

Abstract
Organisms show great variation in ploidy level. For example, chromosome copy number varies among cells, individuals and species. One particularly widespread example of ploidy variation is found in haplodiploid taxa, wherein males are typically haploid and females are typically diploid. Despite the prevalence of haplodiploidy, the regulatory consequences of having separate haploid and diploid genomes are poorly understood. In particular, it remains unknown whether epigenetic mechanisms contribute to regulatory compensation for genome dosage. To gain greater insights into the importance of epigenetic information to ploidy compensation, we examined DNA methylation differences among diploid queen, diploid worker, haploid male and diploid male Solenopsis invicta fire ants. Surprisingly, we found that morphologically dissimilar diploid males, queens and workers were more similar to one another in terms of DNA methylation than were morphologically similar haploid and diploid males. Moreover, methylation level was positively associated with gene expression for genes that were differentially methylated in haploid and diploid castes. These data demonstrate that intragenic DNA methylation levels differ among individuals of distinct ploidy and are positively associated with levels of gene expression. Thus, these results suggest that epigenetic information may be linked to ploidy compensation in haplodiploid insects. Overall, this study suggests that epigenetic mechanisms may be important to maintaining appropriate patterns of gene regulation in biological systems that differ in genome copy number.
Keywords: DNA methylome, dosage compensation, epigenetics, gene regulation, haplodiploid, social insect
1. Introduction
Organisms display a remarkable diversity in ploidy level [1–5]. For example, all sexual organisms show variation in ploidy during their life cycle. In addition, members of different species sometimes vary in ploidy number. Such ploidy variation shapes molecular evolution, genetic interactions and gene function [5–9]. Thus, variation in ploidy fundamentally affects evolutionary and developmental processes.
A prime example of variation in ploidy is embodied by the haplodiploid genetic system. Haplodiploid species are typically characterized by having unfertilized eggs develop into haploid males and fertilized eggs develop into diploid females [3,4]. The haplodiploid genetic system has arisen at least 17 independent times during the course of animal evolution [4], and is the ancestral genetic system of the order Hymenoptera (ants, bees and wasps) [3]. Consequently, as many as 20% of all animal species may be haplodiploid [10]. Despite the taxonomic prevalence of haplodiploidy, the regulatory consequences of ploidy differences between sexes remain largely unknown (but see [6,9,11]). This lack of information represents a gap in our understanding of how biological systems respond to ploidy variation.
Epigenetic modifications to chromatin are prime candidates for regulating gene function in haplodiploid taxa. Epigenetic marks are heritable and make fundamental contributions to gene regulation [12]. One of the most important types of epigenetic marks is the methylation of DNA. DNA methylation is found in all three domains of life, suggesting a role in the common ancestor of all metazoa [13,14].
Recently, DNA methylation and histone modifications have been implicated in the regulation of social insect caste differences [15–19]. In addition, global sex chromosome dosage compensation is achieved in Drosophila and mammals by epigenetic mechanisms [20,21], demonstrating that distinct epigenetic states can achieve transcriptional compensation associated with ploidy variation. However, the contributions of epigenetic inheritance to regulatory mechanisms that compensate for ploidy differences in haplodiploids have not been investigated. In this study, we attempted to gain insights into whether epigenetic information was associated with gene regulation in haplodiploid taxa.
In order to assess the epigenetic states of haploid and diploid genomes, we compared single nucleotide resolution DNA methylation profiles (DNA methylomes) of haploid and diploid individuals of the red imported fire ant, Solenopsis invicta. Sex in S. invicta and many other hymenopteran insects is determined by complementary sex determination [3]. Under single-locus complementary sex determination, sex is controlled by zygosity at a single genetic locus. In this case, heterozygous individuals develop into females and hemizygous (haploid) individuals develop into males.
Interestingly, diploid individuals that are homozygous at the sex-determining locus develop into diploid males. Diploid males are generally rare in hymenopteran populations. However, diploid males are produced at high frequency in invasive S. invicta owing to loss of variation at the sex-determining locus [22]. S. invicta diploid males are larger than haploid males, but otherwise have highly similar morphologies and behaviours to haploid males. Moreover, haploid and diploid males differ substantially in phenotype from diploid queens and workers [23,24]. Importantly, the common production of haploid and diploid males makes S. invicta well suited to investigating epigenetic gene regulation in the context of ploidy differences while simultaneously controlling for sex differences.
Our analyses uncovered striking differences in DNA methylation between haploid and diploid individuals in S. invicta. The link between DNA methylation and ploidy variation suggests that haploid and diploid genomes in S. invicta exhibit distinct epigenetic states. These results provide support for the hypothesis that epigenetic mechanisms are associated with genomic dosage compensation of haplodiploid organisms. More broadly, our results suggest that epigenetic information may influence the evolution of ploidy differences among cells, organisms and species.
2. Material and methods
(a). Whole-genome bisulfite-sequencing
Sample collection, DNA extraction, bisulfite conversion, sequencing, quality control and read mapping were performed as described elsewhere [25]. Briefly, all samples were taken from a single S. invicta colony. Male ploidy was confirmed by DNA microsatellite analysis at three to four highly variable loci. Genomic DNA was separately pooled from whole bodies of haploid males, diploid males, alate queens and workers, comprising one sample per caste. We obtained between seven and nine times mean coverage of genomic CpG sites per sample [25]. Solenopsis invicta SI2.2.3 gene models were used for analysis of genes, exons and introns [26]. Solenopsis invicta whole-genome bisulfite-sequencing data are available online from Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo; GSE39959).
(b). DNA methylation targets and levels
Significantly methylated CpG sites were assessed using a binomial test, implemented using the Math::CDF module in Perl, which incorporated deamination rate (from our unmethylated control) as the probability of success, and assigned a significance value to each CpG site related to the number of unconverted reads (putatively methylated Cs) compared with the expected number from control [15]. Resulting p-values were then adjusted for multiple testing [27]. Only sites with false discovery rate (FDR)-corrected binomial p-values <0.01 and ≥3 reads were considered ‘methylated’. Fractional methylation values were calculated as described previously [25] for each CpG site or for each genomic feature (exons and introns).
(c). Hierarchical clustering and dendrogram generation
The pvclust package in the R statistical computing environment was used to generate clustering and dendrogram diagrams of fractional methylation values of exons and introns [28]. We used the ‘average’ linkage agglomeration method, the ‘correlation’ distance measure and 1000 bootstrap replications. Only those genomic features (exons and introns) targeted by DNA methylation in at least one caste, according to FDR-corrected binomial tests, were included in hierarchical clustering analysis. Fractional DNA methylation values of a given exon or intron in castes that did not exhibit significant DNA methylation were set to zero prior to hierarchical clustering in order to minimize noise contributed by unconverted, putatively unmethylated cytosines.
(d). Differential DNA methylation
Significantly differentially methylated features (exons and introns) were assessed for each pairwise comparison between castes using generalized linear models (GLMs), implemented in the R statistical computing environment [28], where methylation levels for features were modelled as functions of ‘caste’ and ‘CpG position’. If caste contributed significantly (chi-square test of GLM terms) to the methylation status of a feature (after adjustment for multiple testing using the method of [27]), then it was considered differentially methylated between castes [15]. Only CpG sites that were methylated in one or both castes and covered by at least four reads in both libraries were used in these comparisons, and only features with at least three such CpG sites were considered in further analyses.
Once exons and introns were assigned differential methylation status using the above GLM, each significantly differentially methylated exon or intron was called as elevated in the caste with higher fractional methylation status of that feature. These features were then combined by gene, and each gene was called as a unidirectional differentially methylated gene (DMG) if more than two-thirds of the gene's differentially methylated features were elevated in the same direction.
(e). Gene ontology
Gene ontology (GO) annotations were assigned using Blast 2GO [29]. Significant enrichment was assessed with a Fisher's exact test and corrected for multiple testing with a Benjamini–Hochberg FDR [27]. The ‘generic GO slim’ subset of GO terms was used to assess significantly enriched terms (FDR, p < 0.05).
(f). Gene expression
Solenopsis invicta whole-body cDNA microarray data [30,31] were mapped to S. invicta gene models as described previously [25]. Expression ratios between queen, worker and haploid male castes [30] were calculated as 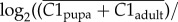
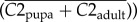 , where C1 is the expression value estimated by BAGEL [32] for the first caste, and C2 is the estimated expression value for the second caste.
, where C1 is the expression value estimated by BAGEL [32] for the first caste, and C2 is the estimated expression value for the second caste.
For each gene, we assessed the coefficient of variation (standard deviation/mean; CV) of expression values as the mean of CV values calculated separately for whole body S. invicta adult and pupal workers, queens and haploid males (median of five biological replicates per morph) [30].
For array data from haploid and diploid males (Gene Expression Omnibus accession: GSE42786 and GSE35217) [33], the Limma R package [34] was used to perform background correction (method = ‘normexp’), and within- and between-array normalization (method = ‘printtiploess’ and method = ‘Rquantile’, respectively), followed by generation of gene expression ratios between haploid and diploid male arrays.
(g). Coding sequence evolution
We used OrthoDB [35] 12-insect orthology data to assign single-copy orthologues between the ants S. invicta, Pogonomyrmex barbatus and Linepithema humile. Non-synonymous substitutions per non-synonymous site and synonymous substitutions per synonymous site were determined for the S. invicta lineage using codeml in PAML as described previously [36]. Genes with aligned sequence length ≤100, dS ≥ 4 or dN/dS ≥ 4 were filtered out prior to analysis.
3. Results
(a). DNA methylation is associated with ploidy in Solenopsis invicta
We observed significant differences in methylation level in one or more pairwise comparisons between castes for 3478 exons (32.7% of 10 628 exons methylated in one or more caste) and 577 introns (23.3% of 2479 introns methylated in one or more caste) in S. invicta. Ultimately, we classified any gene with a significant difference in the methylation level of at least one exon or intron in at least one pairwise comparison between castes as a DMG.
We found that DNA methylation levels in all libraries derived from diploid individuals were more similar to one another than to the library derived from haploid males (figure 1a). Diploid males, queens and workers all showed methylation profiles that were highly diverged from haploid males. In particular, the majority of significantly differential methylation occurred between the haploid and diploid castes (figure 1b; electronic supplementary material, figure S1). The pairwise comparison with the greatest number of DMGs was that between haploid and diploid males. This is particularly noteworthy given the high degree of morphological and behavioural similarity between haploid and diploid males in S. invicta [23,24]. The pairwise comparison with the fewest differences was that between queens and workers, both of which are diploid females (figure 1b; electronic supplementary material, figure S1). We note that these findings are unlikely to be the result of bisulfite conversion efficiency, as the queen library exhibited the highest unmethylated cytosine non-conversion rate, and haploid and diploid males had the most similar unmethylated cytosine non-conversion rate among all libraries (electronic supplementary material, table S1).
Figure 1.
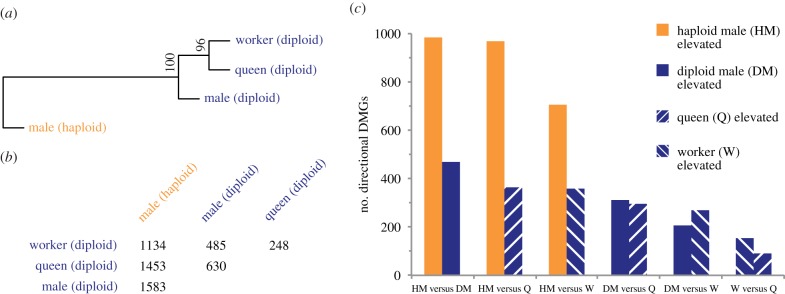
DNA methylation differs between haploid and diploid castes in S. invicta. (a) Dendrogram produced by hierarchical clustering of fractional methylation levels representing all introns and exons targeted by DNA methylation in at least one library (n = 10 560 genetic features); bootstrap probability values are shown. (b) Number of differentially methylated genes (DMGs) detected between castes. (c) Number of directional DMGs from panel (b) that exhibit pairwise elevated methylation in haploid and diploid castes. (Online version in colour.)
We next defined directional DMGs as those wherein at least two-thirds of differentially methylated features (exons and introns) were more highly methylated in one caste of a given pairwise comparison. For example, if three of four differentially methylated features were more highly methylated in haploid males, then the gene would be categorized as having elevated methylation in haploid males relative to diploid males. By contrast, if two of four differentially methylated features were more highly methylated in haploid males (with the other two more highly methylated in diploid males), then the gene would not be characterized as a directional DMG. Analysis of directional DMGs provided insights into the castes that most frequently exhibited elevated DNA methylation levels. In each comparison between haploid and diploid castes, we observed considerably more DMGs with elevated methylation levels biased to the haploid caste (figure 1c; electronic supplementary material, figure S1).
(b). Differentially methylated genes in Solenopsis invicta have unique characteristics
We conducted enrichment analysis of GO annotations for DMGs relative to methylated non-DMGs. We found that DMGs in S. invicta were enriched for annotations including ‘nucleotide binding’ and ‘developmental process’ (table 1; electronic supplementary material, table S2). By contrast, non-DMGs were enriched for terms related to core cellular functions such as ‘translation’ (table 1; electronic supplementary material, table S3), as is typical of methylated genes in general in S. invicta and other insects [25,37].
Table 1.
Enrichment of gene ontology (GO) annotations associated with differentially methylated genes (DMGs) and non-DMGs in S. invicta.
| term | categorya | accession number | no. genes | fold enrichmentb | FDR p-value |
|---|---|---|---|---|---|
| differentially methylated genes | |||||
| binding | F | GO:0005488 | 1434 | 1.13 | 0.0001 |
| nucleotide binding | F | GO:0000166 | 464 | 1.28 | 0.0277 |
| developmental process | P | GO:0032502 | 278 | 1.36 | 0.0435 |
| chromosome | C | GO:0005694 | 122 | 1.67 | 0.0435 |
| non-differentially methylated genes | |||||
| structural molecule activity | F | GO:0005488 | 64 | 1.92 | 0.0082 |
| cytoplasmic part | C | GO:0000166 | 447 | 1.26 | 0.0103 |
| ribosome | C | GO:0032502 | 111 | 1.51 | 0.0427 |
| translation | P | GO:0005694 | 138 | 1.42 | 0.0488 |
aP, biological process; F, molecular function; C, cellular component.
bEnrichment determined for DMGs or non-DMGs relative to all other methylated genes.
We further tested whether there were significant differences between DMGs and non-DMGs in a number of gene characteristics in order to better understand which types of genes are variably methylated. Specifically, we determined whether DMGs and non-DMGs differed in overall DNA methylation level (all castes combined), gene length, gene expression variability among samples as measured by the coefficient of variation [30] and rates of protein coding sequence evolution.
We found that DMGs exhibited substantially lower DNA methylation levels, and were substantially longer in terms of both coding sequence and gene body, than non-DMGs (table 2; p < 0.0001 in each case). DMGs were also modestly, but significantly, more variable in expression, and more highly conserved at the sequence level, than non-DMGs (table 2; p < 0.01 in each case).
Table 2.
Characteristics of differentially methylated genes (DMGs) and non-DMGs in S. invicta.
| trait | DMGs (mean ± s.e.; gene count) | non-DMGs (mean ± s.e.; gene count) | Mann–Whitney U-test p-value |
|---|---|---|---|
| fractional coding sequence methylation | 0.186 ± 0.003; 2518 | 0.255 ± 0.005; 1705 | <0.0001 |
| fractional gene body (exons + introns) methylation | 0.174 ± 0.002; 2521 | 0.240 ± 0.004; 1718 | <0.0001 |
| coding sequence length | 1888.09 ± 31.07; 2518 | 1254.34 ± 29.27; 1705 | <0.0001 |
| gene body length | 3869.21 ± 74.75; 2521 | 2483.83 ± 67.44; 1718 | <0.0001 |
| gene expression coefficient of variation | 0.167 ± 0.002; 1068 | 0.160 ± 0.002; 836 | 0.0022 |
| non-synonymous substitutions per non-synonymous site (dN) | 0.033 ± 0.001; 1728 | 0.037 ± 0.001; 1012 | 0.0046 |
| synonymous substitutions per synonymous site (dS) | 0.344 ± 0.004; 1727 | 0.375 ± 0.005; 1012 | <0.0001 |
| dN/dS | 0.101 ± 0.002; 1725 | 0.102 ± 0.003; 1012 | 0.7962 |
We next investigated whether variation in DNA methylation was associated with variation in gene expression among castes. In order to investigate the regulatory significance of differential DNA methylation, we integrated available microarray gene expression data from S. invicta haploid males, diploid queens and diploid workers [30], as well as from a separate comparison of haploid and diploid males [33].
Our analyses revealed that directional DMGs with elevated methylation in haploid castes versus diploid castes were significantly more highly expressed in haploid castes than in diploid castes (figure 2; electronic supplementary material, figure S1). This finding is consistent with the observed association between DNA methylation and active gene expression in insects [25,38,39]. Intriguingly, however, we found no significant association between differential methylation and gene expression bias when examining genes differentially methylated between worker and queen castes (both diploid; figure 2b).
Figure 2.
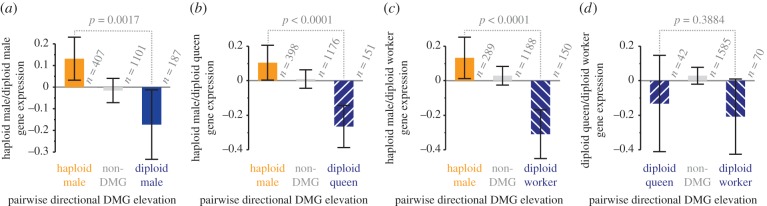
Gene expression bias is associated with directional differentially methylated genes (DMGs) in S. invicta. DMGs that exhibit elevated methylation in haploid males are more highly expressed in haploid males, whereas DMGs that exhibit elevated methylation in diploid (a) males, (b) queens or (c) workers are more highly expressed in diploid males, queens or workers, respectively. By contrast, there is no significant difference between the ratio of expression for DMGs that exhibit elevated methylation in (d) a pairwise comparison of queens and workers. Expression ratio data were standardized (mean zero, unit variance) following log2-transformation; p-values denote the results of Mann–Whitney U-tests. (Online version in colour.)
Finally, we determined whether directional DMGs between males of different ploidy were enriched for distinct GO annotations. Our goal with this analysis was to determine whether elevated methylation in haploid males, which may reflect an epigenetic state associated with haploid gene upregulation (figure 2a–c), was targeted to genes associated with distinct functions, when compared with other DMGs.
We found that genes with elevated methylation in haploid males relative to diploid males were enriched for several metabolic process terms, as well as the terms ‘nucleotide binding’ and ‘chromosome’ (electronic supplementary material, table S4). By contrast, there were no significantly enriched terms below the FDR cut-off (p < 0.05) for genes with elevated methylation in diploid males relative to haploid males. Nevertheless, several terms related to growth and development, including ‘developmental process’, were enriched among genes with elevated methylation in diploid males prior to FDR correction (p < 0.05; electronic supplementary material, table S5). Together, these data suggest a marked difference between the gene classes that exhibit elevated methylation in haploid and diploid males. Elevated methylation in haploid males appears to preferentially target genes associated with basal cellular processes, whereas elevated methylation in diploid males may be associated with a larger number of genes implicated in development.
(c). Orthologues of genes implicated in Drosophila dosage compensation exhibit differential DNA methylation and expression in Solenopsis invicta
We assessed patterns of DNA methylation and gene expression for S. invicta orthologues of genes associated with dosage compensation in Drosophila. Our goal was to provide initial insights into whether common molecular machinery may underlie dosage compensation for sex chromosomes in Drosophila and regulatory compensation for haploidy versus diploidy in S. invicta. Interestingly, we found that orthologues of four of eight genes (with data) related to dosage compensation in D. melanogaster were differentially methylated between haploid and diploid males in S. invicta (electronic supplementary material, table S6 and figure S2). Moreover, three of four of these genes (with data) were differentially expressed between haploids and diploids (electronic supplementary material, table S6). Thus, several genes involved in Drosophila dosage compensation are differentially methylated and differentially expressed between haploids and diploids in S. invicta.
4. Discussion
The purpose of this investigation was to gain a greater understanding of the molecular mechanisms that regulate gene function among individuals that differ in ploidy. Our analysis of DNA methylation patterns among S. invicta castes uncovered strong associations between levels of DNA methylation and ploidy. These methylation differences were further found to be related to gene expression differences among castes.
We found most DMGs arose between haploid and diploid castes, and therefore that the number of DMGs was not related to the overall morphological similarity of the castes being compared (figure 1). In particular, our comparison of haploid and diploid males produced more DMGs than our comparison of haploid males and diploid queens, which are sexually dimorphic, and produced many more DMGs than were observed between diploid queens and diploid workers, which are a classical example of insect polyphenism. Thus, in S. invicta, differences in DNA methylation more closely track differences in ploidy than differences in morphology, behaviour or physiology associated with distinct queen and worker castes [40].
The DNA methylomes of haploid males and diploid females were sequenced previously in the ants Camponotus floridanus and Harpegnathos saltator [18]. When we assessed directional DMGs between adult castes of C. floridanus and H. saltator, we found that, in four of six comparisons between haploid and diploid castes, more DMGs were elevated in haploids than in diploids (three of three comparisons in H. saltator and one of three comparisons in C. floridanus; electronic supplementary material, figure S3). Thus, the data of Bonasio et al. [18] further suggest that haploids may be prone to elevated DNA methylation relative to diploids.
Intriguingly, we found that DMGs, as a whole, exhibited several distinguishing characteristics in S. invicta. DMGs were enriched relative to non-DMGs for the GO annotations ‘nucleotide binding’ and ‘developmental process’ (table 1), consistent with important regulatory roles for differential DNA methylation in S. invicta, as in the honeybee [15,16,19]. Furthermore, DMGs differed significantly from other methylated genes in methylation level, gene length, expression variability and substitution rate (table 2), suggesting key architectural and regulatory differences between DMGs and non-DMGs.
In S. invicta, differential methylation events were also associated with ploidy-specific gene expression bias (figure 2), suggesting that DMGs are associated with regulatory differences between haploid and diploid genomes. Interestingly, the association of intragenic DNA methylation with active gene expression in insects suggests DNA methylation may be a useful marker of active chromatin states [25,37,38]. In support of this idea, the presence of DNA methylation has recently been linked to the presence of several active histone modifications in insect genomes [25,41]. We speculate that elevated haploid DNA methylation may be indicative of regulatory pressures associated with the single-copy state of haploid loci.
Notably, our data cannot directly address whether changes in DNA methylation are the cause or consequence of changes in gene expression. However, experimental investigations in model systems indicate the DNA methylation can cause changes in gene function through interactions with other components of chromatin. For example, DNA methylation has been shown to affect alternative splicing through its interaction with RNA polymerase II [42]. In addition, DNA methylation has been shown to alter the positioning of certain histone variants, which ultimately influence gene expression [43]. Experimental changes in levels of DNA methylation have also been found to lead to changes in levels of gene expression in Arabidopsis [43,44], suggesting that intragenic methylation has functional effects.
The suggestion that epigenetic gene regulation plays a role in genome-wide chromosomal dosage compensation is consistent with the observation that epigenetic marks play key roles in sex chromosome dosage compensation [20,21,45]. Intriguingly, we found that S. invicta orthologues of several genes implicated in D. melanogaster dosage compensation were differentially regulated between haploid and diploid castes (electronic supplementary material, table S6 and figure S2), raising the prospect of some degree of molecular convergence. Although the genome of D. melanogaster is not substantially methylated, previous studies have revealed that, in species that harbour functional DNA methylation systems, DNA methylation interacts with histone modifications associated with dosage compensation in D. melanogaster [25,41]. Regardless, we note that the mechanisms by which intragenic methylation affect gene function remain poorly understood [39], and direct connections between mechanisms of sex chromosome dosage compensation and ploidy compensation remain speculative at present.
Given the evidence for different epigenetic states in haploid and diploid S. invicta, it is important to consider why one may expect different regulatory requirements for genes in haploid genomes when compared with diploid genomes. For example, there may be increased metabolic requirements placed on loci in haploid, relative to diploid, genomes [1]. Our results agree with this notion, as several metabolic process GO annotations were enriched among genes with elevated DNA methylation in haploid males (electronic supplementary material, table S4). One additional reason for epigenetic states to differ between haploid and diploid genomes may be related to the amelioration of haploid gene expression noise, particularly at genes essential to cellular function. Indeed, gene expression variability is negatively associated with dosage in yeast, where diploid cells exhibit less expression variability than haploid cells [46], and where overall gene expression variability can lower organismal fitness [47]. We previously found that DNA methylation is negatively associated with the coefficient of variation of gene expression among replicate S. invicta samples [25], potentially implying a role for DNA methylation in the stabilization of gene expression [48]. We speculate that, if DNA methylation plays a role in reducing gene expression stochasticity [48], the variable expression of haploid loci may itself provide an impetus for elevated levels of DNA methylation in haploid males.
Overall, our results suggest that epigenetic mechanisms are associated with regulatory response to global differences in dosage in haplodiploid hymenopterans. However, we must emphasize that these results are preliminary in nature, requiring additional study to resolve whether epigenetic information is functionally implicated in ploidy-associated regulatory compensation. One important consideration is that haploid males in Hymenoptera are known to compensate for lower genomic content relative to diploid females through endoreplication [6,9], wherein cells increase their genomic content without dividing [1]. Our results raise the possibility that epigenetic information similarly contributes to haploid regulatory compensation, particularly given that endoreplication is not ubiquitous among tissues [6]. An alternative, but presently unexplored possibility is that endoreplication itself is associated with epigenetic changes.
We have shown that differential DNA methylation is more closely linked to ploidy variation than to queen and worker castes in the fire ant S. invicta. We observed elevated DNA methylation in haploids, and a positive association between ploidy-biased DNA methylation and gene expression, which together demonstrate the existence of distinct epigenetic states for haploid and diploid genomes. Overall, our results highlight the prospect that epigenetic mechanisms may be involved in achieving ploidy compensation in haplodiploid taxa.
Acknowledgements
We thank M. Nipitwattanaphon, J. Wang, K. G. Ross, O. Riba-Grognuz, Y. Wurm, C. Khurewathanakul and L. Keller for access to gene expression data.
Funding statement
This work was supported by the US National Science Foundation (grant nos. DEB-1011349, DEB-0640690, IOS-0821130 and MCB-0950896) and the Georgia Tech–Elizabeth Smithgall Watts endowment.
References
- 1.Edgar BA, Orr-Weaver TL. 2001. Endoreplication cell cycles: more for less. Cell 105, 297–306. ( 10.1016/S0092-8674(01)00334-8) [DOI] [PubMed] [Google Scholar]
- 2.Sassone-Corsi P. 2002. Unique chromatin remodeling and transcriptional regulation in spermatogenesis. Science 296, 2176–2178. ( 10.1126/science.1070963) [DOI] [PubMed] [Google Scholar]
- 3.Heimpel GE, de Boer JG. 2008. Sex determination in the Hymenoptera. Annu. Rev. Entomol. 53, 209–230. ( 10.1146/annurev.ento.53.103106.093441) [DOI] [PubMed] [Google Scholar]
- 4.Otto SP, Jarne P. 2001. Haploids: hapless or happening? Science 292, 2441–2443. ( 10.1126/science.1062890) [DOI] [PubMed] [Google Scholar]
- 5.Galitski T, Saldanha AJ, Styles CA, Lander ES, Fink GR. 1999. Ploidy regulation of gene expression. Science 285, 251–254. ( 10.1126/science.285.5425.251) [DOI] [PubMed] [Google Scholar]
- 6.Aron S, de Menten L, Van Bockstaele DR, Blank SM, Roisin Y. 2005. When hymenopteran males reinvented diploidy. Curr. Biol. 15, 824–827. ( 10.1016/j.cub.2005.03.017) [DOI] [PubMed] [Google Scholar]
- 7.Adams KL, Wendel JF. 2005. Novel patterns of gene expression in polyploid plants. Trends Genet. 21, 539–543. ( 10.1016/j.tig.2005.07.009) [DOI] [PubMed] [Google Scholar]
- 8.Otto SP. 2007. The evolutionary consequences of polyploidy. Cell 131, 452–462. ( 10.1016/j.cell.2007.10.022) [DOI] [PubMed] [Google Scholar]
- 9.Rasch E, Cassidy J, King R. 1977. Evidence for dosage compensation in parthenogenetic Hymenoptera. Chromosoma 59, 323–340. ( 10.1007/BF00327973) [DOI] [PubMed] [Google Scholar]
- 10.Evans J, Shearman DCA, Oldroyd BP. 2004. Molecular basis of sex determination in haplodiploids. Trends Ecol. Evol. 19, 1–3. ( 10.1016/j.tree.2003.11.001) [DOI] [PubMed] [Google Scholar]
- 11.Scholes DR, Suarez AV, Paige KN. 2013. Can endopolyploidy explain body size variation within and between castes in ants? Ecol. Evol. 3, 2128–2137. ( 10.1002/ece3.623) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonasio R, Tu S, Reinberg D. 2010. Molecular signals of epigenetic states. Science 330, 612–616. ( 10.1126/science.1191078) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klose RJ, Bird AP. 2006. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97. ( 10.1016/j.tibs.2005.12.008) [DOI] [PubMed] [Google Scholar]
- 14.Suzuki MM, Bird A. 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476. ( 10.1038/nrg2341) [DOI] [PubMed] [Google Scholar]
- 15.Lyko F, Foret S, Kucharski R, Wolf S, Falckenhayn C, Maleszka R. 2010. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 8, e1000506 ( 10.1371/journal.pbio.1000506) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foret S, Kucharski R, Pellegrini M, Feng S, Jacobsen SE, Robinson GE, Maleszka R. 2012. DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees. Proc. Natl Acad. Sci. USA 109, 4968–4973. ( 10.1073/pnas.1202392109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simola DF, Ye C, Mutti NS, Dolezal K, Bonasio R, Liebig J, Reinberg D, Berger SL. 2013. A chromatin link to caste identity in the carpenter ant Camponotus floridanus. Genome Res. 23, 486–496. ( 10.1101/gr.148361.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonasio R, et al. 2012. Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr. Biol. 22, 1755–1764. ( 10.1016/j.cub.2012.07.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kucharski R, Maleszka J, Foret S, Maleszka R. 2008. Nutritional control of reproductive status in honeybees via DNA methylation. Science 319, 1827–1830. ( 10.1126/science.1153069) [DOI] [PubMed] [Google Scholar]
- 20.Conrad T, Akhtar A. 2012. Dosage compensation in Drosophila melanogaster: epigenetic fine-tuning of chromosome-wide transcription. Nat. Rev. Genet. 13, 123–134. ( 10.1038/nrg3124) [DOI] [PubMed] [Google Scholar]
- 21.Payer B, Lee JT. 2008. X chromosome dosage compensation: how mammals keep the balance. Annu. Rev. Genet. 42, 733–772. ( 10.1146/annurev.genet.42.110807.091711) [DOI] [PubMed] [Google Scholar]
- 22.Ross KG, Vargo EL, Keller L, Trager JC. 1993. Effect of a founder event on variation in the genetic sex-determining system of the fire ant Solenopsis invicta. Genetics 135, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ross KG, Fletcher DJC. 1985. Genetic origin of male diploidy in the fire ant, Solenopsis invicta (Hymenoptera: Formicidae), and its evolutionary significance. Evolution 39, 888–903. ( 10.2307/2408688) [DOI] [PubMed] [Google Scholar]
- 24.Krieger MJB, Ross KG, Chang CWY, Keller L. 1999. Frequency and origin of triploidy in the fire ant Solenopsis invicta. Heredity 82, 142–150. ( 10.1038/sj.hdy.6884600) [DOI] [Google Scholar]
- 25.Hunt BG, Glastad KM, Yi SV, Goodisman MAD. 2013. Patterning and regulatory associations of DNA methylation are mirrored by histone modifications in insects. Genome Biol. Evol. 5, 591–598. ( 10.1093/gbe/evt030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wurm Y, et al. 2011. The genome of the fire ant Solenopsis invicta. Proc. Natl Acad. Sci. USA 108, 5679–5684. ( 10.1073/pnas.1009690108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Royal Stat. Soc. B (Methodological) 57, 289–300. [Google Scholar]
- 28.R Development Core Team. 2013. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 29.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. ( 10.1093/bioinformatics/bti610) [DOI] [PubMed] [Google Scholar]
- 30.Ometto L, Shoemaker D, Ross KG, Keller L. 2011. Evolution of gene expression in fire ants: the effects of developmental stage, caste, and species. Mol. Biol. Evol. 28, 1381–1392. ( 10.1093/molbev/msq322) [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Jemielity S, Uva P, Wurm Y, Graff J, Keller L. 2007. An annotated cDNA library and microarray for large-scale gene-expression studies in the ant Solenopsis invicta. Genome Biol. 8, R9 ( 10.1186/gb-2007-8-1-r9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Townsend J, Hartl D. 2002. Bayesian analysis of gene expression levels: statistical quantification of relative mRNA level across multiple strains or treatments. Genome Biol. 3, research0071 ( 10.1186/gb-2002-3-12-research0071) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Wurm Y, Nipitwattanaphon M, Riba-Grognuz O, Huang Y-C, Shoemaker D, Keller L. 2013. A Y-like social chromosome causes alternative colony organization in fire ants. Nature 493, 664–668. ( 10.1038/nature11832) [DOI] [PubMed] [Google Scholar]
- 34.Smyth GK. 2005. Limma: linear models for microarray data. In Bioinformatics and computational biology solutions using R and bioconductor (eds Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W.), pp. 397–420. New York, NY: Springer. [Google Scholar]
- 35.Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV. 2011. OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Res. 39, D283–D288. ( 10.1093/nar/gkq930) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunt BG, Ometto L, Wurm Y, Shoemaker D, Yi SV, Keller L, Goodisman MAD. 2011. Relaxed selection is a precursor to the evolution of phenotypic plasticity. Proc. Natl Acad. Sci. USA 108, 15 936–15 941. ( 10.1073/pnas.1104825108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glastad KM, Hunt BG, Yi SV, Goodisman MAD. 2011. DNA methylation in insects: on the brink of the epigenomic era. Insect Mol. Biol. 20, 553–565. ( 10.1111/j.1365-2583.2011.01092.x) [DOI] [PubMed] [Google Scholar]
- 38.Foret S, Kucharski R, Pittelkow Y, Lockett G, Maleszka R. 2009. Epigenetic regulation of the honey bee transcriptome: unravelling the nature of methylated genes. BMC Genomics 10, 472 ( 10.1186/1471-2164-10-472) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunt BG, Glastad KM, Yi SV, Goodisman MAD. 2013. The function of intragenic DNA methylation: insights from insect epigenomes. Integr. Comp. Biol. 53, 319–328. ( 10.1093/icb/ict003) [DOI] [PubMed] [Google Scholar]
- 40.Smith CR, Toth AL, Suarez AV, Robinson GE. 2008. Genetic and genomic analyses of the division of labour in insect societies. Nat. Rev. Genet. 9, 735–748. ( 10.1038/nrg2429) [DOI] [PubMed] [Google Scholar]
- 41.Nanty L, Carbajosa G, Heap GA, Ratnieks F, van Heel DA, Down TA, Rakyan VK. 2011. Comparative methylomics reveals gene-body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res. 21, 1841–1850. ( 10.1101/gr.121640.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. 2011. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74–79. ( 10.1038/nature10442) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S. 2008. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456, 125–129. ( 10.1038/nature07324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. 2007. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 39, 61–69. ( 10.1038/ng1929) [DOI] [PubMed] [Google Scholar]
- 45.Gelbart ME, Kuroda MI. 2009. Drosophila dosage compensation: a complex voyage to the X chromosome. Development 136, 1399–1410. ( 10.1242/dev.029645) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Z, Zhang J. 2011. Impact of gene expression noise on organismal fitness and the efficacy of natural selection. Proc. Natl Acad. Sci. USA 108, E67–E76. ( 10.1073/pnas.1100059108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Talia SD, Skotheim JM, Bean JM, Siggia ED, Cross FR. 2007. The effects of molecular noise and size control on variability in the budding yeast cell cycle. Nature 448, 947–951. ( 10.1038/nature06072) [DOI] [PubMed] [Google Scholar]
- 48.Huh I, Zeng J, Park T, Yi S. 2013. DNA methylation and transcriptional noise. Epigenet. Chromat. 6, 9 ( 10.1186/1756-8935-6-9) [DOI] [PMC free article] [PubMed] [Google Scholar]