

Abstract
Multiple sclerosis (MS) is an inflammatory, demyelinating disease of the central nervous system (CNS). Although its etiology remains unknown, pathogenic T cells are thought to underlie MS immune pathology. We recently showed that MS patients harbor CNS-specific CD8+ Tregs that are deficient during disease relapse. We now demonstrate that CNS-specific CD8+ Tregs were cytolytic and could eliminate pathogenic CD4+ T cells. These CD8+ Tregs were present primarily in terminally differentiated (CD27−, CD45RO−) subset and their suppression was IFNγ, perforin and granzyme B-dependent. Interestingly, MS patients with acute relapse displayed a significant loss in terminally differentiated CD8+ T cells, with a concurrent loss in expression of perforin and granzyme B. Pre-treatment of exacerbation-derived CD8+ T cells with IL-12 significantly restored suppressive capability of these cells through upregulation of granzyme B. Our studies uncover immune-suppressive mechanisms of CNS-specific CD8+ Tregs, and may contribute to design of novel immune therapies for MS.
Keywords: Multiple Sclerosis, CD8, T cells, Regulatory, IL12
Introduction
Multiple Sclerosis (MS) is a chronic debilitating neurological disorder that affects an estimated 400,000 individuals in the United States alone [1]. The disease is most common in individuals aged 20–40 years and leads to substantial disability through deficits of sensation of motor, autonomic, and neurocognitive function [2]. The clinical presentation of the disease can vary form clearly defined relapses and remissions (relapsing-remitting MS or RRMS) to slow accumulation of disability with or without punctuated exacerbation [3]. Currently, the exact etiology of the disease and its relapses remains unknown, but ultimately, the disease process leads to demyelination in areas of the central nervous system (CNS) that may result in axon dysfunction, and neuronal death [4].
Over the past decades, a considerable body of evidence primarily stemming from work in the murine model of MS, experimental autoimmune encephalomyelitis (EAE), has pointed to an autoimmune-based pathology. The chronic inflammatory disease of the CNS that underlies MS is thought to be mediated by pathogenic autoimmune T cells [5–7]. In healthy individuals, these pathogenic T cells are kept in check by regulatory T cells (Tregs) that maintain peripheral tolerance and prevent chronic inflammatory diseases [8, 9]. Increasing evidence suggest that in relapsing remitting multiple sclerosis (RRMS) patients, the Tregs are defective and cannot prevent the damage caused by the pathogenic T cells. Several laboratories, including ours, have observed diminished regulatory potential of conventional CD4+CD25+ Tregs in RRMS patients [10–12]. In contrast, less attention has been given to suppressive CD8+ T cells.
In spite of some previous controversy, the importance of CD8+ Tregs to the maintenance of immunological homeostasis has been confirmed recently in several human autoimmune diseases and their murine models. In both rheumatoid arthritis and type 1 diabetes mellitus, the induction and function of CD8+ Tregs is reduced among patients with active disease and in the experimental murine models of the diseases respectively [13–17]. Similar disease suppressing roles for CD8+ Tregs have been reported in systemic lupus erythematosus, systemic sclerosis, Grave’s disease, ankylosing spondylitis, myasthenia gravis and experimental autoimmune uveoretinitis [18].
Recently, our laboratory demonstrated a novel and unexpected regulatory potential for CNS-targeted CD8+ cells in MS. We demonstrated that both MS and healthy subjects harbor neuroantigen-specific CD8+ Tregs which appear to be selectively deficient in MS patients during disease exacerbation [19, 20]. Using antigen-specific CD8+ and CD4+ T cell lines, we have shown that while neuroantigen-specific CD8+ T cells can suppress CD4+ T cells of multiple specificities, cognate antigen recognition of neuroantigen is required for the suppressive activity of these cells [20]. The disease-alleviating function of CD8+ Tregs was further validated in murine models where adoptive transfer of neuroantigen-specific CD8+ Tregs drastically ameliorated EAE disease severity [21, 22]. In the current study, we sought to further characterize these novel neuroantigen-specific CD8+ T cells and examine if they can directly target neuroantigen-specific CD4+ T cells. We hypothesized that these neuroantigen-specific CD8+ Tregs were harbored in a unique CD8+ T cell subset, and that a deficit in that population underlies the impaired CD8+ Treg suppressive ability during acute disease exacerbation Importantly, we demonstrate that the suppressive function of the exacerbation-derived CD8+ T cells can be restored by enhancing the production of its regulatory mediator. To our knowledge, the current study is the first to clearly define the subset of CD8+ T cells that harbor neuroantigen-specific regulatory properties and adds substantial credence to the protective role of CD8+ T cells in autoimmune diseases, which can be harnessed for the design of future immunotherapeutic strategies.
Materials and Methods
Subject characteristics
MS patients were recruited and gave written informed consent at the UT Southwestern Clinical Center for Multiple Sclerosis. Table 1 summarizes patient characteristics. 7 adult clinically definite RRMS patients (McDonald criteria) with disease exacerbation were recruited. 3 months post-exacerbation samples were collected from the same patients during disease quiescence. History of disease-modifying immunomodulatory therapy prior to and post exacerbation is described. 5 additional MS patients were recruited during an active acute clinical episode for the IL-12 supplementation study. At the time of exacerbation, 5 patients were treatment naïve, 3 patients were on Copaxone therapy, 2 patients were on natalizumab therapy, one patient was on interferon-beta1a therapy and one patient was on rituximab. At the quiescence phase of the disease, two patients were on no therapy, 2 patients were on Copaxone therapy, one patient was on fingolimod, one patient was on rituximab, and one patient on natalizumab. 57 healthy controls samples were obtained from Carter Blood Care. Exclusion criteria included neurodegenerative conditions, clinical relapse or corticosteroid treatment for MS patients. All studies were approved by the UT Southwestern institutional review board (IRB) according to Declaration of Helsinki principles.
Table 1.
Summary of patient characteristics
| Healthy Controls (HC) | RRMS Patients (MS) | |
|---|---|---|
| Number of Subjects | 57 | 12 |
| Average age, y (Range) | 42 (21–65) | 36 (24–50) |
| Sex (M/F) | 32/25 | 3/9 |
Cell preparation and bead sorting
Using Ficoll Hypaque (GE Healthcare Biosciences, Pittsburgh, PA) density gradient, PBMCs were isolated from whole blood and frozen on day of collection. Longitudinal specimens were assayed in parallel to minimize assay-based variation, as validated previously [19, 20]. Enrichment of CD8+ T cells was performed by using CD8+ microbeads positive selection kit (Miltenyi Biotec, Auburn, CA). Isolation of CD4+ T cells was carried out using CD4 negative selection kits (Miltenyi Biotech). CD25+ T cells were depleted from the purified CD4+ using CD25 microbeads (Miltenyi Biotec). Purity of enriched populations were >98% as measured by flow cytometric analysis. The irradiated CD4− and CD8− T cell depleted PBMC population was used as APCs. For isolation of marker specific-CD8+ T cells, CD8 mutli-sort isolation kits (Miltenyi Biotech) were used followed by additional positive selections for either CD25, CD27, CD28, CD45RO, CD62L or NKG2D using microbeads positive selection kits (Miltenyi Biotech). For flow cytometry based cytotoxic assays, monocytes, B cells and myeloid dendritic cells were isolated using CD14+, CD19+ and BDCA1+ microbeads (Miltenyi Biotech).
Flow cytometry-based suppression assay
Responses to antigenic challenge were assessed by staining cells with carboxyfluorescein diacetate succinimidyl ester (CFSE) (Invitrogen Molecular Probes, Eugene, OR) described previously (19). Briefly, CD4+CD25− cells were suspended at 1 x106 cells/mL and incubated for 7 min at 37°C with 0.25 mM CFSE, followed by two wash cycles with media containing 5% human serum. 106 CFSE-stained CD4+CD25− T cells were used as responders in a 1 mL culture. 106 CD4− and CD8-depleted PBMC were irradiated with 3000 rad and used as APCs. In replicate cultures, varying ratios of suppressors were added and cultured with various antigenic stimuli for 7 days in complete RPMI 1640 media containing 5% human serum, 100 U/mL penicillin, 100 mg/mL sptreptomycin, and 0.92 mg/mL L-glutamine. Cells were washed and stained for flow cytometry, as described below. Antigenic stimulus was provided with pools of 15-mer peptides, overlapping by 10, spanning entire neuroantigenic proteins, as described previously (19). These were used at 1 μg/mL final concentration for each peptide and included myelin basic protein (MBP) and proteolipid protein (PLP). In addition, 1 μg/mL anti-CD3 monoclonal antibody (OKT3) was used for mitogenic stimulation. Unless indicated otherwise, all suppression assays were set up at a responder: suppressor ratio (1:0.25) with two different neuroantigen-stimulus (PLP and MBP) being used for each patient sample. For all analyses, background proliferation from the “No antigen” cultures was subtracted from the antigen-stimulated cultures having no suppressors, as published earlier [20]. A “positive” T-cell response to antigen was defined as having (1) a response index (RI) greater than or equal to 1.5 and (2) a %CD25+CFSElow response of the antigen-stimulated cells at least 1% greater than the %CD25+CFSElow response of the cells in the no antigen tube (ΔPF), following previously published calculations [20]. If these criteria were unmet, absence of T-cell response was indicated. These criteria are based on our prior analysis of reproducibility and interassay variability [19]. By using both the parameters, we avoided over-interpreting SI values associated with low background and ΔPF values in case of high background. Positive responses were observed both with low and high background proliferations; eg., ranging from a 0.18% mean background with a corresponding MBP response of 5.75% (p<0.001) and a PLP response to of 5.20% (p=0.002) to a background of 4.12% with a MBP response of 7.25% (p=0.02). For suppression assays, % response was calculated by normalizing the ‘responder only’ proliferation to 100%. %Suppression was 100 minus %response. Stimulation index of the CD8 response was defined as the percentage of CD25+ cells with antigenic stimulus divided by percentage with no antigen [20].
Blocking, inhibition and transwell assays
Blocking antibodies included anti-HLAI, anti-IFNγ, anti-NKG2D, anti-TNFα, anti-PD1, anti-IL-10, anti-TGFβ (1,2,3), anti-CTLA-4, anti-FasL and isotype IgG (Biolegend). Antibodies were used at 4 μg/ml 1 hr prior to addition of antigenic stimulus to cultures. For inhibition assays, the degranulation inhibitor, concanamycin A (CMA; Sigma, St. Louis, MO, USA) was used at 20 nM in suppression assays [23]. Transwell experiments were performed in 14 mL polypropelene round-bottom tubes (BD Falcon) using 0.4 μm Maxicell cell culture inserts (TPP/MIDSCI, St Louis, MO).
Surface and Intracellular staining
For suppression assays, cells were washed with 0.1% (w/v) sodium azide/phosphate-buffered saline (Mediatech Cellgro) on day 7 of in vitro stimulation and were stained with anti-CD4 PECy5 (BD Pharmingen), anti-CD8 Pacific Blue (Biolegend), and anti-CD25 APCCy7 (BD Pharmingen. For surface phenotyping of cells, bulk PBMCs and enriched CD8+ T cells were stained with anti-CD3 Alexa 700 (BD Pharmingen), anti-CD8 AmCyan (BD Biosciences), anti-CD27 APCCy7 (Biolegend), anti-CD28 APC (BD Pharmingen), CD45RO Pacific Blue (Biolegend), anti-CD62L PECy5 (BD Pharmingen), and anti-CD57 PE (Southern Biotech). For intracellular staining of cytokines, cells were initially activated with 1 μL of leukocyte activation cocktail (BD Pharmingen) for 5 hours. Cells were surface stained with anti-CD8 APC (BD Biosciences), anti-CD4 PECy5 (BD Pharmingen) and anti-CD25 APCCy7 (BD Pharmingen) and permeabilized as described previously. Intracellular staining was performed using anti-IFNγ PECy7 (BD Pharmingen), anti-IL-17A PE (Ebioscience), anti-Granzyme B Alexa 700 (BD Pharmingen) and anti-Perforin Pacific Blue (BD Pharmingen). All cells were resuspended in 1% paraformaldehyde (Electron Microscopy Sciences, Hatfiled, PA) for FACS analysis. Flow cytometric data were acquired on a 4-Laser, 17-color LSRII using FACSDiva software (Becton Dickinson). CFSE was detected in the FITC channel on the LSR.
Flow cytometry cytotoxic assays
These assays were adapted from previously published methodologies [24, 25]. CD8+ T cells, CD4+CD25− T cells, monocytes (CD14+), B cells (CD19+) and myeloid dendritic cells (BDCA1+) were enriched from healthy donors PBMCs. CD8+ T cells were incubated with CD4+CD25− responder T cells and with individuals APC subsets for 7 days with either neuroantigen stimulus or vehicle control. Anti-CD3 stimulus was used as a positive control. Cells were collected at 72hrs time point and stained with individual antibody panels consisting of anti-CD3-Alexa 700, anti-CD4 PECy5, anti-CD8 AmCyan and anti-CD19/BDCA-1/CD14 Pacific Blue. Following surface staining cells were further stained with for Propidium Iodide (PI) and Annexin V using the FITC Annexin V Apoptosis detection kit (BD Pharmingen). % of PI+/Annexin V+ cells was assessed for each cell type.
IL-12 pretreatment of CD8+ T cells
Neuroantigen-specific CD8+ T cells were stimulated by culturing bulk PBMCs at 30 × 106 cells at 10 × 106 /mL for 7 days in 6 well plates. Culture medium was either left untreated or supplemented with 25ng/mL of IL-12 or IL-23(BD Pharmingen). All cultures were supplemented with 1 μg/mL of neuroantigen peptide pool described above. One week post in vitro PBMC stimulation, CD8+ T cells were isolated by magnetic bead sorting and used with autologous APCs and CD4+CD25− responder cells, as described above.
Data analysis
Linear uncompensated data was transferred as FCS 3.0 files and analyzed after compensation and transformation using FlowJo version 9.4.1 (TreeStar, Ashland, OR). Using Flowjo software (Treestar), putative CD8+ Tregs were gated out from flow cytometric analysis of CFSE-stained cells. T cell activation and proliferation were quantified by the percentage of CD25 (high) and CFSE (low) events among gated CD4+ T cells. Cut-offs for positive populations were determined by using either fluorescence minus one (FMO) staining for polychromatic flow cytometry, no stimulus background CFSE staining, or isotype control staining, as appropriate. Response index (RI) and % Suppression were determined as described previously [20].
Statistical analyses
Statistical tests were performed using Prism 5 (Graphpad Software, La Jolla, CA). Paired t-tests were used to compute a two-tailed P value assuming a 95% confidence interval. P values >0.05 were not significant a “ns” notation was applied on the figures. Likewise P values <0.05 were significant and notated with “*”.
Results
CD8-mediated suppression is contact-dependent and requires MHC Class I, IFNγ, perforin and granzyme B
We previously demonstrated the regulatory properties of neuroantigen-specific CD8+ T cells in their ability to suppress the proliferation of CD4+CD25− T cells (Fig. 1A). The mechanisms used by CD8+ Tregs to mediate their suppressive effects may include the production of soluble immunosuppressive factors and/or cell–cell contact with CD4+CD25− T cells. In vitro transwell culture assays were used to determine whether suppression by neuroantigen-specific CD8+ Tregs was contact-dependent or mediated through soluble factors [26, 27]. Separation of CD8+ Tregs and CD4+CD25− T cells with transwell membranes in the co-cultures resulted in a significant reduction in Treg-mediated suppression compared with co-cultures with no separation between the populations, suggesting that neuroantigen-specific CD8+ Tregs primarily operated via a contact-dependent mechanism (Fig. 1B). To determine which molecular mediators were required by the CD8+ Tregs, blocking Abs (anti–HLAI, anti-IFNγ, anti–NKG2D, anti–TNFα, anti-PD1, anti-IL-10, anti–TGF-β, anti–CTLA-4 and anti–FasL) were added to the co-culture assays containing CD8+ Tregs, CD4+CD25− responder T cells and antigen-presenting cells (APCs). As shown in Fig. 1C, neuroantigen-specific CD8+ T cell-mediated suppression required MHC class I, NKG2D, and IFNγ. To further examine if CD8+ Treg-mediated suppression was granzyme B/perforin-dependent, CD8+ T cells were treated with concanamycin A and were used in the suppression assays. As seen in Fig. 1C, inhibition of the granzyme B/perforin mechanism significantly impeded CD8+ T cell-mediated suppression.
Figure 1. CD8+ Treg-mediated suppression is contact-dependent and requires MHC Class I, NKG2D, IFNγ, perforin and granzyme B.

(A) Representative dotplots from healthy subjects displaying CD8+ T cell-mediated suppression in the presence of the neuroantigens (MBP and PLP). CFSE expression is represented on the X-axis and CD25 expression on the Y-axis. Indicated in red at the top of each dot plot is the gated percentage of CD25+/CFSE-low cells (activated and proliferating). Shown in black in the lower left is the calculated % suppression based on normalizing CD4+CD25− neuroantigen-specific T cell responses in the absence of suppressors (leftmost column). (B) CD8+ T cell contact-dependent suppression was assessed in a transwell culture system. CD8+ T cells from healthy controls were either directly co-cultured with autologous CFSE-labeled ex-vivo-purified CD4+CD25− T cells or added to the top chamber of transwell culture tubes with autologous CD4+CD25− T cells added to the bottom chamber. Autologous APCs were included in all chambers and cells were stimulated with either neuroantigen or anti-CD3 for 7 days. A 0.4-μm pore sized membrane separated the two chambers. Four independent experiments were performed using autologous CD8+ T cells, CD4+CD25− T cell responders and APCs using PBMCs from 4 independent healthy subjects. (C) Ab blockade of membrane molecules and inhibition of granzyme B release significantly curtails CD8+ T cell-mediated suppression. CD8+ T cells from 6 healthy controls were incubated with autologous CD4+CD25− T cells and APCs in co-cultures to which various blocking antibodies and inhibitory agent were added. Suppression mediated by CD8+ T cells was assessed via percentage of CD25+/CFSE-low cells. *p < 0.05; Four independent experiments were performed with each assay using the inhibitory and blocking agents for a total of 6 healthy controls.
CD8+ Tregs eliminate neuroantigen-specific CD4+ T cells
Since CD4+ T cells have been deemed to be the classical mediators of MS pathogenesis, we sought to examine if they were selectively targeted by the CD8+ Tregs. First, we examined if the CD4+CD25− T cells were altered by their interaction with CD8+ Tregs. CD4+CD25− T cells were stimulated with neuroantigen for 7 days with or without CD8+ T cells in the presence of APCs. On day 7, bulk CD4+ T cells were isolated from the cultures and restimulated with neuroantigen in the presence of APCs during a recall response. Interestingly, CFSE-low CD4+ T cells originating from CD8+ T cell supplemented cultures showed a significant decrease in the frequency of IFNγ-expressing cells (29.8% vs 22.1%; p<0.05). However, no significant change was observed in the percentages of IL-17 producing CD4+ T cells (Fig. 2A). Additionally, CD4+ T cells that had been co-cultured with CD8+ T cells also displayed significantly lower responder indexes (24.7 vs 12.9; p<0.05) (Fig. 2B). Alteration in the phenotype of the CD4+ T cells implied that either the cells were being eliminated by apoptosis or being stably altered though anergy. In order to differentiate between these outcomes and to determine whether the CD4+ T cells were specifically undergoing apoptosis, annexin V/propidium iodide (PI) staining was used. CD4+CD25− T cells were co-cultured with CD8+ T cells and distinct APC subsets including monocytes (CD14+), B cells (CD19+) and dendritic cells (BDCA1+). The staining scheme described in Fig. S1 with annexin V/PI revealed that the CD4+ T cells were primarily being targeted for apoptosis by the neuroantigen-specific CD8+ T cells with the neuroantigen supplemented cultures showing a 38% increase in annexin V+/PI+ CD4+ T cells compared to vehicle controls (5.28% vs 7.3% annexin V+/PI+ cells; p<0.05) (Fig. 2C). In contrast, monocytes (CD14+), B cells (CD19+) and myeloid dendritic cells (BDCA1+) did not appear to be targeted by neuroantigen-specific CD8+ T cells (Fig. S2A–C).
Figure 2. CD8+ Tregs modulate neuroantigen-specific CD4+ T cells.
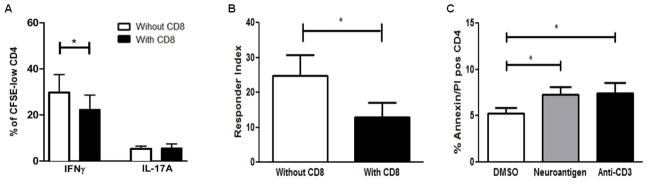
Decreased cytokine secretion and proliferative responses by suppressed CD4+ T cells. (A) 7 days post stimulation with neuroantigen, bulk CD4+ T cells were collected from co-cultures with and without CD8+ T cells. Both sets of CD4+ T cells were stained with CFSE, and restimulated with neuroantigen and autologous APCs for an additional 7 days. Cultures were subsequently stimulated for 5 hrs with leucocyte activation factor (LAF) (BD Pharmingen), followed by Intracellular staining for IFNγ and IL-17A. Expression of the cytokines was examined in proliferating CFSE-low CD4+ T cells. (B) The responder index i.e. proliferative capacity of the CD4+ T cells was similarly examined post incubation with CD8+ T cells. The experiments in A and B were performed 7 times with PBMCs from 7 independent healthy subjects. (C) CD4+CD25− T cells are targeted for apoptosis by CD8+ T cells. CD8+ T cells, CD4+CD25− T cells, monocytes (CD14+), B-cells (CD19+) and myeloid dendritic cells (BDCA1+) were isolated from healthy PBMCs. CD4+CD25− T cells were co-cultured with CD8+ T cells and individual subsets of APCs in the presence and absence of indicated antigenic stimulus. At 72hrs, cultures were collected and frequency of annexin V/PI positive CD4+ T cells was evaluated. *p < 0.05; Five independent experiments were performed with PBMCs from 5 healthy subjects.
Neuroantigen-specific CD8+ Tregs are terminally differentiated
To identify which subset of CD8+ T cells harbored the neuroantigen-specific CD8+ Tregs, CD8+ T cells were sorted using an array of surface markers. Bulk CD8+ T cells were initially isolated through positive selection using the multisort CD8+ T cell isolation kit after which the magnetic beads were cleaved. A second set of positive selections were performed using magnetic bead selection kits for the markers CD27, CD28, CD45RO, CD62L and NKG2D. Suppression assays were set up using bulk CD8+ T cells (the positive and negative fractions), CD4+CD25− T cells and APCs either in the presence or absence of neuroantigens. The maximal neuroantigen specific suppressive ability was harbored in the CD27− (Bulk CD8+ = 36.1%; CD27+ CD8+ = −28%: CD27− CD8+ = 52.5%) (Fig. 3A), CD28− (Bulk CD8+ = 21.5%; CD28+ CD8+ = 6.9%: CD28− CD8+ = 55.13%) (Fig. 3B), CD45RO− (Bulk CD8+ = 48.0%; CD45RO+ CD8+ = 27.0%: CD45RO− CD8+ = 61.3%) (Fig. 3C), CD62L− (Bulk CD8+ = 9.9%; CD62L+ CD8+ = −14.0%: CD62L− CD8+ = 30.7%) (Fig. 3D) and NKG2D+ (Bulk CD8+ = 54.2%; NKG2D+ CD8+ = 79.2%: NKG2D− CD8 = 44.0%) (Fig. 3E) fractions. These data suggested that the terminally differentiated subset of CD8+ T cells harbored the neuroantigen-specific CD8+ Tregs. Anti-CD3 was used to further ascertain that the differences in suppressive ability were in the neuroantigen-specific CD8 T cells. Global activation abrogated the difference in suppressive potential between the various subsets (Fig. S3 A–E). To further hone in on the neuroantigen-specific CD8+ Treg population, a triple selection was performed using the markers CD27, CD28, and CD45RO. The choice of the markers to be used was made based on cell isolation schemes that would allow sufficient cell yield to set up suppression assays. As shown in Fig. 3F, the CD27−/CD28−/CD45RO− subset harbored the maximal suppressive ability (CD27−CD45RO−CD28− CD8+ = 52.7%; CD27−CD45RO−CD28+ CD8+ = 18.3%; CD27-CD45RO+ CD8+ = 12.9%; CD27+ CD8+ = 3.8%; Bulk CD8+ = 30.1%). Similarly, the enhanced suppressive potential was restricted to neuroantigen-specific CD8+ T cells and was not seen in anti-CD3-induced suppression assays (Fig. S3F). Overall the results suggest that, unlike polyclonally activated CD8+ T cells, neuroantigen-specific CD8+ T cells are a phenotypically distinct suppressive population which is present predominantly in terminally differentiated pool.
Figure 3. Neuroantigen-specific CD8+ Tregs are terminally differentiated.

Bulk CD8+ T cells were isolated from healthy controls and sorted into (A) CD27+/−, (B) CD28+/−, (C) CD45RO+/−, (D) CD62L+/− and (E) NKG2D+/− CD8+ T cell fractions using magnetic bead-based positive selection kits. Individual CD8+ T cell fractions along with bulk CD8+ T cells were used in suppression assays with autologous APCs and CFSE-stained CD4+CD25− T cells. (F) Positively enriched CD8+ T cells were collected and successively enriched for CD27, CD45RO and CD28 respectively. Individual fractions were used in separate suppression assays with autologous APCs and responders. *p < 0.05; Four or more independent experiments were performed with PBMCs from 4 healthy control donors.
Phenotypic differences of neuroantigen-specific CD8+ Tregs between exacerbation and quiescence
We recently showed that the neuroantigen-specific CD8+ Tregs in MS patients are deficient during disease relapse. In this study, to further validate our findings, we collected paired blood samples from MS patients undergoing exacerbation and 3 months later when they entered disease remission/quiescence. Paired specimens of peripheral blood mononuclear cells (PBMCs) from 8 such MS-patients were tested in suppression assays with autologous responders and APCs. As shown in Fig. 4A, exacerbation MS patients showed a decrease in their neuroantigen-specific CD8+ T cell suppressive ability. Based on our observation that neuroantigen-specific suppressive ability was concentrated in the terminally differentiated (CD27−CD45RO−) CD8+ T cell pool, we wanted to further determine if a change in the abundance of this population could underlie the loss in suppressive ability of the CD8+ T cells during disease exacerbation. Accordingly, we evaluated the frequency of the terminally differentiated CD8+ T cells in the exacerbation vs quiescent phase of disease. We observed a significant increase in the percentage of CD27-CD45RO− CD8+ T cells in all MS patients tested during disease quiescence as compared to exacerbation (16.9% during disease relapse vs 24.2% during quiescence; p<0.05) (Fig. 4B). To further confirm that the terminally differentiated subset was the one being depleted during disease exacerbation, we used a different set of markers (CD62L and CD57) and observed a similar increase in the CD62L−CD57+ CD8+ T cell population during disease quiescence vs. exacerbation in all the patients (10.8% during exacerbation vs 16.9% during quiescence p<0.05) (Fig. 4C).
Figure 4. Phenotypic differences of neuroantigen-specific CD8+ Tregs during exacerbation vs quiescence.

(A) CD8+ T cells isolated from PBMCs of MS patients during disease exacerbation and quiescence were used in suppression assays with autologous APCs and responder CD4+CD25− T cells from disease exacerbation and quiescence respectively. (B) The frequency of terminally differentiated (Ter), CD27-CD45RO− CD8+ T cells was assessed in peripheral blood of MS during the two phases of disease and a significant increase in the percentage of Ter CD8+ T cells was observed in MS patients as they remitted. (C) MS patients showed a significant increase in the expression of CD57+CD62L− CD8+ T cells from exacerbation to quiescence. (D–F) Enriched CD8+ T cells from MS patients during either disease exacerbation or quiescence were stimulated for 5 hrs with leucocyte activation factor (LAF) (BD Pharmingen), followed by Intracellular staining for granzyme B (D), perforin (E), and IFNγ (F). A significant increase in perforin and granzyme B expression was observed in remitting MS patients while no significant change in IFNγ expression was seen (ns). *p < 0.05; PBMCs were collected from 7 RRMS patients during both disease quiescence and relapse.
We further assessed if the deficit in regulatory potential could also be due to a decrease in the expression of suppressive mediators used by the neuroantigen-specific CD8+ Tregs. Bulk CD8+ T cells isolated from PBMCs of MS patients during both phases of the disease were stimulated for 5 hrs with leukocyte activation factor (LAF) (BD Pharmingen), followed by intracellular staining for granzyme B, perforin and IFNγ. Interestingly, the percentage of granzyme B expressing CD8+ T cells increased significantly from disease exacerbation to disease quiescence (15.2% vs 23.5%; p<0.05) (Fig. 4D). Similarly, the percentage of perforin expressing CD8+ T cells increased significantly from 7.0% during disease relapse to 11.7% during disease quiescence (Fig. 4E). However, no change in the frequency of IFNγ-expressing CD8+ T cells was seen (Fig. 4F).
Loss of CD8-mediated suppressive ability is explained by intrinsic deficit in CD8+ T cells as well as suppressive resistance by CD4+ T cells
The loss in suppressive ability observed during disease relapse may have two origins. First, it might be the result of the decline in the number and function of terminally differentiated CD8+ T cells described above, that leads to a loss in the suppressive ability of the CD8+ Tregs. Alternatively, it might result from the CD4+CD25− T cells being refractory to CD8+ Treg suppression during disease exacerbation. To test this hypothesis, “criss-cross” experiments were set up as outlined in Fig. 5 where CD8+ Tregs from exacerbation were co-cultured with CD4+CD25−T cells and APC derived from disease quiescence and vice-versa. As shown in Fig. 5A, the exacerbation-derived neuroantigen-specific CD8+ Tregs appear to have no difference in suppressive ability when co-cultured with either exacerbation or quiescent responders and APCs. In contrast, the quiescence-derived neuroantigen-specific CD8+ Tregs display significantly enhanced mean suppressive ability from 11.9% with exacerbation-derived responders and APCs to 49.1% with quiescence-derived responders and APCs (Fig. 5B). These results suggested that quiescence-derived CD8+ T cells do have enhanced suppressive ability compared to the exacerbation-derived CD8+ T cells, but at the same time also implied that the exacerbation-derived CD4+CD25− T cells may be more refractory to Treg-mediated suppression. To further address this question, the suppressibility of the responders was analyzed. As shown in Fig. 5C, there was no difference in the suppressibility of the exacerbation-derived CD4+CD25− T cell responders by either the exacerbation or the quiescence-derived suppressors. In contrast, the quiescence-derived CD4+CD25− T cell responders showed a significant difference in suppressibility, with quiescence-derived CD8+ T cells having higher suppressive potential against them compared to exacerbation-derived CD8+ T cells (19.6% vs 45.5%) (Fig. 5D). Proliferative responses of CD4+ T cells to neuroantigens were comparable between exacerbation and quiescence (means of 2.4% vs. 2.7%, respectively). Thus, differential susceptibility to antigen-induced cell death is unlikely to explain these results.
Figure 5. Loss of CD4+ T cell suppressibility is intrinsic to CD8+ Tregs and CD4+ T cells.

(A) Exacerbation-derived CD8+ T cells were isolated from MS patients and used in suppression assays with either exacerbation or quiescence-derived autologous CD4+CD25− T cell responders and APCs. No significant difference in exacerbation-derived CD8+ T cell suppressive ability was seen between the two responder sets (ns). (B) Quiescent CD8+ T cells display enhanced suppressive ability. Quiescence-derived CD8+ T cells were used in suppression assays with either exacerbation or quiescence-derived autologous CD4+CD25− T cell responders and APCs. Quiescence-derived CD8+ T cells showed significantly increased suppressive ability against quiescence-derived autologous CD4+CD25− T cell responders. (C) Exacerbation-derived CD4+CD25− T cells and APCs were used in suppression assays with either exacerbation or quiescence-derived autologous CD8+ T cells. No significant difference in exacerbation-derived CD4+CD25− T cell suppressibility was seen by between the two sets of CD8+ T cells (ns). (D) Quiescence-derived CD4+CD25− T cells and APCs were used in suppression assays with either exacerbation or quiescence-derived autologous CD8+ T cells. Quiescence-derived CD4+CD25− T cells were significantly more amenable to quiescence-derived CD8+ T cell-mediated suppression. *p < 0.05; PBMCs were obtained from 7 RRMS patients during both disease quiescence and relapse.
No differences in the differentiation status of CD4+ T cells between exacerbation and quiescence
Based on the observation that the loss of CD8+ Treg suppressibility is also associated with refractory neuroantigen-specific CD4+CD25− T cell responders, we wanted to assess whether these CD4+ T cells also had differences in differentiation status. Bulk PBMCs collected from MS patients from both exacerbation and quiescent states were and stained ex vivo to assess the various CD4+ T cell subsets. No differences in the frequencies of naïve (CD27+CD45RO−) (Fig. 6A), central memory (CD27+CD45RO+) (Fig. 6B), effector memory (CD27−CD45RO+) (Fig. 6C), and terminally differentiated (CD27−CD45RO−) (Fig. 6D) CD4+ T cells were observed between the quiescent and exacerbation states. Similarly, no differences were observed in the activation status of the global CD4+ T cells based on CD25 and CD28 expression between the CD4+ T cell subsets (Fig. 6E and Fig. 6F). Finally, we also did not see differences in the percentages of neuroantigen-specific CD4+ Th1 cells (Fig. 6H) and Th17 cells (Fig. 6G) between the two disease states. These results would suggest that for the CD4+CD25− T cell responders neither their lineage commitment nor memory status determine their susceptibility to CD8+ Treg-mediated suppression.
Figure 6. Phenotypic differences of neuroantigen-specific CD4+ T cells during disease exacerbation vs quiescence.
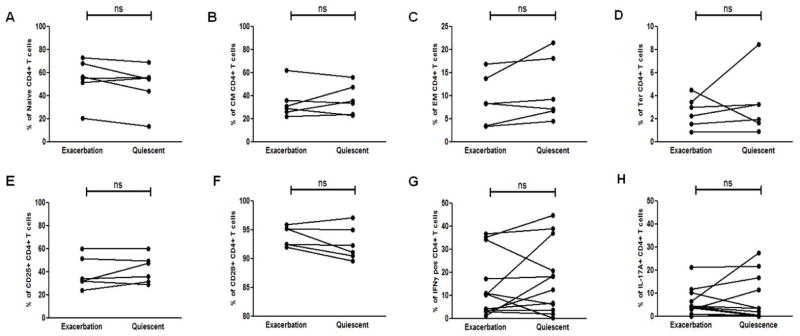
PBMCs from MS patients during disease exacerbation and quiescence were examined for the frequencies of (A) Naïve (CD27+CD45RO−), (B) Central Memory (CM - CD27+CD45RO−), (C) Effector Memory (EM - CD27+CD45RO−) and (D) Terminally differentiated (Ter - CD27−CD45RO−) CD4+ T cells. The activation status of the CD4+ T cells between the two disease states was similarly assessed ex vivo by examining the surface expression of (E) CD25 and (F) CD28. Th differentiation was examined by stimulating cells with neuroantigen for 7 days, stimulating the cells for 5 hrs with LAF on day 7, followed by Intracellular staining for indicated cytokines. Expression of (G) IFNγ and (H) IL-17A was evaluated in proliferating CFSE-low CD4+ T cells. No significant difference in the CD4+ T cells between the two disease states were found with the parameters evaluated (ns). PBMCs used in the assay were collected from 7 RRMS patients during both disease quiescence and relapse.
IL-12 enhances suppressive ability of neuroantigen-specific CD8+ Tregs
To gauge whether the loss of CD8+ Treg suppressive ability during disease exacerbation could be restored, we used the cytokine IL-12, which has been shown to significantly enhance the CD8+ T cell’s effector ability [28–30]. First, bulk PBMCs from healthy subjects were cultured with either no cytokine, IL-12 (25 μg/mL) or IL-23 (25 μg/mL) for seven days after which CD8+ T cells were isolated and used in suppression assays with autologous CD4+CD25− T cell responders isolated from freshly thawed PBMCs. As shown in Fig. 7A, pre-treatment of CD8+ T cells with IL-12 significantly enhanced the suppressive ability of the CD8+ T cells from 49.5% in the untreated group to 75.7% in the IL-12-pretreated CD8+ T cells. No significant enhancement in CD8+ T cell suppressive ability was observed in IL-23-treated CD8+ T cells. Accompanying the enhanced suppressive ability was a significant increase in the expression of granzyme B by CD8+ T cells (Fig. 7B). To test whether exacerbation-derived CD8+ T cells could similarly be potentiated, bulk PBMCs from MS exacerbation patients were cultured with and without IL-12. Seven days post incubation, CD8+ T cells were similarly isolated and suppression assays set up. As demonstrated in Fig. 7C, IL-12-treated CD8+ T cells had significantly increased suppressive ability from −14.87% (non-suppressive) to 44.97%. This suggested that CD8+ Treg suppressive ability could be restored and possibly harnessed to suppress disease in MS patients.
Figure 7. IL-12 enhances suppressive ability of neuroantigen-specific CD8+ Tregs.
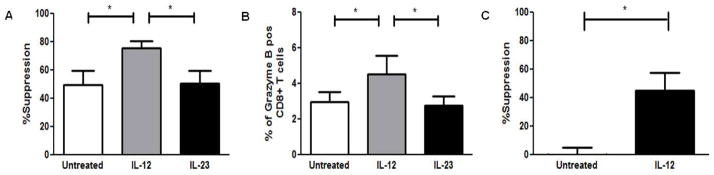
(A) Neuroantigen-specific CD8+ T cells were stimulated by culturing 30 × 106 cells bulk PBMCs from healthy subjects at 10 × 106 /mL for 7 days in 6 well plates. Culture medium was either left untreated or supplemented with 25ng/mL of IL-12 or IL-23. All cultures were supplemented with 1 mg/mL of one of two different neuroantigen peptides (MBP and PLP). 7 days post in vitro PBMC stimulation, CD8+ T cells were isolated by magnetic bead sorting and used in suppression assays with freshly thawed autologous APCs and CD4+CD25− T cells. (B) CD8+ T cells from IL-12-treated cultures expressed significantly higher levels of granzyme B. Enriched CD8+ T cells from cytokine-treated co-cultures were stimulated for 5 hrs with LAF on day 7, followed by Intracellular staining for granzyme B. (C) Suppressive ability of exacerbation derived CD8+ T cells was restored through IL-12 treatment. Exacerbation PBMCs from five RRMS patients were cultured 10 × 106 /mL for 7 days in 6 well plates. Culture medium was either left untreated or supplemented with 25ng/mL of IL-12. All cultures were supplemented with 1 μg/mL of one of two different neuroantigen peptides. 7 days post in vitro stimulation, exacerbation-derived CD8+ T cells were isolated by magnetic bead sorting and used in suppression assays with autologous exacerbation-derived freshly thawed APCs and CD4+CD25− T cells. *p < 0.05; Five independent experiments were performed with PBMCs from 5 healthy control donors for (A) and (B). Five independent experiments were performed with PBMCs from 5 RRMS donors during disease exacerbation for (C).
Discussion
The role of CD8+ T cells in the pathogenesis of MS remains a controversial topic. Initially thought to be pathogenic because of their preponderance in an oligoclonally expanded state in CNS lesions [31] and their ability to target oligodendrocytes in vitro [32, 33], a considerable body of evidence has established their protective role in the pathogenesis of MS. Several studies using the murine model of the disease have shown that CD8+ T cells are required for the amelioration of EAE [21, 22, 34–36]. In human studies, several reports have similarly demonstrated the regulatory properties of CD8+ T cells in MS [20, 37–42]. Additionally, a recent report further suggested that the preponderance of CD8+ T cells in MS lesions might be explained by the concomitant upregulation of HLA-E in the white lesions of multiple sclerosis patients [43]. The authors suggested that the increase in HLA-E may have a protective effect through the activation of HLA-E-restricted regulatory CD8+ T cells, albeit ultimately unsuccessful in MS patients. Interestingly, recent observations indicate perturbed CD8+ T cell homeostasis with increasing age in MS patients [44].
The current study provides significant inisghts into our previous novel and unexpected observation that neuroantigen-specific CD8+ T cells are regulatory in nature and deficient during disease exacerbation of MS [20]. We demonstrate here that the CD8+ Treg-mediated suppression is contact-dependent and requires expression of MHCI, NKG2D, IFN-γ, perforin and granzyme B. While the requirement for perforin, granzyme B and MHC class I is expected, it is interesting to note the need for IFN-γ. IFNγ has been shown to induce Indoleamine 2,3-dioxygenase (IDO) expression on APCs and convert CD4+CD25− T cells into CD4+Foxp3+ regulatory cells [41, 45]. We have recently shown the requirement of IDO in generating regulatory CD8+ T cells in the mouse model of MS [46]. Furthermore, we have also shown IFN-γ to be an absolute requirement in mediating the effects of CNS-specific and therapeutically induced CD8+ Tregs [36, 46].
The decreased IFNγ production and impaired proliferative capability of neuroantigen-specific CD4+ T cells (that were pre-cultured with CD8+ Tregs) during recall responses also suggests that the CD4+ T cells might be preferentially targeted by the CD8+ Tregs in a manner that is similar to what we have observed in the EAE animal model [22, 36]. The annexin V/PI-based cytotoxic assay lends support to this hypothesis by showing that the CD4+ T cells were directly targeted for apoptosis by the CD8+ Tregs. However, the differences in annexin V/PI staining between the groups are modest and we think that CD8+ Tregs use multiple pathways to target neuroantigen specific CD4+ T cells. Interestingly, it appears that no significant subsets of APCs are cytotoxically targeted by the neuroantigen-specific CD8+ Tregs in our ass ay. If this observation is reflected in the CNS, significant bystander damage could be averted.
The loss in suppressive ability of CD8+ Tregs during relapses that we described previously could primarily be explained by the loss of a distinct subset of neuroantigen-specific CD8+ Tregs. We found that indeed the most potent neuroantigen-specific CD8+ Tregs were harbored in the terminally differentiated subset (CD27−CD45RO−) and that this population was significantly diminished during disease exacerbation compared to disease quiescence. Additionally, the expression of the regulatory molecules notably granzyme B and perforin required by the CD8+ Tregs to mediate their function were significantly altered. Nonetheless, the deficit in neuroantigen-specific CD8+ Treg suppression could alternatively be explained by the prevalence of a refractory CD4+ effector T cell population during relapse [47]. Through crisscross studies, we show that the quiescence-associated CD8+ Tregs indeed have enhanced suppressive ability when compared to exacerbation-derived CD8+ Tregs when the targets of the suppression are quiescence-derived CD4+CD25− T cells. In contrast, when the targets to be suppressed are exacerbation-derived CD4+ T cells, neither CD8+ Treg populations have an advantage in terms of suppressive potential. Instead, it appears that the exacerbation-derived CD4+ T cells are refractory to quiescence- and exacerbation-associated CD8+ T cells. Thus, our results would suggest that the defect seen in CD8+ T cells appear to be a mix of intrinsic Treg dysreguation and responder resistance to suppression. However, it appears that exacerbation-derived CD8+ T cells are not irreparably impaired and agents such as IL-12 that upregulate the expression of the regulatory molecule, granzyme B can significantly enhance and restore the suppressive ability of the CD8+ Tregs, possibly to quiescence levels. The factor(s) that contributes to refractory nature of the CD4+ T cells however remains to be elucidated. It appears that the activation levels, memory status and lineage commitment of the CD4+ T cells do not dictate their suppresibility. Unless this refractory property is intrinsic to the CD4+ T cells, exacerbation-derived APCs might also be a potential culprit which needs to be dissected in future studies. Indeed, ongoing investigations in our laboratory examining the difference between quiescent and exacerbation APCs suggest significant differences in the two populations that might confer resistance to the CD4+ T cells.
From a clinical stand point, the current observations provide very interesting insight into the biology of MS exacerbation and possible therapeutic modalities to curtail relapses. For instance, it is tempting to speculate that one could predict a potential relapse by tracking the levels of the terminally differentiated CD8+ T cells, perhaps helping with pre-emptive treatment. It is also interesting to note that the same terminally differentiated population is predominantly expanded and activated by Copaxone treatment [37]. Based on the observation that CD8+ T cells from patients with MS can still be potentiated, adoptive transfer of in vitro potentiated CD8+ Tregs could also be considered as a viable therapeutic modality for MS patients. Collectively, our studies provide significant novel insights into the biology of CD8 T cell regulation during MS, with multiple potential strategies of harnessing this knowledge in clinical management of the disease.
Supplementary Material
Highlights.
Neuroantigen-specific CD8+ T cells are terminally differentiated
Neuroantigen-specific CD8 suppression is IFNγ, perforin and granzyme B-dependent
Acute relapse in MS is associated with loss of terminally differentiated CD8 cells
Suppressive potential of neuroantigen-specific CD8 cells can be restored with IL12
Acknowledgments
The authors are indebted to all the patients who participated in this study. We also thank Wallace Baldwin, Thomas Lee, Thomas Abraham, Stephanie Taylor, Yolanda Rodriguez, Samuel Hughes, and Gina Remington for technical support and help with patient recruitment.
These studies were supported, in part, by grant awards (to NJK) from the NIH and National MS Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kantarci O, Wingerchuk D. Epidemiology and natural history of multiple sclerosis: new insights. Curr Opin Neurol. 2006;19:248–54. doi: 10.1097/01.wco.0000227033.47458.82. [DOI] [PubMed] [Google Scholar]
- 2.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–17. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 3.Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–69. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- 4.Ontaneda D, Hyland M, Cohen JA. Multiple sclerosis: new insights in pathogenesis and novel therapeutics. Annu Rev Med. 2012;63:389–404. doi: 10.1146/annurev-med-042910-135833. [DOI] [PubMed] [Google Scholar]
- 5.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 6.Bhat R, Steinman L. Innate and adaptive autoimmunity directed to the central nervous system. Neuron. 2009;64:123–32. doi: 10.1016/j.neuron.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 7.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–9. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 8.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–32. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowther DE, Hafler DA. Regulatory T cells in the central nervous system. Immunol Rev. 2012;248:156–69. doi: 10.1111/j.1600-065X.2012.01130.x. [DOI] [PubMed] [Google Scholar]
- 10.Venken K, Hellings N, Liblau R, Stinissen P. Disturbed regulatory T cell homeostasis in multiple sclerosis. Trends Mol Med. 2010;16:58–68. doi: 10.1016/j.molmed.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–9. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venken K, Hellings N, Broekmans T, Hensen K, Rummens JL, Stinissen P. Natural naive CD4+CD25+CD127low regulatory T cell (Treg) development and function are disturbed in multiple sclerosis patients: recovery of memory Treg homeostasis during disease progression. Journal of immunology. 2008;180:6411–20. doi: 10.4049/jimmunol.180.9.6411. [DOI] [PubMed] [Google Scholar]
- 13.Seo SK, Choi JH, Kim YH, Kang WJ, Park HY, Suh JH, et al. 4–1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med. 2004;10:1088–94. doi: 10.1038/nm1107. [DOI] [PubMed] [Google Scholar]
- 14.Wang R, Han G, Song L, Wang J, Chen G, Xu R, et al. CD8+ regulatory T cells are responsible for GAD-IgG gene-transferred tolerance induction in NOD mice. Immunology. 2009;126:123–31. doi: 10.1111/j.1365-2567.2008.02884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davila E, Kang YM, Park YW, Sawai H, He X, Pryshchep S, et al. Cell-based immunotherapy with suppressor CD8+ T cells in rheumatoid arthritis. Journal of immunology. 2005;174:7292–301. doi: 10.4049/jimmunol.174.11.7292. [DOI] [PubMed] [Google Scholar]
- 16.Bisikirska B, Colgan J, Luban J, Bluestone JA, Herold KC. TCR stimulation with modified anti-CD3 mAb expands CD8+ T cell population and induces CD8+CD25+ Tregs. J Clin Invest. 2005;115:2904–13. doi: 10.1172/JCI23961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klimiuk PA, Goronzy JJ, Weyand CM. IL-16 as an anti-inflammatory cytokine in rheumatoid synovitis. Journal of immunology. 1999;162:4293–9. [PubMed] [Google Scholar]
- 18.Filaci G, Fenoglio D, Indiveri F. CD8(+) T regulatory/suppressor cells and their relationships with autoreactivity and autoimmunity. Autoimmunity. 2011;44:51–7. doi: 10.3109/08916931003782171. [DOI] [PubMed] [Google Scholar]
- 19.Crawford MP, Yan SX, Ortega SB, Mehta RS, Hewitt RE, Price DA, et al. High prevalence of autoreactive, neuroantigen-specific CD8+ T cells in multiple sclerosis revealed by novel flow cytometric assay. Blood. 2004;103:4222–31. doi: 10.1182/blood-2003-11-4025. [DOI] [PubMed] [Google Scholar]
- 20.Baughman EJ, Mendoza JP, Ortega SB, Ayers CL, Greenberg BM, Frohman EM, et al. Neuroantigen-specific CD8+ regulatory T-cell function is deficient during acute exacerbation of multiple sclerosis. J Autoimmun. 2011;36:115–24. doi: 10.1016/j.jaut.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee YH, Ishida Y, Rifa’i M, Shi Z, Isobe K, Suzuki H. Essential role of CD8+CD122+ regulatory T cells in the recovery from experimental autoimmune encephalomyelitis. Journal of immunology. 2008;180:825–32. doi: 10.4049/jimmunol.180.2.825. [DOI] [PubMed] [Google Scholar]
- 22.York NR, Mendoza JP, Ortega SB, Benagh A, Tyler AF, Firan M, et al. Immune regulatory CNS-reactive CD8+T cells in experimental autoimmune encephalomyelitis. J Autoimmun. 2010;35:33–44. doi: 10.1016/j.jaut.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barber A, Zhang T, Megli CJ, Wu J, Meehan KR, Sentman CL. Chimeric NKG2D receptor-expressing T cells as an immunotherapy for multiple myeloma. Exp Hematol. 2008;36:1318–28. doi: 10.1016/j.exphem.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langhans B, Ahrendt M, Nattermann J, Sauerbruch T, Spengler U. Comparative study of NK cell-mediated cytotoxicity using radioactive and flow cytometric cytotoxicity assays. J Immunol Methods. 2005;306:161–8. doi: 10.1016/j.jim.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 25.Thiery J, Walch M, Jensen DK, Martinvalet D, Lieberman J. Isolation of cytotoxic T cell and NK granules and purification of their effector proteins. Curr Protoc Cell Biol. 2010;Chapter 3(Unit 3):37. doi: 10.1002/0471143030.cb0337s47. [DOI] [PubMed] [Google Scholar]
- 26.Smith TR, Kumar V. Revival of CD8+ Treg-mediated suppression. Trends Immunol. 2008;29:337–42. doi: 10.1016/j.it.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Konya C, Goronzy JJ, Weyand CM. Treating autoimmune disease by targeting CD8(+) T suppressor cells. Expert Opin Biol Ther. 2009;9:951–65. doi: 10.1517/14712590903020759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chowdhury FZ, Ramos HJ, Davis LS, Forman J, Farrar JD. IL-12 selectively programs effector pathways that are stably expressed in human CD8+ effector memory T cells in vivo. Blood. 2011;118:3890–900. doi: 10.1182/blood-2011-05-357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCarron MJ, Reen DJ. Neonatal CD8+ T-cell differentiation is dependent on interleukin-12. Hum Immunol. 2010;71:1172–9. doi: 10.1016/j.humimm.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–51. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niland B, Banki K, Biddison WE, Perl A. CD8+ T cell-mediated HLA-A*0201-restricted cytotoxicity to transaldolase peptide 168–176 in patients with multiple sclerosis. Journal of immunology. 2005;175:8365–78. doi: 10.4049/jimmunol.175.12.8365. [DOI] [PubMed] [Google Scholar]
- 33.Jurewicz A, Biddison WE, Antel JP. MHC class I-restricted lysis of human oligodendrocytes by myelin basic protein peptide-specific CD8 T lymphocytes. Journal of immunology. 1998;160:3056–9. [PubMed] [Google Scholar]
- 34.Koh DR, Fung-Leung WP, Ho A, Gray D, Acha-Orbea H, Mak TW. Less mortality but more relapses in experimental allergic encephalomyelitis in CD8−/− mice. Science. 1992;256:1210–3. doi: 10.1126/science.256.5060.1210. [DOI] [PubMed] [Google Scholar]
- 35.Jiang H, Zhang SI, Pernis B. Role of CD8+ T cells in murine experimental allergic encephalomyelitis. Science. 1992;256:1213–5. doi: 10.1126/science.256.5060.1213. [DOI] [PubMed] [Google Scholar]
- 36.Ortega SB, Kashi VP, Tyler AF, Cunnusamy K, Mendoza JP, Karandikar NJ. The disease-ameliorating function of autoregulatory CD8 T cells is mediated by targeting of encephalitogenic CD4 T cells in experimental autoimmune encephalomyelitis. Journal of immunology. 2013;191:117–26. doi: 10.4049/jimmunol.1300452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ratts RB, Lovett-Racke AE, Choy J, Northrop SC, Hussain RZ, Karandikar NJ, et al. CD28−CD57+ T cells predominate in CD8 responses to glatiramer acetate. J Neuroimmunol. 2006;178:117–29. doi: 10.1016/j.jneuroim.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Antel J, Bania M, Noronha A, Neely S. Defective suppressor cell function mediated by T8+ cell lines from patients with progressive multiple sclerosis. Journal of immunology. 1986;137:3436–9. [PubMed] [Google Scholar]
- 39.Balashov KE, Khoury SJ, Hafler DA, Weiner HL. Inhibition of T cell responses by activated human CD8+ T cells is mediated by interferon-gamma and is defective in chronic progressive multiple sclerosis. J Clin Invest. 1995;95:2711–9. doi: 10.1172/JCI117973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Correale J, Villa A. Isolation and characterization of CD8+ regulatory T cells in multiple sclerosis. J Neuroimmunol. 2008;195:121–34. doi: 10.1016/j.jneuroim.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. Ann Neurol. 2010;67:625–38. doi: 10.1002/ana.21944. [DOI] [PubMed] [Google Scholar]
- 42.Tennakoon DK, Mehta RS, Ortega SB, Bhoj V, Racke MK, Karandikar NJ. Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. Journal of immunology. 2006;176:7119–29. doi: 10.4049/jimmunol.176.11.7119. [DOI] [PubMed] [Google Scholar]
- 43.Durrenberger PF, Webb LV, Sim MJ, Nicholas RS, Altmann DM, Boyton RJ. Increased HLA-E expression in white matter lesions in multiple sclerosis. Immunology. 2012;137:317–25. doi: 10.1111/imm.12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pender MP, Csurhes PA, Pfluger CM, Burrows SR. CD8 T cell deficiency impairs control of Epstein--Barr virus and worsens with age in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2012;83:353–4. doi: 10.1136/jnnp-2011-300213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z, Hong J, Sun W, Xu G, Li N, Chen X, et al. Role of IFN-gamma in induction of Foxp3 and conversion of CD4+ CD25− T cells to CD4+ Tregs. J Clin Invest. 2006;116:2434–41. doi: 10.1172/JCI25826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tyler AF, Mendoza JP, Firan M, Karandikar NJ. CD8 T Cells Are Required For Glatiramer Acetate Therapy in Autoimmune Demyelinating Disease. PLoS One. 2013;8:e66772. doi: 10.1371/journal.pone.0066772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneider A, Long SA, Cerosaletti K, Ni CT, Samuels P, Kita M, et al. In Active Relapsing-Remitting Multiple Sclerosis, Effector T Cell Resistance to Adaptive Tregs Involves IL-6-Mediated Signaling. Sci Transl Med. 2013;5:170ra15. doi: 10.1126/scitranslmed.3004970. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.