

Abstract
Objectives
To test the hypothesis that late potentials and fractionated electrogram activity are due to delayed depolarization within the anterior aspects of right ventricular (RV) epicardium in experimental models of Brugada syndrome (BrS).
Background
Clinical reports have demonstrated late potentials in SAECG recorded in patients with BrS. Recent studies report the appearance of late potentials and fractionated activity in bipolar electrograms recorded from the epicardium of the RV outflow tract in patients with BrS.
Methods
Action potential (AP) and bipolar electrograms were recorded from epicardial and endocardial sites of coronary-perfused canine RV wedge preparations together with a pseudo-electrocardiogram (ECG). The Ito agonist NS5806 (5 μM) and Ca2+ channel blocker verapamil (2 μM) were used to pharmacologically mimic BrS genotypes.
Results
Fractionated electrical activity was observed in RV epicardium but not endocardium as a consequence of heterogeneities in the appearance of the second upstroke of the epicardial AP and discrete high frequency spikes developed as a result of concealed phase-2-reentry. In no case did we observe primary conduction delay as the cause of the BrS ECG phenotype or of late potential or fractionated electrogram activity. Quinidine (10 μM) or phosphodiestaerase-3-inhibitors milrinone (2.5 μM) and cilostazol (10 μM) restored electrical homogeneity, thus abolishing all late potential and fractionated electrical activity.
Conclusions
Our data point to an alternative pathophysiological basis for late potentials and fractionated electrograms recorded from the RV in the setting of BrS. We demonstrate association of such activity with abnormal repolarization and not with abnormal depolarization or structural abnormalities.
Keywords: Cardiac arrhythmias, electrophysiology, pharmacology
Introduction
Brugada syndrome (BrS) is a disease that causes vulnerability to ventricular tachycardia (VT) and sudden cardiac death in young adults with structurally normal hearts. The ECG pattern of BrS is characterized by the appearance of prominent J waves, often appearing as ST segment elevation in the right precordial leads (1–3), which are often concealed, but can be unmasked by vagal stimulation and potent sodium channel blockers (3,4). BrS has been linked to mutations causing decreased inward currents such as peak sodium channel current (INa) or L-type calcium channel current (ICa) or increased outward currents, especially the transient outward potassium current (Ito) (5,6). The net outward shift of current during phase 1 of the epicardial (Epi) action potential (AP) leads to accentuation of the spike-and-dome morphology, most prominently in the epicardium of the right ventricular outflow tract (RVOT) (3). Loss of the dome (phase 2 of the AP) and consequent development of dispersion of repolarization within epicardium and between epicardium and endocardium create the substrate for phase 2 reentry and polymorphic VT (7).
Previous clinical reports have demonstrated the presence of late potentials on the signal average ECG (SAECG) of patients with BrS (8). These investigators also recorded delayed potentials using a unipolar electrogram (EG) lead introduced into the conus branch through the right coronary artery. This activity was hypothesized to be due to a “myocardial abnormality” in the epicardium but not endocardium of the RVOT. Other investigators documenting late potentials in SAECG of BrS patients (9,10) have attributed this activity to structural abnormalities and delayed conduction within the RV. Using bipolar electrograms (EG), Nademanee et al. demonstrated late potentials and fractionated activity in RVOT epicardium of patients with BrS and hypothesized that these represent regions of delayed conduction or depolarization defects (11). A recent publication by Sacher et al. demonstrated such activity in BrS patients challenged with potent sodium channel blockers and likewise hypothesized that these are due to depolarization defects (12).
The present study provides a test of this hypothesis using experimental models that closely recapitulate the ECG and arrhythmic manifestations of BrS. The principal focus of the study is to elucidate the cellular mechanisms responsible and test the hypothesis that late potentials and fractionated EG activity recorded from the Epi surface of experimental models of BrS are associated with development of abnormal repolarization rather than abnormal depolarization.
Methods
Wedge Preparations
All experiments were carried out in compliance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health (NIH publication No 85–23, Revised 1996) and was approved by the Institutional Animal Care and Use Committee. Detailed methods for isolation and recording of transmembrane activity from coronary-perfused canine right ventricle (RV) wedge preparations have been reported previously (13,14). Briefly, adult mongrel dogs (20–35 kg) of either sex were used. Transmural wedge preparations were dissected (1.9×0.9×0.9 to 3.2×1.6×1.3 cm) from the RV free wall of dogs. The preparations were cannulated via the marginal branch of the right coronary artery and perfused with cardioplegic solution (Tyrode’s containing 12 mmol/L KCl). Unperfused tissue was carefully removed using a razor blade. The preparations were then placed in a tissue bath and perfused with oxygenated Tyrode’s solution (mM): NaCl 129, KCl 4, NaH2PO4 0.9, NaHCO3 20, CaCl2 1.8, MgSO4 0.5, glucose 5.5, pH 7.4. The perfusate was delivered using a roller pump (Cole Parmer Instrument Co., Niles, Illinois) at a constant flow rate at 8–10 mL/min warmed to 37±0.5°C.
The preparations were equilibrated in the tissue bath until electrically stable, usually 1 hour, while stimulated at a basic cycle length of 1000 ms using bipolar silver electrodes insulated except at the tips, applied to the endocardial surface. A transmural ECG was recorded using two electrodes consisting of AgCl half cells placed in the tissue bath, 1.0 to 1.5 cm from the Epi and endocardial (Endo) surfaces of the preparation, along the same axis as the transmembrane recordings (Epi electrode is connected to the positive input of the ECG amplifier).
Transmembrane APs were simultaneously recorded from two Epi (Epi 1 [distal] and Epi 2 [proximal]; Epi 1-Epi 2 distance was approx. 5–10 mm) and one Endo site with the use of floating microelectrodes (DC resistance=10 to 20 MΩ) filled with 2.7 mol/L KCl, each connected to a high-input impedance amplifier. Impalements were obtained from the Epi and Endo surfaces of the preparation at positions approximating the transmural axis of the ECG recording. Two unipolar electrograms were placed in the epicardium or endocardium. Virtual bipolar electrograms were derived as the difference of two unipolar EGs.
Spike 2 for Windows (Cambridge Electronic Design, Cambridge, UK) was used to record and analyze the ECG, EGs and the AP. NS5806, verapamil, quinidine, cilostazol and milrinone were dissolved in dimethyl sulphoxide (10 mM stock).
Results
Using coronary-perfused canine right ventricular wedge preparations, we induced the Brugada phenotype by addition of 5 μM NS5806 (Ito activator) and 2 μM verapamil (Ca2+ channel blocker) to the coronary perfusate. NS5806 has previously been shown to increase Ito in isolated canine cardiomyocytes, resulting in augmentation of the notched appearance of the RV AP, most notably in the epicardium (15). NS5806 (5 μM) and verapamil (2 μM) accentuated the AP notch in RV Epi, leading to the development of a prominent J point and ST segment elevation, characteristic of the Brugada phenotype. Late potentials and fractionated activity were often observed in the bipolar EGs comparable to those recorded in the clinical cases reported by Nademanee et al. (11) (Fig. 1). The accentuation of the AP notch in epicardium but not endocardium generates a transmural voltage gradient responsible for the accentuated J waves in the ECG. The fractionated EG activity was observed to result from heterogeneity in the appearance of phase 2 of the AP. In no case did we observe any major delays in phase 0 depolarization or any type of conduction problem of the primary depolarization wave.
Figure 1. Heterogeneities in the appearance of the epicardial action potential second upstroke gives rise to fractionated epicardial electrogram (EG) activity in the setting of Brugada syndrome (BrS).
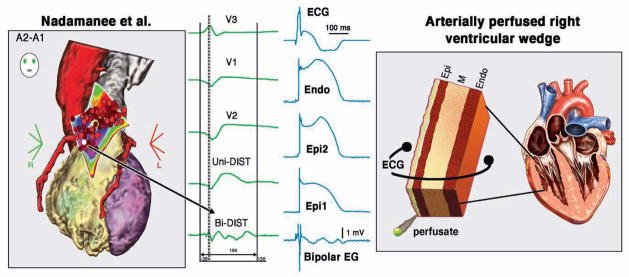
Left panel: Shown are right precordial lead recordings, unipolar and bipolar EGs recorded form the right ventricular outflow tract of a BrS patient (from Nademanee et al. (11), with permission). Right panel: ECG, action potentials from endocardium (Endo) and two epicardial (Epi) sites, and a bipolar epicardial EG (Bipolar EG) all simultaneously recorded from a coronary-perfused right ventricular wedge preparation treated with NS5806 (5 μM) and verapamil (2 μM) to induce the Brugada phenotype. Basic cycle length=1000 ms.
With longer exposure to the provocative agents, we observed loss of the AP dome at some Epi sites but not others. This further accentuated the transmural voltage gradient, thus contributing to the manifestation of J waves, often appearing as ST segment elevation. The dispersion of repolarization generated as a result of abbreviation of the Epi AP at some sites but not others or in endocardium created a vulnerable window for the development of reentrant arrhythmias. Propagation of the dome from regions at which it was maintained to regions at which it was lost, caused local re-excitation via a phase 2 re-entrant mechanism, leading to the development of very closely coupled re-excitation of epicardium. More often than not, because of its early appearance, the phase 2 re-entrant beat was unable to propagate out of the Epi focus in which it was generated, thus resulting in “concealed phase 2 reentry”, which was not manifest as an extrasystole in the ECG. This local activity however accentuated the inverted T wave and caused late potentials and fractionated activity in EGs recorded from epicardium but not endocardium, very similar to those observed in the clinical cases reported by Nademanee et al. (16) (Figs. 2, 3 and 8). Concealed phase 2 reentry associated with the appearance of late potential activity was observed in 7/7 wedge preparations. The average coupling interval of concealed phase 2 reentrant beat and the delay in the appearance of the associated high frequency late potentials in the bipolar EG were 117.5 ± 8.2 (range:81–138 ms) and 121.1 ± 8.9 ms, respectively (mean±SEM, n=7).
Figure 2. Concealed phase 2 reentry as the basis for late potential and fractionated bipolar epicardial (Epi) electrogram (Bipolar EG) activity in an experimental model of Brugada syndrome.
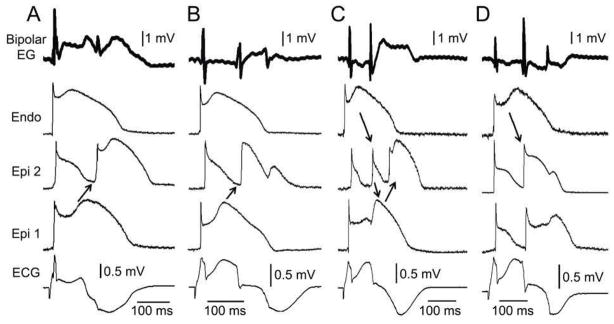
Each panel shows (from top to bottom) a Bipolar EG, action potentials recorded from endocardium (Endo) and two Epi sites and an ECG all simultaneously recorded from a coronary-perfused right ventricular wedge preparation exposed to NS5806 (5 μM) and verapamil (2 μM) to induce the Brugada phenotype. Heterogeneous loss of the dome at epicardium caused local re-excitation via a ‘concealed’ phase 2 re-entry mechanism, leading to the development of late potentials and fractionated bipolar epicardial EG activity. No major delays in conduction of the primary beat were ever observed. Each panel shows results from a different preparation. Basic cycle length=1000 ms.
Figure 3.
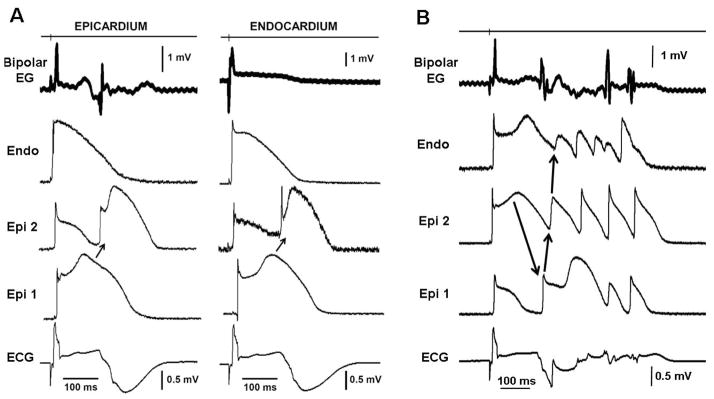
A: Concealed phase 2 reentry gives rise to late potentials and fractionated bipolar electrogram (Bipolar EG) activity recorded from epicardium but not endocardium (Endo) in an experimental model of Brugada syndrome. Each panel shows (from top to bottom) a bipolar epicardial (Epi) EG, action potentials recorded from Endo and two Epi sites and an ECG all simultaneously recorded from a coronary-perfused right ventricular (RV) wedge preparation exposed to NS5806 (5 μM) and verapamil (2 μM) to induce the Brugada phenotype. Heterogeneous loss of the dome at epicardium caused local re-excitation via a concealed phase 2 re-entry mechanism, leading to the development of late potentials and fractionated bipolar epicardial EGs. Basic cycle length = 1000 ms. B: Phase 2 Reentry-induced ventricular fibrillation. All traces were simultaneously recorded from a coronary-perfused RV wedge preparation exposed to NS5806 (5 μM) and verapamil (2 μM). The phase 2 reentrant beat produced a closely coupled extrasystole that precipitated an episode of polymorphic tachycardia.
Figure 8. Comparison of the late potential and fractionated bipolar electrogram (EG) activity recorded form the epicardial (Epi) surface of the right ventricular outflow tract (RVOT) of a patient with Brugada syndrome (BrS) with similar activity recorded from coronary perfused right ventricular (RV) wedge models of BrS.
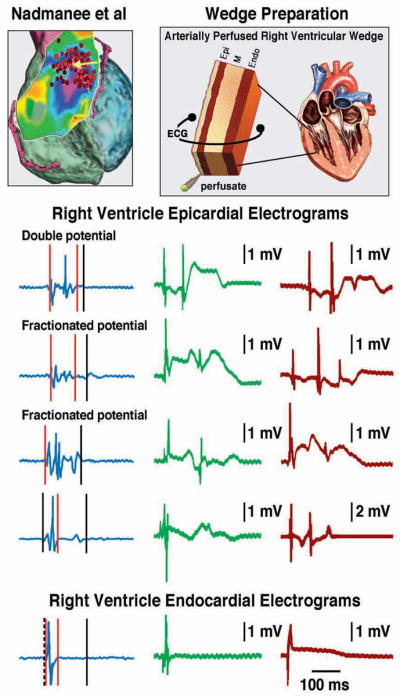
Left: Bipolar RV Epi and endocardial (Endo) EGs recorded from the RVOT of BrS patient from the study of Nademanee et al. (11). Right: Bipolar EGs recorded from the Epi surface of coronary-perfused canine RV wedge models of BrS. All recordings were obtained from preparations displaying concealed phase 2 reentry. In all cases late potentials and fractionated EG activity was the result of repolarization defects created by a inward shift in the balance currents active during the early phases of the Epi action potential. In no case did we observe primary impulse conduction delays. In both clinical and experimental models, late potentials or fractionated activity was not observed in the Endo EGs.
When the phase 2 re-entrant beat was able to exit the Epi focus in which it was generated, it produced a closely-coupled extrasystole capable of initiating a VT/ventricular fibrillation (VF), as illustrated in Figure 3B. When the temperature of the coronary perfusate was reduced to 30°C, verapamil (3 μM) alone was able to induce concealed phase 2 reentry and delayed potentials (Fig. 4).
Figure 4. Alternans of concealed phase 2 reentry gives rise to alternans of late potential and T wave.
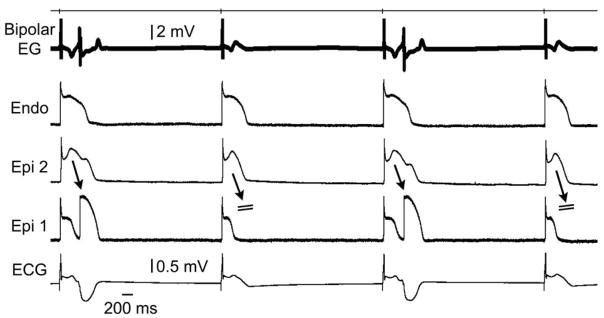
Traces are as in the previous figures. All traces were simultaneously recorded from a coronary-perfused right ventricular wedge preparation exposed to verapamil (3 μM) and hypothermia (30 °C) to induce a Brugada phenotype. EG: electrogram; Endo: endocardium; Epi: epicardial.
Figure 4 is illustrates an example of 2:1 alternans of concealed phase 2 reentry, giving rise to marked T wave alternans and 2:1 alternation in the appearance of the epicardial EG late potential. In the presence of 5 μM NS5806 and 2 μM verapamil, the introduction of a stimulated premature beat (S2) restored homogeneity of the transmural action potentials and abolished the late potential and fractionated epicardial EG activity (Fig. 5).
Figure 5. Stimulated premature beat (S2) restores homogeneity of action potentials and abolishes fractionated bipolar epicardial (Epi) electrogram (Bipolar EG) in an experimental model of Brugada syndrome.

Traces are as in the previous figures. All traces were simultaneously recorded from a coronary-perfused right ventricular wedge preparation exposed to NS5806 (5 μM) and verapamil (2 μM) to induce a Brugada phenotype. BCL=1000 ms. S1–S2=240 ms. Endo: endocardium.
Quinidine and cilostazol have been shown to be capable of suppressing arrhythmogenesis associated with Brugada syndrome. Quinidine and cilostazol have been reported to eliminate SAECG late potentials in patients with BrS (17,18). In another series of experiments we tested the hypothesis that the ameliorative effects of these agents are due to and effect of these two agents to reverse the repolarization defect, thus suppressing concealed phase 2 re-entry and late potential activity. We tested the effect of quinidine and two phosphodiesterase III inhibitors, cilostazol and milrinone. As illustrated in Figures 6 and 7, quinidine (10 μM), cilostazol (10 μM) and milrinone (2.5 μM) all promptly restored the AP dome throughout epicardium, thus dramatically reducing Epi and transmural dispersion of repolarization (TDR), normalizing the ECG, abolishing concealed phase 2 reentry and eliminating all late potential and fractionated epicardial EGs activity.
Figure 6. Effect of quinidine (10 μM) to reverse the repolarization defects responsible for phase 2 reentry and associated late potentials and fractionated epicardial (Epi) electrogram (EG) activity.

Traces are as in the previous figures. All traces were simultaneously recorded from a coronary-perfused right ventricular wedge preparation exposed to NS5806 (5 μM) and verapamil (2 μM) to induce a Brugada phenotype. The addition of quinidine (10 μM) to the coronary perfusate reversed the repolarization defects, restored action potential homogeneity, normalized the ECG and abolished phase 2 reentry and associated late potentials on the bipolar electrogram (Bipolar EG). Endo: endocardium
Figure 7. Effect of milrinone (2.5 μM) and cilostazol (10 μM) to reverse repolarization defects responsible for phase 2 reentry and associated late potential and fractionated bipolar epicardial (Epi) electrogram (EG) activity.

Traces are as in the previous figures. All traces were simultaneously recorded from a coronary-perfused right ventricular wedge preparation exposed to NS5806 (5 μM) and verapamil (2 μM) to induce a Brugada phenotype. Milrinone and cilostazol reversed the repolarization defects induced by NS5806 and verapamil, thus restoring the Epi action potential dome throughout, normalizing the ECG and abolishing phase 2 reentrant activity and associated late potential and fractionated bipolar Epi EG. Bipolar EG: bipolar electrogram; Endo: endocardium.
Discussion
It is well established that mutations leading to a decrease in inward currents (INa or ICaL) or increase in outward currents (Ito and IK-ATP) are capable of causing BrS in humans (5,19–22). We used NS5806 (Ito agonist) and verapamil (ICa antagonist) to pharmacologically model the BrS genotypes responsible for a loss of function of ICa (BrS 3,4 and 9) (23) and a gain of function of Ito (BrS5, 6 and 10) (5,6,24,25) so as to induce the Brugada phenotype in coronary-perfused canine RV wedge preparations. The NS5806-induced increase in Ito caused an outward shift in the balance of current in the early phase of the Epi AP leading to a moderate increase in notch in epicardium, but little change in endocardium. The greater accentuation of the AP notch in epicardium than endocardium gives rise to an increase in transmural voltage gradient causing an increase in the magnitude of the J wave (Figs. 6 and 7). The addition of verapamil to the coronary perfusate caused a further outward shift in the balance of current leading to further accentuation of these parameters. Longer exposure caused all-or-none repolarization at the end of the Epi AP phase 1, leading to loss or delay of the AP dome at some Epi sites. In some preparations, the increased repolarization forces caused variable delay in the appearance of the second epicardial AP upstroke leading to the appearance of low voltage fractionated activity in the bipolar EG recording, as previously demonstrated in clinical recordings reported by Nademanee and co-workers (11) (Fig. 1).
Loss of the dome and marked AP duration abbreviation at some epicardial sites but not others caused a prominent increase in epicardial and TDR, thus creating the substrate for the development of phase 2 reentry and VT (Figs. 2 and 3). Conduction of the AP dome, from sites at which it was maintained to sites at which it was lost, caused local re-excitation via a phase 2 re-entry mechanism. In the majority of cases, phase 2 reentry was concealed because the extra-beat was unable to exit the focus in which it was generated. However, the concealed phase 2 reentrant beat was able to produce high frequency late potentials and fractionated bipolar EGs in epicardium but not in endocardium (Figs. 2, 3 and 8) comparable to clinical observations reported by Nademanee et al. and Sacher et al. (11,12).
The hypothesis proposed by Nademanee et al. (11) and Sacher et al. (12) that late potentials and fractionated activity recorded in the RVOT of patients with BrS are due to depolarization defects is not supported by our results, which provide an alternative explanation for the appearance of such activity, based on the development of primary repolarization defects. Our observations suggest that without the benefit of AP recordings it is difficult to discern between repolarization and depolarization defects. It is noteworthy that recordings of monophasic APs from the epicardium and endocardium of the RVOT of patients with BrS have demonstrated AP activity very similar to that recorded from the canine coronary–wedge preparations, with no major transmural conduction delay (26,27).
One way to distinguish between the two mechanisms in the clinic might be to examine the effect of rate or atrial premature responses. When due to delayed conduction, the notched appearance should become accentuated with acceleration of rate or prematurity, because delayed conduction almost always becomes more accentuated at faster rates or with prematurity. When due to repolarization problems the amplitude of the J wave should diminish due to insufficient time for Ito to reactivate, as in the example illustrated in Figure 5. A word of caution needs to be raised here because in the presence of drugs with use-dependent actions to inhibit sodium or calcium channel current, an opposite response may be observed following an increase in rate or even following a single premature beat. Indeed in the presence of verapamil (Fig. 5A), the use-dependent inhibition of ICa leads to accentuation of the repolarizations defects. The reduced repolarization defect with a single premature beat is due to the fact that the reduction of Ito was greater than the reduction of ICa following a single premature beat.
Another way to discern between depolarization and repolarization defects as a cause of the BrS phenotype is by observing the response to quinidine. The INa inhibitory effect of quinidine is expected to accentuate late potential and fractionated EG activity if due to a depolarization defect. If on the other hand, the BrS phenotype is due to a repolarization defect, quinidine via its action to inhibit Ito would be expected to reduce or abolish late potential and fractionated EG activity, which is what we observed in our preparations (Fig. 6).
Quinidine and cilostazol have been reported to eliminate SAECG late potentials in patients with Brugada syndrome (17,18), and to exert antiarrhythmic effects in this setting (7,28–32). Our results provide support for the hypothesis that the ameliorative effects of these agents are due to and effect of these two agents to reverse the repolarization defect, thus suppressing concealed phase 2 reentry and late potential activity (Figs. 6 and 7). In addition, we demonstrate that milrinone, another phosphodiesterase (PDE) III inhibitor, is a potent agent capable of exerting ameliorative actions in the setting of BrS syndrome (Fig. 7). The ameliorative action of cilostazol and milrinone are likely attributable to the effect of these PDE inhibitors to increase cAMP and thus boost ICa, thus leading to a reversal of the repolarization defect permitting the development of the ECG manifestations of BrS. Quinidine, and cilostazol at higher concentrations (33), exert a direct effect to block Ito.
Quinidine is used today for primary and secondary prevention and as adjunct therapy for BrS. Ameliorative effects of the PDE inhibitors (especially cilostazol) and quinidine to suppress the BrS phenotype is consistent with clinical findings (28,30,31,34–37).
Finally, if the late potentials recorded in the anterior surface of the RVOT do not reflect delayed conduction, why does ablation of these sites produce an ameliorative response as reported by Nademanee et al. (11). Our hypothesis is that eliminating the cells a high density of Ito, known to reside in the RVOT, prevents the development of the repolarization defects responsible for the Brugada ECG, phase 2 reentry and VT/VF. A lower level of Ito is the basis for why female carriers are protected from developing the electrocardiographic and arrhythmic manifestations of BrS (38) and is the basis for the actions of quinidine and cilostazol. We are endeavoring to test this hypothesis by ablating the regions of concealed phase 2 reentry and late potentials in coronary-perfused RV wedge models of BrS. Our preliminary results provide strong support for the hypothesis, showing that ablation of the sites of abnormal repolarization normalizes the ECG and prevents the development of phase 2 reentry and VT/VF (Patocskai and Antzelevitch, unpublished data).
Conclusion
The present study explores the alternate hypothesis that the late potentials observed on the SAECG and fractionated bipolar epicardial electrograms are not due abnormal conduction or structural abnormalities in the right ventricular epicardium, but are associated with development of abnormal repolarization giving rise to concealed phase 2 reentry in epicardium. Our results might prove helpful in better understanding the cellular mechanisms underlying late potential and fractionated electrograms activity in a variety of pathophysiological conditions.
Study limitations
As with all data derived from experimental animal models, extrapolation of the data from in vitro models to the clinic must be done with due caution.
Acknowledgments
Funding sources: This study was supported by grant HL47678 from NHLBI (CA), NYSTEM grant # C026424 (CA) and the Masons of New York, Florida, Massachusetts, Connecticut, Maryland, Delaware, New Hampshire and Wisconsin.
We are grateful José Di Diego M.D. for continuous support and personal guidance and to Istvan Koncz M.D., Ph.D. and Serge Sicouri M.D. for helpful discussions and support. We also gratefully acknowledge the technical assistance of Robert Goodrow, Judy Hefferon, and Rebecca Warren.
Abbreviations and Acronyms
- AP
action potential
- BrS
Brugada syndrome
- Endo
endocardial
- EG
electrogram
- Epi
epicardial
- ICa
L-type calcium channel current
- INa
sodium channel current
- Ito
transient outward potassium current
- LV
left ventricle
- RVOT
right ventricular outflow tract
- SAECG
signal average ECG
- TDR
transmural dispersion of repolarization
Footnotes
Conflicts of interest: There are no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome: a multicenter report. J Am Coll Cardiol. 1992;20:1391–6. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 2.Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93:372–9. doi: 10.1161/01.cir.93.2.372. [DOI] [PubMed] [Google Scholar]
- 3.Antzelevitch C. Brugada syndrome. PACE. 2006;29:1130–59. doi: 10.1111/j.1540-8159.2006.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brugada R, Brugada J, Antzelevitch C, et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101:510–5. doi: 10.1161/01.cir.101.5.510. [DOI] [PubMed] [Google Scholar]
- 5.Delpón E, Cordeiro JM, Núñez L, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209–18. doi: 10.1161/CIRCEP.107.748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giudicessi JR, Ye D, Kritzberger CJ, et al. Novel mutations in the KCND3-encoded Kv4. 3 K+ channel associated with autopsy-negative sudden unexplained death. Hum Mutat. 2012;33:989–97. doi: 10.1002/humu.22058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST segment elevation. Circulation. 1999;100:1660–6. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 8.Nagase S, Kusano KF, Morita H, et al. Epicardial electrogram of the right ventricular outflow tract in patients with the Brugada syndrome: using the epicardial lead. J Am Coll Cardiol. 2002;39:1992–5. doi: 10.1016/s0735-1097(02)01888-0. [DOI] [PubMed] [Google Scholar]
- 9.Nosaka K, Morita H, Nishii N, et al. Detection of conduction abnormality within QRS complex and risk stratification by wavelet transform of SAECG in Brugada syndrome. Heart Rhythm. 2009;6:S179. [Google Scholar]
- 10.Tatsumi H, Takagi M, Nakagawa E, et al. Risk stratification in patients with Brugada syndrome: analysis of daily fluctuations in 12-lead electrocardiogram (ECG) and signal-averaged electrocardiogram (SAECG) J Cardiovasc Electrophysiol. 2006;17:705–11. doi: 10.1111/j.1540-8167.2006.00508.x. [DOI] [PubMed] [Google Scholar]
- 11.Nademanee K, Veerakul G, Chandanamattha P, et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation. 2011;123:1270–9. doi: 10.1161/CIRCULATIONAHA.110.972612. [DOI] [PubMed] [Google Scholar]
- 12.Sacher F, Jesel L, Jais P, et al. Insight into the mechanism of Brugada syndrome: epicardial substrate and modification during ajmaline testing. Heart Rhythm. 2013 May 31; doi: 10.1016/j.hrthm.2013.05.023. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 13.Fish JM, Welchons DR, Kim YS, et al. Dimethyl lithospermate B, an extract of danshen, suppresses arrhythmogenesis associated with the Brugada syndrome. Circulation. 2006;113:1393–400. doi: 10.1161/CIRCULATIONAHA.105.601690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Diego JM, Sicouri S, Myles RC, et al. Optical and electrical recordings from isolated coronary-perfused ventricular wedge preparations. J Mol Cell Cardiol. 2013;54:53–64. doi: 10.1016/j.yjmcc.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calloe K, Cordeiro JM, Di Diego JM, et al. A transient outward potassium current activator recapitulates the electrocardiographic manifestations of Brugada syndrome. Cardiovasc Res. 2009;81:686–94. doi: 10.1093/cvr/cvn339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barajas-Martinez H, Hu D, Pfeiffer R, et al. Loss-of-function cardiac L-type calcium channel mutation identifies CACNA2D1 as a new Brugada syndrome susceptibility gene. Heart Rhythm. 2011;8(5S):S105. [Google Scholar]
- 17.Yodogawa K, Morita N, Kobayashi Y, et al. High-frequency potentials developed in wavelet-transformed electrocardiogram as a novel indicator for detecting Brugada syndrome. Heart Rhythm. 2006;3:1436–44. doi: 10.1016/j.hrthm.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe H, Chinushi M, Osaki A, et al. Elimination of late potentials by quinidine in a patient with Brugada syndrome. J Electrocardiol. 2006;39:63–6. doi: 10.1016/j.jelectrocard.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–9. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanisms for idiopathic ventricular fibrillation. Nature. 1998;392:293–6. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe H, Koopmann TT, Le Scouarnec S, et al. Sodium channel b1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008;118:2260–8. doi: 10.1172/JCI33891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.London B, Michalec M, Mehdi H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–8. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burashnikov E, Pfeiffer R, Barajas-Martinez H, et al. Mutations in the cardiac L-type calcium channel associated J wave sydnrome and sudden cardiac death. Heart Rhythm. 2010;7:1872–82. doi: 10.1016/j.hrthm.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu D, Barajas-Martinez H, Medeiros-Domingo A, et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na(v)1.5 and K(v)4. 3 channel currents. Heart Rhythm. 2012;9:760–9. doi: 10.1016/j.hrthm.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakajima T, Wu J, Kaneko Y, et al. KCNE3 T4A as the genetic basis of brugada-pattern electrocardiogram. Circ J. 2012;76:2763–72. doi: 10.1253/circj.cj-12-0551. [DOI] [PubMed] [Google Scholar]
- 26.Kurita T, Shimizu W, Inagaki M, et al. The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. J Am Coll Cardiol. 2002;40:330–4. doi: 10.1016/s0735-1097(02)01964-2. [DOI] [PubMed] [Google Scholar]
- 27.Antzelevitch C, Brugada P, Brugada J, et al. Brugada syndrome: a decade of progress. Circ Res. 2002;91:1114–9. doi: 10.1161/01.res.0000046046.53721.90. [DOI] [PubMed] [Google Scholar]
- 28.Belhassen B, Viskin S. Pharmacologic approach to therapy of Brugada syndrome: quinidine as an alternative to ICD therapy? In: Antzelevitch C, Brugada P, Brugada J, et al., editors. The Brugada Syndrome: From Bench to Bedside. Oxford: Blackwell Futura; 2004. pp. 202–211. [Google Scholar]
- 29.Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110:1731–7. doi: 10.1161/01.CIR.0000143159.30585.90. [DOI] [PubMed] [Google Scholar]
- 30.Belhassen B. Is quinidine the ideal drug for Brugada syndrome? Heart Rhythm. 2012;9:2001–2. doi: 10.1016/j.hrthm.2012.08.037. [DOI] [PubMed] [Google Scholar]
- 31.Viskin S, Wilde AA, Tan HL, et al. Empiric quinidine therapy for asymptomatic Brugada syndrome: time for a prospective registry. Heart Rhythm. 2009;6:401–4. doi: 10.1016/j.hrthm.2008.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimizu W, Aiba T, Antzelevitch C. Specific therapy based on the genotype and cellular mechanism in inherited cardiac arrhythmias. Long QT syndrome and Brugada syndrome. Curr Pharm Des. 2005;11:1561–72. doi: 10.2174/1381612053764823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao GS, Liao YH. Effect of colostazol on transient outward potassium current in human atrial myocytes. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2004;20:238–41. [PubMed] [Google Scholar]
- 34.Belhassen B, Viskin S, Fish R, et al. Effects of electrophysiologic-guided therapy with Class IA antiarrhythmic drugs on the long-term outcome of patients with idiopathic ventricular fibrillation with or without the Brugada syndrome. J Cardiovasc Electrophysiol. 1999;10:1301–12. doi: 10.1111/j.1540-8167.1999.tb00183.x. [DOI] [PubMed] [Google Scholar]
- 35.Kanlop N, Shinlapawittayatorn K, Sungnoon R, et al. Cilostazol attenuates ventricular arrhythmia induction and improves defibrillation efficacy in swine. Can J Physiol Pharmacol. 2010;88:422–8. doi: 10.1139/y09-127. [DOI] [PubMed] [Google Scholar]
- 36.Szel T, Koncz I, Antzelevitch C. Cellular mechanisms underlying the effects of milrinone and cilostazol to supress arrhythmogenesis associated with Brugada syndrome. Heart Rhythm. 2013;10:1720–7. doi: 10.1016/j.hrthm.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsuchiya T, Ashikaga K, Honda T, et al. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 2002;13:698–701. doi: 10.1046/j.1540-8167.2002.00698.x. [DOI] [PubMed] [Google Scholar]
- 38.Di Diego JM, Cordeiro JM, Goodrow RJ, et al. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation. 2002;106:2004–11. doi: 10.1161/01.cir.0000032002.22105.7a. [DOI] [PubMed] [Google Scholar]