals in a large population-based registry. Am J Med Genet. 1998;77:431–438. [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vis JC, Duffels MG, Winter MM, Weijerman ME, Cobben JM, Huisman SA, Mulder BJ. Down syndrome: a cardiovascular perspective. J Intellect Disabil Res. 2009;53:419–425. doi: 10.1111/j.1365-2788.2009.01158.x. [DOI] [PubMed] [Google Scholar]
- Williams AD, Mjaatvedt CH, Moore CS. Characterization of the cardiac phenotype in neonatal Ts65Dn mice. Dev Dyn. 2008;237:426–435. doi: 10.1002/dvdy.21416. [DOI] [PubMed] [Google Scholar]
- Yin Z, Haynie J, Yang X, Han B, Kiatchoosakun S, Restivo J, Yuan S, Prabhakar NR, Herrup K, Conlon RA, Hoit BD, Watanabe M, Yang YC. The essential role of Cited2, a negative regulator for HIF-1alpha, in heart development and neurulation. Proc Natl Acad Sci U S A. 2002;99:10488–10493. doi: 10.1073/pnas.162371799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Li Z, Jia Z, Clapcote SJ, Liu C, Li S, Asrar S, Pao A, Chen R, Fan N, Carattini-Rivera S, Bechard AR, Spring S, Henkelman RM, Stoica G, Matsui S, Nowak NJ, Roder JC, Chen C, Bradley A, Yu YE. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet. 2010;19:2780–2791. doi: 10.1093/hmg/ddq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Bradley A. Engineering chromosomal rearrangements in mice. Nat Rev Genet. 2001;2:780–790. doi: 10.1038/35093564. [DOI] [PubMed] [Google Scholar]
- Yu YE, Morishima M, Pao A, Wang DY, Wen XY, Baldini A, Bradley A. A deficiency in the region homologous to human 17q21.33-q23.2 causes heart defects in mice. Genetics. 2006;173:297–307. doi: 10.1534/genetics.105.054833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Fu D, Belichenko PV, Liu C, Kleschevnikov AM, Pao A, Liang P, Clapcote SJ, Mobley WC, Yu YE. Genetic analysis of Down syndrome facilitated by mouse chromosome engineering. Bioeng Bugs. 2012;3:8–12. doi: 10.4161/bbug.3.1.17696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Zhang, Meng Kai, Jiang Xiaoling, Liu Chunhong, Pao Annie, Belichenko Pavel V, Kleschevnikov Alexander, Josselyn Sheena, Liang Ping, Ye Ping, Mobley William C, Yu Y Eugene. Human chromosome 21 orthologous region on mouse chromosome 17 is a major determinant for Down syndrome-related developmental cognitive deficits. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt446. (Epub ahead of print) http://hmg.oxfordjournals.org/content/early/2013/09/24/hmg.ddt446.long. [DOI] [PMC free article] [PubMed]
- Zhang Z, Huynh T, Baldini A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development. 2006;133:3587–3595. doi: 10.1242/dev.02539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.