

Abstract
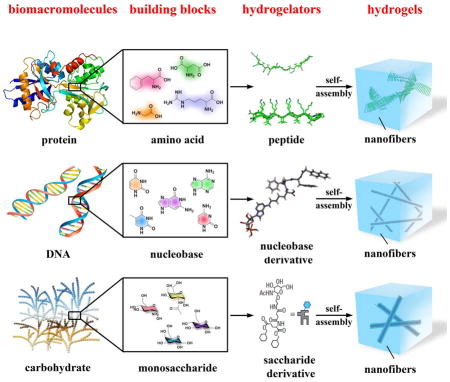
As a consequence of the self-assembly of small organic molecules in water, supramolecular hydrogels are evolving from serendipitous events during organic synthesis to become a new type of materials that promise increased applications in biomedicine. In this focus review, we describe the recent development on the use of basic biological building blocks for creating molecules that act as hydrogelators and the potential applications of the corresponding hydrogels. After introducing the concept of supramolecular hydrogels and defining the scope of this review, we briefly describe the methods for making and characterizing supramolecular hydrogels. Then, we discuss representative hydrogelators according to the categories of their building blocks, such as amino acids, nucleobases, and saccharides, and highlight the applications of the hydrogels when necessary. Finally, we offer our perspectives and outlooks on this fast-growing field at the interface of organic chemistry, materials, biology, and medicine. By providing a snapshot for chemists, engineers, and medical scientists, we hope that this focus review will contribute to the development of multidisciplinary research on supramolecular hydrogels for a wide range of applications in different fields.
Keywords: supramolecular, hydrogels, biomedicine, peptides, nucleobase saccharides
1. Introduction
This review aims to provide the updates on the development of supramolecular hydrogels based on fundamental biological molecular building blocks of biomacromolecules, such as amino acids, nucleobases, and saccharides. As a type of versatile soft materials, hydrogels consist of a three-dimension, continuous three-dimensional network that encapsulates a large amount of water. The versatility of the hydrogels largely stems from the gel matrices, a three-dimensional solid-like network that consists of cross-linked polymers[1] or fibrous superstructures resulted from self-assembled small molecules.[2] Usually, the polymers are referred as gellants and the small molecules as gelators, as defined by Weiss.[3] Because they share prominent physical features, soft and wet, with soft tissues, hydrogels made of biological or synthetic polymers have received extensive exploration in last several decades and achieved considerable successes in biomedical engineering and pharmaceutical formulation.[1b, 4] For example, Peppas et al. have pioneered the use of polymeric hydrogels for drug delivery,[5] which stimulated many other applications of hydrogels.[6] Mooney’s group has developed alginate based hydrogels as the mimic of extracellular matrices[7] for a wide range of applications, from tissue engineering[8] to cancer immunotherapy.[9] Hoffman and coworkers have pioneered stimuli-responsive polymeric hydrogels for drug release.[10] Gong et al. have engineered exceptionally strong hydrogels based a double-network (DN) structure.[11] Although these representative results and developments highlight the tremendous potentials of hydrogel materials, polymeric hydrogels still have certain limitations. For example, most of them are passive and conformational random. Though it is possible to attach biological functional groups to the polymers, the precise molecular control is rather difficult because polymers inherently are a mixture of chains with different lengths. In addition, the immunogenicity and elimination of the polymers in vivo continues to be a challenging problem.[12]
Unlike polymeric hydrogels, a supramolecular hydrogel contains a network of nanofibers formed by the self-assembly of small molecules (i.e., hydrogelators) as the solid phase to encapsulate water. Though containing up to more than 97% water, supramolecular hydrogels share similar rheological properties as polymeric hydrogels, such as solid-like and responsiveness to an external stimulus. In addition, supramolecular hydrogelators share common features, such as amphiphilicity and non-covalent interactions (e.g., π-π interactions, hydrogen bonding, and charge interactions), with certain self-assembled structures in biology. These non-covalent interactions drive the formation of nanostructures to result in the three-dimensional networks as the matrices of supramolecular hydrogels. Self-assembly and non-covalent interactions also enable supramolecular hydrogels to show rapid responses to a variety of external stimuli, including pH, temperature, ionic strength, and ligand-receptor interactions.
Being composed of small molecules, supramolecular hydrogels have several unique advantages that are complementary to polymeric hydrogels. For example, when the application involves in vivo use of a polymer, the cellular toxicity of the polymer becomes critical issues, especially for applications involving intracellular delivery of biomolecular drugs such as peptides, proteins and nucleic acid drugs, which mainly act within cells.[12] Being resulted from self-assembly of small molecules driven by non-covalent interactions, supramolecular hydrogels are easily biodegradable and biocompatible. Since the molecular weights of the small molecules usually are less than 2000 Da, the renal clearance of hydrogelators would be more efficient than the polymers of which the molecular weights are above about 30,000 Da. More importantly, the well-established organic synthesis and characterization allow the hydrogelators to be produced as pure compounds, thus eliminating the impurities and cellular toxicity associated with the cross-linking reactions in the synthesis of polymeric hydrogels.[13] In polymeric hydrogels, the frequency and content of the functional group along the backbone can vary from one chain to the next, and the groups may not be evenly spaced due to differences in monomer reactivity.[14] The formation of supramolecular hydrogels likely eliminate this problem since self-assembly of hydrogelators usually result in ordered structures consisting of well-defined or regular repeats.[15]
Although in the early stage of the development of supramolecular hydrogels, most of the small molecule hydrogelators are organic molecules, such as urea derivatives,[16] serine-based amphiphiles,[17] and derivatives of aroyl-L-cystines,[18] the use of bioactive small molecules (e.g., vancomycin[19]) as hydrogelators is very attractive for biomedical applications, especially after the demonstration of the use of supramolecular hydrogels for applications such as three-dimensional cell cultures,[20] screening biomolecules,[21] wound healing,[22] and drug delivery.[21, 23] These promising results naturally lead to the exploration of fundamental biological building blocks for making hydrogelators. As biological systems have widely relied on the self-assembly of three classes of biomolecules, nucleic acids, proteins, and polysaccharides, for performing biological functions, the self-assembly of small molecules derived from basic biological building blocks in water offers an excellent starting point for mimicking the structures and functions of biomacromolecules. Therefore, supramolecular hydrogels made of basic biological building blocks, ultimately, may lead to soft materials with designed or unexpected biological functions.
This focus review discusses the supramolecular hydrogels made of amino acids, nucleobases, saccharides, and the conjugates of them. Since there are several excellent reviews on supramolecular hydrogels,[22a, 24] this review will focus on the works published after 2004. Due to the fast development of the field, the choice of examples for this review inevitably is arbitrary and incomplete. Interested readers are also referred to additional review articles.[15, 20, 25] We arrange this review in the following way. After introducing briefly the methods for generating and characterizing supramolecular hydrogels, we discuss representative hydrogelators according to their categories, such as amino acids, nucleobases, and saccharides, as well as highlight the potential applications of the hydrogels. Finally, we offer our perspectives and outlooks on this field. By providing a snapshot for chemists, engineers, and medical scientists, we hope this focus review to contribute to the multidisciplinary research on supramolecular hydrogels for a wide range of applications in different fields.
2. Methods for the formation of supramolecular hydrogels
The formation of a supramolecular hydrogel, or supramolecular hydrogelation, originates from the balance of two seemingly paradoxical behavior—dissolution and precipitation—of the hydrogelators in water. It requires adequate intermolecular interactions to form a molecular network, yet meanwhile the chains of the network must interact with large amount of water molecules. Therefore, hydrogelators require both hydrophobic and hydrophilic moieties for achieving the essential balance for hydrogelation. Additionally, to produce supramolecular hydrogels, an appropriate perturbation of a physical or chemical state is necessary for triggering a phase transition from solution to hydrogel. Such perturbation can be a physical, chemical, or enzymatic process, as briefly discussed in the following three sections.
2.1. Formation of supramolecular hydrogels by a physical process
The change of temperature is the most common physical stimulus to trigger the formation of a supramolecular hydrogel. Many supramolecular hydrogels undergo a sol-gel transition in response to the decrease of temperature, which results in thermal reversible hydrogels. In the other words, supramolecular hydrogelation occurs in water due to weak interactions (e.g., hydrophobic interactions or hydrogen bonds) of which the strength of the interactions is temperature dependent. Besides temperature, ultrasound is another kind of commonly used physical stimuli for tuning the behavior of the supramolecular assembly in water.[26a] Usually being used as a tool in laboratories for speeding up dissolution or dispersion of molecules, ultrasound provides adequate energy to disrupt weak intermolecular interactions to favor the formation of molecular networks, thus resulting in hydrogelation. Recently, the reports from several labs have renewed the interest of using sonication to rearrange intramolecular hydrogen bonds or π-π stacking into intermolecular interactions for making hydrogels.[26b, 26c] In addition, light triggered hydrogelation is a very useful process and becoming popular for modulating the properties of the hydrogels.[27] Physical processes, due to their convenience and simplicity, will still be the first choice of perturbation for testing supramolecular hydrogelators.
2.2. Formation of supramolecular hydrogels by a chemical process
Unlike the physical processes, a chemical process involves the interconversion of two or more molecular species. The most common and convenient chemical process for making supramolecular hydrogels is the change of pH (i.e., protonation and deprotonation) if a hydrogelator bears acid or amine groups. This property likely popularizes the use of amino acids for making supramolecular hydrogelators. The change of the pH of aqueous phase alters the degree of ionization of the amino acid groups, thus controlling sol-gel or gel-sol transition. While most of works simply use this process, several studies have provided interesting and noteworthy insights on pH-responsive self-assembly of small molecules during hydrogelation. For example, Ulijn et al. suggested that the self-assembly of highly hydrophobic molecules could induce dramatic pKa shifts.[28] Kaler et al. reported a pH-induced structural transition of self-assemblies in aqueous mixtures of anionic surfactant and amino acid groups such as histidine and lysine.[29] Hassan’s group introduced a structural evolution from small globular micelles to long worm-like assemblies because the increase in pH will decrease the surface charge of the micelles.[30] The use of pH to control the morphology of the self-assembled nanostructures is convenient and likely will continue to evolve.
Since hydrogelation is a kinetically trapped state, the rate of the change of the pH is critical. Generally, the slow change of pH results in more ordered nanostructures because of the sufficient equilibrium process. Notably, Adams et al. has recently reported the use of the hydrolysis of glucono-δ-lactone (GdL) to gluconic acid as a means for adjusting the pH. By applying the process to a range of naphthalene-dipeptide based hydrogelators, they demonstrated the formation of highly reproducible hydrogels.[31] The simplicity of this elegant process likely will make it be useful for generating many other hydrogels. In addition, this method can prepare supramolecular hydrogels without a shear force that often dominates the outcome of the gelation process.
Another important and increasingly explored chemical process is ligand–receptor interactions for the formation of supramolecular gels. Although ligand-receptor pairs of small molecules are rather limited,[19b, 32] metal–ligand interactions as a form of non-covalent interaction or alternative weak driving force commonly lead to gelation.[33] An excellent recent review has discussed the use of metal-ligand interaction for the gelation of metal complexes.[34]
2.3. Formation of supramolecular hydrogels by an enzymatic process
Enzymatic reactions are ubiquitous in biological systems. Obviously, the hydrogels (e.g., cornea) in living organism are the products of sophisticated enzymatic reactions. Thus, enzymatic reactions, as a special kind of chemical process, can trigger hydrogelation. For example, Messersmith et al. demonstrated the use of enzymes for cross-linking polymers to cause rapid hydrogelation.[35] Recognizing that the use of enzymes to convert a precursor to a hydrogelator can catalytically trigger and regulate molecular self-assembly, Xu et al. developed a simple process that uses phosphatase to catalyze the bond-cleavage reaction for generating supramolecular hydrogels.[21, 36] Based on enzyme catalyzed bond formation, Ulijn et al. used thermolysin to catalyze reverse hydrolysis to form oligopeptides for hydrogelation.[37] An important aspect of enzymatic process is that it permits the control of the delivery, function, and response of a hydrogel according to a specific biological cue or environment. Although enzymatic process provides an accessible route to create sophisticated materials for biomedicine, its exploration only reaches a small set of enzymes, such as phosphatase,[38] thermolysin,[39] β-lactamase,[40] β-glucuronidase,[41] matrix metalloproteases[42], and phosphatase/kinase switch[36a]. Considering the diversity of enzymes and the high efficiency of enzymatic reactions, the continuous exploration of enzymatic process for supramolecular hydrogelation will certainly be fruitful and rewarding.
3. Methods for characterization of supramolecular hydrogels
With the increase of the number of hydrogelators, the characterization of the supramolecular hydrogels at both nanoscale and molecular levels is providing more and more useful information. Thus, in this focus review, before discussing the various classes of supramolecular hydrogels, we briefly introduce the techniques currently used to evaluate supramolecular hydrogels, and give particular attention to those methods that provide the highest resolution of the nanostructures and accurate description of molecular self-assembly processes. Generally, techniques that can retain the native state (e.g., solvated state) during characterization are more desirable than the ones that require drying or staining of the sample or both. The ultimate aim of all the characterization studies is to develop a better understanding of the molecular organization in the matrices of a hydrogel, which eventually contributes to rational design and development of functional supramolecular hydrogels.
3.1. Visual inspection
The easiest way to assay a supramolecular hydrogel is still the “invert-vial” method.[43] As a “zeroth-order” characterization, visually inspecting the hydrogel upon flipping the vial upside down provides researchers an intuitive assessment of the strength of a supramolecular hydrogel. So one can category the materials easily into solution, viscous liquid, half-gel, or solid-like gel, as the starting point to choose more elaborated methods for characterizing the hydrogels.
3.2. AFM/TEM/SEM
The rapid advance and increased accessibility of microscopic instruments have provided powerful tools to determine the morphology of the micro- and nanostructures that serve as the matrices of supramolecular hydrogels. Atomic force microscopy (AFM), as one type of scanning probe microscopies, is a high-resolution technique that has the potential to image hydrated samples in situ under high humidity conditions or without dehydration.[2c, 44] Electron microscopy techniques, both transmission (TEM)[45] and scanning (SEM)[46], allow imaging of features with resolution up to nanometer, which therefore provides valuable information about the morphology of the aggregates/nanofibrils that result in hydrogelation. However, both TEM and SEM require complete drying of the samples under the operating conditions (e.g., high vacuum), sometimes the dehydration results in artifacts that cause difficulties for interpretation. In addition, TEM usually requires staining agents (e.g., phosphotungstate, osmium tetroxide, or urnayl acetate) to increase the electron density of the samples for improving the quality/contrast of the images. The staining procedure also has the potential of introducing artifacts because the staining agents may interact with the molecular assemblies to change their morphology. Fortunately, the tremendous advancements of TEM techniques for imaging biological samples have already established many methods to reduce or to eliminate the artifacts. For example, now it is relatively common to use cryogenic techniques on supramolecular hydrogels, which makes nanometer resolution images of the native gel state feasible. Several groups have already successfully used cryoTEM to visualize the structures of supramolecular hydrogels and related fibrous assemblies.[2b, 47] The development of environmental scanning electron microscope (ESEM)[48] also offers a new technique to image hydrogels under certain humidity,[49] but at the expense of resolution.
3.3. Rheology and DSC
As a soft and wet material, supramolecular hydrogels exhibit viscoelasticity. Being a comprehensive technique to characterize viscoelastic materials, oscillatory rheometry of supramolecular hydrogels is becoming a routine measurement. Rheology focuses on the flow of supramolecular hydrogels and provides information about the type of network (as a form of tertiary structure), which is responsible for the observed hydrogelation.[23] Particularly, rheological data allow the inference of the type, number, and strength of cross-links. There are several kinds of setups (parallel plates, concentric cylinders, cone-and-plate, etc.) used for the measurement of hydrogels, which measures a thin layer of hydrogel between a stationary and a movable component. By measuring how hydrogels respond to an applied oscillatory stress, several elastic properties can be determined simultaneously, such as G* (complex modulus), G′ (storage or elastic modulus), and G″ (loss modulus or viscosity). Moreover, plotting these variables against the oscillatory frequency, the imposed stress, temperature, and hydrogelator concentration, is able to ascertain some key characteristics (e.g., critical strains, thermodynamic nature of gelation) of the supramolecular hydrogels. If there is a sharp phase transition associated with hydrogelation, differential scanning calorimetry (DSC)[50] would be a useful characterization method. One of the most often reported characterizations of a gel is the temperature of gelation (Tgel), which corresponds to the breaking of non-covalent cross-links or global molecular rearrangements by heat. However, the information about the bulk properties of supramolecular hydrogel gives little insight into the atomistic molecular organization, which relates to the superstructure of the self-assembly of the hydrogelators and requires additional techniques for inferring such information.
3.4. XRD and other methods
SAXS (small-angle X-ray scattering), whose resolution closes to that of TEM, is a useful technique to characterize supramolecular hydrogels.[51] While TEM provides local information of the morphology of gel matrices, SAXS offers a spatial averaging measurement of the matrices, which represents the global information of the matrices of supramolecular hydrogels. Since molecular self-assembly usually leads to ordered structures or repeats in at least one dimension, wide-angle X-ray powder diffraction (XRD) also help elucidate the molecular arrangement of supramolecular hydrogels, particularly if the hydrogelation is resulted from the formation of microcrystals in water.[52] The most useful information from a powder pattern is the long d spacing which corresponds to the longest repeat distance in the ordered structure and contributes to determining the packing of molecules in either extended or bent conformation. Moreover, by combining the XRD pattern of a supramolecular hydrogel and modeling, one can derive a possible model of the molecular packing in the supramolecular hydrogel.
Several other techniques, such as CD, UV/Vis, fluorescence, IR, or NMR, can provide certain atomistic information on the molecular organization in supramolecular hydrogels since they detect supramolecular arrangement or intermolecular interactions in primary or secondary structures. For example, circular dichroism (CD), which is a useful tool to study the secondary structures of proteins, is able to provide further molecular information about the self-assembled superstructures in the gel phase or at the gel-to-sol transition.[53] However, it remains a challenge to interpret the CD spectra precisely from the CD data alone. UV/vis techniques can easily identify π-π stacking or metal coordination that might result in hydrogelation. IR is useful for confirming the presence of hydrogen bonds and determining the protonation state of carboxylic acids. Fluorescent probes help evaluate aggregation of the aromatic groups and identify the formation of hydrophobic pockets inside hydrogels. The incorporation of spectroscopic probes, such as fluorescent groups, into hydrogelators, which can generate large, flat aromatic surfaces that promote aggregation, are emerging as an effective design strategy to study the gelation process and understanding the functions of supramolecular hydrogels in biological environment.[54] Solution-state NMR can identify chemical shift changes from the solution spectra to the gelled ones, thus indicating a change in aggregation.[55] The development of solid-state magic angle spinning NMR (MAS-NMR) for elucidating the structures of protein or peptide aggregates may lead to more extensive use of MAS-NMR for characterizing supramolecular hydrogels. Data collected via these methods are complementary to each other. In general, it is always beneficial to collect data via multiple methods so one can compare structurally diverse hydrogelators and evaluate the potential applications of a given supramolecular hydrogel.
4. Supramolecular hydrogels made of aminoacids
The well-established solid phase peptide synthesis (SPPS),[56] which immensely reduces the burden of the synthesis of long-sequence peptides, allows the desired peptides to be made either quickly or automatically by the researchers without formal training in chemistry. Therefore, amino acids have become the favorite building blocks for supramolecular hydrogelators in comparison with nucleobases and saccharides. On the other hand, the increased understanding of the functions of proteins through structural biology has revealed many peptide epitopes for serving as the functional motif on supramolecular hydrogelators for a wider range of biological applications. In fact, there are several excellent reviews on supramolecular hydrogels made of peptides.[20, 24c, 57] So, in this section, after selectively discussing a few examples on hydrogelators made of peptides, we mainly focus on the supramolecular hydrogels that stem from the self-assembly of peptide derivatives or their mixtures.
4.1. Peptides
The 22 proteinogenic amide acids for producing proteins naturally provide a large molecular space for generating peptides. Unlike most proteins that have well-defined tertiary structures, oligopeptides usually possess considerable conformation flexibility to favor intermolecular non-covalent interactions. Thus, it is not surprise that researchers observed unintended hydrogelation of oligopeptides even the goals of their studies might be something else. For example, even the famous insulin (51 amino acids) forms hydrogels during the infusion,[58] which likely stems from the multiple intermediates and aggregates of insulin.[59] Certainly, other oligopeptides that are shorter than insulin can form hydrogels. For example, Namba et al., during the study of the conformation adaptability of the terminal regions of flagellin, observed that two oligopeptides (consisting of 25 amino acids) from the terminal regions of flagellin adapt β-sheet structures at high concentration (>15 mg/mL) and form hydrogels.[60] This rather overlooked report implies that it is probably common for oligopeptides to self-assemble in water and to form hydrogels at high concentrations. This notion turned out to be valid, and the exploration of oligopeptide-based hydrogels became intensive after the demonstration that hydrogels of oligopeptides are cell compatibles and may serve as an alternative culture medium to expensive Matrigel[2c, 61] for cell culture. Since the beginning of this century, there are many reports on supramolecular hydrogels made of oligopeptides, thus it would be impossible to enumerate all of them in the review so that we select a few recent, representative examples to illustrate the applications of supramolecular hydrogels based on peptides.
While most of the oligopeptide hydrogels consisting of βsheets or β-sheet like assemblies, Woolfson et al. reported of peptide hydrogels based on α-helical peptides that contain high percentage of alanine and glutamine in the heptada sequence repeats (Figure 1A).[62] Being mixed in pairs, these peptides form spanning networks of helical fibrils to result in hydrogels of >99% water content. According to the authors, these are the first example of peptide hydrogels consisting of purely helical structures. Interestingly, the underlying mechanism of hydrogelation depends on the peptide sequences. For example, the mixture of 1 and 2 forms hydrogels through hydrophobic fibril–fibril interactions that become strengthened when being warmed. The mixture of 3 and 4 relies more on hydrogen-bonded networks to form a hydrogel that is only stable at low temperature. By replacing alanine with tryptophan to enhance the hydrophobic interaction between the coils, the authors obtained more stable hydrogels from the mixture of 5 and 6 and demonstrated that, like Matrigel, the hydrogel of 5 and 6 is elastically strong enough to support both growth and differentiation of rat adrenal pheochromocytoma cells for certain periods in culture (Figure 1B, C). This elegant design, again, demonstrates the importance of aromatic-aromatic interactions for enabling molecular self-assembly in water.[19a, 63]
Figure 1.
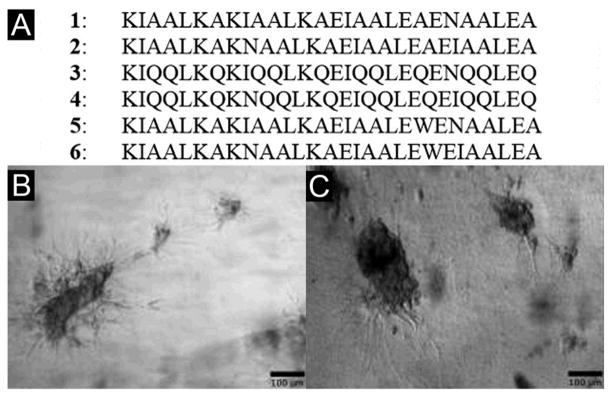
(A) Amino-acid sequences of the responsive α-helical peptide hydrogels. Optical images of differentiating rat adrenal pheochromocytoma (PC12) cells culture on (B) Matrigels and (C) the hydrogels of 5 and 6. (Adapted from © 2009 Macmillan Publishers with permission.[62])
In the recent decade, Schneider and Pochan have pioneered the design of de novo β-hairpin peptides for creating supramolecular hydrogels.[20] One most impressive class among the β-hairpin peptides is the MAX sequences with 20 amino acids (7–14, Figure 2).[64] Upon the change of their state of folding, the molecules of these peptides self-assemble to form β-sheet based nanofibers as the matrices of hydrogels. That is, as random coils, the peptides exhibit excellent solubility in water, but they respond to a stimulus (e.g., ionic strength, pH, temperature, light, or shear forces) to switch to a β-hairpin conformation. These β-hairpin peptides self-assemble into a highly cross-linked network of fibrils, which afford mechanically rigid hydrogels. For example, the addition of salt at physiological pH or the increase of the pH at low ionic strength triggers intramolecular folding of MAX1 to result in the consequent self-assembly and hydrogelation.[65] The replacement of the valine residue by a threonine residue at position 16 of MAX1 affords a new sequence, MAX2, which folds and self-assembles to form hydrogels upon the change of temperature.[64c] Recently, Schneider et al. have developed a simple light-activated hydrogelation peptide that employs a cysteine at position 16 (e.g., MAX7)[64a] or the cysteine modified with a acetic acid group (e.g., MAX7CNB).[66] UV light causes intramolecular folded conformational state to result in its self-assembly and the formation of hydrogels.[64a]
Figure 2.

Representative peptide sequences of β-hairpin peptides that form hydrogels.
One advantage of MAX1 is that DMEM cell culture media can initiate the folding and consequent self-assembly of MAX1 and the hydrogel of MAX1 is cell compatible towards mammalian cells (e.g., NIH 3T3, murine fibroblasts, Figure 3). Besides cell compatibility, the MAX1 hydrogels enable the fibroblast cells to attach to the hydrogel scaffold in the absence or presence of serum proteins. In addition, this hydrogel is able to support fibroblast proliferation to confluence with little effect on the rheological properties of the scaffold.[65] Impressively, a de novo designed β-hairpin peptide (MAX8), also undergoing intramolecular folding and consequently self-assembly to form a cytocompatible hydrogel, is able to serve as an excellent 2D or 3D scaffold for the growth and proliferation of MC3T3 cell[67] or C3H10t1/2 mesenchymal stem cell.[20] Besides the MAX β-hairpin peptides, Schneider et al. developed a three-stranded β-sheet (TSS1, 15) peptide that undergoes temperature-induced folding and self-assembly to afford a network of β-sheet rich nanostructures to constitute a mechanically rigid hydrogel. This hydrogel allows the adhesion and migration of C3H10t1/2 mesenchymal stem cells in 24 h.[68] Moreover, the replacement of the valine in MAX type peptides by a leucine results in a new class of simple, linear, amphiphilic peptides, such as LK13 (16). LK13 undergoes triggered self-assembly to form self-supporting hydrogels that also can act as a scaffold for culturing murine C3H10t1/2 mesenchymal stem cells.[69]
Figure 3.

Cell loading density assay measuring cell proliferation level after 48 h incubation. Degradations per minute (DPM) for 3H-thymidine incorporated into NIH 3T3 cells during DNA synthesis after the cells have been incubated on the surface for 48 h. (Adapted from © 2005 Elsevier Ltd. with permission.[65])
Besides serving as a culture medium for cell adhesion and proliferation, the hydrogel of MAX1 exhibits inherent antibacterial activity due to the high density of lysine residues. This antibacterial effect is particularly attractive because its potential applications in wound healing through directly treating accessible wounds to prevent infection or kill existing bacteria. Schneider et al. reported that the surface of MAX1 hydrogel exhibits broad-spectrum antibacterial activity against both Gram-positive and Gram-negative bacteria (Figure 4).[70] According to the investigation of the authors, the surface of MAX1 hydrogels likely disrupts the inner and outer membranes of Gram-negative bacteria (e.g., E. coli ML-35) to kill the bacteria. Interestingly, the surface of MAX1 hydrogels shows little haemolytic activity toward human erythrocytes, which makes the hydrogels attractive candidates for use in tissue regeneration, even in non-sterile environments.[70] Based on the properties of MAX1, an optimized hydrogel of another β-hairpin peptide, PEP6R (17), shows potent activity for killing both gram-positive and gram-negative bacteria, without comprising their cytocompatibility toward human erythrocytes as well as mammalian mesenchymal stem cells.[71] Another 20-residue peptide, MARG1 (18), containing 6 lysines and 2 arginines, is able to kill methicillin-resistant staphylococcus aureus (MRSA).[72]
Figure 4.
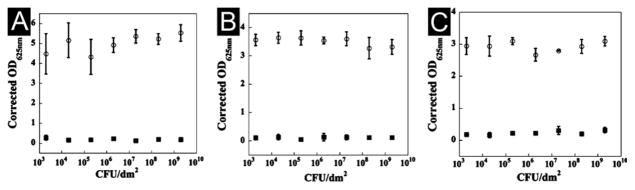
Tissue culture treated polystyrene (TCTP) control surface (○) and 2 wt % MAX1 hydrogel surface (■) challenged with an increasing number of CFUs of Gram-positive bacteria for 48 h. (A) S. aureus. (B) S. epidermidis. (C) S. pyogenes. N = 3. (Adapted from © 2007 American Chemical Society with permission.[70])
Recently, Schneider et al. reported that the self-assembling peptide hydrogel, MAX8 (14), is able to act as an injectable drug delivery vehicle (Figure 5). For example, the encapsulation of curcumin, a hydrophobic polyphenol that have antioxidant, anti-inflammatory, and anti-tumorigenic properties, can occur concurrently with the self-assembly of the peptide. This simple process overcomes the major limitation for the therapeutic use of curcumin due to the lack of water solubility and relatively low bioavailability of curcumin.[74] In addition, it is easy to use MAX8 hydrogels to encapsulate proteins (e.g. lysozyme, lactoferrin, myoglobin, human IgG, α-lactalbumin, and bovine serum albumin (BSA)) for delivering the proteins by injection. According to the authors, the structure of protein is able to control the subsequent release of the protein, likely via electrostatic interactions.[75] Moreover, MAX8 or HLT2 (19) also can encapsulate cells for the delivery of cells (e.g., C3H10t1/2 mesenchymal stem cells,[76] MG63 cells,[77] or chondrocyte[78]) without affecting cell viabilities. More detailed in vitro cell study indicates that the β-hairpin peptide-based hydrogels (e.g., MAX1 and MAX8) barely elicit macrophage activation, which makes these materials promising candidates for in vivo assessment in appropriate animal models, as suggested by the authors.[79]
Figure 5.

Effect of released curcumin from 0.5 wt%, 1 wt% and 2 wt% MAX8 gels prepared with 0 mM, 2mM and 4 mM curcumin on DAOY cells. Cell rounding and detachment as indicator of cell death where found only in the presence of curcumin-encapsulated hydrogels but not with curcumin-free control gels. Scale bar represents 200 μm. (Adapted from © 2011 Elsevier Ltd. with permission.[73])
The function and application of the peptide hydrogels certainly depends on the proteolytic stability of the peptides. For example, Schneider et al. has developed a series of degradable peptides (DP peptides, 20–24, Figure 6B) that have varying proteolytic susceptibilities towards matrix metalloproteinase-13 (MMP-13) (Figure 6A). These peptides undergo environmentally triggered folding and self-assembly under physiologically relevant conditions (e.g., 150 mM NaCl, pH 7.6) to form self-supporting hydrogels. In the presence of the enzyme, gels prepared from distinct peptides degrade at the different rates according to the primary sequences of the peptide. In addition, the proteolysis of the parent peptides occurs at the specific cleavage sites of MMP-13. MMP-3, another common enzyme in tissue injury only minimally cleaves these DP peptides. In vitro migration assays of SW1353 cells show that migration rates through each gel differs according to the peptide sequences, coinciding with the proteolysis studies using exogenous MMP-13.[80] This study illustrates an effective way to control the biodegradation of β-hairpin peptide hydrogels.
Figure 6.
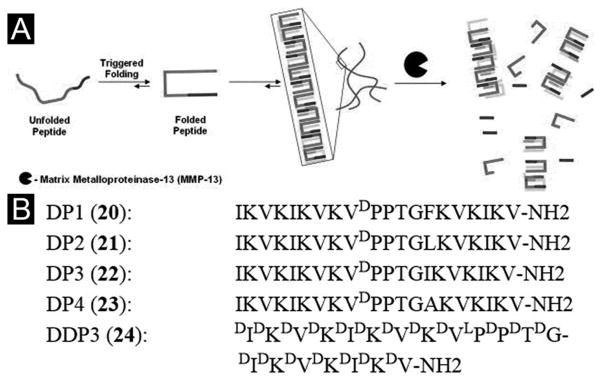
(A) Environmentally triggered folding and self-assembly leading to hydrogelation. Subsequent biodegradation of β-hairpin hydrogels. (B) Sequences of MMP-13 susceptible β-hairpin peptides. (Adapted from © 2011 Elsevier Ltd. with permission.[80])
Besides the work of Schneider and Pochan, there are other reports on peptide hydrogels. For example, Hamachi et al. developed a zwitterionic amino acid tethered amphiphilic molecule, which gave rise to a remarkably stiff hydrogel comparable with polymer-based agarose gel. They described that the zwitterionic amino acid moiety and the C-C double bond unit of the hydrogelator can function as a pH-responsive unit and a light-responsive unit, playing crucial roles in the mechanical reinforcement and the multi-stimuli responsiveness, respectively. Meanwhile, the stiff and multi-stimuli-responsive supramolecular hydrogel is able to be molded to fabricate a collagen gel for spatial patterning, culturing, and differentiation of live cells.[81] Several additional works on peptide hydrogels can be found in the references.[82]
4.2. Derivatives of amino acids or peptides
In addition to the exploration of the self-assembly of oligopeptides in water, the exploration of the derivatives of amino acids or peptides for hydrogel materials has emerged as an active area of research. The most impressive peptide derivatives is peptide amphiphiles[83] that consist of a peptide segment and a hydrophobic tail, especially the one forms a hydrogel and directs differentiation of neuronal progenitor cells.[84] Since there are several excellent reviews on that class of compounds,[85] this section focuses on the discussion of the derivatives of amino acids or peptides other than peptide amphiphiles. We start with the simplest supramolecular hydrogelator made of a derivative of amino acid and move to the derivatives with more elaborated structures.
Except the early report that aroyl L-cystine (25) self-assembles in water to form a hydrogel,[18] the simplest peptide derivatives probably are the phenylalanine derivatives,[86] resulted from the addition of aromatic moieties (phenyl, naphthyl, fluorenyl, and pyrenyl) to the phenylalanine via a simple amide bond. As shown in Figure 7, the aromatic groups in the hydrogelators greatly enhance the intermolecular interaction to result in a series of low molecular weight hydrogelators. Though it is less obvious and less investigated, aromatic–aromatic interaction likely also stabilizes the intermolecular hydrogen bonding in water. Among these supramolecular hydrogelators, the hydrogelator (26) made from a phenylalanine and a cinnamoyl group represented the smallest (MW = 295.34 g/mol) peptide-based hydrogelator prepared to date. The minimum gelation concentration (mgc) of 26 is 1.0 wt%.[87] This result indicates that the cinnamoyl group is able to act as the minimum structure motif to provide sufficient aromatic–aromatic interactions for self-assembly in water. The conversion of phenylalanine to a hydrogelator by the addition of a cinnamoyl group, undoubtedly, support the use of more delocalized aromatic motifs for converting amino acids or peptides to more effective hydrogelators. The naphthalene group (Nap), which is a quite common fragment in clinically approved drug molecules (e.g., propranolol, naphazoline, nafronyl, etc.), can act as an aromatic motif to provide the hydrophobic and aromatic-aromatic interactions for hydrogelation. For example, the attachment of naphthyl group to phenylalanine results two hydrogelators, 27 and 28, which form hydrogels at 0.5 and 0.7 wt%, respectively.[87] Moreover, Xu et al. also reported a class of molecular hydrogelators based on the conjugates of Nap and dipeptides, among which, Nap-Gly-Ala (29) was able to self-assemble and form hydrogel efficiently with helical nanofibers at the lowest concentration of 0.07 wt%.[88] The link of a fluorenyl group to phenylalanine affords a hydrogelator (30) that forms hydrogel at 0.3 wt%.[87]
Figure 7.

The molecular structures of the derivatives of small peptides or amino acids.
Until the serendipitous observation of Fmoc-D-Ala-D-Ala as a hydrogelator,[89] the ability of fluorenyl group (e.g., Fmoc) to enable the self-assemble of peptides in water to form hydrogels might just be an overlooked observation in the synthesis of Fmoc protected peptides. The commercial availability of Fmoc-protected amino acids or small peptides makes it possible for many groups to explore the hydrogelators of Fmoc-protected peptides, especially after Ulijn et al. demonstrated the hydrogels made of Fmoc-peptides can culture cells such as chondrocytes.[90] Recently, Donald et al. designed a peptide derivative (31, Fmoc-Tyr). The addition of glucono-δ-lactone gradually decreases the pH over several hours to produce a particularly transparent hydrogel of 31.[91] Banerjee et al. reported an N-terminally Fmoc-protected dipeptide (32, Fmoc-Val-Asp) to form a transparent, stable hydrogel with a minimum gelation concentration of 0.2% w/v. This simple hydrogel can encapsulate silver ions for producing fluorescent silver nanoclusters under sunlight at physiological pH (7.46), presumably via reducing silver ions with the carboxylate group of the aspartic acid residue.[92] Moreover, they also developed another small peptide based hydrogel (33, Fmoc-Tyr-Asp), which successfully incorporated reduced graphene oxide (RGO). The authors suggested that the aromatic-aromatic interaction between the hydrogelator and the graphene sheets make a stable hybrid hydrogel containing well-dispersed RGO.[93] Ulijn et al. reported the effect on pH by the self-assembly of Fmoc-diphenylalanine (34, Fmoc-FF) to form fibrils consisting of antiparallel β-sheets, which shows that it results in apparent pKa shifts.[28, 94] All the designed Fmoc-dipeptides self-assemble in water, which is consistent with that the main driving force behind the self-assembly process is a combination of the aromatic-aromatic interactions of the fluorenyl moieties and multiple intermolecular hydrogen bonds from the peptidic components.[89] Thus, it is not surprise that the nature of the peptidic tail exhibits a pronounced effect on the type of self-assembled structures.[95] For example, the Hamley group reported the influence of sequence on self-assembled fibrillar structure and the moduli of the hydrogels. They found that two Fmoc-tripeptides (35, Fmoc-VLK(Boc) and 36, Fmoc-K(Boc)LV), both containing a lysine residue that has Nε-tert-butyloxycarbonate (Boc) protected side chain, displayed a remarkable difference in self-assembly properties in borate buffer. That is, hydrogelator 35 forms well-defined β-sheets with a cross-β X-ray diffraction pattern, while hydrogelator 36 forms un-oriented assemblies with multiple stacked sheets.[96] The switch of the K and V residues in 35 and 36 not only changes the peptidic backbones, but also alter the distribution of the hydrophobic groups. It is possible that the latter has a more pronounced effect on the self-assembled structures. By synthesizing fluorinated Fmoc-Phe derivatives, Fmoc-3-F-Phe-OH (37) and Fmoc-F5-Phe-OH (38), Nilsson et al. studied the effect of C-terminal modification on the self-assembly and hydrogelation of Fmoc-Phe derivatives. They demonstrated that the C-terminal carboxylic acid plays a crucial role on the self-assembly and hydrogelation since the C-terminal amide analogues self-assemble more rapidly than the two parent peptides (i.e., 37 and 38) at both acidic and neutral pH,[97] and the methyl ester analogues fail to form a hydrogel.[98]
The ability of Fmoc-Tyr (31) to self-assemble in water also leads the first demonstration of enzyme catalyzed self-assembly of small molecules and supramolecular hydrogelation.[99] Specifically, an alkaline phosphatase (ALP), which is a component of the canonical kinase/phosphatase switch that regulate protein activities, dephosphorylates the PO43- from Fmoc-Tyr-phosphate (Fmoc-pY) under basic conditions, which creates a small molecular hydrogelator, Fmoc-Tyr (31), and results in the formation of a supramolecular hydrogel. Recently, Ulijn et al. studied the mechanism of ALP triggered self-assembly and hydrogelation of Fmoc-Tyr (31).[100] Their study suggests that the phosphorylated Fmoc-Tyr (Fmoc-pY) undergoes rapid and complete dephosphorylation, followed by formation of temporary aggregates, which reorganize into nanofibers and consequently lead to gelation. Their results show a remarkable enhancement of catalytic activity during the early stages of the self-assembly process, which provides evidence for enhancement of enzymatic activation by the formed supramolecular structures and offers new insights in understanding biocatalytic self-assembly.[100] Also using phosphatase, Yang et al. reported enzymatic dephosphorylation as the sole pathway for forming supramolecular hydrogels that have good stability in aqueous solutions at room temperature (Figure 8A). That is, dephosphorylation catalyzed by ALP produces hydrophobic 40 in homogeneous modes and assists the formation of three-dimensional networks (Figure 8B, C). According to the authors, the gels are two-component hydrogels that contains mainly hydrophobic compound 40 doped with hydrophilic compound 39.[98] Besides phosphatase, esterase also can catalyze the formation of a supramolecular hydrogel that is stable over wide pH range.[101]
Figure 8.
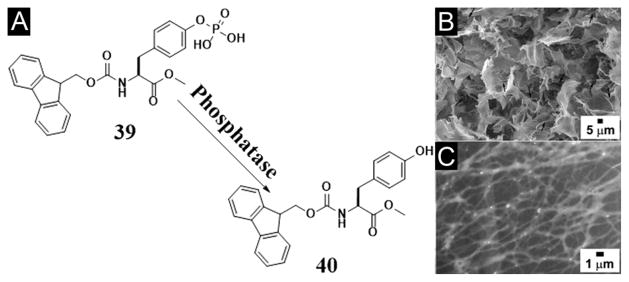
(A) Chemical structures of the enzymatic conversion. (B) SEM and (C) TEM images of hydrogel made by the mixture of 39 and alkaline phosphatase after 1 day. (Adapted from © 2009 American Chemical Society with permission.[98])
Due to its much larger delocalized aromatic group, pyrene group (Pyr) has a strong tendency to form dimers or oligomers in the aqueous phase, which enables many peptides to become hydrogelators. As shown in Figure 9, the molecular hydrogelator based on the conjugate of pyrene and a dipeptide, Pyr-D-Ala-D-Ala (41), is able to form a mechanically weak hydrogel with the mgc of 1.3 wt% at a pH value of 9.5.[19b] Banerjee et al. reported a particularly effective pyrene based hydrogelator, Pyr-Phe-OH (42). 42 not only self-assembles to form a hydrogel in a wide range of aqueous solutions of pH 7.46–14, but also exhibits an exceptionally low mgc (0.037% (w/v)) in phosphate buffer.[102] In addition, hydrophobic and aromatic-aromatic interactions of pyrene groups also induce the self-assembly of other peptide derivatives in water. In fact, a recent study indicates that pyrenyl group renders a variety of pentapeptides (43–46, Figure 9)[102] to be hydrogelators, which form supramolecular hydrogels at mgc from 0.5 to 2.8 wt%.
Figure 9.

The molecular structures of the derivatives of small peptides or amino acids containing a pyrenyl group.
While most of the peptides made of L-amino acids are susceptible to amide bond hydrolysis catalyzed by proteolytic enzymes (e.g., proteases), D-peptides usually are proteolytic resistant. The conjugation of a naphthyl group with D-Phe-D-Phe results in a hydrogelator (47, Nap-D-Phe-D-Phe, Figure 10A) that self-assembles in water to form nanofibers (Figure 10B) and resists proteolytic hydrolysis to offer long-term biostability. As proved by the pharmacokinetics of encapsulated 125I tracers and the SPECT imaging of the hydrogel-encapsulated 131I tracers, the hydrogel of 47 is able to release drugs in vivo in a sustained manner (Figure 10C). As the first in vivo imaging investigation of the drug release properties of the supramolecular hydrogel, isotope encapsulation serves as a valid, useful assay for characterizing the controlled release properties of supramolecular hydrogels in vivo. This work suggests that supramolecular hydrogel is a useful class of biomaterials to complement the well-established drug release systems based on biodegradable polymers.[103]
Figure 10.
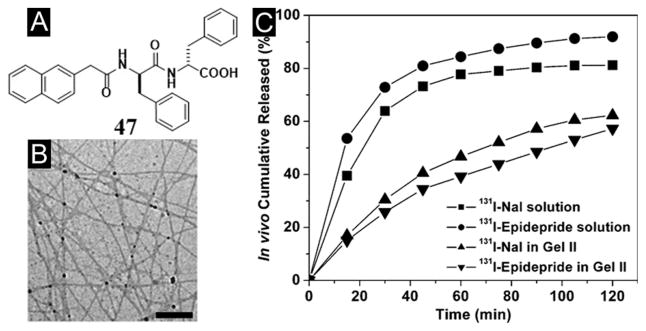
(A) Molecular structure of the hydrogelator. (B) TEM image of the nanofiber matrices in the cryo-dried hydrogel of 47. (C) Cumulative percent in vivo release profiles: (■) 131I-NaI solution, (●) 131I-epidepride solution, (▲) 131I-NaI in gel II, and (▼) 131I-epidepride in hydrogel. (Adapted from © 2009 American Chemical Society with permission.[103])
The study of the both enantiomers of Nap-phe-Phe (abbreviated as NapFF) reveals that NapFF is an exceptional effective hydrogelator, which leads to its use as a general self-assembly motif (termed “samogen”) to enable other non-self-assembling biofunctional molecules to self-assemble in water for the formation of supramolecular nanofibers/hydrogels (Figure 11). The simplicity of this motif allows the conceptual demonstration of many potential applications (e.g., wound healing, drug delivery, controlling cell fate, typing bacteria, and catalysis) of supramolecular hydrogelators in chemistry, materials science, and biomedicine.[104] For example, the design and synthesis of a new hydrogelator NapFF-GEY (49) confirms that it is feasible to use an enzymatic switch for regulating a supramolecular hydrogel. Since 49 is a substrate of the kinase/phosphatase enzyme switch,[105] the phosphorylation and dephosphorylation catalyzed by kinase/phosphatase control sol-gel and gel-sol phase transition (Figure 12A). That is, adding a kinase to the hydrogel induces a gel-sol phase transition because kinase catalyzes the conversion of the tyrosine residue to tyrosine phosphate to give a more hydrophilic molecule of Nap-FFGEY-P(O)(OH)2 (48). Meanwhile, treating the resulting solution with a phosphatase transforms 48 back to 49 and restores the hydrogel. Notably, subcutaneous injection of 48 in mice shows that most of 48 turns into 49 and results in the formation of the supramolecular hydrogel of 49 in vivo (Figure 12B, C). The strategy established in this work may allow minimal invasive drug delivery and promise a new means to engineer biomaterials.[36a] Recognizing the ability of diphenylalanine for self-assembly,[83d] Ulijn et al. attached different aromatic stacking ligands (e.g. Fmoc, Nap, and Cbz) to diphenylalanine. Those hydrogelators self-assemble to form nanofibrils via interlocked β-sheets/π-stacks with varying curvatures, branches and diameters, having similar dimensions to fibrous components of the extracellular matrix, thus the resulting hydrogels support 2D and 3D cell cultures of chondrocyte.[90]
Figure 11.
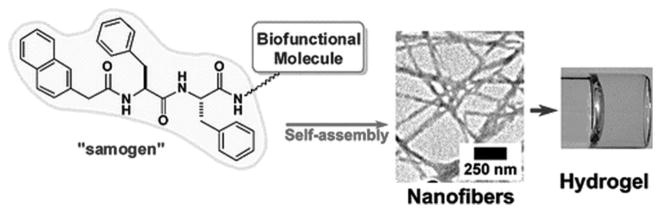
Illustration of the modes of linking bioactive molecules to the self-assembly motif (Nap-phe-Phe). (Adapted from © 2010 American Chemical Society with permission.[104])
Figure 12.
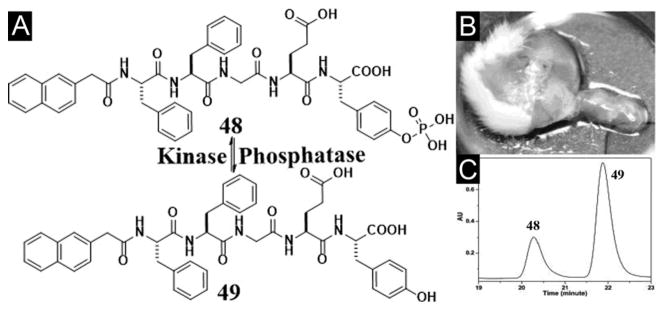
(A) Chemical structures of the enzymatic conversion. (B) Optical image of the hydrogel formed subcutaneously 1 h after injecting 48 into the mice. (C) HPLC trace of the hydrogel shown in Figure 12B. (Adapted from © 2006 American Chemical Society with permission.[36a])
On the basis of the concept illustrated in Figure 12, the covalent connection of taxol, an important anticancer drug, with NapFF through a lysine linkage generates a precursor (50) and a hydrogelator (51) (Figure 13A).[106] The precursor improves the solubility of taxol and becomes the hydrogelators upon the addition of phosphatases (Figure 13B). The hydrogelator (51) not only retains the activity of taxol (Figure 13C), but also is able to release itself from the hydrogel in a sustained or controlled manner. In a recent study,[107] D-amino acids replace L-amino acid in 50 and 51 for conjugating with taxol. In vivo study suggests that the D-peptide-taxol conjugate not only is more effective than taxol and the L-peptide-taxol conjugate, but also is less toxic than taxol.[106] Along the same notion, Yang et al. reported a simpler example that a hydrogelator, consisting of folic acid and taxol, self-assembles to form nanospheres as the matrices of the hydrogel for drug delivery.[108] These works demonstrate that the integration of enzymatic reaction and self-assembly provides a powerful method for engineering functional hydrogels or soft materials for drug delivery.
Figure 13.

(A) Chemical structures of the enzymatic conversion. (B) Optical image and TEM image of the hydrogel 51. (C) Cytotoxicity (y-axis in log10 scale) of taxol, 50, and 51 after incubated with HeLa cells for 48 h. (Adapted from © 2010 American Chemical Society with permission.[106])
Recently, after demonstrating a method to image enzyme-triggered self-assembly of small molecules inside live cells,[54, 109] Xu et al. examined how the self-assembly of small molecules dictates the spatiotemporal profiles of the small molecules in cellular environment. For example, the use of fluorophores to replace taxol in 50 affords several precursors (52–55) and their corresponding hydrogelators (56–58) (Figure 14A) that allow one to study enzyme-triggered self-assembly of small molecules in cellular environment. As shown in Figure 14B, precursor 52, as monomers, diffuses inside the cell and turns to hydrogelator 56 inside the cell via dephosphorylation catalyzed by PTP1B (a potent phosphatase on the outer membrane of ER).[109] Existing mostly in monomeric form, precursor 53 undergoes dephosphorylation to form hydrogelator 57, which self-assembles and largely localizes in the cell membrane. Before enzymatic dephosphorylation, precursor 54 self-assembles significantly to form the nanofibers, its corresponding hydrogelator (58), generated by slow dephosphorylation, localizes mainly outside the cells. Since 59, as the dephosphorylation product of 55, fails to behave as a hydrogelator, it distributes uniformly inside cells. These results, collectively, provide profound insights on the self-assembly of small molecules in cellular environment.[110]
Figure 14.

(A) The chemical structures of the fluorescent phosphorylated precursors and the enzymatic dephosphorylation. (B) Illustration of the distinct spatial distribution of the small molecules in cellular environment due to their different propensity of self-assembly before or after dephosphorylation. (Adapted from © 2013 American Chemical Society with permission.[110])
By inserting a glycine residue between naphthyl group and Phe-Phe motif, Yang et al. reported a new class of hydrogelator, Nap-GFFYGGKO (60, Figure 15). On the basis of the chemical structure of collagen and the repeating tripeptide of glycine-Xaa-4R-hydroxyproline (detonated as GXO; Xaa is any one of the natural amino acids), they designed and synthesized 60. According to the authors, the peptides in the hydrogels have random coil conformation. The authors also suggested that the hydrogel of 60 is the best hydrogel having properties that are similar to those of collagen for 3T3 cell culture.[111] Using the similar approach to generate a supramolecular hydrogelator that has a longer collagen mimic sequence, they demonstrated that its corresponding supramolecular hydrogel supported the differentiation of murine ES cells.[112] These findings illustrate the effectiveness of aromatic-aromatic interactions for generating supramolecular hydrogel scaffolds that mimic nature for tissue engineering.
Figure 15.
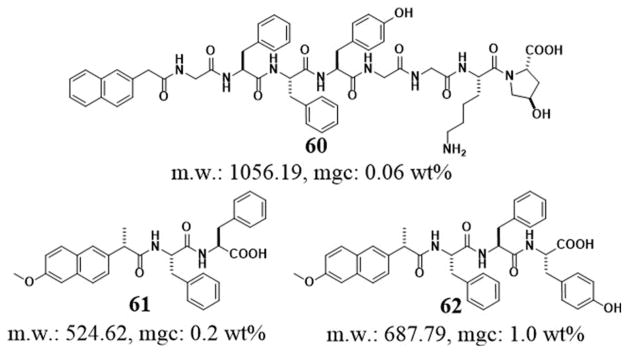
Chemical structures of the hydrogelator 60 and the NSAIDs based peptides hydrogelators 61, 62.
Based on the naphthyl motif in naproxen,[113] a nonsteroidal anti-inflammatory drugs (NSAID), Xu et al. reported the supramolecular hydrogelators made of the conjugates of naproxen and small peptides (e.g. Phe-Phe or Phe-Phe-Tyr) (Figure 15).[114] For example, one resulting hydrogelator, 61, forms a hydrogel at the mgc of 0.2 wt % at pH 7.0. Moreover, hydrogelator 61, also acting as a general motif, enables enzymatic hydrogelation in which the precursor turns into a hydrogelator 62 upon hydrolysis catalyzed by a phosphatase. Surprisingly, after the conjugation with the small peptides made of D-amino acids, the NSAID component in 62 exhibits improved selectivity to their targets. This result implies the attachment of a D-amino acid to a drug candidate may reduce the adverse drug reaction (ADR), a subject that warrants further exploration. In addition, based on the hydrogelator of 62, Xu et al. developed hydrogels that are both anti-inflammatory and anti-HIV.[115] Besides demonstrating common NSAIDs for creating aromatic-aromatic interaction in water to form hydrogels, these works illustrate a feasible way for developing functional molecules that have dual or multiple roles.
The versatility and ability of peptides to self-assemble in water have led to many successful and useful supramolecular hydrogelators (Figure 16). For example, the dipeptide Fmoc-Leu-Gly (63) self-assembles to form surface-supported hydrogel thin films and gap-spanning hydrogel membranes that have controlled thickness (from tens of nanometers to millimeters). SEM and TEM confirm that the molecules of 63 self-assemble to form fibers, with the membranes resembling a dense “mat” of entangled fibers. The films could be reversibly dried and collapsed, then re-swollen to regain the gel structure.[116] Yang et al. reported a small molecular hydrogel of Ac-YYYY-OMe (64) and demonstrated the use of two enzymes, a phosphatase and a tyrosinase, to trigger gel-sol phase transition. By incorporating Congo red as a model drug in the hydrogel, they illustrated that 64 has potentials to controlled release anti-cancer drugs for the treatment of malignant melanoma since it can respond to the tyrosinase over-expressed in malignant melanoma.[117] Recently Yang et al. reported on a supramolecular hydrogel mainly formed by an ester bond hydrolysis process from a taxol derivative (65, Taxol-SA-GSSG). The intratumoral administration of this rather simple hydrogel precursor dramatically inhibit the growth and metastasis of tumors, suggesting a potential application for reducing side effects in taxol chemotherapy.[118] Yang et al. reported a hydrogel precursor (Nap-GFFYE-ss-EE (66)) that contains a disulfide bond. Being reduced by glutathione (GSH), 66 results in a class of supramolecular hydrogels of which the concentration and structure of the hydrogelators regulate the mechanical property and zeta potential of the resulting gels, respectively. Their study suggests that the mechanical property of the hydrogels, but not the zeta potential of self-assembled structures, has big influences on the spreading and proliferation of mouse fibroblast 3T3 cells.[119] Similarly, Yang et al. reported a class of responsive small molecular hydrogels based on the conjugate of adamantane and peptides (67). Due to the supramolecular interaction of adamantine and β-cyclodextrin (β-CD), the addition of β-CD dimers (linked by a disulphide bond) triggers the sol-gel phase transition. 3T3 cells attach and grow well at the surface of the hydrogel of 67 and dimers of β-CD. Furthermore, the addition of a monomeric β-CD derivative competes adamantane-β-CD interactions to result gel-sol transition, thus releasing 3T3 cells after cell culture.[120] Another more elegant example on trigged hydrogelation for cell culture is the photo reactive peptide amphiphiles, demonstrated by Stupp et al. The cleavage of a 2-nitrobenzyl group in a bioactive peptide amphiphile containing the Arg-Gly-Asp (RGD) epitope triggers a sol-to-gel transition as the nanospheres formed by the precursor to become the nanofibers of the hydrogelators (68). As reported, the hydrogel of 68 is able to enhance the bioactivity of the nanostructures surrounding cells in 3D cell cultures.[121] In addition, Stupp et al. also linked cyclic-RGD motif at the side chain of the peptide to construct branched architectures in the monomers (69). They found that the lower packing efficiency and additional space for epitope motion improves cell adhesion, spreading, and migration.[122] The design principles illustrated in these works are valuable for exploring other hydrogelators based on the use peptide epitopes.
Figure 16.

Chemical structures of some peptide derivative hydrogelators.
4.3. Mixtures of peptide-based molecules
One unique advantage of supramolecular hydrogels is that the mixture of different molecular building blocks can result in a hydrogel, providing that there are significant intermolecular interactions. In addition, even if each pure substance fails to act as a hydrogelator alone, the non-covalent complex of two or more different substances may act as hydrogelators to form supramolecular hydrogels. For example, neither Fmoc-leucine (70) nor Fmoc-ε-lysine (71) (Figure 17A) is a hydrogelator, but the mixture of 70 and 71 form a supramolecular hydrogel.[123] This kind of approach greatly expands the pool of hydrogelators and the functions of supramolecular hydrogels. Taking advantage that 70 is an antiinflammatory drug candidates,[124] Xu et al. incorporated another therapeutic agent, pamidronate, into the hydrogel made of 70 and 71 and demonstrated the application of the hydrogel for reducing the toxicity of uranyl oxide at the wound sites on mice model (Figure 17B).[125] This work shows that two types of therapeutic agents, which have discrete yet complementary functions, self-assemble into nanofibers in water to formulate a new supramolecular hydrogel as a self-delivery, drug release biomaterial.
Figure 17.

(A) Molecular structures of the components of the nanofibers as the matrices of the hydrogel. (B) The number of mice that are alive after being wounded in each group. (−) Wound was made on the back of the mice, but was not contaminated with uranyl nitrate; (+) wound was made on the back of the mice and was contaminated with uranyl nitrate; (healing) the uranyl nitrate contaminated wound was treated with gel. (Adapted from © the Royal Society of Chemistry 2005 with permission.[125])
Using the concept of mixing peptide derivatives, after they demonstrated that Fmoc-diphenylalanine (34) provided a suitable matrix for two-dimensional (2D) or three-dimensional (3D) culture of primary bovine chondrocytes,[126] Ulijn et al. investigated whether the introduction of chemical functionality, such as NH2, COOH or OH, could enhance compatibility with different cell types. They designed a series of hydrogel consisting of combinations of Fmoc-FF and N-protected Fmoc amino acids, lysine (K, with side chain R = (CH2)4NH2), glutamic acid (D, with side chain R = CH2COOH), and serine (S, with side chain R = CH2OH), all of which produced fibrous scaffolds. Among the three hydrogels, the hydrogel consisting of Fmoc-FF/S (34+73, Figure 18) is the only hydrogel to support the growth of all three cell types tested (e.g., bovine chondrocytes, human dermal fibroblasts (HDF), and mouse fibroblast 3T3 cells). In addition, the Fmoc-FF/S gel maintains cell morphology in 3D culture of bovine chondrocytes.[127] Expanding the choice of Fmoc-protected peptides, Ulijn et al. reported a peptide-derivatized bioactive hydrogel formed via the self-assembly of a mixture of two aromatic short peptide derivatives: 34 and Fmoc-RGD (74) (Figure 18A). Despite of the simplicity of the self-assembling moieties, the resulted hydrogel also presents RGD at the surface of the nanofibers for mimicking the extracellular matrix. Particularly, the RGD sequence as part of the Fmoc-RGD building block plays a dual role, as a hydrogelator and as a biological ligand. According to the study carried out by the authors, the cylindrical nanofibers interwoven within the hydrogel, which allows the densities of RGDs on the fiber surfaces to be tunable. Through specific RGD–integrin binding, the hydrogel of 34 and 74 promotes the adhesion and proliferation of encapsulated dermal fibroblasts (Figure 18B).[128] These results demonstrate that mixing is a facile method to tailor the mechanical, chemical, and biological properties of supramolecular hydrogels.
Figure 18.

(A) Chemical structures of the hydrogel systems. (B) 2D cell culture on Fmoc-F2/S (1:1) hydrogel. LDH assay results for day 1 (black), 3 (grey) and 7 (white). (Adapted from © 2009 Acta Materialia Inc. Published by Elsevier Ltd. with permission.[127])
Besides serving as the medium for cell culture, the hydrogel made of the mixture of two peptide derivatives can act as a medium for catalytic chemical reactions. For example, the nanofibers formed by the self-assembly of 71 and Fmoc-Phe (75) (Figure 19A) confine heme proteins and luminol to show enhanced chemiluminescence. Specifically, the supramolecular hydrogel formed by the mixture of Fmoc peptides (71+75) is able to mimic the cellular environment of bioluminescence, which improves the quantum yield of chemiluminescence by about an order of magnitude. This simple chemiluminescent system can detect trace levels of blood with 4–6 times enhancement of intensity and better sensitivity for the highly diluted samples (Figure 19B),[129] indicating that this approach might be extended to other applications of chemiluminescent analysis. Moreover, Xu et al. demonstrated that nanofibers in supramolecular hydrogels (71+75) could function as the skeleton of the artificial enzyme and serve as the immobilizing carrier to enhance the catalytic activity of hemin chloride for peroxidation in water or in organic media. The nanofibers in the hydrogel protect the hemin monomers by preventing their dimerization and degradation and facilitate the catalytic reaction by providing nanoporous diffusion channels, which possess unique flexibility to allow the transport of substrates.[130]
Figure 19.

(A) Chemical structures of the molecules. (B) The numbers of photons emitted from the chemiluminescence (CL) reactions of 0.025 mM Gel71+75[76] (Black) or free [76] (Grey) with 0.25 mM H2O2 catalyzed by the blood at various dilutions (from an initial concentration of 11.9 mg Hb mL−1). (Adapted from © 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim with permission.[129])
5. Supramolecular hydrogels made of nucleobases
Despite the well-established intermolecular hydrogen bonds between base pairs (A-T and C-G) of nucleic acids are a source of inspiration for supramolecular chemistry, the number of hydrogelators made of nucleobases, nucleosides, or nucleotide are rather limited. Based on the ability of guanine to form G-quadruplex,[132] Lehn et al. has reported that the guanosine-5′-hydrazide (77) yields a stable supramolecular hydrogel in the presence of metal cations (e.g., K+).[131] As shown in Figure 20A, potassium ion stabilizes the formation of a guanine quartet (G-quartet) of 77 to result a hydrogel (Figure 20B, C). Moreover, the G-quartet of 77 can reversibly react with various aldehydes to form acylhydrazone to result in highly viscous dynamic hydrogels.[131] Moreover, the hydrogel of guanosine-5′-hydrazide 77 can entrap drug molecules such as acyclovir, vitamin C, and vancomycin into its matrices for controlled release (Figure 20D).[133] Besides demonstrating the ability of hydrogels as a promising medium for selective, controlled release of bioactive substances, this work also pioneered the dynamic self-assembly for making dynamic hydrogels.
Figure 20.

(A) Guanine derivatives self-assemble to G-quartets in the presence of metal ions K+. Hydrogel formed from 77. (B) Picture showing that the sample does not flow when the vial is inverted at 15 mM, 23°C in 0.5 M sodium acetate buffer at pH 6.0. (C) TTEM images of fibers forming the gel. (D) Controlled release profiles from the hydrogel formed by 77 as determined by 1H NMR spectroscopy for (■) Acyclovir (553%), (○) Vitamin C (553%), (▲) Vancomycin (393%), (×) Guanosine-5′-hydrazide 77 (73%). (Adapted from © 2005 by the National Academy of Sciences of the USA with permission.[131])
By developing a convenient four-step synthetic route, Barthelemy et al. reported a new, neutral DNA hydrogelator derived from uridine featuring two oleyl chains and one glucose for DNA binding (78, Figure 21A). This amphiphile-nucleic acid complex formation is a consequence of the amphiphilic character of 78, which provides nucleobase-nucleobase interaction and phosphate-sugar interactions when it forms the amphiphile/polyA-polyU complex, according to quasi-elastic light scattering (QELS), TEM, and gel electrophoresis studies (Figure 21B). Fluorescence probing methods indicate the critical aggregation concentration of 78 to be 10 μM (about 0.01 mg/mL). In addition, the author reports that the hydrogelator (78) also stabilizes double helix. This work demonstrates that a glyco-nucleobase-amphiphile is capable of binding efficiently to the nucleic acid double helix structure, which may offer an alternative approach to form stable supramolecular assemblies of nucleic acids. This seminal work likely will expand the current repertoire of amphiphiles for complexing with DNA and provide useful insights for the design and evaluation of new amphiphiles of nucleobases.[134]
Figure 21.
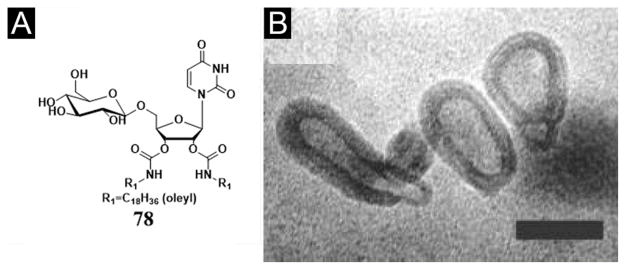
(A) Chemical structures of 78, 1-(2′,3′-dioleylcarbamoyluridine-5′)-β-d-glucopyranoside (DOUGluc). (B) TEM images of small unilamellar vesicles obtained after extrusion of 78 through a 50 nm filter (bar = 50 nm). (Adapted from © 2005 American Chemical Society with permission.[134])
Based on the concept of bolaamphiphiles,[136] Shimizu et al. reported a class of nucleotide hydrogelators, in which a long oligomethylene spacer connects two 3′-phosphorylated thymidine moieties. They investigated the gelling ability of the hydrogelators and found that the longer homologue 79 (Figure 22A) with the C20 oligomethylene spacers gels water very effectively (>253000 water molecules per molecule). Although the bolaamphiphiles spontaneously form a fibrous network of which the gelation behavior strongly depends on the pH and temperature of the aqueous solutions (Figure 22B). The gel-to-sol transition temperature of 79 is approximately 85 °C. XRD measurement of a freeze-dried hydrogel from 79 suggests the presence of lamellar organization consisting of monolayer sheets. According to the authors, the hydrogen bonds involving the 5′-hydroxyl group of the deoxyribose moiety, hydrophobic interaction between the long oligomethylene chains, and π-π stacking of the thymine residues are responsible for the effective hydrogelation.[135]
Figure 22.
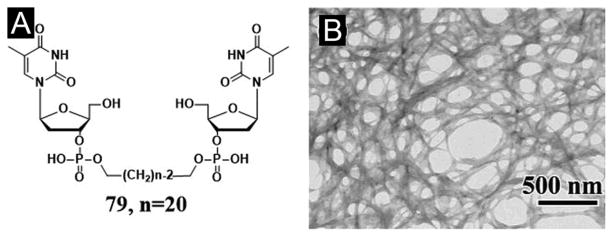
(A) Chemical structure of the nucleotide bolaamphiphile 79. (B) EF-TEM image of the hydrogel from 79. (Adapted from © 2002 American Chemical Society with permission.[135])
Using now the widely applied “click-chemistry”, Schafer et al. synthesized a class of nucleoside hydrogelators based on benzyl azide to ‘click’ with 8-aza-7-deaza-2′-deoxyadenosine bearing 7-ethynyl, 7-octa-(1,7-diynyl), and 7-tri-prop-2-ynyl-amine side chains. Among these nucleoside hydrogelators, the cycloaddition adduct with the shortest linker (80, Figure 23A) yields the most effective hydrogelator that forms a stable hydrogel at a concentration of 0.3 wt% of 80 in water (i.e., one hydrogelator molecule immobilizing 7500 water molecules). In another case, cycloaddition of the 8-aza-7-deaza-7-azido-2′-deoxyadenosine and 3-phenyl-1-propyne leads to 81, an isomeric of 80. 81, with a C–N connectivity between the nucleobase and triazole moiety, self-assembles to form a hydrogel that is less stable than that of 80. Both hydrogels show a similar stability over a wide pH range (4.0–10.0) and consist of with long nanofibers as the matrices. Interestingly, the equal molar mixture of 80 and 81 fail to form a hydrogel (Figure 23B, C, D).[137]
Figure 23.

(A) The benzyltriazole appended nucleosides 80, 81. (B) Tube-inversion test depicting gelation: Gelation test for click adduct 80, click adduct 81, and equimolar mixture of 80 and 81. SEM images of (C) compound 80 and (D) compound 81 with the scale bars 20 μm. (Adapted from © 2011 Elsevier Ltd. with permission.[137])
Kim et al. reported that a simple adenosine-based hydrogelator (82, Figure 24A), having an octyl hydrocarbon tail with a urea linker, self-assembles to form a hydrogel in water under ultrasonic radiation (Figure 24B, C). The formation of hydrogel is possible because the oxidized species of the adenosine derivative produce proper hydrophilic–hydrophobic balance to form a gel network. FTIR and SEM results show that the molecular self-assembly depends on whether the heat or ultrasound is the energy source.[138]
Figure 24.
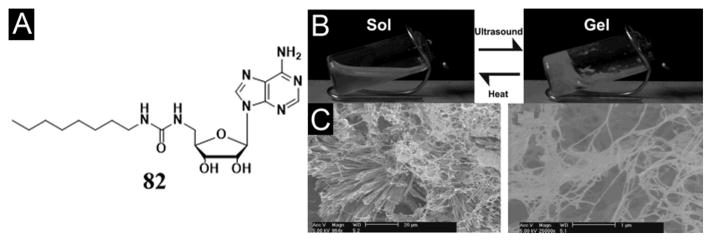
(A) Chemical structure of adenosine-based hydrogelator 82. Gelation of 1.37 × 10−2 M solution of 82 in water at room temperature. (B) Solution of 82 before and after sonication (47 kHz, 30–60 s). (C) SEM images of dried 82 solution (20 μm scale bar) and gel (1 μm scale bar). (Adapted from © the Royal Society of Chemistry 2008 with permission.[138])
6. Supramolecular hydrogels made of saccharides
Although it is quite common for polysaccharides to act as gellant in living organisms,[13] the use of saccharides for supramolecular hydrogels remain less explored, partly due to the difficulty in the synthesis of saccharides. Wang et al. reported a class of methyl benzylidene-α-D-glucopyranoside based low molecular weight gelators (Figure 25). Being able to self-assemble in water, these hydrogelators exhibit different gelation efficiency depending on the structures of their acyl chains. For example, the N-linked carbamates (83, 84), in which the nitrogen atom of the carbamate group is directly attached to the sugar ring, self-assemble to form gels in pure water with long, narrow, uniform fibrous networks at the mgc about 0.5 wt%.[139] The replacement of N-linked to the O-linked carbamates or change in the position of the side chains on the sugar ring still affords excellent hydrogelators (e.g., 85, 86, 87, and 88),[140] which form self-assembled network structures. Although the gelation properties depend on the acyl chain length and the structures of the head groups, these results indicate that the aromatic-aromatic interactions among the gelators and intermolecular hydrophobic interactions contribute to the hydrogelation. Based on the correlation between structures and gelation properties of these molecules, the authors also prepared certain esters and carbamates of 4,6-o-benzylidene-α-D-methyl-glucopyranoside, which consist of amide and urea moieties for generating robust hydrogels.[141]
Figure 25.

Chemical structures of some hydrogelators based on monosaccharide.
Using cyclohexyl groups to enhance intermolecular hydrophobic interactions, Hamachi et al. reported several new photo-responsive glycolipid-based supramolecular hydrogelators bearing fumaric amide as a trans–cis photo switching module. For example, UV light can trigger the gel–sol transition of the hydrogel of 89, and visible light plus bromine can restore the sol of 89 to the hydrogel of 89.[142] According to NMR spectroscopy and various microscopy investigation, the authors suggested that the trans–cis photo isomerization of the double bond of the fumaric amide unit effectively causes assembly or disassembly of the self-assembled supramolecular nanofibers to yield the macroscopic hydrogel or the corresponding sol (Figue 26). Confocal laser scanning microscopy observation shows that the entanglement of the supramolecular nanofibers of 89 produce nanomeshes with pore sizes of about 250 nm. By exploiting the photo-responsive property of the supramolecular nanomeshes, the authors succeeded in off/on switching of bacterial movement and rotary motion of bead-tethered F1-ATPase, a biomolecular motor protein, in the supramolecular hydrogel. Furthermore, photolithographic technique successfully produces sol spots within the gel matrix. The fabricated gel–sol pattern not only regulates bacterial motility in a limited area, but also off/on switches the rotary motion of F1-ATPase at the single-molecule level. These promising results illustrate a unique advantage of photo-responsive supramolecular hydrogels and the resulting nanomeshes.[142]
Figure 26.

Scheme of trans/cis photo isomerization of 89 and a schematic illustration of the pseudo reversible gel–sol transition of hydrogel 89 induced by UV/Vis irradiation. (Adapted from © 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim with permission.[142])
Using the similar concept that N-acetyl glucosamine acts as the hydrophilic head and methyl cyclohexyl groups serve as the hydrophobic tails,[144] Hamachi et al. developed a new glyco-lipid (91) containing muconic amide as the spacer (Figure 27A).[143] Acting as a supramolecular hydrogelator, 91 self-assembles to form helical, stacked nanofibers to result in a stable hydrogel at 0.05 wt3% (Figure 27B). Interestingly, the addition of polystyrene nanobeads (100–500 nm in diameter) facilitates the homogeneous distribution of the supramolecular nanofibers of 91, which leads to use the hydrogel of 91 for efficiently encapsulate and distribute live Jurkat cells in three dimensions.[143] Moreover, they also designed another glyco-lipid (90) as the analogue of 91. Containing 90 at relatively high concentration (5–10 wt%), the hydrogels of 90 exhibit mechanical toughness comparable to that of a polymer hydrogel. The mechanical toughness and thermal reversibility of the hydrogel of 90 allow the fabrication of a supramolecular hydrogel capsule for easy handling in aqueous and cell culture media. The hydrogel also shows response to prostate specific antigen for potential sensing and targeting prostate cancer cells.[145] Recently, Hamachi et al. also demonstrated this kind of glycolipid-based supramolecular hydrogelators (89 and 90) for sophisticated sensing materials, intelligent drug delivery systems, or other biomaterials.[146]
Figure 27.

(A) Chemical structure of the glyco-lipid 90 and 91. (B) TEM image (Left) and AFM image (Right) of hydrogel 91 (0.10 wt3%, distilled water). (Adapted from © 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim with permission.[143])
By introducing a perfluorinated alkyl chain to a glycosyl-nucleosyl motif, Chassande et al. obtained a new type of thermosensitive hydrogel of 92 (Figure 28A) and described the biological properties of the hydrogel. As a low-molecular weight glycosyl-nucleosyl-fluorinated (GNF) compound, 92 forms a hydrogel upon temperature decreasing (Figure 28B, C). Although the incorporation of the perfluorinated chain likely reduces the degradation of 92 in vitro and in vivo, this hydrogelator induces moderate chronic inflammation and prevents the adhesion of adult mesenchymal stem cells derived from adipose tissue (ASC). Interestingly, this hydrogel stimulates osteoblast differentiation of ASC in the absence of osteogenic factors and prolong the survival of cell aggregates. According to the authors, the hydrogel of 92 may find an application as osteogenic materials.[147]
Figure 28.

(A) Developed formula of a GNF compound (92) synthesized by double click chemistry. The fluorinated carbon chain and the glucose moiety are connected to the thymidine by propargyl groups. (B) Time course of elastic (G′) and viscous (G″) moduli of the GNF-based solution immediately after solubilisation at 65 °C and transfer of the liquid GNF solution (included picture, left) into the rheometer tank at room temperature. Note that the curves reach a plateau after 1,500 s (25 min). At this time, gel is completely formed (included picture, right). (C) Elastic (G′) and viscous (G″) moduli of GNF-based hydrogels measured at the plateau at different stimulation frequencies. (Adapted from © 2012 AO Research Institute Davos with permission.[147])
From renewable resources and using enzyme catalysis, John et al. illustrated a novel approach for making hydrogelators containing disaccharide (93, Figure 29).[148] Using curcumin, a known chemopreventive and anti-inflammatory agent, as a model drug, they studied enzyme-triggered drug release via the degradation of the hydrogel of 93. According to their study, the enzyme concentration and/or temperature controls the drug release rate at physiological temperature. The work illustrates a cost-effective drug delivery vehicles from renewable resources.[148] By introducing aromatic ring into glucopyranoside, Zhu et al. synthesized and characterized a new saccharide-derived hydrogelator, 94, as a chemoresponsive hydrogel (Figure 30A). The hydrogel of 94 not only responds to the change of temperature, but also exhibits response to the addition of cysteine and the change of pH due to the presence of aldehyde and acetal groups in 94. In a model study with the presence of cysteine and serine, the hydrogel of 94 quickly release fluorescein disodium (95), but sustainably releases 96, indicating that the release of 95 is controlled by the diffusion, while the release of 96 by the presence of cysteine (Figure 30B).[149]
Figure 29.
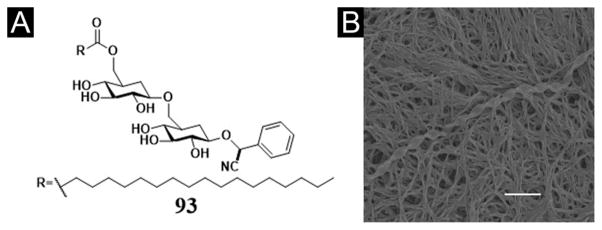
(A) Chemical structure of the amygdalin-based amphiphile 93. (B) SEM micrograph of the hydrogel 93. (Adapted from © 2006 American Chemical Society with permission.[148])
Figure 30.
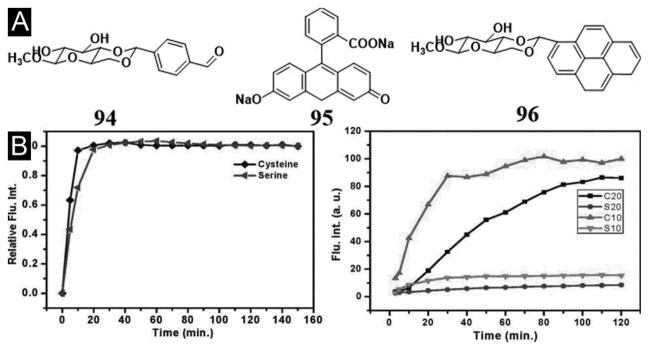
(A) Chemical structure of low molecular weight gelator 94 and guest molecules 95, 96. (B) (Left) Plots of the relative fluorescence intensity at 512 nm of compound 95 (5.0 × 10−5 M) released from the hydrogel of 94 (20 mg/mL, incubated at 37 °C) vs the release time in presence of cysteine and serine (0.5 M). (Right) Plots of the fluorescence intensity at 375 nm of compound 96 (1.0 × 10−5 M) released from the hydrogel of 94 (incubated at 37 °C) vs the release time in presence of cysteine and serine (0.5 M) under different hydrogel concentrations: 20 mg/mL and 10 mg/mL. (Adapted from © 2009 American Chemical Society with permission.[149])
7. Supramolecular hydrogels made of the hybrids of basic biological building blocks
In nature, the functions of living organisms have relied on three classes of biomolecules, nucleic acid, protein, and polysaccharides, which are composed of three fundamental biological building blocks, nucleobases, amino acids, and glycosides, respectively. Despite that most of biomacromolecules consist of a single class of the building blocks (i.e., glycosides form polysaccharides, and amino acids form proteins), it is rather common for biomacromolecules to employ more than one class of the building blocks for achieving diverse functionalities (e. g., glycoproteins consisting of amino acids and saccharides). Therefore, it is logical to explore hydrogelators that consist of more than one class of biological building blocks. This approach provides a unique opportunity for not only expanding the scope of the candidates of hydrogelators, but also integrating multiple bioactivities to the hydrogelators. Although this research is still at its beginning, it already exhibits considerable promises and offer a new class of targets for chemists who wish to develop multifunctional bionanomaterials. The following section discusses some notable examples.
7.1. Supramolecular hydrogels made of aminoglycides
Based on clinical used antibiotics, kanamycin A, Xu et al. have developed the first example of hydrogelator derived from aminoglycosides.[150] For example, hydrogelator 97 (Figure 31A) forms a transparent hydrogel at 0.5 wt%. When Boc-Phe-Phe-Phe-Phe replaces Nap-phe-Phe in 97, the resulting compound (98) also act as a hydrogelator to form a hydrogel at 1.0 wt%. More importantly, the hydrogel of kanamycin (97) is able to sequester 16S rRNA selectively from of the lysate of bacteria (Figure 31B). Although the selectivity of the gel towards 16S remains to be improved, possibly by designing and screening new kanaymicin containing hydrogelators, this work is the first demonstration of a feasible approach for exploring and identifying the potential targets of supramolecular nanofibers/hydrogels. The principle illustrated in this work would help identify new targets of supramolecular nanostructures, which have already led to the development of supramolecular hydrogel pull-down assay for identifying the targets of supramolecular nanofibers.[151]
Figure 31.
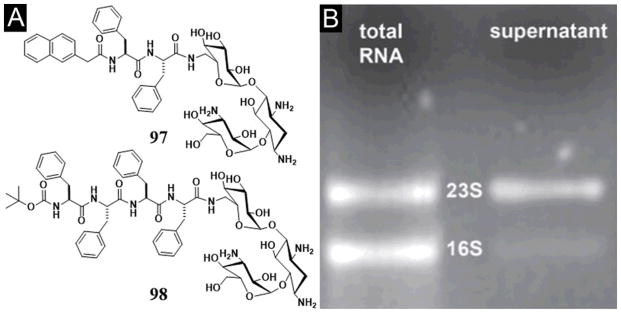
(A) Chemical structure of the hydrogelator 97 and 98 based on kanamycin A. (B) Gel electrophoresis of 16S rRNA and 23S rRNA in the extract before and after incubation with hydrogel of 97 for 4 h. (Adapted from © the Royal Society of Chemistry 2012 with permission.[150])
7.2. Supramolecular hydrogels made of nucleopeptides
Consisting of nucleobases and small peptides, naturally occurring nucleopeptides usually bear considerable biological and biomedical importance.[153] For example, willardiine, a nucleopeptide, acts as antibiotics against microorganisms.[154] As a unnatural nucleic acids containing peptides, peptide nucleic acids (PNA) already have found successful applications in biology and biomedicine, such as serving as an analog of DNA.[155] Therefore, it is quite natural to integrate nucleobases with small peptides for generating supramolecular hydrogelators based on nucleopeptides. Here, besides some studies of nucleosides based peptides,[153, 156] we will introduce some works to use nucleopeptides to serve as building blocks for developing novel class of supramolecular hydrogels.
Recently, Xu et al. demonstrated a new type of hydrogelators (Figure 32A) based on the conjugates of nucleobases (B = thymine, adenine, cytosine, and guanine) and short peptides. Upon a pH- or enzymatic trigger, these nucleopeptides self-assemble in water to afford a novel type of supramolecular hydrogels that are biocompatible (Figure 32B). In addition, both 100 and 101 exhibit significant resistance to proteinase K, a powerful digestive enzyme, which makes nucleopeptides a very attractive molecular platform for developing supramolecular hydrogels that require long-term biostability in vivo. Since these hydrogelators, after self-assembly, are able to interact with the nucleic acids through Watson–Crick H-bonding, this work not only illustrates the first example of nucleopeptides as hydrogelators made by an enzymatic reaction, but also provides a facile way to explore the potential applications of nucleopeptides as biomaterials to interact with DNA or RNA. Changing the nucleobase to nucleoside, Xu et al. developed a precursor of hydrogelators made of adenosine (Figure 33A). Upon the action of phosphatase, the precursor (102) becomes the hydrogelator (103) to self-assemble in water to form a supramolecular nanofibers/hydrogels (Figure 33B).[157] Clearly, more extensive development of hydrogelators of nucleopeptides will lead to more new materials since nucleopeptides are inherently biologically active.
Figure 32.

(A) Molecular structures of the hydrogelators 99, 101, and the corresponding precursors 100 based on nucleopeptides. (B) Transmission electron micrographs of the hydrogels formed by 99, 101 and the solution of 101C (scale bar=100 nm). (Adapted from © 2011 Wiley-VCH with permission.[152])
Figure 33.

(A) Molecular structure of the precursor 102 and hydrogelator 103 of adenosine. (B) TEM images, obtained by negative staining, of the solution of 102 and the gel formed by the addition of the phosphatase. (Adapted from © the Royal Society of Chemistry 2012 with permission.[157])
7.3. Supramolecular hydrogels made of glycopeptides
While vancomycin derivative is likely the most complicated glycopeptide to form a supramolecular hydrogel,[19a] the smallest glycopeptide to form a hydrogel probably is a conjugate of D-phenylalanine and D-glucosamine, 104 (Figure 34A, B, C). More importantly, the supramolecular hydrogel of 104 not only is biocompatible, but also promotes wound healing in vivo (e.g., on mice) (Figure 34D). This result[158] promises a new way to develop new biomaterials for applications such as wound healing. Xu et al. also reported the first example of using β-galactosidase (β-gal) to trigger molecular self-assembly in water to form ordered nanostructure of 106 (Figure 35) and to result in a hydrogel. Given the prevalence of the glycosidases in living organisms, this work, by using β-gal to generate cell compatible, supramolecular nanofibers, may open a new venue to generate nanostructures via the action of enzymes for glycosides.[159]
Figure 34.

(A) Chemical structure of the hydrogelator, Nap-D-Phe-D-glucosamine (104). (B) Optical image of Gel 104. (C) TEM image of Gel 104. (D) Gross appearance of the dorsal skins of Balb/C mice on day 6 after wounding. Left is the negative control and right is Gel 104 treated immediately after the incision was made. (Adapted from © the Royal Society of Chemistry 2007 with permission.[158])
Figure 35.
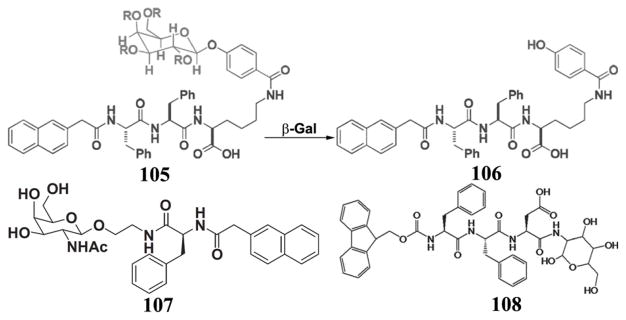
Chemical structure of the precursor 105 and the transformation from 105 to 106 by β-gal, and the molecular structures of the glycopeptide 107 and 108.
Connecting the Nap-phe motif to the anomeric oxygen of N-acetyl-glucosamine (GlcNac), Yang et al. designed and synthesized three novel saccharide-based compounds and tested their hydrogelation and the prospect of the hydrogels for cell culture. They found that one of the glycopeptides, 107, self-assembles to form nanofibers to cause hydrogelation. According to fluorescence spectroscopy, the authors suggested that the extensive hydrogen bonds between sugar rings enhance the formation of efficient aromatic-aromatic interactions between the naphthalene groups in 107, which facilitates hydrogelation. By seeding cells on the surface of the hydrogel, the authors showed that cells adhere to the hydrogel and proliferate, thus demonstrating the biocompatibility of the hydrogel of 107.[160] Recently, Zhang et al. designed and synthesized a biocompatible glycopeptide comprised of an N-fluorenyl-9-methoxycarbonyl phenylalanine-phenylalanine-aspartic acid (Fmoc-Phe-Phe-Asp) sequence and glucosamine (108). Similarly to the cases of 104 and 107, hydrogen bonding interactions stem from the peptide backbones and saccharide, and aromatic-aromatic interactions derive from the Phe and Fmoc groups, which make 108 to form a hydrogel in PBS solution (pH 7.4) via the self-assembly. Impressively, the authors showed the wound healing effect of the hydrogel of 108 for glaucoma-filtering surgery on rabbit animal model.[161] The authors also suggested that these kind of biocompatible hydrogels may serve as an alternative to avoid the cytotoxicity of traditional antiproliferative drug administrated after glaucoma-filtering surgery. This result not only confirms the early observation of the wound healing effect of 104,[158] but also supports that the hydrogels of glycopeptides have a potential to be developed into useful materials in biomedicine.
7.4. Supramolecular hydrogels made of the hybrids of nucleobases, aminoacids, and saccharides
Drawn the inspiration that tRNA is a sophisticated hybrid of nucleobases, amino acids, and saccharides, Xu et al. developed likely the simplest conjugates of nucleobases, amino acids, and saccharides. For example, the covalent connection of three fundamental biological building blocks—nucleobases (e.g., thymine, adenine, cytosine, or guanine), amino acids (e.g., phenylalanine), and a saccharide (e.g., D-glucosamine)—generates a series of novel hydrogelators, 109 and 110 (Figure 36A). These hydrogelators self-assemble in water to yield ordered nanostructures (Figure 36B) and supramolecular hydrogels that possess multifunctional properties, such as biocompatibilities and biostabilities. One unexpected observation is that the inclusion of saccharide at the C-terminal of the hydrogelators significantly enhances their resistance to proteases, which should greatly expand the use of this kind of hydrogels for applications in vivo. Moreover, the nanofibers of these hydrogelators exhibit significant interbase interactions with nucleic acids, which facilitate single strand oligonucleic acids entering cells and the nuclei of cells.[162] This feature is particularly attractive and warrants further exploration by incorporating different biofunctional peptides or molecular recognition motifs to achieve a wide range of biological functions, such as receptor targeting, nucleic acids condensation, blocking metabolism, endosomal escape, and nuclear localization.
Figure 36.
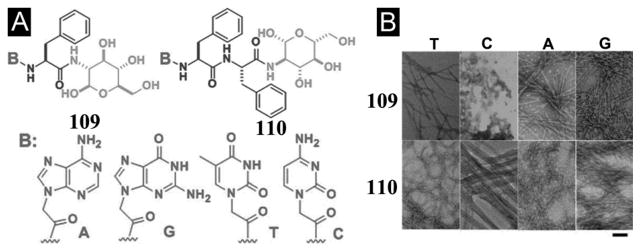
(A) Chemical structures of the hydrogelators 109 and 110 consisting of nucleobase, amino acid, and glycoside. (B) Transmission electron micrographs of the negatively stained hydrogels of 109 and 110, and solution 109C. Scale bar = 100 nm. (Adapted from © 2011 American Chemical Society with permission.[162])
The recent work on sugar–amino acid–nucleoside as potential glycosyltransferase inhibitors,[163] in fact, supports the notion that the integration of saccharides, amino acids, and nucleobases into hydrogelators will lead to multifunctional and bioactive soft materials. Despite the potential applications of this kind of molecular hybrids (or conjugates), their exploration is just at the beginning. For example, by integrating nucleobases, Arg-Gly-Asp (RGD) peptides, and glucosamine in a single molecule, Xu et al. also generated a new kind of biostable and biofunctional hydrogels, 111 and 112 (Figure 37), which are suitable for applications that require long-term biostability.[164] The incorporation of RGD into hydrogelators affords the hydrogels with inherent functionality to bind with live cells through αvβ3 and αvβ5 integrin receptors. The attachment of glycoside at the C-terminal not only improves the biostability of the hydrogels, but also allows the self-assembled glycoside to mimic the functions of glycoproteins and/or proteoglycan. Hydrogelator 111A (i.e., B is adenine) or 111G (i.e., B is is guanine) can self-assemble to form stable hydrogels under physiological conditions (pH = 7.4), which promise a variety of potential applications, including 3D cell culture, tissue engineering, and drug delivery. In addition, the conjugation of different nucleobases, FRGD (a tetrapeptide), and glucosamine generates stable supramolecular hydrogels with fair biostability to resist proteinase K digestion. A recent study, in fact, indicates that 111A is able to promote the proliferation of mouse embryonic stem cells (mES). These results underscore a new opportunity for systematic exploration of the self-assembly of small biomolecules by varying any individual segment to generate a large array of supramolecular hydrogels for biological functions and for biomedical applications.[165]
Figure 37.
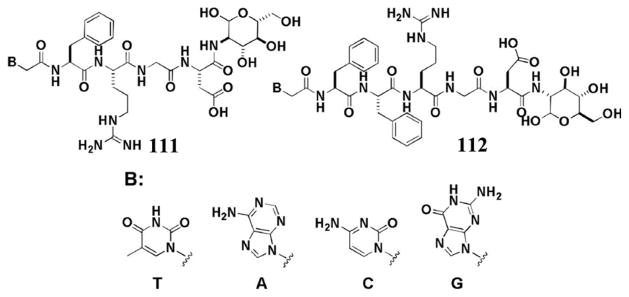
(A) The molecular structures of the hydrogelators 111 and 112 consisting of nucleobases, RGD peptides, and glycoside.(Adapted from © the Royal Society of Chemistry 2012 with permission.[164])
8. Perspective and outlooks
As eloquently stated by Whitesides “Gels are also used throughout complex organisms: in joints, in the eye, in thrombi, on the surface of cells and organs. Combining a deep understanding of the complex physical characteristics of gels with sophisticated, designed biological function is just beginning”[166], the exploration of supramolecular hydrogels consisting of basic biological building block will provide a rich molecular foundation for this endeavor. As illustrated in this review, considerable progresses in the development of the supramolecular hydrogels have validated many concepts on their applications, ranging from 3D cell cultures, screening biomolecules, wound healing, to drug delivery. The self-assembly of small molecules derived from fundamental biological building blocks (e.g., nucleobases, amino acids, and glycosides) offers an excellent starting point for organic chemists who are interested to develop soft biomaterials that mimic, regulate, and complement the functions of biomacromolecules. Since it is a largely unexplored area, the exploration of supramolecular hydrogels made of basic biological building blocks may lead to soft materials with not only designed, but also unexpected biological functions, and the latter probably is more exciting and likely will be more rewarding. While the design, synthesis, and characterization remain an important integral part of this type of research, the key focus should be on the understanding and exploration of the interactions of the supramolecular assemblies of the hydrogelators with cells. Therefore, one should use the most effective known synthetic methodology to generate the hydrogelators and spend major efforts to elucidate the interaction of the hydrogels with the complex biological processes in cellular environment.
As there will be more nanostructured supramolecular hydrogels from basic biological building blocks for achieving specific functionalities, considerable challenges remain to be solved. Currently, there are three widely acknowledged hurdles for supramolecular assemblies: prediction of properties, identification of targets, and determination of atomistic structures. Although there are certain guiding principles for promoting molecular self-assembly in water, it is still impossible from the molecular structures to judge a priori whether a molecule acts as a hydrogelator. While it is difficult to develop a theory for predicting molecular self-assembly, the easiness for making small molecule hydrogelators and the relative abundant of existing supramolecular hydrogelators may provide a comprehensive molecular set for verifying the theories. Since most of biological receptors or epitopes are discovered for purposes that are unrelated to supramolecular self-assembly of small molecules, it is necessary to identify and to verify the cellular targets of the supramolecular assemblies of hydrogelators. Despite the recent developments on this front,[109, 151b] a more effective method is still in great need. Because gel phase is a kinetically trapped state, the determination of the atomistic structures of the nanostructure in a supramolecular hydrogel is a great challenge, which makes the synthesis and experimental evaluation of the supramolecular hydrogels as the main approach to evaluate the correlation between molecular structures and the biological properties of the hydrogels. Hopefully, the fast development in the characterization of microcrystals and other nanoscale structures will one day offer more reliable and fast way to determine the structures of non-crystalline supramolecular assemblies.
Despite the challenges listed above, supramolecular hydrogelators promise a significant advantage over polymeric materials because the molecular structures, degradation profiles, and the purity of supramolecular hydrogelators are more predictable or manageable. However, the successful exploration of hydrogelators made of the basic biological building blocks likely will need cross-disciplinary efforts from chemists, engineers, and medicinal scientists. We envision that the collaboration between scientists in different disciplines and communication across the boundary of subfields of science will address current challenges and discover exciting opportunities in the applications of supramolecular hydrogels made of the basic biological building blocks, and ultimately lead to an innovative class of de novo biological soft materials.
Acknowledgments
This work was partially supported by NSF (DMR 0820492), an HFSP grant (RGP0056/2008), and an NIH grant (R01CA142746).
References
- 1.a) Refojo MF. J Polym Sci A1. 1967;5:3103. doi: 10.1002/pol.1967.150051211. [DOI] [PubMed] [Google Scholar]; b) Peppas NA, Bures P, Leobandung W, Ichikawa H. Eur J Pharm Biopharm. 2000;50:27–46. doi: 10.1016/s0939-6411(00)00090-4. [DOI] [PubMed] [Google Scholar]
- 2.a) Yoshida R, Uchida K, Kaneko Y, Sakai K, Kikuchi A, Sakurai Y, Okano T. Nature. 1995;374:240–242. [Google Scholar]; b) Schneider JP, Pochan DJ, Ozbas B, Rajagopal K, Pakstis L, Kretsinger J. J Am Chem Soc. 2002;124:15030–15037. doi: 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]; c) Yokoi H, Kinoshita T, Zhang SG. Proc Natl Acad Sci USA. 2005;102:8414–8419. doi: 10.1073/pnas.0407843102. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Estroff LA, Hamilton AD. Chem Rev. 2004;104:1201–1217. doi: 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]; e) Terech P, Weiss RG. Chem Rev. 1997;97:3133–3159. doi: 10.1021/cr9700282. [DOI] [PubMed] [Google Scholar]
- 3.George M, Weiss RG. Acc Chem Res. 2006;39:489–497. doi: 10.1021/ar0500923. [DOI] [PubMed] [Google Scholar]
- 4.a) Lee KY, Mooney DJ. Chem Rev. 2001;101:1869–1879. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]; b) Hoffman AS. Adv Drug Deliver Rev. 2002;54:3–12. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 5.Korsmeyer RW, Peppas NA. J Membrane Sci. 1981;9:211–227. [Google Scholar]
- 6.a) Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Adv Mater. 2006;18:1345–1360. [Google Scholar]; b) Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA. Adv Mater. 2009;21:3307–3329. doi: 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rowley JA, Madlambayan G, Mooney DJ. Biomaterials. 1999;20:45–53. doi: 10.1016/s0142-9612(98)00107-0. [DOI] [PubMed] [Google Scholar]
- 8.Lee KY, Peters MC, Anderson KW, Mooney DJ. Nature. 2000;408:998–1000. doi: 10.1038/35050141. [DOI] [PubMed] [Google Scholar]
- 9.Kim J, Mooney DJ. Nano Today. 2011;6:466–477. doi: 10.1016/j.nantod.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Dong LC, Hoffman AS. J Control Release. 1991;15:141–152. [Google Scholar]; b) Chen GH, Hoffman AS. Nature. 1995;373:49–52. doi: 10.1038/373049a0. [DOI] [PubMed] [Google Scholar]
- 11.a) Gong JP, Katsuyama Y, Kurokawa T, Osada Y. Adv Mater. 2003;15:1155–1158. [Google Scholar]; b) Gong JP. Soft Matter. 2010;6:2583–2590. [Google Scholar]
- 12.Hoffman AS. Adv Drug Deliver Rev. 2013;65:10–16. doi: 10.1016/j.addr.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Lee KY, Mooney DJ. Prog Polym Sci. 2012;37:106–126. doi: 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffman AS, Stayton PS. Prog Polym Sci. 2007;32:922–932. [Google Scholar]
- 15.Estroff LA, Hamilton AD. Chem Rev. 2004;104:1201–1217. doi: 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]
- 16.a) Yamanaka M, Fujii H. J Org Chem. 2009;74:5390–5394. doi: 10.1021/jo900894q. [DOI] [PubMed] [Google Scholar]; b) Hanabusa K, Yamada M, Kimura M, Shirai H. Angew Chem Int Edit. 1996;35:1949–1951. [Google Scholar]; c) Estroff LA, Hamilton AD. Angew Chem Int Edit. 2000;39:3447–3450. doi: 10.1002/1521-3773(20001002)39:19<3447::aid-anie3447>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 17.Boettcher C, Schade B, Fuhrhop JH. Langmuir. 2001;17:873–877. [Google Scholar]
- 18.a) Menger FM, Caran KL. J Am Chem Soc. 2000;122:11679–11691. [Google Scholar]; b) Gortner RA, Hoffman WF. J Am Chem Soc. 1921;43:2199–2202. [Google Scholar]
- 19.a) Xing BG, Yu CW, Chow KH, Ho PL, Fu DG, Xu B. J Am Chem Soc. 2002;124:14846–14847. doi: 10.1021/ja028539f. [DOI] [PubMed] [Google Scholar]; b) Zhang Y, Yang ZM, Yuan F, Gu HW, Gao P, Xu B. J Am Chem Soc. 2004;126:15028–15029. doi: 10.1021/ja044401g. [DOI] [PubMed] [Google Scholar]
- 20.Branco MC, Schneider JP. Acta Biomater. 2009;5:817–831. doi: 10.1016/j.actbio.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Z, Liang G, Xu B. Acc Chem Res. 2008;41:315–326. doi: 10.1021/ar7001914. [DOI] [PubMed] [Google Scholar]
- 22.a) Ulijn RV, Bibi N, Jayawarna V, Thornton PD, Todd SJ, Mart RJ, Smith AM, Gough JE. Mater Today. 2007;10:40–48. [Google Scholar]; b) Yang Z, Xu B. J Mater Chem. 2007;17:2385–2393. [Google Scholar]
- 23.Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, Deming TJ. Nature. 2002;417:424–428. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- 24.a) Sangeetha NM, Maitra U. Chem Soc Rev. 2005;34:821–836. doi: 10.1039/b417081b. [DOI] [PubMed] [Google Scholar]; b) Araki K, Yoshikawa I. Top Curr Chem. 2005;256:133–165. doi: 10.1007/b107173. [DOI] [PubMed] [Google Scholar]; c) Shimizu T. Polym J. 2003;35:1–22. [Google Scholar]
- 25.a) Ikeda M, Ochi R, Kurita Y-s, Pochan DJ, Hamachi I. Chem-Eur J. 2012;18:13091–13096. doi: 10.1002/chem.201201670. [DOI] [PubMed] [Google Scholar]; b) Suzuki M, Hanabusa K. Chem Soc Rev. 2009;38:967–975. doi: 10.1039/b816192e. [DOI] [PubMed] [Google Scholar]; c) Tamaru S-i, Hamachi I. Struct Bond. 2008;129:95–125. [Google Scholar]; d) Tomatsu I, Peng K, Kros A. Adv Drug Deliver Rev. 2011;63:1257–1266. doi: 10.1016/j.addr.2011.06.009. [DOI] [PubMed] [Google Scholar]; e) Ulijn RV. J Mater Chem. 2006;16:2217–2225. [Google Scholar]; f) Wu ZL, Gong JP. NPG Asia Mater. 2011;3:57–64. [Google Scholar]
- 26.a) Yu XD, Liu QA, Wu JC, Zhang MM, Cao XH, Zhang S, Wang Q, Chen LM, Yi T. Chem-Eur J. 2010;16:9099–9106. doi: 10.1002/chem.201000187. [DOI] [PubMed] [Google Scholar]; b) Maity S, Kumar P, Haldar D. Soft Matter. 2011;7:5239–5245. [Google Scholar]; c) Roy B, Bairi P, Nandi AK. Soft Matter. 2012;8:2366–2369. [Google Scholar]
- 27.a) Ajayaghosh A, Praveen VK, Vijayakumar C. Chem Soc Rev. 2008;37:109–122. doi: 10.1039/b704456a. [DOI] [PubMed] [Google Scholar]; b) He MT, Li JB, Tan S, Wang RZ, Zhang Y. J Am Chem Soc. 2013;135:18718–18721. doi: 10.1021/ja409000b. [DOI] [PubMed] [Google Scholar]
- 28.Tang C, Smith AM, Collins RF, Ulijn RV, Saiani A. Langmuir. 2009;25:9447–9453. doi: 10.1021/la900653q. [DOI] [PubMed] [Google Scholar]
- 29.Gonzalez YI, Kaler EW. Langmuir. 2005;21:7191–7199. doi: 10.1021/la0503698. [DOI] [PubMed] [Google Scholar]
- 30.Verma G, Aswal VK, Hassan P. Soft Matter. 2009;5:2919–2927. [Google Scholar]
- 31.a) Adams DJ, Butler MF, Frith WJ, Kirkland M, Mullen L, Sanderson P. Soft Matter. 2009;5:1856–1862. [Google Scholar]; b) Chen L, Morris K, Laybourn A, Elias D, Hicks MR, Rodger A, Serpell L, Adams DJ. Langmuir. 2010;26:5232–5242. doi: 10.1021/la903694a. [DOI] [PubMed] [Google Scholar]
- 32.a) Rao JH, Lahiri J, Isaacs L, Weis RM, Whitesides GM. Science. 1998;280:708–711. doi: 10.1126/science.280.5364.708. [DOI] [PubMed] [Google Scholar]; b) Lahiri J, Isaacs L, Tien J, Whitesides GM. Anal Chem. 1999;71:777–790. doi: 10.1021/ac980959t. [DOI] [PubMed] [Google Scholar]; c) Zhang Y, Gu HW, Yang ZM, Xu B. J Am Chem Soc. 2003;125:13680–13681. doi: 10.1021/ja036817k. [DOI] [PubMed] [Google Scholar]
- 33.a) Xing BG, Choi MF, Xu B. Chem-Eur J. 2002;8:5028–5032. doi: 10.1002/1521-3765(20021104)8:21<5028::AID-CHEM5028>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]; b) Xing BG, Choi MF, Xu B. Chem Commun. 2002:362–363. doi: 10.1039/b111245g. [DOI] [PubMed] [Google Scholar]
- 34.Steed JW. Chem Soc Rev. 2009;38:506–519. doi: 10.1039/b810364j. [DOI] [PubMed] [Google Scholar]
- 35.a) Sanborn TJ, Messersmith PB, Barron AE. Biomaterials. 2002;23:2703–2710. doi: 10.1016/s0142-9612(02)00002-9. [DOI] [PubMed] [Google Scholar]; b) Douglas TEL, Messersmith PB, Chasan S, Mikos AG, de Mulder ELW, Dickson G, Schaubroeck D, Balcaen L, Vanhaecke F, Dubruel P, Jansen JA, Leeuwenburgh SCG. Macromol Biosci. 2012;12:1077–1089. doi: 10.1002/mabi.201100501. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hu BH, Messersmith PB. J Am Chem Soc. 2003;125:14298–14299. doi: 10.1021/ja038593b. [DOI] [PubMed] [Google Scholar]
- 36.a) Yang ZM, Liang GL, Wang L, Xu B. J Am Chem Soc. 2006;128:3038–3043. doi: 10.1021/ja057412y. [DOI] [PubMed] [Google Scholar]; b) Wang QG, Yang ZM, Gao Y, Ge WW, Wang L, Xu B. Soft Matter. 2008;4:550–553. doi: 10.1039/b715439a. [DOI] [PubMed] [Google Scholar]
- 37.McDonald TO, Qu HL, Saunders BR, Ulijn RV. Soft Matter. 2009;5:1728–1734. [Google Scholar]
- 38.Yang ZM, Liang GL, Ma ML, Gao Y, Xu B. Small. 2007;3:558–562. doi: 10.1002/smll.200700015. [DOI] [PubMed] [Google Scholar]
- 39.Toledano S, Williams RJ, Jayawarna V, Ulijn RV. J Am Chem Soc. 2006;128:1070–1071. doi: 10.1021/ja056549l. [DOI] [PubMed] [Google Scholar]
- 40.Yang ZM, Ho PL, Liang GL, Chow KH, Wang QG, Cao Y, Guo ZH, Xu B. J Am Chem Soc. 2007;129:266–267. doi: 10.1021/ja0675604. [DOI] [PubMed] [Google Scholar]
- 41.Li AA, Shen F, Zhang T, Cirone P, Potter M, Chang PL. J Biomed Mater Res B. 2006;77B:296–306. doi: 10.1002/jbm.b.30342. [DOI] [PubMed] [Google Scholar]
- 42.Tauro JR, Gemeinhart RA. Bioconjugate Chem. 2005;16:1133–1139. doi: 10.1021/bc0501303. [DOI] [PubMed] [Google Scholar]
- 43.a) Chung YI, Lee SY, Tae G. Colloid Surface A. 2006;284:480–484. [Google Scholar]; b) Brettar I, Hofle MG. Mar Ecol-Prog Ser. 1993;94:253–265. [Google Scholar]
- 44.Wang SF, Chen T, Zhang ZL, Shen XC, Lu ZX, Pang DW, Wong KY. Langmuir. 2005;21:9260–9266. doi: 10.1021/la050947k. [DOI] [PubMed] [Google Scholar]
- 45.Willcox PJ, Howie DW, Schmidt-Rohr K, Hoagland DA, Gido SP, Pudjijanto S, Kleiner LW, Venkatraman S. J Polym Sci Pol Phys. 1999;37:3438–3454. [Google Scholar]
- 46.Chen J, Park H, Park K. J Biomed Mater Res. 1999;44:53–62. doi: 10.1002/(sici)1097-4636(199901)44:1<53::aid-jbm6>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 47.Krysmann MJ, Castelletto V, Kelarakis A, Hamley IW, Hule RA, Pochan DJ. Biochemistry. 2008;47:4597–4605. doi: 10.1021/bi8000616. [DOI] [PubMed] [Google Scholar]
- 48.Gibson P, Schreuder-Gibson H, Rivin D. Colloid Surface A. 2001;187:469–481. [Google Scholar]
- 49.Xia YQ, Guo TY, Song MD, Zhang BH, Zhang BL. Biomacromolecules. 2005;6:2601–2606. doi: 10.1021/bm050324l. [DOI] [PubMed] [Google Scholar]
- 50.Siepmann J, Peppas NA. Adv Drug Deliver Rev. 2001;48:139–157. doi: 10.1016/s0169-409x(01)00112-0. [DOI] [PubMed] [Google Scholar]
- 51.a) Moreau L, Barthelemy P, El Maataoui M, Grinstaff MW. J Am Chem Soc. 2004;126:7533–7539. doi: 10.1021/ja039597j. [DOI] [PubMed] [Google Scholar]; b) Anderson KM, Day GM, Paterson MJ, Byrne P, Clarke N, Steed JW. Angew Chem Int Edit. 2007;47:1058–1062. doi: 10.1002/anie.200703785. [DOI] [PubMed] [Google Scholar]
- 52.Roy S, Banerjee A. Soft Matter. 2011;7:5300–5308. [Google Scholar]
- 53.a) Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang ZW, Fletterick RJ, Cohen FE, Prusiner SB. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tarentin Al, Maley F. J Biol Chem. 1974;249:811–817. [PubMed] [Google Scholar]
- 54.Gao Y, Berciu C, Kuang Y, Shi J, Nicastro D, Xu B. ACS nano. 2013;7:9055–9063. doi: 10.1021/nn403664n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim H, Ralph J. Org Biomol Chem. 2010;8:576–591. doi: 10.1039/b916070a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Merrifield RB. J Am Chem Soc. 1963;85:2149. [Google Scholar]
- 57.a) Ryan DM, Nilsson BL. Polymer Chemistry. 2012;3:18–33. [Google Scholar]; b) Das D, Kar T, Das PK. Soft Matter. 2012;8:2348–2365. [Google Scholar]; c) Altunbas A, Pochan DJ. Peptide-Based Materials. 2012;310:135–167. doi: 10.1007/128_2011_206. [DOI] [PubMed] [Google Scholar]
- 58.Hutchison KG. J Pharm Pharmacol. 1985;37:528–531. doi: 10.1111/j.2042-7158.1985.tb03060.x. [DOI] [PubMed] [Google Scholar]
- 59.Jimenez JL, Nettleton EJ, Bouchard M, Robinson CV, Dobson CM, Saibil HR. Proc Natl Acad Sci USA. 2002;99:9196–9201. doi: 10.1073/pnas.142459399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vonderviszt F, Sonoyama M, Tasumi M, Namba K. Biophys J. 1992;63:1672–1677. doi: 10.1016/S0006-3495(92)81751-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang SG. Biotechnol Adv. 2002;20:321–339. doi: 10.1016/s0734-9750(02)00026-5. [DOI] [PubMed] [Google Scholar]
- 62.Banwell EF, Abelardo ES, Adams DJ, Birchall MA, Corrigan A, Donald AM, Kirkland M, Serpell LC, Butler MF, Woolfson DN. Nat Mater. 2009;8:596–600. doi: 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma ML, Kuang Y, Gao Y, Zhang Y, Gao P, Xu B. J Am Chem Soc. 2010;132:2719–2728. doi: 10.1021/ja9088764. [DOI] [PubMed] [Google Scholar]
- 64.a) Haines LA, Rajagopal K, Ozbas B, Salick DA, Pochan DJ, Schneider JP. J Am Chem Soc. 2005;127:17025–17029. doi: 10.1021/ja054719o. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pochan DJ, Schneider JP, Kretsinger J, Ozbas B, Rajagopal K, Haines L. J Am Chem Soc. 2003;125:11802–11803. doi: 10.1021/ja0353154. [DOI] [PubMed] [Google Scholar]; c) Rajagopal K, Ozbas B, Pochan DJ, Schneider JP. Eur Biophys J Biophy. 2006;35:162–169. doi: 10.1007/s00249-005-0017-7. [DOI] [PubMed] [Google Scholar]
- 65.Kretsinger JK, Haines LA, Ozbas B, Pochan DJ, Schneider JP. Biomaterials. 2005;26:5177–5186. doi: 10.1016/j.biomaterials.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 66.Chang CY, Niblack B, Walker B, Bayley H. Chem Biol. 1995;2:391–400. doi: 10.1016/1074-5521(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 67.Hule RA, Nagarkar RP, Altunbas A, Ramay HR, Branco MC, Schneider JP, Pochan DJ. Faraday Discuss. 2008;139:251–264. doi: 10.1039/b717616c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rughani RV, Salick DA, Lamm MS, Yucel T, Pochan DJ, Schneider JP. Biomacromolecules. 2009;10:1295–1304. doi: 10.1021/bm900113z. [DOI] [PubMed] [Google Scholar]
- 69.Geisler IM, Schneider JP. Adv Funct Mater. 2012;22:529–537. [Google Scholar]
- 70.Salick DA, Kretsinger JK, Pochan DJ, Schneider JP. J Am Chem Soc. 2007;129:14793–14799. doi: 10.1021/ja076300z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Veiga AS, Sinthuvanich C, Gaspar D, Franquelim HG, Castanho MARB, Schneider JP. Biomaterials. 2012;33:8907–8916. doi: 10.1016/j.biomaterials.2012.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salick DA, Pochan DJ, Schneider JP. Adv Mater. 2009;21:4120. [Google Scholar]
- 73.Altunbas A, Lee SJ, Rajasekaran SA, Schneider JP, Pochan DJ. Biomaterials. 2011;32:5906–5914. doi: 10.1016/j.biomaterials.2011.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Altunbas A, Lee SJ, Rajasekaran SA, Schneider JP, Pochan DJ. Biomaterials. 2011;32:5906–5914. doi: 10.1016/j.biomaterials.2011.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Branco MC, Pochan DJ, Wagner NJ, Schneider JP. Biomaterials. 2010;31:9527–9534. doi: 10.1016/j.biomaterials.2010.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ, Schneider JP. Proc Natl Acad Sci USA. 2007;104:7791–7796. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yan C, Mackay ME, Czymmek K, Nagarkar RP, Schneider JP, Pochan DJ. Langmuir. 2012;28:6076–6087. doi: 10.1021/la2041746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sinthuvanich C, Haines-Butterick LA, Nagy KJ, Schneider JP. Biomaterials. 2012;33:7478–7488. doi: 10.1016/j.biomaterials.2012.06.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haines-Butterick LA, Salick DA, Pochan DJ, Schneider JP. Biomaterials. 2008;29:4164–4169. doi: 10.1016/j.biomaterials.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Giano MC, Pochan DJ, Schneider JP. Biomaterials. 2011;32:6471–6477. doi: 10.1016/j.biomaterials.2011.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Komatsu H, Tsukiji S, Ikeda M, Hamachi I. Chem-Asian J. 2011;6:2368–2375. doi: 10.1002/asia.201100134. [DOI] [PubMed] [Google Scholar]
- 82.a) Ruan L, Zhang H, Luo H, Liu J, Tang F, Shi YK, Zhao X. Proc Natl Acad Sci USA. 2009;106:5105–5110. doi: 10.1073/pnas.0900026106. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Anzini P, Xu C, Hughes S, Magnotti E, Jiang T, Hemmingsen L, Demeler B, Conticello VP. J Am Chem Soc. 2013;135:10278–10281. doi: 10.1021/ja404677c. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dublin SN, Conticello VP. J Am Chem Soc. 2008;130:49. doi: 10.1021/ja0775016. [DOI] [PubMed] [Google Scholar]; d) Lu K, Guo L, Mehta AK, Childers WS, Dublin SN, Skanthakumar S, Conticello VP, Thiyagarajan P, Apkarian RP, Lynn DG. Chem Commun. 2007:2729–2731. doi: 10.1039/b701029j. [DOI] [PubMed] [Google Scholar]; e) Zimenkov Y, Dublin SN, Ni R, Tu RS, Breedveld V, Apkarian RP, Conticello VP. J Am Chem Soc. 2006;128:6770–6771. doi: 10.1021/ja0605974. [DOI] [PubMed] [Google Scholar]
- 83.a) Yu YC, Berndt P, Tirrell M, Fields GB. J Am Chem Soc. 1996;118:12515–12520. [Google Scholar]; b) Kunitake T. Angew Chem Int Edit. 1992;31:709–726. [Google Scholar]; c) Ihara H, Yoshikai K, Takafuji M, Hirayama C, Yamada K. Kobunshi Ronbunshu. 1991;48:327–334. [Google Scholar]; d) Reches M, Gazit E. Science. 2003;300:625–627. doi: 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- 84.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 85.a) Matson JB, Zha RH, Stupp SI. Curr Opin Solid St M. 2011;15:225–235. doi: 10.1016/j.cossms.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hirst AR, Escuder B, Miravet JF, Smith DK. Angew Chem Int Edit. 2008;47:8002–8018. doi: 10.1002/anie.200800022. [DOI] [PubMed] [Google Scholar]; c) de Loos M, Feringa BL, van Esch JH. Eur J Org Chem. 2005:3615–3631. [Google Scholar]; d) Mann S. Nat Mater. 2009;8:781–792. doi: 10.1038/nmat2496. [DOI] [PubMed] [Google Scholar]
- 86.Sutton S, Campbell NL, Cooper AI, Kirkland M, Frith WJ, Adams DJ. Langmuir. 2009;25:10285–10291. doi: 10.1021/la9011058. [DOI] [PubMed] [Google Scholar]
- 87.Shi J, Gao Y, Yang Z, Xu B. Beilstein J Org Chem. 2011;7:167–172. doi: 10.3762/bjoc.7.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang ZM, Liang GL, Ma ML, Gao Y, Xu B. J Mater Chem. 2007;17:850–854. [Google Scholar]
- 89.Zhang Y, Gu HW, Yang ZM, Xu B. J Am Chem Soc. 2003;125:13680–13681. doi: 10.1021/ja036817k. [DOI] [PubMed] [Google Scholar]
- 90.Jayawarna V, Smith A, Gough JE, Ulijn RV. Biochem Soc T. 2007;35:535–537. doi: 10.1042/BST0350535. [DOI] [PubMed] [Google Scholar]
- 91.Aufderhorst-Roberts A, Frith WJ, Donald AM. Soft Matter. 2012;8:5940–5946. [Google Scholar]
- 92.Adhikari B, Banerjee A. Chem-eur J. 2010;16:13698–13705. doi: 10.1002/chem.201001240. [DOI] [PubMed] [Google Scholar]
- 93.Adhikari B, Banerjee A. Soft Matter. 2011;7:9259–9266. [Google Scholar]
- 94.Smith AM, Williams RJ, Tang C, Coppo P, Collins RF, Turner ML, Saiani A, Ulijn RV. Adv Mater. 2008;20:37–41. [Google Scholar]
- 95.Tang C, Ulijn RV, Saiani A. Langmuir. 2011;27:14438–14449. doi: 10.1021/la202113j. [DOI] [PubMed] [Google Scholar]
- 96.Cheng G, Castelletto V, Moulton CM, Newby GE, Hamley IW. Langmuir. 2010;26:4990–4998. doi: 10.1021/la903678e. [DOI] [PubMed] [Google Scholar]
- 97.Ryan DM, Doran TM, Anderson SB, Nilsson BL. Langmuir. 2011;27:4029–4039. doi: 10.1021/la1048375. [DOI] [PubMed] [Google Scholar]
- 98.Gao J, Wang H, Wang L, Wang J, Kong D, Yang Z. J Am Chem Soc. 2009;131:11286–11287. doi: 10.1021/ja9042142. [DOI] [PubMed] [Google Scholar]
- 99.Yang ZM, Gu HW, Fu DG, Gao P, Lam JK, Xu B. Adv Mater. 2004;16:1440. [Google Scholar]
- 100.Thornton K, Abul-Haija YM, Hodson N, Ulijn RV. Soft Matter. 2013;9:9430–9439. [Google Scholar]
- 101.Zhao F, Gao Y, Shi J, Browdy HM, Xu B. Langmuir. 2011;27:1510–1512. doi: 10.1021/la103982e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nanda J, Biswas A, Banerjee A. Soft Matter. 2013;9:4198–4208. [Google Scholar]
- 103.Liang G, Yang Z, Zhang R, Li L, Fan Y, Kuang Y, Gao Y, Wang T, Lu WW, Xu B. Langmuir. 2009;25:8419–8422. doi: 10.1021/la804271d. [DOI] [PubMed] [Google Scholar]
- 104.Zhang Y, Kuang Y, Gao Y, Xu B. Langmuir. 2011;27:529–537. doi: 10.1021/la1020324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yurke B, Turberfield AJ, Mills AP, Simmel FC, Neumann JL. Nature. 2000;406:605–608. doi: 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- 106.Gao Y, Kuang Y, Guo ZF, Guo Z, Krauss IJ, Xu B. J Am Chem Soc. 2009;131:13576–13577. doi: 10.1021/ja904411z. [DOI] [PubMed] [Google Scholar]
- 107.Li JY, Gao Y, Kuang Y, Shi JF, Du XW, Zhou J, Wang HM, Yang ZM, Xu B. J Am Chem Soc. 2013;135:9907–9914. doi: 10.1021/ja404215g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang H, Yang C, Wang L, Kong D, Zhang Y, Yang Z. Chem Commun. 47:4439–4441. doi: 10.1039/c1cc10506j. [DOI] [PubMed] [Google Scholar]
- 109.Gao Y, Shi J, Yuan D, Xu B. Nat Commun. 2012;3:1033. doi: 10.1038/ncomms2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gao Y, Kuang Y, Du X, Zhou J, Chandran P, Horkay F, Xu B. Langmuir. 2013;29:15191–15200. doi: 10.1021/la403457c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hu Y, Wang H, Wang J, Wang S, Liao W, Yang Y, Zhang Y, Kong D, Yang Z. Org Biomol Chem. 2010;8:3267–3271. doi: 10.1039/c002609c. [DOI] [PubMed] [Google Scholar]
- 112.Liu H, Hu Y, Wang H, Wang J, Kong D, Wang L, Chen L, Yang Z. Soft Matter. 2011;7:5430–5436. [Google Scholar]
- 113.Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, Day R, Ferraz MB, Hawkey CJ, Hochberg MC, Kvien TK, Schnitzer TJ, Weaver A, Grp VS. New Engl J Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- 114.Li J, Kuang Y, Shi J, Gao Y, Zhou J, Xu B. Beilstein J Org Chem. 2013;9:908–917. doi: 10.3762/bjoc.9.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li J, Li X, Kuang Y, Gao Y, Du X, Shi J, Xu B. Adv Health Mater. 2013;2:1586–1590. doi: 10.1002/adhm.201300041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Johnson EK, Adams DJ, Cameron PJ. J Am Chem Soc. 2010;132:5130–5136. doi: 10.1021/ja909579p. [DOI] [PubMed] [Google Scholar]
- 117.Gao J, Zheng W, Kong D, Yang Z. Soft Matter. 2011;7:10443–10448. [Google Scholar]
- 118.Wang H, Wei J, Yang C, Zhao H, Li D, Yin Z, Yang Z. Biomaterials. 2012;33:5848–5853. doi: 10.1016/j.biomaterials.2012.04.047. [DOI] [PubMed] [Google Scholar]
- 119.Lv L, Liu H, Chen X, Yang Z. Colloid Surface B. 2013;108:352–357. doi: 10.1016/j.colsurfb.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 120.Yang C, Li D, Liu Z, Hong G, Zhang J, Kong D, Yang Z. J Phys Chem B. 2011;116:633–638. doi: 10.1021/jp209441r. [DOI] [PubMed] [Google Scholar]
- 121.Muraoka T, Koh CY, Cui H, Stupp SI. Angew Chem Int Edit. 2009;48:5946–5949. doi: 10.1002/anie.200901524. [DOI] [PubMed] [Google Scholar]
- 122.Storrie H, Guler MO, Abu-Amara SN, Volberg T, Rao M, Geiger B, Stupp SI. Biomaterials. 2007;28:4608–4618. doi: 10.1016/j.biomaterials.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 123.Yang ZM, Gu HW, Zhang Y, Wang L, Xu B. Chem Commun. 2004:208–209. doi: 10.1039/b310574a. [DOI] [PubMed] [Google Scholar]
- 124.Burch RM, Weitzberg M, Blok N, Muhlhauser R, Martin D, Farmer SG, Bator JM, Connor JR, Ko C, Kuhn W, McMillan BA, Raynor M, Shearer BG, Tiffany C, Wilkins DE. Proc Natl Acad Sci USA. 1991;88:355–359. doi: 10.1073/pnas.88.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang ZM, Xu KM, Wang L, Gu HW, Wei H, Zhang MJ, Xu B. Chem Commun. 2005:4414–4416. doi: 10.1039/b507314f. [DOI] [PubMed] [Google Scholar]
- 126.Jayawarna V, Ali M, Jowitt TA, Miller AE, Saiani A, Gough JE, Ulijn RV. Adv Mater. 2006;18:611–614. [Google Scholar]
- 127.Jayawarna V, Richardson SM, Hirst AR, Hodson NW, Saiani A, Gough JE, Ulijn RV. Acta Biomater. 2009;5:934–943. doi: 10.1016/j.actbio.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 128.Zhou M, Smith AM, Das AK, Hodson NW, Collins RF, Ulijn RV, Gough JE. Biomaterials. 2009;30:2523–2530. doi: 10.1016/j.biomaterials.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 129.Wang Q, Li L, Xu B. Chem-eur J. 2009;15:3168–3172. doi: 10.1002/chem.200801653. [DOI] [PubMed] [Google Scholar]
- 130.Wang Q, Yang Z, Wang L, Ma M, Xu B. Chem Commun. 2007:1032–1034. doi: 10.1039/b615223f. [DOI] [PubMed] [Google Scholar]
- 131.Sreenivasachary N, Lehn JM. Proc Natl Acad Sci USA. 2005;102:5938–5943. doi: 10.1073/pnas.0501663102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.a) Parkinson GN, Lee MPH, Neidle S. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]; b) Zahler AM, Williamson JR, Cech TR, Prescott DM. Nature. 1991;350:718–720. doi: 10.1038/350718a0. [DOI] [PubMed] [Google Scholar]
- 133.Sreenivasachary N, Lehn JM. Chem-asian J. 2008;3:134–139. doi: 10.1002/asia.200700041. [DOI] [PubMed] [Google Scholar]
- 134.Arigon J, Prata CAH, Grinstaff MW, Barthelemy P. Bioconjugate Chem. 2005;16:864–872. doi: 10.1021/bc050029y. [DOI] [PubMed] [Google Scholar]
- 135.Iwaura R, Yoshida K, Masuda M, Yase K, Shimizu T. Chem Mater. 2002;14:3047–3053. [Google Scholar]
- 136.Gao XY, Matsui H. Adv Mater. 2005;17:2037–2050. doi: 10.1002/adma.200401849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Seela F, Pujari SS, Schaefer AH. Tetrahedron. 2011;67:7418–7425. [Google Scholar]
- 138.Park SM, Kim BH. Soft Matter. 2008;4:1995–1997. [Google Scholar]
- 139.Wang GJ, Cheuk S, Yang H, Goyal N, Reddy PVN, Hopkinson B. Langmuir. 2009;25:8696–8705. doi: 10.1021/la804337g. [DOI] [PubMed] [Google Scholar]
- 140.Wang GJ, Cheuk S, Williams K, Sharma V, Dakessian L, Thorton Z. Carbohyd Res. 2006;341:705–716. doi: 10.1016/j.carres.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 141.Goyal N, Cheuk S, Wang GJ. Tetrahedron. 2010;66:5962–5971. [Google Scholar]
- 142.Matsumoto S, Yamaguchi S, Ueno S, Komatsu H, Ikeda M, Ishizuka K, Iko Y, Tabata KV, Aoki H, Ito S, Noji H, Hamachi I. Chem-Eur J. 2008;14:3977–3986. doi: 10.1002/chem.200701904. [DOI] [PubMed] [Google Scholar]
- 143.Ikeda M, Ueno S, Matsumoto S, Shimizu Y, Komatsu H, Kusumoto K-i, Hamachi I. Chem-Eur J. 2008;14:10808–10815. doi: 10.1002/chem.200801144. [DOI] [PubMed] [Google Scholar]
- 144.Kiyonaka S, Sada K, Yoshimura I, Shinkai S, Kato N, Hamachi I. Nat Mater. 2004;3:58–64. doi: 10.1038/nmat1034. [DOI] [PubMed] [Google Scholar]
- 145.a) Ikeda M, Ochi R, Wada A, Hamachi I. Chem Sci. 2010;1:491–498. [Google Scholar]; b) Tamaru S, Kiyonaka S, Hamachi I. Chem-eur J. 2005;11:7294–7304. doi: 10.1002/chem.200500666. [DOI] [PubMed] [Google Scholar]
- 146.a) Koshi Y, Nakata E, Yamane H, Hamachi I. J Am Chem Soc. 2006;128:10413–10422. doi: 10.1021/ja0613963. [DOI] [PubMed] [Google Scholar]; b) Wada A, Tamaru S-i, Ikeda M, Hamachi I. J Am Chem Soc. 2009;131:5321–5330. doi: 10.1021/ja900500j. [DOI] [PubMed] [Google Scholar]
- 147.Ziane S, Schlaubitz S, Miraux S, Patwa A, Lalande C, Bilem I, Lepreux S, Rousseau B, Le Meins JF, Latxague L, Barthelemy P, Chassande O. Eur Cells Mater. 2012;23:147–160. doi: 10.22203/ecm.v023a11. [DOI] [PubMed] [Google Scholar]
- 148.Vemula PK, Li J, John G. J Am Chem Soc. 2006;128:8932–8938. doi: 10.1021/ja062650u. [DOI] [PubMed] [Google Scholar]
- 149.Chen Q, Lv Y, Zhang D, Zhang G, Liu C, Zhu D. Langmuir. 2010;26:3165–3168. doi: 10.1021/la903102z. [DOI] [PubMed] [Google Scholar]
- 150.Yang Z, Kuang Y, Li X, Zhou N, Zhang Y, Xu B. Chem Commun. 2012;48:9257–9259. doi: 10.1039/c2cc34935c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.a) Gao Y, Long MJC, Shi J, Hedstrom L, Xu B. Chem Commun. 2012;48:8404–8406. doi: 10.1039/c2cc33631f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kuang Y, Yuan D, Zhang Y, Kao A, Du X, Xu B. Rsc Advances. 2013;3:7704–7707. doi: 10.1039/C3RA41523F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li X, Kuang Y, Lin HC, Gao Y, Shi J, Xu B. Angew Chem Int Edit. 2011;50:9365–9369. doi: 10.1002/anie.201103641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.a) Roviello GN, Musumeci D, Bucci EM, Pedone C. Mol Biosyst. 2011;7:1073–1080. doi: 10.1039/c0mb00214c. [DOI] [PubMed] [Google Scholar]; b) Roviello GN, Di Gaetano S, Capasso D, Cesarani A, Bucci EM, Pedone C. Amino Acids. 2010;38:1489–1496. doi: 10.1007/s00726-009-0361-5. [DOI] [PubMed] [Google Scholar]
- 154.a) Azzam ME, Algranat Id. Proc Natl Acad Sci USA. 1973;70:3866–3869. doi: 10.1073/pnas.70.12.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hector RF, Zimmer BL, Pappagianis D. Antimicrob Agents Ch. 1990;34:587–593. doi: 10.1128/aac.34.4.587. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Itaya M, Yamaguchi I, Kobayashi K, Endo T, Tanaka T. J Biochem. 1990;107:799–801. doi: 10.1093/oxfordjournals.jbchem.a123128. [DOI] [PubMed] [Google Scholar]
- 155.a) Haaima G, Lohse A, Buchardt O, Nielsen PE. Angew Chem Int Edit. 1996;35:1939–1942. [Google Scholar]; b) Kool ET. Acc Chem Res. 2002;35:936–943. doi: 10.1021/ar000183u. [DOI] [PubMed] [Google Scholar]; c) Kool ET, Morales JC, Guckian KM. Angew Chem Int Edit. 2000;39:990–1009. doi: 10.1002/(sici)1521-3773(20000317)39:6<990::aid-anie990>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]; d) Nielsen PE, Egholm M, Berg RH, Buchardt O. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]; e) Pradeepkumar PI, Hobartner C, Baum DA, Silverman SK. Angew Chem Int Edit. 2008;47:1753–1757. doi: 10.1002/anie.200703676. [DOI] [PubMed] [Google Scholar]
- 156.a) Roviello GN, Ricci A, Bucci EM, Pedone C. Mol Biosyst. 2011;7:1773–1778. doi: 10.1039/c1mb05007a. [DOI] [PubMed] [Google Scholar]; b) Wong OY, Pradeepkumar PI, Silverman SK. Biochemistry. 2011;50:4741–4749. doi: 10.1021/bi200585n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Du X, Li J, Gao Y, Kuang Y, Xu B. Chem Commun. 2012;48:2098–2100. doi: 10.1039/c2cc16723a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yang Z, Liang G, Ma M, Abbah AS, Lu WW, Xu B. Chem Commun. 2007:843–845. doi: 10.1039/b616563j. [DOI] [PubMed] [Google Scholar]
- 159.Zhao F, Weitzel CS, Gao Y, Browdy HM, Shi JF, Lin HC, Lovett ST, Xu B. Nanoscale. 2011;3:2859–2861. doi: 10.1039/c1nr10333d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang W, Wang H, Ren C, Wang J, Tan M, Shen J, Yang Z, Wang PG, Wang L. Carbohyd Res. 2011;346:1013–1017. doi: 10.1016/j.carres.2011.03.031. [DOI] [PubMed] [Google Scholar]
- 161.Xu XD, Liang L, Cheng H, Wang XH, Jiang FG, Zhuo RX, Zhang XZ. J Mater Chem. 2012;22:18164–18171. [Google Scholar]
- 162.Li X, Kuang Y, Shi J, Gao Y, Lin HC, Xu B. J Am Chem Soc. 2011;133:17513–17518. doi: 10.1021/ja208456k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Vembaiyan K, Pearcey JA, Bhasin M, Lowary TL, Zou W. Bioorgan Med Chem. 2011;19:58–66. doi: 10.1016/j.bmc.2010.11.044. [DOI] [PubMed] [Google Scholar]
- 164.Li XM, Du XW, Gao Y, Shi JF, Kuang Y, Xu B. Soft Matter. 2012;8:7402–7407. doi: 10.1039/C2SM25725D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li XM, Du XW, Li JY, Gao Y, Pan Y, Shi JF, Zhou N, Xu B. Langmuir. 2012;28:13512–13517. doi: 10.1021/la302583a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Whitesides GM, Wong AP. MRS Bull. 2006;31:19–27. [Google Scholar]