

Abstract
The golden age of antimicrobial drug development is a distant memory, and the likelihood of there being another seems slim. In part, this is because the pharmaceutical industry, which has now adopted an unsustainable business model, abandoned the anti-infective sector, and the pipeline is almost empty. The contribution to this crisis of national governments, health agencies and funders also merits discussion. Much of the basis for drug discovery is funded by the public sector, thereby generating intellectual property and leads for drug development that are often not pursued owing to funding gaps. In particular, the cost of testing drug efficacy in clinical trials is beyond the means of most companies and organizations. Lack of a concerted international effort to develop new antimicrobials is particularly alarming at a time when multidrug-resistant bacteria threaten all areas of human medicine globally. Here, the steps that led to this situation are retraced, and some possible solutions to the dilemma are proposed.
Keywords: antibiotics, drug resistance, drug discovery, pharmaceutical industry, biotechnology
1. Introduction
The era of chemotherapy began in the early twentieth century, and Paul Ehrlich is rightly considered as its father because he promoted a scientific approach to the treatment of infectious diseases of humans that culminated in the organoarsenical compound, salvarsan, which was introduced into the clinic in 1910. In the decades that followed, chemotherapy gained considerable momentum, thanks to the ground-breaking achievements of Fleming, Domagk, Dubos, Waksman and their many co-workers [1,2]. The discovery of antibiotics, natural products produced by microorganisms that are able to prevent the growth of bacteria and thus cure infectious diseases, transformed medical practice and saved countless millions of lives. In the 1940s, for the first time, life-threatening and often fatal diseases such as pneumonia or tuberculosis (TB) could be controlled and cured with antibiotics such as penicillin and streptomycin, respectively. It is thus incontestable that antibiotics have changed the course of infectious diseases and, in so doing, radically improved human medicine and extended life expectancy [1,2].
Not only did antibiotics save lives, but they also created wealth, and in the latter half of the twentieth century spawned the modern pharmaceutical industry, a highly lucrative sector of commerce with its roots firmly embedded in scientific research. Following the Second World War, many pharmaceutical companies such as Bayer, Merck and Pfizer blossomed and expanded to become household names, thanks to the success of their products in the clinic and on the market. In the 1950s–1960s, research into antibiotic-producing organisms, identified by screening soil samples for antimicrobial activity, led to the discovery of bioactive molecules, including antibiotics belonging to the tetracycline, rifamycin, quinolone and aminoglycoside families, to name just a few. In addition, some antibiotics were found to be active in other areas of medicine such as cancer and immunosuppression leading to new therapeutic or prophylactic interventions and additional means to create wealth. At this time, because pharmaceutical companies regularly generated huge profits, they were considered a safe investment and underpinned the economies of the industrialized world to a large extent. Such multinational companies are often referred to as ‘big pharma’.
From the 1970s, the situation began to change. Despite intensive investigation into the traditional microbial sources of antibiotics, the actinomycete bacteria and filamentous fungi, the number of new molecules and activities discovered had markedly declined giving rise to the notion that perhaps all the major classes of antibiotics had been found. Developing agents to treat bacterial infections is often considered more difficult than for other therapeutic areas for a variety of reasons. These include poor penetration into bacterial cells, innate resistance mechanisms such as efflux pumps and drug-inactivating enzymes, and the requirement for relatively high concentrations that occasionally provokes side effects [1,2].
Synthetic organic chemistry and screening played a bigger part in the drug discovery process, but therapeutic priorities also began to change. For instance, successive Surgeons General of the United States made statements such as ‘it is time to close the book on infectious diseases, declare the war against pestilence won, and shift national resources to such chronic problems as cancer and heart disease’. Admittedly, there were good grounds for this misplaced optimism as many battles had indeed been won in the war against infectious diseases. Thanks to vaccination, smallpox had been eradicated, and other viral diseases such as polio were close to being eliminated [3]. Previously life-threatening diseases could now be cured by antibiotic treatment, and the range of therapeutic options offered by antibiotics for the treatment of bacterial infections was still considered to be sufficiently large, thereby inducing a certain complacency that such options would persist indefinitely.
Partly as a result of these successes against infectious diseases but also owing to pressure from market forces to maintain double-digit profits, the major pharmaceutical companies now intensified their research into the non-communicable diseases that increasingly affected humans. Their prominence had grown considerably as the incidence of infectious diseases decreased and living standards rose in the industrialized world, meaning life expectancy had never been so great. Cancer, diabetes, metabolic disease, Parkinson's disease, hypertension and other cardiovascular conditions could also be controlled by chemotherapy and, compared with most infectious diseases, afforded the additional economic advantages of chronicity and an expanding market. Thus, in contrast to a pneumococcal infection that could be cured within a week by a course of antibiotics, a non-communicable disease such as type II diabetes will last a lifetime, and the patient, requiring regular medication, represents a captive market of long duration initially for big pharma and subsequently for other drug manufacturers. Hence, these considerations also influenced the choice between investing in antibacterials which are more difficult to produce and potentially less profitable, or in other more lucrative therapeutic areas.
2. What we have gained through antibiotics and what we will lose
Foremost among the gains to have been made from the introduction of antibiotics into clinical practice are the ability to treat effectively and, above all, to cure both acute bacterial infections as well as chronic infectious diseases such as TB. As stated previously, antibiotics have reduced immeasurably the suffering and loss of human life to infectious diseases and changed medical practice in countless ways. For instance, in many surgical interventions that are nowadays considered routine, such as joint replacements, or more challenging procedures such as organ transplants, antibiotics are administered to the patient prior to surgery in order to reduce the risk of infection. Likewise, antibiotics protect prematurely born babies and cancer patients as well as those suffering from chronic conditions, including asthma, cystic fibrosis, diabetes, rheumatoid arthritis and HIV/AIDS.
All of these surgical procedures and therapeutic interventions will be severely compromised or even of high risk if the levels of resistance to the current antibiotics continue to rise and no new anti-infective agents are developed to replace them. In essence, if nothing is done, then we are in jeopardy of returning to the pre-antibiotic era. Unlike in other therapeutic areas, e.g. cancer treatment, drug resistance in infectious disease is considerably more dangerous owing to its often transferable nature [2].
3. The current state of drug resistance in infectious diseases
Infectious diseases remain a leading cause of morbidity and mortality, claiming an estimated 14 million lives in 2011 [4]. Because of globalization, it is clear that the risk of disease spreading rapidly throughout the world today is greater than ever before owing to the ease of intercontinental travel. Additionally, the displacement of populations as a result of famine, poverty or war also contributes to disease spread, and the likelihood of drug resistance arising in these settings is considerable. Furthermore, new diseases will also emerge as a result of environmental and demographic change. Drug-resistant forms of disease arise both within communities and hospital settings where drug use is intensive and constant. Today, drug resistance is common among all the major pathogens and to all classes of antibiotics, even to those that are no longer in clinical use. Infections caused by multidrug-resistant bacteria are associated with substantial extra costs owing to reduced treatment options. Table 1 presents the findings of recent surveys of drug resistance and its impact in Europe and the USA.
Table 1.
Estimates of morbidity, mortality and economic factors associated with drug resistance (DR). Note that healthcare costs are considerably higher in the USA so comparisons are difficult and probably unreliable. Sources [5–7].
| Europe: 2007a | USA: 2008–2013 | |
|---|---|---|
| incidence of nosocomial infections | 4 100 000 | 2 000 000 |
| number of DR in selected infections | 386 100 | 2 000 000 |
| deaths from nosocomial infections (of which number due to DR) | 37 000 (25 000) | 99 000 (36 000) |
| levels of DR bacteria | >25% | 70% |
| extra healthcare costs due to DR | €937.8 million | $20 billion |
| additional days in hospital owing to DR | 2.5 million | 8 million |
| total costs | €1.534 billion | $55 billion |
aEurope, EU Member States, Iceland and Norway (http://www.ecdc.europa.eu/en/healthtopics/Healthcare-associated_infections/pages/index.aspx).
(a). Europe
Resistance to antibiotics is high among Gram-positive and Gram-negative bacterial pathogens, although levels vary widely between countries in the EU owing to differences in antibiotic use. There were over four million nosocomial or hospital-acquired infections in the EU in 2007 and 386 100 of these were attributable to drug-resistant bacteria (table 1). In some countries, resistance levels exceed 25%, and recent estimates indicate that approximately 25 000 patients die annually in the EU from an infection with multidrug-resistant (MDR) bacteria [5]. In addition to mortality, the MDR-bacterial infections cost the EU over €1.5 billion annually in extra healthcare costs and productivity losses.
Of particular concern were blood infections owing to methicillin resistant Staphylococcus aureus, (MRSA); vancomycin-intermediate and vancomycin-resistant S. aureus; vancomycin-resistant Enterococcus spp. such as Enterococcus faecium (VRE); penicillin-resistant Streptococcus pneumoniae; third-generation cephalosporin-resistant Enterobacteriaceae (e.g. Escherichia coli, Klebsiella pneumoniae); carbapenem-resistant Enterobacteriaceae and Pseudomonas aeruginosa.
In 2007, the most common, single, MDR bacterium in the EU was MRSA. The sum of the cases of antibiotic-resistant Gram-positive bacteria (mostly MRSA and VRE) was comparable to that of antibiotic-resistant Gram-negative bacteria (third-generation cephalosporin-resistant E. coli and K. pneumoniae, and carbapenem-resistant P. aeruginosa). Resistance is increasing among certain Gram-negative bacteria, as exemplified by carbapenem-resistant Enterobacteriaceae producing the so-called New Delhi metallo-beta-lactamase 1 (NDM-1) that is often encoded by a conjugative plasmid [8]. As its name implies, NDM-1 producers are associated with the Indian subcontinent. In a microbiological and geographical survey, it was found that many NDM-1 positive patients in the UK had travelled to India or Pakistan within the past year, or had links with these countries, thus illustrating the ease with which drug resistance can spread between countries and continents.
(b). USA
Drug resistance levels in the USA are comparable to those in the EU, and two recent surveys from the Infectious Diseases Society of America (IDSA) and the Centers for Disease Control (CDC) provide valuable information about their prevalence (table 1). IDSA estimates that there are two million cases of nosocomial infections in the USA annually, resulting in around 99 000 deaths. According to CDC, 36 000 deaths are attributable to drug-resistant infections [6]. As in Europe, MRSA is the leading single killer claiming more lives each year than emphysema, HIV/AIDS, Parkinson's disease and homicide combined. Sepsis and pneumonia alone, both resulting from nosocomial infections, accounted for nearly 50 000 lives and cost the US healthcare system more than $8 billion in 2006. IDSA has also emphasized the importance of developing new drugs to treat the ‘ESKAPE’ pathogens, which are the bacteria described in §2a, plus drug-resistant Acinetobacter baumannii, a growing menace in clinical settings [9].
(c). Zoonoses and drug resistance
Owing to increased demand for animal products in the past three decades, there has been an intensification of agriculture and a concomitantly higher density of livestock. This has led to a rise in zoonotic diseases, 56 of which cause 2.5 billion cases in humans annually, thereby claiming an estimated 2.7 million lives [10]. Zoonotic infections are difficult to treat as there are often few, if any, therapeutic options. The situation has been exacerbated by the use of antibiotics in agriculture, notably as antimicrobial growth promoters in feed, where the selective pressure created leads to the emergence of antibiotic-resistant bacteria in livestock that can subsequently colonize or infect humans [11]. There are many examples of this spread, but I cite the use of nourseothricin in pigs. No resistance had been reported to this antibiotic in Enterobacteriaceae from humans and animals prior to its use as an antimicrobial growth promoter in 1983, but two years later, resistant E. coli were detected in the gut of pigs and in meat products. By 1990, nourseothricin-resistant E. coli were found in the gut flora of pig farmers, their families and neighbouring communities as well as in patients suffering from urinary tract infections [11]. Thus, not only did the strain spread from pigs to humans, but this also took place without any apparent selective pressure. Fortunately, the use of antimicrobial growth promoters has been banned in the EU in order to reduce the risk of cross-resistance to antibiotics used in human medicine [12].
4. The current state of anti-infective research in the pharmaceutical industry
Almost all of the antibiotics used to treat infections with the pathogens discussed in §2 were derived from a limited number of chemical scaffolds, mostly based on those of natural products, and these were discovered over 40 years ago. Since then, only two new broad-spectrum chemical entities (NCEs) have been developed and approved for use in humans: the oxazolidinones represented by linezolid and the lipopeptide antibiotic, daptomycin [13]. A few NCEs for specific diseases, such as TB (see §6), have been found however. The paucity of NCEs and the steadily dwindling number of new drug registrations (figure 1) reflects the innovation gap that continues to widen as more pharmaceutical companies disengage from this sector and limited financial means become ever scarcer.
Figure 1.
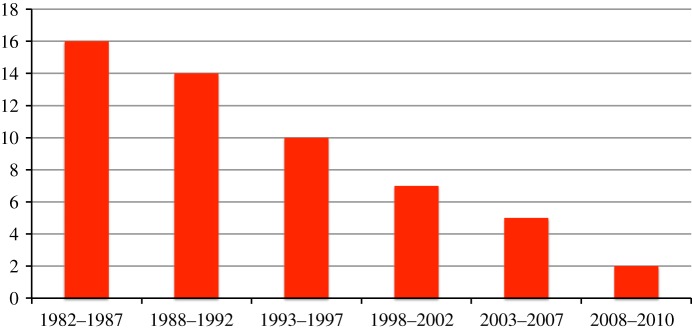
Number of new antibacterials approved for human use in the past 30 years. Figures were updated from those presented previously [6,14,15]. (Online version in colour.)
The lack of innovation in the 1970s and 1980s saw numerous ‘me-too’ drugs saturating a slow-growing market, leading in turn to a low return on investment. Furthermore, the often unfeasible and unpredictable approval pathway at regulatory agencies such as the Food and Drugs Administration (FDA) caused some companies to leave the antibiotics market completely. The subsequent development of blockbuster drugs for chronic care and non-communicable human diseases has further eroded the skill base and led to a career in antibiotic research becoming unattractive in industry. Unsurprisingly, greater resources were made available for research on the more lucrative therapeutic areas by the major pharmaceutical companies, and scientists who developed blockbusters received richer rewards. In recent times, the leading companies have undergone several rounds of mergers and fusions accompanied by consolidation and staff reduction. Consequently, the ability and expertise required to discover and develop a new antibacterial agent in big pharma has become exceedingly rare.
5. The role of biotechnology companies
There was once hope that the leaner and more agile biotechnology companies would compensate for this decline in research and development activity by closing the innovation gap and discovering novel NCEs that could later be acquired by, or developed in partnership with, big pharma; rightly so, as small biotechnology companies could almost certainly not bear the cost of phase II and III clinical trials alone. In 2005, a highly experienced drug expert, the late John F. Barrett of Merck Research Laboratories, analysed the drug discovery landscape and made some predictions of the success of potential products emerging from the biotechnology sector [16]. On the basis of public information and publications about drugs in development, his forecast included several new beta-lactam, glycopeptide and quinolone antibiotics, as well as next-generation oxazolidinones, together with new targets and drug leads derived from genomics such as inhibitors of the essential enzyme, peptidyl deformylase (PDF). Table 2 presents an update of this forecast [16] together with the subsequent outcome of the compound or target. As may be seen, although some of these new antibacterials are now on the market, a number have failed, others have received restricted regulatory approval or progressed far more slowly than anticipated. It is particularly disheartening to note the complete lack of NCEs in table 2 and the preponderance of ‘me-too’ drugs.
Table 2.
Development status of new antibacterials with respect to Barrett's predictions. Updated from [16].
| year expected on market | drug | class | status—comments |
|---|---|---|---|
| 2013 | genomics-derived inhibitors | ? | none yet |
| 2013 | PDF inhibitors | ? | none yet |
| 2012 | sutezolid | oxazolidinones | phase II |
| 2011 | omadacycline | tetracycline | phase II, stalled |
| 2010 | EP-013420 | bicyclic ketolide | phase I |
| 2010 | ceftaroline fosamil | beta-lactams | FDA approved for restricted use |
| 2009 | tebipenem | beta-lactams | progressing from phase II |
| 2008 | telavancin | lipoglycopeptide | approved for restricted use |
| 2008 | iclaprim | diaminopyridine DHFR inhibitor | phase II |
| 2008 | faropenem | beta-lactams | not approved by FDA |
| 2007 | ceftobripole | beta-lactams | approved but intellectual property issues |
| 2007 | oritivancin | glycopeptide | withdrawn |
| 2006 | garenoxacin | quinolone | withdrawn |
| 2006 | ramoplanin | glycopeptide | phase III |
| 2005 | doripenem | beta-lactams | on market |
| 2005 | dalbavancin | lipoglycopeptide | phase III |
| 2005 | tygacil | glycylcycline | injectable, on market with warnings |
| 2004 | telithromycin | ketolide (macrolide) | on market with black box warning |
| 2003 | daptomycin | lipopeptide | on market |
In 2009, a joint report from two European agencies presented their assessment of the antibacterial pipeline [5]. Fifteen systemically administered antibacterial agents with new mechanisms of action or targets were described as being in development. Although these compounds could potentially overcome multidrug resistance, most of them were in early phases of development and primarily targeting bacteria for which treatment options were already available. The lack of new agents with novel targets or mechanisms of action against MDR Gram-negative bacteria was emphasized as being of particular concern [5].
6. Success stories
A stunning example of how successful the pharmaceutical industry can be, when sufficient means and financial resources are assembled, is provided by the history of the development of antiretroviral agents (ARVs) for the treatment of HIV/AIDS. Within a decade of HIV being recognized as the aetiological agent of AIDS, not only had several individual ARVs been developed and approved, but a highly active combination ARV therapy (HAART) was also successfully implemented [17]. Today, there are 23 ARVs with five different mechanisms of action that can be combined in various forms, with the result that a diagnosis of AIDS has been transformed from a death sentence in the 1980s to a chronic and manageable condition from the mid-1990s onwards. A major factor in this chemotherapeutic breakthrough was accelerated approval from the regulatory agencies. The role of treatment activist groups in ensuring patients gained rapid access to new ARVs and in lobbying for HIV/AIDS research should also be highlighted.
On a more modest scale, good progress has been made to develop new drugs to treat TB, because the existing combination regimen is increasingly threatened by resistance to both first- and second-line drugs [18]. A fairly robust drug development pipeline has been established comprising NCEs and some repurposed drugs, which were first designed to treat other bacterial infections but are also active against Mycobacterium tuberculosis. Two NCEs, bedaquiline and delamanid are in late stage clinical trials [19,20] and both have been recently approved for the treatment of MDR-TB by regulatory agencies in Europe and the USA.
An important innovation here has been the proposal to develop a treatment comprising at least three NCEs rather than to combine them individually with existing drugs whose future is already compromised heavily by the spread of resistance [21]. However, the levels of funding available are inadequate to ensure progression of three NCEs through the development pipeline, let alone to progress a new combination therapy through clinical trials. This is highly regrettable, because unless better TB therapy becomes available all the progress made in controlling and reversing the HIV/AIDS pandemic will be lost given the extensive overlap between these two diseases [18].
7. Cause for hope
The examples from the TB and HIV fields provide us with two important reasons to be hopeful.
First, the TB drug discovery work convincingly demonstrates that not only are there more novel drug targets to be discovered but also that NCEs may still be found. Together, this augurs well for new treatments for other bacterial diseases. For instance, one lesson that can be learned is to adhere less strictly to Lipinski's ‘rule of five’ [22] a widely used filter proposed for drug-like molecules. These rules appear to be too stringent when applied to antibacterials, where penetration is more difficult and complex than in eukaryotic cells. Likewise, it is instructive to note that two of the most important TB drug candidates, delamanid and PA-824, are nitroaromatic molecules. Such compounds are typically discarded by big pharma companies because of perceived safety issues.
Second, given sufficient financial support and political and societal motivation, there is no overarching scientific reason why new drugs could not be developed to treat other MDR bacterial infections. Whether this will be via broad- or narrow-spectrum antibiotics remains to be seen. Again, as illustrated by the HIV-TB field, the importance of maintaining a high profile for the corresponding therapeutic area is essential in order to retain the attention of the decision makers and funders.
8. Conclusions and perspectives
The serious nature of drug resistance is periodically noted by our leaders and policymakers but soon loses their attention as other emergencies appear. Drug development is a lengthy process whose time frame is incompatible with government election cycles. It is heartening to note that in some industrialized countries, such as the UK, the Chief Medical Officer has raised political awareness of the grave risk posed by antimicrobial resistance to national and international health. There is thus hope of assertive action being taken. In addition to national governments, it is critical to obtain a long-lasting and sustainable commitment from other key stakeholders such as the international health agencies, the pharmaceutical and diagnostics industries, various healthcare providers, including insurance companies, the drug regulatory agencies, the public sector research funders, philanthropists and patient advocacy groups. Table 3 introduces these stakeholders and their motivations.
Table 3.
Potential stakeholders in global initiative to develop new antibacterial agents.
| stakeholder | motivation |
|---|---|
| national governments | ensure wellbeing of population, improve quality of life and economic output |
| international health agencies (WHO, Bill & Melinda Gates Foundation, etc.) | core activity, improve global health |
| pharmaceutical and diagnostics industries | generate new products, ensure profitability, increase market share, improve global health |
| national health systems and private health insurers | reduce insurance payments and increase profitability, improve services |
| drug regulatory agencies | speed access to market while ensuring patient safety, reform operating procedures |
| research funding agencies | support science of direct relevance to society, improve return on translational science, satisfy taxpayers |
| patient advocacy groups | ensure patients’ voices are heard and needs met, raise public awareness |
Most governments in the industrialized world dedicate approximately 1% of their GDP to research and development. Some of these public funds underpin discovery science and generate leads for drug development that give rise to intellectual property. This is a laudable starting point, but one which could be strengthened by dedicating a fixed percentage of the research spending to a global fund for new antibacterial drug development, because this is vital for our globalized society. Other stakeholders should also be encouraged to contribute. For instance, in the long term, private health insurance providers could make substantial economies if their clients spent less time in hospital or recovering from MDR infections.
Publically funded patents for new NCEs often remain unexploited, as documented above, because there is insufficient funding available to support the development phase. Engaging earlier with industry may remove this obstacle and more public–private partnerships are to be welcomed, such as Europe's Innovative Medicines Initiative that aims to improve the drug development process by supporting more efficient discovery and development of better and safer medicines (http://www.imi.europa.eu). The emerging economies of Brazil, Russia, India, China and South Africa (the BRICS) could also contribute in this arena, because they are all home to excellent pharmaceutical companies that produce off-patent, generic medicines as well as providing services to big pharma. Access to external intellectual property would boost innovation in the BRICS while allowing medicines to be produced at more competitive rates for use worldwide.
No commercial company will knowingly engage in a potentially loss-making drug production activity. Incentives therefore need to be made available in order to ensure that products reach the marketplace and remain there sufficiently long to be profitable. Various forms of encouragement could be provided, including interest-free loans and tax reductions from national governments, or extended patent life for products deemed of strategic importance to the healthcare sector. Cash rewards could be also be paid by an international fund or governments to companies whose products progress through the different phases of clinical trials, especially if they address important medical needs such as killing MRSA or combatting MDR-TB [23].
It is essential to reduce the costs of drug development and the risks to companies that engage therein. According to the Boston Consulting Group, it not only takes more than 10 years to bring a new drug to market now, but the costs incurred are well in excess of one billion dollars [24]. Phase III clinical trials account for over half the cost of drug development, so there are grounds for seeking economies in this sector without compromising patient safety [25]. The truly necessary studies for safety and efficacy are essentially complete by the end of phase II, whereas in a phase III, clinical trial the efficacy of a new drug is compared with that of the standard treatment, on a far broader scale than in phase II, but looking for non-inferiority. There is thus a sound case for replacing phase III trials, by larger, more extensive phase IIb clinical trials in order to provide patients with much needed drugs sooner and at lower cost [25]. It is sobering to note that in the midst of the Second World War, Merck & Co. was not only able to establish an industrial-scale manufacturing process for streptomycin [26], but also to begin curing patients of TB and other bacterial infections less than two years after the antibiotic's discovery! In the same vein, we should recall that the HIV/AIDS pandemic was slowed and reversed in part owing to the accelerated approval of ARV. Fast-track evaluation of drugs for conditions with no approved cures, as typified by many life-threatening MDR bacterial infections, should be envisioned whenever possible.
Developing new drugs for bacterial infections is of global importance and needs to be supported and intensified, but with the knowledge that microbial evolution will continue unabated and drug resistance will always emerge eventually as a result of natural selection. Past practice has shown that careful antibiotic management can prolong their active life and ensure profits for their manufacturers. These opportunities should be grasped as it is incumbent upon us to ensure that future generations benefit from antibiotics as ours has done.
Acknowledgements
Many thanks to the MM4TB Consortium for encouragement.
Funding statement
I thank the European Community's Seventh Framework Programme (grant no. 260872) and the Swiss National Science Foundation (31003A-140778) for supporting my work.
References
- 1.Hopwood DA. 2007. Streptomyces in nature and medicine: the antibiotic makers. Oxford, UK: Oxford University Press. [Google Scholar]
- 2.Walsh C. 2003. Antibiotics: actions, origins, resistance. Washington, DC: ASM Press. [Google Scholar]
- 3.Henderson DA. 1987. Principles and lessons from the smallpox eradication programme. Bull. World Health Organ. 65, 535–546. [PMC free article] [PubMed] [Google Scholar]
- 4.WHO 2012. See http://www.who.int/mediacentre/factsheets/fs310/en/index2.html.
- 5.ECDC/EMEA 2009. The bacterial challenge: time to react. London, UK: European Centre for Disease Control & European Medicines Agency. [Google Scholar]
- 6.CDC 2013. Antibiotic resistance threats in the United States, 2013 1–114 See http://www.cdc.gov/drugresistance/threat-report-2013/.
- 7.IDSA 2013. See http://www.idsociety.org/AR_Facts/.
- 8.Kumarasamy KK, et al. 2010. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect. Dis. 10, 597–602. ( 10.1016/S1473-3099(10)70143-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12. ( 10.1086/595011) [DOI] [PubMed] [Google Scholar]
- 10.Karesh WB, et al. 2012. Ecology of zoonoses: natural and unnatural histories. Lancet 380, 1936–1945. ( 10.1016/S0140-6736(12)61678-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witte W. 1998. Medical consequences of antibiotic use in agriculture. Science 279, 996–997. ( 10.1126/science.279.5353.996) [DOI] [PubMed] [Google Scholar]
- 12.European Commission 2005. Ban on antibiotics as growth promoters in animal feed enters into effect 2005. IP/05/1687 22/12/2005 See http://europa.eu/rapid/press-release_IP-05-1687_en.htm.
- 13.Fischbach MA, Walsh CT. 2009. Antibiotics for emerging pathogens. Science 325, 1089–1093. ( 10.1126/science.1176667) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Nussbaum F, Brands M, Hinzen B, Weigand S, Habich D. 2006. Antibacterial natural products in medicinal chemistry: exodus or revival? Angew. Chem. 45, 5072–5129. ( 10.1002/anie.200600350) [DOI] [PubMed] [Google Scholar]
- 15.Demain AL. 2011. Antibiotic discovery: a step in the right direction. Chem. Biol. 18, 939 ( 10.1016/j.chembiol.2011.08.002) [DOI] [PubMed] [Google Scholar]
- 16.Barrett JF. 2005. Can biotech deliver new antibiotics? Curr. Opin. Microbiol. 8, 498–503. ( 10.1016/j.mib.2005.08.007) [DOI] [PubMed] [Google Scholar]
- 17.Palmisano L, Vella S. 2011. A brief history of antiretroviral therapy of HIV infection: success and challenges. Ann. Istituto Superiore Sanita 47, 44–48. [DOI] [PubMed] [Google Scholar]
- 18.Zumla A, Nahid P, Cole ST. 2013. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 12, 388–404. ( 10.1038/nrd4001) [DOI] [PubMed] [Google Scholar]
- 19.Diacon AH, et al. 2009. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 360, 2397–2405. ( 10.1056/NEJMoa0808427) [DOI] [PubMed] [Google Scholar]
- 20.Gler MT, et al. 2012. Delamanid for multidrug-resistant pulmonary tuberculosis. N. Engl. J. Med. 366, 2151–2160. ( 10.1056/NEJMoa1112433) [DOI] [PubMed] [Google Scholar]
- 21.Diacon AH, et al. 2012. 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: a randomised trial. Lancet 380, 986–993. ( 10.1016/S0140-6736(12)61080-0) [DOI] [PubMed] [Google Scholar]
- 22.Lipinski C. 2000. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 44, 235–249. ( 10.1016/S1056-8719(00)00107-6) [DOI] [PubMed] [Google Scholar]
- 23.Médecins Sans Frontières/Doctors without Borders. 2014. Accelerating innovation and access to medicines for tuberculosis through open collaboration: a push, pull, pool approach (“the 3P Project”).
- 24.BCG 2011. Life sciences R&D: changing the innovation equation in India 2011 See http://www.bcg.com/expertise_impact/biopharma_summit.aspx.
- 25.Lachmann PJ. 2012. The penumbra of thalidomide, the litigation culture and the licensing of pharmaceuticals. Q. J. Med. 105, 1179–1189. ( 10.1093/qjmed/hcs148) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pringle P. 2012. Experiment eleven. Deceit and betrayal in the discovery of the cure for tuberculosis. London, UK: Bloomsbury. [Google Scholar]